
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption of toxic gases on borophene: surface deformation links to chemisorptions†
Luong Thi Ta abc,
Ikutaro Hamadaad,
Yoshitada Morikawa*ade and
Van An Dinh*bf
abc,
Ikutaro Hamadaad,
Yoshitada Morikawa*ade and
Van An Dinh*bf
aDepartment of Precision Engineering, Graduate School of Engineering, Osaka University, 2-1, Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: morikawa@prec.eng.osaka-u.ac.jp
bNanotechnology Program, VNU Vietnam Japan University, Luu Huu Phuoc Str., My Dinh I, Nam Tu Liem, Hanoi, 100000, Vietnam. E-mail: dv.an@vju.ac.vn
cDepartment of Chemistry, Institute of Environment, Vietnam Maritime University, Le Chan, Haiphong, 18000, Vietnam
dElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Goryo-Ohara, Nishikyo-ku, Kyoto 615-8245, Japan
eResearch Center for Precision Engineering, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
fCenter for Atomic and Molecular Technologies, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 20th May 2021
Abstract
β12 borophene has received great attention because of its intriguing mechanical and electronic properties. One of the possible applications of borophene is gas sensing. However, the interaction between common gases and β12 borophene remains to be clarified. In this work, we study the interactions of β12 borophene towards five hazardous gases, namely, CO, NO, NH3, NO2, and CO2 using various non-empirical van der Waals density functionals and provide an insight into the adsorption behavior of borophene. The adsorption mechanism and molecular vibrations are discussed in great detail. Among the gases considered, CO2 is physisorbed while other gases are chemically bonded to β12 borophene. We also demonstrate that the deformation at the ridge of borophene enables its active pz orbital to strongly hybridize with frontier orbitals of the studied polar gases. Consequently, borophene is predicted to interact strongly with CO, NO, NH3, and especially NO2, making it a sensitive sensing material for toxic gases.
1. Introduction
The emission of hazardous industrial gases (COx, NOx, NH3, SO2, etc.) driven by economic growth not only threatens human wellbeing but also damages the environment. These gases may contribute to the worsening of cardiovascular and respiratory diseases, the greenhouse effect, and ozone depletion.1–3 The severe impacts of these gases have raised the need for efficient toxic-gas sensing technology to monitor and remove the harmful culprits.In the trend of constant attempts at sensor minimization, the two-dimensional polymorph of boron β12 (also known as ν1/6) is a promising candidate for an efficient sensing material.4 β12 borophene is the line-defective phase of borophene5–7 and can be stably synthesized by various bottom-up6–9 and top-down10 methods. This material was found to have intriguing electronic properties such as spin gapless Dirac cone, rich band structure, and extraordinary mechanical and optical behavior,11–15 which account for its potential in many practical applications. However, there has been a limited number of theoretical work on β12 borophene for sensing applications, compared to the theoretically proposed buckled borophene, which received more attention15–19 despite the lack of its empirical realization. Thus, to accurately explain or predict the sensing performance of borophene, it is essential to understand the adsorption behaviors of β12 borophene.
The study of gas adsorptions on β12 borophene is still in its infancy.10,20,21 Tan et al. theoretically studied the CO2 adsorption on β12 borophene under an electric field and found that CO2 moderately adsorbed on this material.20 Huang and co-workers analyzed how four inorganic gases (NH3, NO, NO2, and CO) interact with both buckled and β12 borophene by employing density functional theory (DFT) calculations with semiempirical dispersion correction.21 They found that β12 borophene chemically interacts with these gases and is significantly distorted upon adsorption. Experimentally, the realization of borophene as a gas sensor has been ignited. Ranjan and co-workers demonstrated that borophene-based sensor exhibited high sensitivity to NH3.10 Nevertheless, in these works, the gas adsorption mechanism of borophene remains unclear and the dependence of the predicted adsorption performance on exchange–correlation functionals is not considered.
In this work, we investigate the adsorption of five main hazardous industrial gases, namely CO, NO, NH3, NO2, and CO2, on β12 borophene by using van der Waals (vdW) density functionals (vdW-DFs).22–24 These functionals allow us to consider the contributions of non-local vdW interaction to the different toxic gas–β12 borophene systems in a non-empirical fashion, which is lacking in the literature to date. Furthermore, we discuss the bonding nature of these molecules with β12 borophene via electronic structure and vibrational analyses.
2. Computational details
All of the periodic DFT25,26 calculations were performed using the projector augmented wave method27,28 as implemented in the Vienna Ab initio Simulation Package (VASP).29 The vdW-DFs are shown to accurately describe molecular adsorption, but for the current system, there are no benchmark calculations obtained from using highly accurate methods (see for example ref. 30 and 31). We used four vdW-DFs, namely revPBE-vdW (referred to as vdW-DF1 hereafter),32 vdW-DF2,33 optPBE-vdW,34 and rev-vdW-DF2,35 to provide the range of adsorption geometries and energies. In addition, an attempt to apply a recently developed vdW-DF3 functional36 for comparison was made. The generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof functional (PBE)37 was also used for comparison. The convergence threshold for the self-consistent field calculations was set to 10−5 eV per cell, and the geometrical structures were fully optimized until the Hellmann–Feynman forces acting on atoms were less than 0.01 eV Å−1. To eliminate spurious interactions with image borophene sheets, a vacuum of 20 Å thickness was inserted between the neighboring borophene sheets. The wave functions were expanded using a plane-wave basis set with the kinetic energy cutoff of 500 eV. An 11 × 11 k-point mesh of the first Brillouin zone was used in optimizing the geometrical structure of the β12 borophene conventional unit cell. For the adsorption systems, a (4 × 3) borophene supercell was employed and a 3 × 3 k-point mesh was used. The spin polarization was properly taken into account for the systems with molecules having unpaired electrons (i.e., NO and NO2).To seek the most stable geometries of the adsorption systems (gas–borophene), structural optimization was carefully carried out by climbing up the ladder of accuracy. The borophene layer was restricted along the z-direction and the structural optimization was performed first by using the universal force field,38 which was further optimized by DFT. Then, the stable gas–borophene configuration was explored by calculating the interaction energy as a function of spatial coordinate using the computational DFT-based Nanoscope tool39,40 (see ESI S1†), and subsequently, full geometry optimization was performed. Finally, the adsorption energy, amount of charge transfer, and the change of work function were computed for the most favorable configurations.
We define the adsorption energy as
| Ead = Egas+boro − (Egas + Eboro) | (1) |
To understand the hybridization between the gas molecule and borophene orbitals, we performed the crystal orbital overlap population (COOP)44–46 and the projected density of states (PDOS) analyses as implemented in the STATE (Simulation Tool for Atom Technology) code.47,48 The change in molecular internal bonds of the adsorbed state is understood by using COOP analysis among atomic orbitals (AOs) implemented in the LOBSTER package.49 The vibrational modes of molecules were obtained using finite-difference harmonic approximation by diagonalizing the mass-weighted Hessian matrix.
3. Results and discussion
3.1. Geometric and electronic structures of pristine borophene
To investigate the impacts of non-empirical vdW functionals on the adsorption systems, we first employed five different functionals to calculate the lattice constants of pristine borophene (Table 1). These optimized lattice constants converge to the order of pm and agree well with published experimental6–8 and other theoretical works.50–52 Our results suggest that any of the four investigated vdW-DFs can properly describe the pristine borophene.As a sensing material, β12-borophene possesses various high symmetry adsorption sites. This property arises from the presence of hollow hexagons (Fig. 1a) which leads to site-discrimination in the electronic state (Fig. 1b). As in the middle panel of Fig. 1b, the B4 site has more unoccupied pz states compared with the other atop sites. Furthermore, with less spatial and coordinated constraint, B4 is the most likely adsorption site for a compact adsorbate, which agrees with our findings in the optimized gas–borophene configurations as discussed below.
 | ||
| Fig. 1 Geometric and electronic structures of β12 borophene. (a) The (4 × 3) supercell used in the calculation and its high symmetry adsorption sites. (b) Electronic band structure (left), site-projected density of states (middle) and total density of states of β12 borophene (right). In (a) the primitive cell is marked by the dotted line. Bi denotes the i-coordinated boron atom site. | ||
3.2. Adsorption geometries
We investigated the adsorption state of different molecules on β12-borophene. For the four out of five gases (CO, NO, NH3, and NO2) studied, the most favorable adsorption site is B4 (Fig. 2a–d). These four cases are well categorized as chemisorption since their adsorption distances are in the range of chemical interaction (Table 2). Additionally, in these cases, the deformation of β12-borophene is substantial. This flat-to-wrinkled deformation of borophene is consistent with its highly anisotropic mechanical properties53 and its undulation observed when it is supported on a silver substrate.14,54 The underlying mechanism of this deformation is discussed in detail later in the electronic structure analysis. | ||
| Fig. 2 Equilibrium geometries of (a) CO, (b) NO, (c) NH3, (d) NO2, and (e) CO2 on β12 borophene. | ||
| Molecules | Adsorption properties | vdW-DF2 | vdW-DF1 | optPBE-vdW | rev-vdW-DF2 | PBE-D2 |
|---|---|---|---|---|---|---|
| CO2 | Ead (eV) | −0.19 | −0.21 | −0.27 | −0.32 | −0.18 (ref. 20) |
| d (Å) | 3.36 | 3.41 | 3.30 | 3.30 | ||
| Δq (e) | +0.05 | +0.05 | +0.03 | +0.05 | ||
| Δϕ (eV) | +0.03 | +0.05 | +0.05 | +0.05 | ||
| CO | Ead (eV) | −0.49 | −0.64 | −0.86 | −1.18 | −1.19 (ref. 21) |
| d (Å) | 1.49 | 1.49 | 1.49 | 1.49 | ||
| Δq (e) | +0.37 | +0.38 | +0.40 | +0.35 | ||
| Δϕ (eV) | −0.03 | −0.01 | −0.02 | −0.04 | ||
| NH3 | Ead (eV) | −0.41 | −0.52 | −0.83 | −1.15 | −1.11 (ref. 21) |
| d (Å) | 1.68 | 1.66 | 1.64 | 1.63 | ||
| Δq (e) | −0.09 | −0.11 | −0.15 | −0.10 | ||
| Δϕ (eV) | −0.03 | −0.01 | −0.03 | 0.01 | ||
| NO | Ead (eV) | −0.38 | −0.39 | −0.72 | −1.05 | −0.95 (ref. 21) |
| d (Å) | 1.38 | 1.38 | 1.38 | 1.38 | ||
| Δq (e) | +0.52 | +0.70 | +0.63 | +0.65 | ||
| Δϕ (eV) | +0.03 | 0.00 | −0.02 | −0.02 | ||
| NO2 | Ead (eV) | −1.28 | −1.56 | −1.67 | −1.91 | −1.80 (ref. 21) |
| d (Å) | 1.59 | 1.59 | 1.58 | 1.57 | ||
| Δq (e) | +0.82 | +0.83 | +0.80 | +0.83 | ||
| Δϕ (eV) | +0.30 | +0.28 | +0.26 | +0.23 | ||
| Borophene | ϕ (eV) | 5.01 | 4.95 | 5.02 | 5.09 |
In contrast to the other molecules, CO2 is physisorbed at the hollow site with its molecular axis parallel to borophene surface, as shown in Fig. 2e. This configuration offers optimal interaction between the frontier orbitals of CO2 (i.e., 2π and 3π*) and the pz orbital of borophene (see Fig. S3 of ESI†).
3.3. Impacts of vdW functionals on adsorption properties
The adsorption properties obtained using the four non-empirical vdW-DFs are summarized in Table 2, along with those obtained using the semiempirical PBE-D2 dispersion correction. We found that the adsorption energies strongly depend on the vdW-DF used, especially for chemisorption cases (i.e., CO, NO, NH3 and NO2). The magnitude of the energies is sorted in a certain order as vdW-DF2 < vdW-DF1 < optPBE-vdW < rev-vdW-DF2. This consistent prediction trend is in line with the performance of vdW-DFs in literature.55,56 Therein, the vdW-DF1 and vdW-DF2 systematically predict comparatively smaller adsorption energies due to their overestimation of Pauli repulsion.24 Among four functionals, the rev-vdW-DF2 predicts the largest adsorption energies. Interestingly, these values resemble those predicted by the semiempirical PBE-D2 in ref. 21. We further confirm the prediction by using a recent generation of non-local vdW functionals vdW-DF3 (ref. 36) implemented in Quantum ESPRESSO code57–59 (see ESI Table S2†). Notably, comparable predictions of adsorption characteristics obtained by rev-vdW-DF2 and vdW-DF3-opt1 are recorded in the chemisorption cases, whereas the similar values of adsorption energy to those obtained by vdW-DF1 and vdW-DF2 are given by both versions of vdW-DF3 for the physisorption case of CO2.The calculated adsorption energies indicate that borophene interacts more strongly with CO, NH3, NO, and NO2 compared with other 2D materials such as MoS2,60 germanene,61 WS2,62 phosphorene,63 or graphene64 (see Table S3 in ESI for detailed comparison†). Therefore, we expect borophene to be a good toxic-gas sensing material.
In the realization of borophene as a chemiresistive gas sensor, the amount of charge transferred between the adsorbate and borophene is an indicator to predict the sensitivity and selectivity of the sensor. As shown in Table 2, the Bader charge analysis indicates that borophene is impressively sensitive to NO2, NO, and CO due to the remarkably large amount of charge transferred. The results show that NH3 is an electron donor while other gases are electron acceptors. The accepted or donated charge is expected to cause the change in resistivity of the substrate material. Thus, NH3 might be recognized among these gases due to the opposite trend of resistivity change.
Upon adsorption, the amount of either lost or gained charge is expected to influence the work function of borophene. While most gases induce an insignificant change in the work function, NO2 causes a remarkable increase (+0.23 to +0.30 eV depending on the functional). This increase is not surprising since it is consistent with the Bader charge analysis, where borophene donates a great amount of charge to NO2 so that the valence electron has to overcome a greater total potential barrier to escape to the vacuum level. Given the fact that the calculated work function of pristine borophene is quite high (∼5.0 eV), which is consistent with the experiment value of ∼4.9 eV,65 the significant enhancement of work function induced by the NO2 adsorption is interesting.
3.4. Analyses of the adsorption systems
Despite the great effects of vdW functionals on the adsorption energies, the electronic structure and properties are barely affected by the functionals if the geometric structures remain similar.66,67 Since the geometry of each gas–borophene system are analogous among the functionals, we used the optimized geometries obtained using optPBE-vdW for further analyses. In the following subsections, we discuss the vibrational properties and the adsorption mechanisms based on the electronic structures.| CO | NO | NH3 | NO2 | CO2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| νS | νS | νSS | νDS | νDD | νSD | νAS | νSS | νB | νAS | νSS | νIPB | νOPB | |
| a S: stretching, SS: symmetrical stretching, DS: degenerate stretching, DD: degenerate deformation, SD: symmetrical deformation, AS: asymmetrical stretching, B: bending, IPB: in-plane bending, OPB: out-of-plane bending. | |||||||||||||
| Gas phase (Exp.)68–70 | 2149 | 1875 | 3337 | 3444 | 1627 | 950 | 1618 | 1318 | 750 | 2349 | 1333 | 667 | 667 |
| Gas phase (DFT) calculation | 2115 | 1894 | 3379 | 3494 | 1639 | 1022 | 1624 | 1316 | 722 | 2335 | 1306 | 625 | 625 |
| Adsorption state (DFT) | 2030 | 1717 | 3282 | 3372, 3378 | 1611, 1608 | 1338 | 1558 | 1002 | 696 | 2325 | 1304 | 617 | 608 |
| Frequency shifts (DFT) | −85 | −177 | −97 | −122, −116 | −28, −31 | +316 | −66 | −314 | −26 | −10 | −2 | −8 | −17 |
It is noteworthy to elaborate the origin of the frequency shifts of NH3 because of its interesting vibrational behavior (for other molecules, see ESI S6†). Upon the strong interaction with borophene, the normal modes of NH3 are red-shifted, except for the symmetric deformation mode. Counterintuitively, the decreases in the frequencies for the N–H stretching modes are significant although N–H bonds point toward vacuum in the adsorption configuration (Fig. 2c). These shifts can be understood as follows: the lowest unoccupied molecular orbital (LUMO) 3a1 of NH3 has an antibonding character with respect to the N–H bond and is spatially distributed over the hydrogen atoms (inset in Fig. 3). Upon adsorption, this orbital hybridizes with the borophene states and become partially occupied as indicated by COOP between atomic orbitals of N and H atoms in Fig. 3. This occupation results in elongating N–H bonds (ESI Table S1†) and softening the stretching modes.
 | ||
| Fig. 3 Crystal orbital overlap population between atomic orbitals of N and H atoms for NH3 chemisorbed on β12 borophene. | ||
A similar red-shift was observed in the C–H stretching modes of n-alkane molecules adsorbed on metal surfaces48 and cyclohexane molecule on Rh(111)71 and was attributed to the interaction between LUMO and metal substrate states.48 There, the softening is considerable when C–H bonds point to the surface, opposite to our case with NH3 where none of the N–H bonds point to the surface. However, this softening can be understood rather straightforwardly by considering the orbital interactions.
CO. Fig. 4a shows PDOS onto the molecular orbitals (MOs) of CO (details of MOs of isolated CO and other molecules are provided in ESI Fig. S3†), while Fig. 4b shows COOP between MOs of CO and borophene states, in which positive peaks indicate bonding states, while negative ones, antibonding.44 The interaction between CO and borophene substrate can be understood by Blyholder's 5σ donation and 2π* backdonation model.72 By comparing COOP and PDOS of CO (Fig. 4a and b) to PDOS of B atom (Fig. 4c) between EF −9.5 eV and EF −7 eV, we can see positive peaks on both 5σ (1π) orbital of CO and 2s & 2pz (2px,y) orbitals of B in the corresponding energy range. The positive peaks demonstrate bonding states established by the hybridizations of 5σ and 2s & 2pz and of 1π and 2px,y due to the similar symmetries. The bonding states are far below EF and are closer to core states. From EF −6 eV to EF, an important bonding contribution results from the interaction between the filled CO 2π* and B 2px & 2py orbitals, which can be understood by COOP in this energy range. As for the antibonding states, we found that 5σ is present as a delocalized negative peak as seen at wide energy range from EF −8 eV to EF. Above EF, the unoccupied 2π* is dominant, an unoccupied part of 5σ is also significant.
 | ||
| Fig. 4 Electronic structure of CO adsorbed on β12 borophene. (a) Molecular-orbital projected density of states of CO on borophene. (b) Crystal orbital overlap population between CO and borophene. (c) Atomic-orbital projected density of states of the nearest neighbor B atom to CO. (d) Charge density difference induced by the adsorption of CO on β12 borophene. | ||
In Fig. 4d, we plot the charge density difference defined by Δρ = ρgas+boro − ρgas − ρboro, where ρgas+boro, ρgas, and ρboro are the charge densities of the adsorption system, isolated molecule, and isolated borophene, respectively. There is the charge accumulation due to the backdonation to 2π* orbital of CO, as well as the depletion due to the donation from 5σ orbital. More importantly, charge accumulates between CO and B underneath, implying the chemical bond formation between the C and B atoms.
NH3. Fig. 5 shows COOP and PDOS for the doubly degenerate 1e orbitals (HOMO−1 and HOMO−2), lone pair 2a1 orbital (highest occupied molecular orbital (HOMO)),
 orbital (LUMO), and 2e orbital (LUMO+1) of NH3. We found that both bonding and antibonding components of 2a1 orbital are occupied, indicating a repulsive interaction of this orbital with the substrate state. On the other hand, there is a noticeable contribution to the bonding from the
orbital (LUMO), and 2e orbital (LUMO+1) of NH3. We found that both bonding and antibonding components of 2a1 orbital are occupied, indicating a repulsive interaction of this orbital with the substrate state. On the other hand, there is a noticeable contribution to the bonding from the 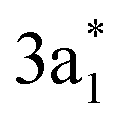 orbital, as indicated by the positive peaks in COOP and PDOS at EF −5 eV. By comparing PDOS onto AOs of the B atom underneath the N atom of NH3, we concluded that the bonding between NH3 and borophene is mainly attributed to the hybridization of the NH3 lone pair 2a1 orbital with B 2pz and 2s orbitals. Therein, the 2a1 orbital donates electrons to the empty pz states of B, leading to the increase in electrons in the substrate. The instant increase in electron as charge carriers in borophene might explain the significant increase in electric current of borophene sensor upon NH3 exposure as observed in the experiment in ref. 10.
orbital, as indicated by the positive peaks in COOP and PDOS at EF −5 eV. By comparing PDOS onto AOs of the B atom underneath the N atom of NH3, we concluded that the bonding between NH3 and borophene is mainly attributed to the hybridization of the NH3 lone pair 2a1 orbital with B 2pz and 2s orbitals. Therein, the 2a1 orbital donates electrons to the empty pz states of B, leading to the increase in electrons in the substrate. The instant increase in electron as charge carriers in borophene might explain the significant increase in electric current of borophene sensor upon NH3 exposure as observed in the experiment in ref. 10.
 | ||
| Fig. 5 Electronic structure of NH3 adsorbed on β12 borophene. (a) Molecular-orbital projected density of states of NH3 on borophene. (b) Crystal orbital overlap population between NH3 and borophene. (c) Atomic-orbital projected density of states of the nearest neighbor B atom to NH3. (d) Charge density difference induced by the adsorption of NH3 on β12 borophene. | ||
The charge density difference plot (Fig. 5d) shows that in addition to the charge depletion around the molecule due to the Pauli repulsion of the occupied MOs, the charge accumulation between B and N as well as those in N–H bonds were found, indicating the chemical bond formation between NH3 and borophene.
NO2. The gas-phase NO2 molecule has an unpaired electron (i.e., spin-polarized) (Fig. S3†). Upon adsorption, NO2 turns to be spin-unpolarized. It results from great hybridizations among MOs of NO2 and between them and borophene as shown in PDOS and COOP analyses (Fig. 6). The 5a1 orbital of NO2 strongly hybridizes with B 2s states, distributes over a wide energy range. Both the bonding and antibonding states are occupied, indicating that interaction between NO2 5a1 and B 2s is repulsive. Likewise, the 1a2 orbital (HOMO−1) is lifted and interacts repulsively with the B 2py states. The split 6a1 orbitals, which form singly occupied and unoccupied MOs in the gas phase, are fully occupied upon adsorption. This orbital forms bonding states with B 2pz and the antibonding counterpart is partially occupied, contributing to the repulsive interaction. On the other hand, the attraction between the molecule and the surface arises from the filled 5b1 of NO2 (LUMO), which is indicated by the partially occupied positive COOP ranging from ∼EF −5.0 eV to EF.
 | ||
| Fig. 6 Electronic structure of NO2 adsorbed onβ12 borophene. (a) Molecular-orbital projected density of states of NO2 on borophene. (b) Crystal orbital overlap population between NO2 and borophene. (c) Atomic-orbital projected density of states of nearest neighbor B atom of NO2. (d) Charge density difference induced by the adsorption of NO2 on β12 borophene. | ||
The PDOS and COOP analyses are consistent with the charge density difference plotted in Fig. 6d. It is found that NO2 gain electrons from borophene through the 6a1 orbital, as indicated by the charge accumulation. The electron filling of the antibonding-type orbital weakens the N–O bond, leading to the considerable elongation of this bond (from 1.21 to 1.36 Å), narrowing of the O–N–O angle (from 133° to 120°), and red-shifts of the vibrational frequencies of NO2.
3.5. Role of the borophene deformation
Finally, we discuss the deformation of the substrate borophene. To clarify the electronic origin of the protrusion of the B4 atom in borophene, we calculated the projected density of states onto atomic orbitals of this atom in both wrinkled and flat borophene structures. Fig. 7a shows that B4 pz electron in the wrinkled borophene is likely to be more localized around EF. In the flat borophene, on the other hand, B orbitals form π band, and therefore its density of states is broadly distributed (Fig. 7b). By wrinkling from the surface, B atomic orbitals tend to form sp3 hybridization rather than the sp2 ones in the flat borophene. Having an sp3-like hybridized state, pz orbital is relatively localized and therefore, it forms a peak at around the Fermi level, leading to a strong bonding with adsorbates. | ||
| Fig. 7 Projected density of states on atomic orbitals of B4 in cases of (a) wrinkled and (b) flat β12 borophene. | ||
We have clarified the important role of the deformation. In particular, we compared the adsorption performance of borophene on the two systems so-called: (i) z-restricted system and (ii) unrestricted system. In (i), we constrained the movement of boron atoms along z axis (i.e., flat borophene). In (ii), there is no constraint in any degree of freedom (i.e., fully relaxed borophene). The adsorption energies of the two systems are compared in Fig. 8. In the z-restricted systems, most of the adsorptions are physisorptions, which are in contrast to the strong chemisorptions in the unrestricted ones. Thus, the deformation is responsible for the great interactions between borophene and the molecules, especially in the cases of CO and NH3 adsorption.
 | ||
| Fig. 8 Adsorption energies of the molecules on the (i) z-restricted and (ii) unrestricted borophene. | ||
4. Conclusions
We presented a systematic study on the interactions of β12 borophene with five hazardous gas molecules, namely CO, NO, NH3, NO2, and CO2 using four different vdW-DFs (vdW-DF1, optPBE-vdW, vdW-DF2, and rev-vdW-DF2). We found that the magnitude of the adsorption energies increases in the sequence vdW-DF2 < vdW-DF1 < optPBE-vdW < rev-vdW-DF2, regardless of the nature of the gas-phase molecules while the adsorption geometries are essentially the same. We also clarified the important role played by the deformation of β12 borophene, especially in the cases of CO and NH3 adsorption. The deformation can strengthen the interaction of molecules and the surface. The results of adsorption energies and charge transfers imply that borophene is most sensitive to NO2 gas and most inert to CO2. Analyzing the electronic structure, we have gained an insight into the adsorption mechanisms of the gas molecules on borophene. Furthermore, our vibrational analyses show the vibrational modes of chemisorbed molecules are significantly softened. Our study consolidates the theoretical21 and experimental10 works on sensing application of β12 borophene and is useful to interpret the results obtained by further experiments in this field.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under Grant Number 103.01-2018.315, and also partly by Grant-in-Aid for Scientific Research (B) (Grant No. JP20H02569 and JP18H01829) and for Transformative Research Areas “Hyperordered Science” (Grant No. JP20H05883) from the Japan Society for the Promotion of Science. The authors also acknowledge the project on the establishment of Master Program of Nanotechnology, Vietnam Japan University, under the contract between Japan Cooperation International Agency (JICA) and Osaka University. L. T. T. acknowledges the Ministry of Education, Culture, Sport, Science and Technology, Japan (MEXT) and the Innovative Asia Program from JICA for financial supports.References
- H. Ritchie, M. Roser, CO2 and Greenhouse Gas Emissions, Our World in Data, 2020, (August), pp. 1–52, Available from: https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions [Online Resource] Search PubMed.
- B. Zhao, S. X. Wang, H. Liu, J. Y. Xu, K. Fu, Z. Klimont, J. M. Hao, K. B. He, J. Cofala and M. Amann, Atmos. Chem. Phys., 2013, 13, 9869–9897 CrossRef.
- H. Wang, W. P. Lustig and J. Li, Chem. Soc. Rev., 2018, 47, 4729–4756 RSC.
- Z. Xie, X. Meng, X. Li, W. Liang, W. Huang, K. Chen, J. Chen, C. Xing, M. Qiu, B. Zhang, G. Nie, N. Xie, X. Yan and H. Zhang, Research, 2020, 2020, 1–23 CrossRef PubMed.
- Z. A. Piazza, H. S. Hu, W. L. Li, Y. F. Zhao, J. Li and L. S. Wang, Nat. Commun., 2014, 5, 1–6 Search PubMed.
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed.
- G. P. Campbell, A. J. Mannix, J. D. Emery, T.-L. Lee, N. P. Guisinger, M. C. Hersam and M. J. Bedzyk, Nano Lett., 2018, 18, 2816–2821 CrossRef CAS PubMed.
- Q. Li, V. S. C. Kolluru, M. S. Rahn, E. Schwenker, S. Li, R. G. Hennig, P. Darancet, M. K. Y. Chan and M. C. Hersam, Science, 2021, 371, 1143–1148 CrossRef CAS PubMed.
- P. Ranjan, T. K. Sahu, R. Bhushan, S. S. Yamijala, D. J. Late, P. Kumar and A. Vinu, Adv. Mater., 2019, 31, 1900353 CrossRef PubMed.
- B. Feng, O. Sugino, R.-Y. Liu, J. Zhang, R. Yukawa, M. Kawamura, T. Iimori, H. Kim, Y. Hasegawa, H. Li, L. Chen, K. Wu, H. Kumigashira, F. Komori, T.-C. Chiang, S. Meng and I. Matsuda, Phys. Rev. Lett., 2017, 118, 096401 CrossRef PubMed.
- N. Gao, X. Wu, X. Jiang, Y. Bai and J. Zhao, FlatChem, 2018, 7, 48–54 CrossRef CAS.
- Y. Huang, S. N. Shirodkar and B. I. Yakobson, J. Am. Chem. Soc., 2017, 139, 17181–17185 CrossRef CAS PubMed.
- A. Brotchie, Nat. Rev. Mater., 2016, 1, 16083 CrossRef.
- H. Cui, X. Zhang and D. Chen, Appl. Phys. A, 2018, 124, 636 CrossRef.
- V. Shukla, J. Wärnå, N. K. Jena, A. Grigoriev and R. Ahuja, J. Phys. Chem. C, 2017, 121, 26869–26876 CrossRef CAS.
- Y. Valadbeigi, H. Farrokhpour and M. Tabrizchi, J. Chem. Sci., 2015, 127, 2029–2038 CrossRef CAS.
- T. Liu, Y. Chen, M. Zhang, L. Yuan, C. Zhang, J. Wang and J. Fan, AIP Adv., 2017, 7, 125007 CrossRef.
- V. Nagarajan and R. Chandiramouli, J. Inorg. Organomet. Polym. Mater., 2018, 28, 920–931 CrossRef CAS.
- X. Tan, H. A. Tahini and S. C. Smith, ACS Appl. Mater. Interfaces, 2017, 9, 19825–19830 CrossRef CAS PubMed.
- C. S. Huang, A. Murat, V. Babar, E. Montes and U. Schwingenschlögl, J. Phys. Chem. C, 2018, 122, 14665–14670 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- D. C. Langreth, B. I. Lundqvist, S. D. Chakarova-Käck, V. R. Cooper, M. Dion, P. Hyldgaard, A. Kelkkanen, J. Kleis, L. Kong, S. Li, P. G. Moses, E. Murray, A. Puzder, H. Rydberg, E. Schröder and T. Thonhauser, J. Phys.: Condens. Matter, 2009, 21, 084203 CrossRef CAS PubMed.
- K. Berland, V. R. Cooper, K. Lee, E. Schröder, T. Thonhauser, P. Hyldgaard and B. I. Lundqvist, Rep. Prog. Phys., 2015, 78, 6 CrossRef PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. G. Brandenburg, A. Zen, D. Alfè and A. Michaelides, J. Chem. Phys., 2019, 151, 164702 CrossRef PubMed.
- J. A. Garrido Torres, B. Ramberger, H. A. Früchtl, R. Schaub and G. Kresse, Phys. Rev. Mater., 2017, 1, 060803 CrossRef.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890 CrossRef CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed.
- I. Hamada, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 121103 CrossRef.
- D. Chakraborty, K. Berland and T. Thonhauser, J. Chem. Theory Comput., 2020, 16, 5893–5911 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef.
- V. O. Vo, T. L. Pham and V. A. Dinh, Mater. Trans., 2020, 61, 1449–1454 CrossRef CAS.
- T. L. Pham, T. L. Ta, V. O. Vo and V. A. Dinh, VNU J. Sci. Math. - Phys., 2020, 36, 95–102 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- J. Neugebauer and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16067–16080 CrossRef CAS PubMed.
- L. Bengtsson, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12301–12304 CrossRef CAS.
- R. Hoffmann, Rev. Mod. Phys., 1988, 60, 601–628 CrossRef CAS.
- H. Aizawa and S. Tsuneyuki, Surf. Sci., 1998, 399, L364–L370 CrossRef CAS.
- Y. Hamamoto, S. A. Wella, K. Inagaki, F. Abild-Pedersen, T. Bligaard, I. Hamada and Y. Morikawa, Phys. Rev. B, 2020, 102, 75408 CrossRef CAS.
- T. N. Pham, Y. Hamamoto, K. Inagaki, D. N. Son, I. Hamada and Y. Morikawa, J. Phys. Chem. C, 2020, 124, 2968–2977 CrossRef CAS.
- Y. Morikawa, H. Ishii and K. Seki, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 041403 CrossRef.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- B. Peng, H. Zhang, H. Shao, Z. Ning, Y. Xu, G. Ni, H. Lu, D. W. Zhang and H. Zhu, Mater. Res. Lett., 2017, 5, 399–407 CrossRef CAS.
- B. Mortazavi, O. Rahaman, A. Dianat and T. Rabczuk, Phys. Chem. Chem. Phys., 2016, 18, 27405–27413 RSC.
- H. Xiao, W. Cao, T. Ouyang, S. Guo, C. He and J. Zhong, Sci. Rep., 2017, 7, 45986 CrossRef CAS PubMed.
- Z. Zhang, Y. Yang, E. S. Penev and B. I. Yakobson, Adv. Funct. Mater., 2017, 27, 9 Search PubMed.
- Z. Zhang, A. J. Mannix, Z. Hu, B. Kiraly, N. P. Guisinger, M. C. Hersam and B. I. Yakobson, Nano Lett., 2016, 16, 6622–6627 CrossRef CAS PubMed.
- L. Spanu, S. Sorella and G. Galli, Phys. Rev. Lett., 2009, 103, 196401 CrossRef PubMed.
- S. Lebègue, J. Harl, T. Gould, J. G. Ángyán, G. Kresse and J. F. Dobson, Phys. Rev. Lett., 2010, 105, 196401 CrossRef PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. De Gironcoli, P. Delugas, R. A. Distasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Kücükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-De-La-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. De Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- S. Zhao, J. Xue and W. Kang, Chem. Phys. Lett., 2014, 595–596, 35–42 CrossRef CAS.
- W. Xia, W. Hu, Z. Li and J. Yang, Phys. Chem. Chem. Phys., 2014, 16, 22495–22498 RSC.
- V. Q. Bui, T.-T. Pham, D. A. Le, C. M. Thi and H. M. Le, J. Phys.: Condens. Matter, 2015, 27, 305005 CrossRef PubMed.
- Y. Cai, Q. Ke, G. Zhang and Y.-W. Zhang, J. Phys. Chem. C, 2015, 119, 3102–3110 CrossRef CAS.
- K. Takeuchi, S. Yamamoto, Y. Hamamoto, Y. Shiozawa, K. Tashima, H. Fukidome, T. Koitaya, K. Mukai, S. Yoshimoto, M. Suemitsu, Y. Morikawa, J. Yoshinobu and I. Matsuda, J. Phys. Chem. C, 2017, 121, 2807–2814 CrossRef CAS.
- X. Liu, L. Wang, B. I. Yakobson and M. C. Hersam, Nano Lett., 2021, 21, 1169–1174 CrossRef CAS PubMed.
- I. Hamada and S. Yanagisawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 153104 CrossRef.
- S. Gautier, S. N. Steinmann, C. Michel, P. Fleurat-Lessard and P. Sautet, Phys. Chem. Chem. Phys., 2015, 17, 28921–28930 RSC.
- G. E. Leroi, G. E. Ewing and G. C. Pimentel, J. Chem. Phys., 1964, 40, 2298–2303 CrossRef CAS.
- E. P. J. Linstrom and W. G. Mallard, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, Coblentz Society, Inc., 2017, p. 20899 Search PubMed.
- T. Shimanouchi, J. Phys. Chem. Ref. Data, 1977, 6, 993–1102 CrossRef CAS.
- K. Fidanyan, I. Hamada and M. Rossi, Adv. Theory Simul., 2021, 2000241 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The details of electronic structure, geometrical profiles, and vibrational modes of molecules in the gas phase; the 2D potential energy surfaces of gases on z-restricted borophene; vdW-DF3 calculations; and analyses of chemical interactions of NO and CO2 with borophene. See DOI: 10.1039/d1ra02738g |
| This journal is © The Royal Society of Chemistry 2021 |