
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical synthesis of quinazolinone via I2-catalyzed tandem oxidation in aqueous solution†
Huiqing Houa,
Xinhua Maa,
Yingying Lina,
Jin Lina,
Weiming Suna,
Lei Wangc,
Xiuzhi Xu*a and
Fang Ke *ab
*ab
aSchool of Pharmacy, Fujian Provincial Key Laboratory of Natural Medicine Pharmacology, Fujian Medical University, Fuzhou 350004, China. E-mail: kefang@mail.fjmu.edu.cn; Fax: +86-591-22862016; Tel: +86-591-22862016
bFaculty of Material and Chemical Engineering, Yibin University, Yibin 644000, China
cSchool of Science, Xuchang University, Xuchang 461000, China
First published on 17th May 2021
Abstract
The development of protocols for synthesizing quinazolinones using biocompatible catalysts in aqueous medium will help to resolve the difficulties of using green and sustainable chemistry for their synthesis. Herein, using I2 in coordination with electrochemical synthesis induced a C–H oxidation reaction which is reported when using water as the environmentally friendly solvent to access a broad range of quinazolinones at room temperature. The reaction mechanism strongly showed that I2 cooperates electrochemically promoted the oxidation of alcohols, then effectively cyclizing amides to various quinazolinones.
The N-heterocycles are key core structures that form the basis of many pharmaceutical, agrochemical and natural products.1 Among them, quinazolinones are an important motif in several biologically relevant pharmacophores,2 such as methaqualone which is famous for its effective sedative and hypnotic effects, luotonin A which is a quinazolinone alkaloid with anti-inflammatory effects, and erlotinib which is an anti-tumour agent, and all these compounds contain a quinazoline bond in their backbone (Fig. 1).3
 | ||
| Fig. 1 Bioactive compounds containing quinazolinone skeleton. | ||
Due to their advantageous structures quinazolinones have been widely explored in numerous syntheses.4 The classical method involves condensation of aldehydes and o-aminobenzamides to give aminal intermediates, which then undergo oxidation to yield the final quinazolinone product.5 Another strategy is to use more benign and readily available alcohols as starting materials.6 The reaction takes place through a two-step oxidation pathway, where the alcohols are first oxidized to aldehydes, followed by coupling with o-aminobenzamides. The catalyst needs to demonstrate high activity and selectivity as the reaction involves dehydrogenation of both the C–H and N–H bonds in one pot. In 2018, Sarma and co-workers7 demonstrated that a magnetically recoverable iron oxide-carbon dot nanocomposite was an effective catalyst for cyclooxidative tandem synthesis of quinazolinones in aqueous medium using alcohols as starting materials. Furthermore, annulation reactions of o-aminoaryl acids may be the most employed strategies, which include the condensation of o-aminoaryl acids with amides, nitriles, or acid derivatives plus a nitrogen source.8 Moreover, the synthesis of quinazolinone involving transition metal catalysed reactions of o-haloarylamides with nitriles, or amines9 and reaction of o-halogenated aryl acid with amides10 have been explored (Scheme 1).
 | ||
| Scheme 1 Methods for the synthesis of quinazolinones. | ||
Although the above approaches solved a lot of practical problems, there are still some limitations such as long reaction time, high temperature and by-products. Hence, development of greener, atom economic, synthetic approaches for the preparation of quinazolinones from inexpensive and easily available starting materials under relatively mild conditions is desirable. On one hand, electrochemical-induced direct functionalization has gained significant attention from the synthetic chemistry community due to it being environmentally friendly, and requiring mild conditions, and low-energy irradiation.11 With electrons as the oxidizing/reducing agent, organic electrosynthesis could offer appropriate alternatives to traditional oxidation or reduction reactions. For example, Zhao and co-workers12 reported an efficient electrochemical-induced C–H methylthiolation of electron-rich aromatics via a three-component cross-coupling strategy. On the other hand, the non-metallic oxidant, iodine, catalysed C–H oxidation has attracted great interest in recent times due to its low toxicity and because it is inexpensive compared with transition metal catalysts.13 Therefore, the combination of electrochemical catalysis and non-metallic oxidant iodine is a very feasible means of organic synthesis and does not require use of the historical large doses of iodine. In this research the possibility of combining the two in a one-pot reaction was explored, thus avoiding the isolation of either aldehyde or amine intermediates leading to quinazolinones formed from alcohols as starting materials.
Furthermore, water as a reaction medium is generally considered as an inexpensive, safe, and environmentally benign alternative to organic solvents.14 Recently, Muthaiah and co-workers15 demonstrated a catalyst system for the dehydrogenative oxidation of alcohols to carbonyl compounds and dehydrogenative lactonization of diols in water catalyzed by a water-soluble bifunctional iridium complex. In continuation of our work to develop new organic transformations,16,11f herein, it is demonstrated that I2 is an efficient catalyst for a novel, one-pot electrochemical-induced tandem reaction in aqueous solution.
The investigation was initially begun by selecting o-aminobenzamide 1a and benzyl alcohol 2a as the model substrate to optimize the reaction conditions shown in Table 1. The reaction was initially heated in water using a catalyst: I2 (0.4 equiv.) and a base: NaOH (4 equiv.) to give the desired product 3aa with a yield of 92% (Table 1, entry 1). The amount of catalyst used was reduced to one-fifth and the separation rate of the corresponding product was only 42% (Table 1, entry 2). When the reaction mixture was placed in other solvents for 6 h, this reaction takes place with a 73% isolated yield (Table 1, entry 3), and for the sake of environmental protection, water was chosen as a green and energy saving solvent. Poor yields were obtained in the absence of I2 (Table 1, entry 4). Removal of NaOH from the reaction system led to a disruption reaction that may be due to the unstable or even interrupted current (Table 1, entry 5). Other catalysts such as CuI, TBAI and KI all gave lower reactivity or poor reactivity (Table 1, entries 6–8). Among the bases investigated, NaOH was found to be the best choice (Table 1, entries 9 and 10). Control experiments revealed that no reaction occurred in the absence of current, indicating that it was essential to the reaction (Table 1, entry 11). Also, no significant improvement in the product yield was observed even when tetrabutylammonium hexafluorophosphate (Bu4NPF6) was added as the electrolyte (Table 1, entry 12). An attempt was made to replace the current by increasing the amount of catalyst so that a large number of imines were produced and less than 30% of 3aa was produced (Table 1, entries 13 and 14). Additionally, other electrode materials such as a graphite anode or Cu cathode have proved less efficient (Table 1, entries 15 and 16). This may be due to the discrepancy in reactivity between the graphite electrode and the Pt electrode which may cause the difference in current density at the anode surface, which results in differing concentrations of electron oxidation products, inhibiting further oxidation of 2a and decreasing the rate of electron transfer. In addition, the overpotential of hydrogen evolution at the cathode (Cu electrode) is higher than that of Pt, and the aldehydes formed by anodic oxidation may be preferentially reduced to alcohols at the cathode.17 It was also observed that the reduction in current also leads to a reduction in yield (Table 1, entry 17).
|
||
|---|---|---|
| Entry | Variations from the standard conditions | Yieldb (%) |
| a Standard conditions: undivided cell, Pt anode, Pt cathode, 1a (0.5 mmol), 2a (0.6 mmol), I2 (0.2 mmol), NaOH (2 mmol), H2O (3 mL), I = 80 mA at room temperature (r.t.) for 6 h.b Isolated yield. | ||
| 1 | None | 92 |
| 2 | 0.1 mmol I2 | 42 |
| 3 | MeCN/H2O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent :1) as solvent |
73 |
| 4 | In the absence of I2 | Trace |
| 5 | In the absence of NaOH | Trace |
| 6 | CuI instead of I2 | 19 |
| 7 | TBAI instead of I2 | 26 |
| 8 | KI instead of I2 | 42 |
| 9 | Cs2CO3 instead of NaOH | 31 |
| 10 | KOH instead of NaOH | 73 |
| 11 | No current | Trace |
| 12 | Addition of Bu4NPF6 as electrolyte | 92 |
| 13 | 0.4 mmol I2 instead of current | 28 |
| 14 | 1.0 mmol I2 instead of current | 30 |
| 15 | C(+)/Pt(−) | 64 |
| 16 | Pt(+)/Cu(−) | 16 |
| 17 | 40 mA | 71 |

The optimized conditions were then applied to various substrates to extend the scope of the method (Table 2). Various substituted alcohols with different electronic properties and functional groups were tested. It was observed that alcohols bearing electron-donating or electron-withdrawing functional groups could be smoothly converted to the desired products with excellent yields of 79–94% (Table 2, 3ab–3aj). Because of the stability of the electron donor to intermediates and free radical electrons, it was found that the yield of electron-donating substrates (–Et, –OMe) was slightly higher than that of electron-withdrawing substrates (–NO2, –F, –Cl, –Br, –OH). The highest yield of 94% was obtained with 4-methoxybenzyl alcohol whereas the lowest yield was 79% with 2-hydroxybenzyl alcohol (Table 2, 3ac and 3aj). It was obvious that heterocyclic alcohols such as pyridinyl, thienyl and methylfuran were well-tolerated in this catalytic system with 78% and 77% yields (Table 2, 3al, 3am and 3az). Notably, long-chain aliphatic alcohols also yielded a corresponding product, but the yield was dramatically lower than that of aromatic alcohols (Table 2, 3ak). Furthermore, highly catalytic activities were found in transformations of naphthalenemethanol to the corresponding products (Table 2, 3ba). When alkenyl alcohol was employed under the same reaction conditions, the product formed only had a 46% yield (Table 2, 3bb).

Subsequently, the substrates of o-aminobenzamide were also expanded (Table 2, 3an–3aw). Similar to the observation with the alcohol substitution compounds, o-aminobenzamides with electron-donating groups (–Me, –OMe) gave relatively higher reactivity than those with electron-withdrawing groups (–NO2, –F, –Cl, –Br). Whereas when heterocyclic amides were used as the starting substrates, yields of 65% and 73% of the corresponding products were also obtained (Table 2, 3ax and 3ay).
In order to demonstrate the practicality of this method, the gram-scale preparation of 1a (5 mmol) and 2a (6 mmol) with H2O (20 mL) under the optimized reaction conditions was performed as shown in Scheme 2, which gave the desired product 3aa with an 83% yield (1.47 g). This reveals that the new procedure has significant advantages over many current methods for further practical applications.
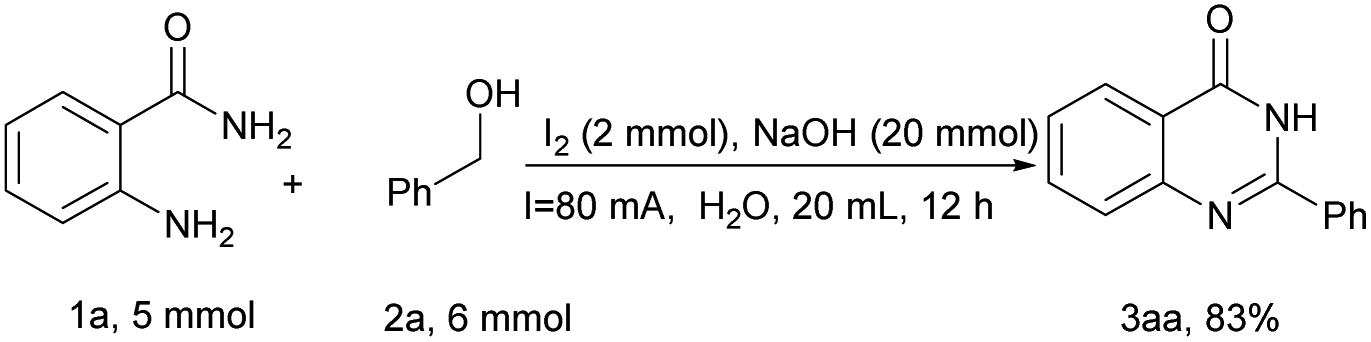 | ||
| Scheme 2 Gram-scale experiment. | ||
To exclude the possibility of other reaction pathways, some control experiments were performed (Scheme 3). Firstly, benzyl alcohol 2a effectively gave benzaldehyde 4a using aerobic oxidation reactions under standard conditions. In contrast, the absence of either base or current lead to the production of trace amounts of benzaldehyde, furthermore, the lack of catalyst lead to the formation of 5a (Scheme 3 a and b) with a yield of 43%. Nevertheless, the yield of 4a was reduced to 90% when nitrogen was substituted for air (Scheme 3c). Then, the reaction of 1a and 4a under the standard conditions effectively gave 3aa with a high yield of 93% (Scheme 3d), whereas the reaction of o-aminobenzamide under a nitrogen atmosphere, resulted in 3aa with a yield of 68% and 32% (Scheme 3e and f).
 | ||
| Scheme 3 Control experiments of alcohol oxidation and synthesis of 3aa. | ||
To gain an understanding of the reaction mechanism, cyclic voltammetry (CV) was then conducted (Fig. 2). Using H2O as the solvent, a glass electrode as the working electrode, platinum wire as the opposite electrode, and SCE as the reference electrode with a 0.1 V s−1 scanning rate. By comparing curves c, f and g, it was observed that the onset potential of the I2 oxidation shifted from 1.57 to 1.07 V vs. SCE in the presence of substrate 2a, which was more than that in the presence of NaOH, indicating that I2 was more likely to react with b than disproportionated with NaOH (Fig. 2, curves c, f and g). However, a reduction peak appears in b at 0.43 V, and the reduction peak of I2 was 0.69 V, indicating that I− is still carrying out the reduction reaction in the presence of b (Fig. 2, curve c and g). Interestingly, the addition of 1a and NaOH further decreases the onset potential of I2 complex oxidation to 0.99 V vs. SCE (Fig. 2, curve e). In contrast, the oxidation potential increased when NaOH was added individually, and this may be evidence that NaOH does not interact with 2a and I2 (Fig. 2, curve d).
 | ||
| Fig. 2 Cyclic voltammetry measurements were performed at room temperature with standard three electrode systems. | ||
By considering the whole of the experimental findings, a plausible mechanistic pathway for the formation of compound 3aa is outlined in Scheme 4. First of all, benzyl alcohol 2a is first oxidized to benzaldehyde 4a by I2, which is generated in situ on the anode.6a,18 The iodine undergoes cathodic reduction by regenerating the iodide ion for the catalytic cycle. Next, 4a can readily react with 1a to obtain the imine D, imine D is then converted to E in the presence of a base, and thus E oxidizes and dehydrogenates to give the desired product 3aa.19 Finally, H+ removed from E combines with O2 to form H2O at the cathode.20
 | ||
| Scheme 4 Proposed mechanism for this transformation. | ||
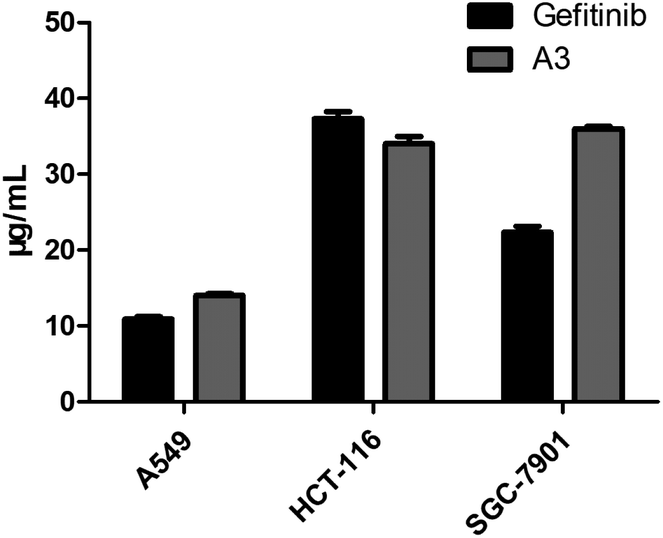
Lots of nuclear nitrogen heterocycles, such as pteridine which is a widely existing aromatic compound, similar to quinazolines, which are tyrosine kinase inhibitors, which have good specificity and inhibitory activity on tumour cells. It is thought that many fluorine-containing drugs have been widely used in clinical situations. The introduction of fluorine atoms and fluorine-containing groups into drugs can improve the clinical efficacy of drugs and reduce their side effects. Based on this, the electrocatalytic system was applied to the synthesis of the structure of the trexine and the synthesis of gefitinib analogues. For example, N-(4-methoxyphenyl)-6-(2,2,2-trifluoroethoxy)pteridin-4-amine A3 was synthesized from 3-amino-6-(2,2,2-trifluoroethoxy) pyrazine-2-carboxamide with a yield of 56% (see Scheme 5). The compounds were then tested using the MTT assay and the results are shown in Fig. 3. The A3 inhibited human non-small-cell lung carcinoma cells, A549, human colon cancer cells, HCT-116, and human gastric cancer cells, SGC-7901 to different degrees. As shown in Fig. 3, the results showed that the IC50 value of A3 in HCT116 cells (IC50 = 14.79 μg mL−1 in A549 cells, IC50 = 34.52 μg mL−1 in HCT116 cells, IC50 = 36.44 μg mL−1 in SCG-7901 cells) was just surpassed by that of gefitinib.
 | ||
| Scheme 5 Synthesis of N-(4-methoxyphenyl)-6-(2,2,2-trifluoroethoxy) pteridin-4-amine (A3). | ||
 | ||
| Fig. 3 In vitro inhibitory data of target compounds against A549, HCT-116 and SGC-7901 cell line. | ||
Conclusions
In summary, it is demonstrated that simple and commercially available I2 could be used as an electrocatalyst for the selective oxidation of a range of activated C–H bonds in alcohols. It is also demonstrated that I2 together with electrochemistry is a mild catalyst to efficiently promote C–H oxidation reactions. Oxidative tandem cyclization is of importance in the synthesis of heteropolycyclic skeletons and has a high atom economy. Furthermore, use of both electrocatalysis as a reaction process and water as a green solvent was consistent with the concept of green synthesis. This catalytic protocol avoids using toxic reagents and shows a broad substrate scope, providing consistently good yields of quinazolinones under constant current at room temperature for 6 h. Further extension of this electrochemical protocol to the synthesis of other heterocycles is underway.Experimental section
General procedure (for Tables 1 and 2)
To a dried 10 mL quartz tube equipped with 80 mA current was added benzamide (0.5 mmol), alcohol (0.6 mmol), I2 (0.2 mmol), NaOH (2.0 mmol) and water (3 mL). The mixture was stirred at room temperature for 6 h in a 80 mA circuit. After the reaction was completed the solution containing the crude product was concentrated under vacuum, and the residue was purified by column chromatography on a silica gel (petroleum ether/ethyl acetate = 3/1) to give the target product as a white solid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fujian Provincial Foundation (Grant No. FJNMP-201902, FJNMP-2020004, 2019R11020034-1, 2020J01622, 2020J01627).Notes and references
- (a) A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Comprehensive heterocyclic chemistry II, Elsevier Oxford, 1996 Search PubMed; (b) Z. Z. Ma, Y. Hano, T. Nomura and Y. J. Chen, Heterocycles, 1997, 46, 541–546 CrossRef CAS; (c) I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036–1045 RSC; (d) D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153–10202 CrossRef CAS; (e) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS.
- (a) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed; (b) N. P. McLaughlin, P. Evans and M. Pines, Bioorg. Med. Chem., 2014, 22, 1993–2004 CrossRef CAS PubMed; (c) T. Hisano, M. Ichikawa, A. Nakagawa and M. Tsuji, Chem. Pharm. Bull., 1975, 23, 1910–1916 CrossRef CAS PubMed; (d) W. Xu and H. Fu, J. Org. Chem., 2011, 76, 3846–3852 CrossRef CAS PubMed; (e) M. R. Ghaleno, M. Ghaffari-Moghaddam, M. Khajeh, A. R. Oveisi and M. Bohlooli, J. Colloid Interface Sci., 2019, 535, 214–246 CrossRef CAS PubMed.
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS PubMed; (b) F. Xie, Q.-H. Chen, R. Xie, H.-F. Jiang and M. Zhang, ACS Catal., 2018, 8, 5869–5874 CrossRef CAS; (c) A. Cagir, S. H. Jones, R. Gao, B. M. Eisenhauer and S. M. Hecht, J. Am. Chem. Soc., 2003, 125, 13628–13629 CrossRef CAS PubMed.
- (a) H. Li, L. He, H. Neumann, M. Beller and X.-F. Wu, Green Chem., 2014, 16, 1336–1343 RSC; (b) X. Liu, H. Fu, Y. Jiang and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 348–351 CrossRef CAS PubMed; (c) L. He, H. Li, J. Chen and X.-F. Wu, RSC Adv., 2014, 4, 12065–12077 RSC; (d) N. Y. Kim and C.-H. Cheon, Tetrahedron Lett., 2014, 55, 2340–2344 CrossRef CAS.
- (a) R. J. Abdel-Jalil, W. Voelter and M. Saeed, Tetrahedron Lett., 2004, 45, 3475–3476 CrossRef CAS; (b) G.-W. Wang, C.-B. Miao and H. Kang, Bull. Chem. Soc. Jpn., 2006, 79, 1426–1430 CrossRef CAS; (c) D. Maitraie, G. V. Reddy, V. V. V. N. S. R. Rao, S. R. Kanth, P. S. Rao and B. Narsaiah, J. Fluorine Chem., 2002, 118, 73–79 CrossRef CAS; (d) Y. Nagasawa, Y. Matsusaki, T. Nobuta, N. Tada, T. Miura and A. Itoh, RSC Adv., 2015, 5, 63952–63954 RSC; (e) D. Qiu, Y. Wang, D. Lu, L. Zhou and Q. Zeng, Monatsh. Chem., 2015, 146, 1343 CrossRef CAS; (f) J.-J. Zhong, W.-P. To, Y. Liu, W. Lu and C.-M. Che, Chem. Sci., 2019, 10, 4883–4889 RSC.
- (a) D.-Z. Lin, Y.-L. Lai and J.-M. Huang, ChemElectroChem, 2019, 6, 4188–4193 CrossRef CAS; (b) T. Song, P. Ren, Z. Ma, J. Xiao and Y. Yang, ACS Sustainable Chem. Eng., 2020, 8, 267–277 CrossRef CAS; (c) Q. Wang, M. Lv, J. Liu, Y. Li, H. Cao, X. Zhang and Q. Xu, ChemSusChem, 2019, 12, 3043–3048 CrossRef CAS PubMed.
- B. Majumdar, D. Sarma, S. Jain and T. K. Sarma, ACS Omega, 2018, 3, 13711–13719 CrossRef CAS PubMed.
- K. Rad-Moghadam and M. S. Khajavi, J. Chem. Res., Synop., 1998, 11, 702–703 RSC.
- (a) N. Yan, C. You and M. Cai, J. Organomet. Chem., 2019, 897, 161–169 CrossRef CAS; (b) M. A. Iqbal, L. Lu, H. Mehmood, D. M. khan and R. Hua, ACS Omega, 2019, 4, 8207–8213 CrossRef CAS PubMed; (c) R. Sharma, R. A. Vishwakarma and S. B. Bharate, Eur. J. Org. Chem., 2016, 2016, 5227–5233 CrossRef CAS; (d) X. Yu, L. Gao, L. Jia, Y. Yamamoto and M. Bao, J. Org. Chem., 2018, 89, 10352–10358 CrossRef PubMed; (e) W. Xu and H. Fu, J. Org. Chem., 2011, 76, 3846–3852 CrossRef CAS PubMed.
- (a) X. Zhang, D. Ye, H. Sun, D. Guo, J. Wang, H. Huang, X. Zhang, H. Jiang and H. Liu, Green Chem., 2009, 11, 1881–1888 RSC; (b) A. V. Dubey and A. V. Kumar, ACS Sustainable Chem. Eng., 2018, 6, 14283–14291 CrossRef CAS.
- (a) P.-S. Gao, X.-J. Weng, Z.-H. Wang, C. Zheng, B. Sun, Z.-H. Chen, S.-L. You and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 15254–15259 CrossRef CAS PubMed; (b) D.-Z. Lin, Y.-L. Lai and J.-M. Huang, ChemElectroChem, 2019, 6, 4188–4193 CrossRef CAS; (c) J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (d) S. Liang and C.-C. Zeng, Curr. Opin. Electrochem., 2020, 24, 31–43 CrossRef CAS; (e) H. Wang, J. Shi, J. Tan, W. Xu, S. Zhang and K. Xu, Org. Lett., 2019, 21, 9430–9433 CrossRef CAS PubMed; (f) L. Yang, H. Hou, L. Li, J. Wang, S. Zhou, M. Wu and F. Ke, Org. Biomol. Chem., 2021, 19, 998–1003 RSC.
- Y. Wu, H. Ding, M. Zhao, Z.-H. Ni and J.-P. Cao, Green Chem., 2020, 22, 4906–4911 RSC.
- (a) F. V. Singh and T. Wirth, Chem.–Asian J., 2014, 9, 950–971 CrossRef CAS PubMed; (b) J.-Y. Chen, C.-T. Zhong, Q.-W. Gui, Y.-M. Zhou, Y.-Y. Fang, K.-J. Liu, Y.-W. Lin, Z. Cao and W.-M. He, Chin. Chem. Lett., 2021, 32, 475–479 CrossRef CAS; (c) Q.-W. Gui, X. He, W. Wang, H. Zhou, Y. Dong, N. Wang, J.-X. Tang, Z. Cao and W.-M. He, Green Chem., 2020, 22, 118–122 RSC.
- (a) R. Ponduri, P. Kumar, L. R. Vadali and N. R. Modugu, ChemistrySelect, 2018, 3, 7766–7770 CrossRef CAS; (b) H. Hikawa, N. Matsuda, H. Suzuki, Y. Yokoyama and I. Azumaya, Adv. Synth. Catal., 2013, 355, 2308–2320 CrossRef CAS; (c) W.-H. Bao, Z. Wang, X. Tang, Y.-F. Zhang, J.-X. Tan, Q. Zhu, Z. Cao, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2259–2262 CrossRef CAS.
- M. Kannan, P. Barteja, P. Devi and S. Muthaiah, J. Catal., 2020, 386, 1–11 CrossRef CAS.
- F. Ke, C. Liu, P. Zhang, J. Xu and X. Chen, Synth. Commun., 2018, 48, 3089–3098 CrossRef CAS.
- (a) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53(1), 72–83 CrossRef CAS PubMed; (b) Y. Imada, Y. Okada, K. Noguchi and K. Chiba, Angew. Chem., Int. Ed., 2019, 58, 125–129 CrossRef CAS PubMed; (c) N. Chen, X. Lai and H. Xu, Chinese Journal of Organic Chemistry, 2020, 40, 2592–2593 CrossRef.
- (a) L. Cao, H. Huo, H. Zeng, Y. Yu, D. Lu and Y. Gong, Adv. Synth. Catal., 2018, 360, 4764–4773 CrossRef CAS; (b) Q. Wang, M. Lv, J. Liu, Y. Li, H. Cao, X. Zhang and Q. Xu, ChemSusChem, 2019, 12, 3043–3048 CrossRef CAS PubMed; (c) M. Abdullaha, S. Mohammed, M. Ali, A. Kumar, R. A. Vishwakarma and S. B. Bharate, J. Org. Chem., 2019, 84, 5129–5140 CrossRef CAS PubMed.
- (a) Y. Hu, S. Li, H. Li, Y. Li, J. Lin, C. Duanmu and B. Li, Org. Chem. Front., 2019, 6, 2744–2748 RSC; (b) M. Kumar, Richa, S. Sharma, V. Bhatt and N. Kumar, Adv. Synth. Catal., 2015, 357, 2862–2868 CrossRef CAS.
- M. Sadahl, J. Conrad, C. Braunberger and U. Beifuss, RSC Adv., 2019, 9, 19549–19559 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02706a |
| This journal is © The Royal Society of Chemistry 2021 |