
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the insertion and addition reactions of group 13 analogues of the six-membered N-heterocyclic carbenes: interplay of electrophilicity, basicity, and aromaticity governing the reactivity†
Zheng-Feng Zhanga and
Ming-Der Su *ab
*ab
aDepartment of Applied Chemistry, National Chiayi University, Chiayi 60004, Taiwan. E-mail: midesu@mail.ncyu.edu.tw
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung 80708, Taiwan
First published on 4th June 2021
Abstract
Three fundamental concepts (aromaticity/basicity/electrophilicity), being heavily used in modern chemistry, have been applied in this work to study the chemical reactivity of six-membered-ring group 13 N-heterocyclic carbenes (G13-6-Rea; G13 = group 13 elements) using density functional theory (BP86-D3(BJ)/def2-TZVP). G13-6-Rea is isolobal to benzene. Two model reactions have been used in the present study: the insertion reaction of G13-6-Rea with methane and the [1 + 2] cycloaddition reaction of G13-6-Rea with ethene. Our theoretical analysis reveals that the chemical reactivity of B-6-Rea, Al-6-Rea, and Ga-6-Rea is governed by their HOMO (the sp2-σ lone pair orbital on the G13 element), and thus they can be considered nucleophiles. Conversely, the chemical behavior of In-6-Rea and Tl-6-Rea is determined by their LUMO (the vacant p-π orbital on the G13 element), and thus they can be considered electrophiles. On the basis of the VBSCD (valence bond state correlation diagram) model and ASM (activation strain model), this theoretical evidence demonstrates that the origin of activation barriers for the above model reactions is due to the atomic radius of the pivotal group 13 element in the six-membered-ring of G13-6-Rea. Accordingly, our theoretical conclusions suggest that the lower the atomic number and the smaller the atomic radius of the G13 atom, the higher the aromaticity of the six-membered-ring of G13-6-Rea and the smaller the singlet–triplet energy splitting ΔEst of this N-heterocyclic carbene analogue, which will facilitate its chemical reactions. The theoretical findings originating from this study allow many predictions in experiments to be made.
I. Introduction
The simple carbene (CH2) is well known to be an electron-deficient two-coordinate carbon molecule, possessing two non-bonding electrons on the central carbon atom.1 For over one hundred years, CH2 and the carbene derivatives were always classified as only transient species since they were notoriously difficult to be experimentally isolated, let alone structurally characterized. Since 1991, the synthesis and isolation of thermally stable N-heterocyclic carbenes (NHCs) by Arduengo and co-workers2 have greatly opened considerable research activity into the various fields of organic, inorganic, and organometallic chemistry.3–21 For instance, many researches demonstrated that NHCs exhibit high tendency to act as strong σ-donors with a weak π-backbonding ability.22 In particular, these NHC analogues exhibit the attractive natures with lower toxicity as well as higher air and thermal stability. As a result, NHCs are creating tremendous interests either in their ability of acting as versatile nucleophilic catalysts in a variety of organic designs23–32 or as key ligands in many organometallic complexes, which mainly turned out to be useful components for broad applications.33–40Through many sophisticated efforts by excellent synthetic chemists, the chemistry of monomeric four-, five-, and six-membered heterocycles of the general form I–III containing a pivotal group 13 element G13 (= B, Al, Ga, In, and Tl; Scheme 1) has been extensively studied in experiments in recent decades and still continues to be an active research area in many laboratories.8–15 In this work, we devote our attention to the reactivity of the neutral six-membered NHC analogues featuring a group 13 element (G13-6-Rea). Although the understanding of the six-membered-ring G13-6-Rea molecules has certainly grown in recent years,41–60 our knowledge concerning their relative chemical reactivity remains primitive compared to that of five-membered-ring NHC analogues bearing a central group 14 element.22–25 In this study, we thus chose two typical chemical reactions (insertion, eqn (1), and cycloaddition, eqn (2)) based on density functional theory (DFT) to investigate the origin of the activation barriers for group 13 analogs of the six-membered NHCs.
 | (1) |
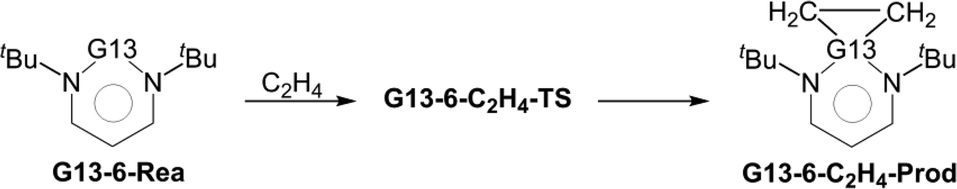 | (2) |
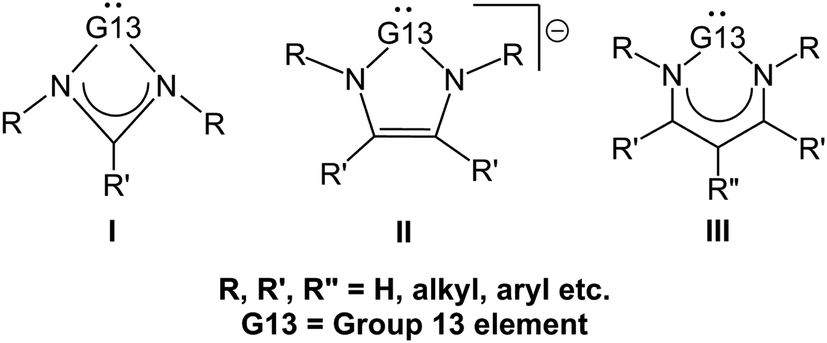 | ||
| Scheme 1 General structures of group 13 element (G13) N-heterocycles. | ||
To gain better understanding of the chemical reactivity of G13-6-Rea, their chemical natures are analyzed in this work using three important fundamental concepts: aromaticity, basicity, and electrophilicity. Moreover, the sources of their activation barriers are studied with the VBSCD model61–63 and the activation strain model (ASM)64–67 method. Hopefully, our theoretical study focusing on the above fundamental concepts and electronic factors that characterize this six-membered-ring G13 family of NHC analogues will provide essential and useful information for understanding and explaining its chemical reactivity.
II. Methodology
A comprehensive computational gas-phase DFT study was performed using the Gaussian 09 E.0 software package68 to optimize all molecular structures. Geometry optimizations were performed using the BP86 functional69,70 including D3 dispersion corrections.71,72 All atoms in this study were described with a def2-TZVP triple-ζ quality basis set.73 We thus refer to the theoretical level as BP86-D3(BJ)/def2-TZVP. Frequency calculations at BP86-D3(BJ)/def2-TZVP were performed to identify either minimum energy geometries (zero imaginary frequencies) or transition states (one imaginary frequency). The pathway involving the initial reactants, transition states, and final products was examined by calculating the intrinsic coordinate (IRC).74–76 Nucleus-independent chemical shift (NICS)77–79 and anisotropy of the current-induced density (ACID)80,81 calculations were performed using the GIAO method at BP86-D3(BJ)/def2-TZVP.III. Results and discussion
(1) The geometries and electronic structures of G13-6-Rea (G13 = group 13 elements)
As mentioned in the Introduction, DFT was first used to study the model reactants G13-6-Rea (G13 = B, Al, Ga, In, and Tl), which feature tert-butyl substituents attached on the nitrogen atoms, by determining their geometrical and electronic structures. Our calculated BP86-D3(BJ)/def2-TZVP results for the key geometrical parameters and some physical properties of G13-6-Rea are shown in Table 1 (also see Fig. S1†).
|
||||||
|---|---|---|---|---|---|---|
| B-6-Rea | Al-6-Rea | Ga-6-Rea | In-6-Rea | Tl-6-Rea | ||
| a See ref. 69–72.b See ref. 73.c Energy relative to the corresponding singlet state. A positive value means that the singlet is the ground state.d NICS(0) stands for the NICS value calculated at the center of the molecular plane.e NICS(1) stands for the NICS value calculated 1.0 Å above the center of the molecular plane.f NICS(1)zz stands for the NICS value calculated at the zz component of the magnetic tensor NICS(1).g The proton affinity (PA) of G13-6-Rea, which is the reaction enthalpy based on the equation: (G13-6-Rea)H+(g) → G13-6-Rea(g) + H+.h The gas-phase basicity (GPB) of G13-6-Rea, which is the Gibbs free energy based on the equation: (G13-6-Rea)H+(g) → G13-6-Rea(g) + H+.i μ stands for the electronic chemical potential. For details see the text and Table S2.j η stands for the chemical hardness. For details see the text and Table S1.k ω stands for the electrophilicity index. For details see the text and Table S1.l fk+ stands for the nucleophilic attack. For details see Table S2.m fk− stands for the electrophilic attack. For details see Table S2. | ||||||
| G13–N1 (Å) | 1.429 | 2.045 | 2.143 | 2.377 | 2.480 | |
| G13–N2 (Å) | 1.429 | 2.045 | 2.143 | 2.377 | 2.480 | |
| ∠N1–G13–N2 (°) | 121.4 | 92.28 | 88.42 | 85.78 | 78.92 | |
| ΔEstc (kcal mol−1) | 4.2 | 27.2 | 46.3 | 50.8 | 51.8 | |
| NICS(0)d (ppm) | −8.540 | −1.476 | −1.365 | −1.185 | −1.135 | |
| NICS(1)e (ppm) | −7.094 | −3.250 | −3.219 | −2.367 | −2.162 | |
| NICS(1)zzf (ppm) | −7.002 | −4.004 | −3.841 | −3.713 | −2.735 | |
| PAg (kcal mol−1) | 308.6 | 268.0 | 248.5 | 229.0 | 206.7 | |
| GPBh (kcal mol−1) | 291.8 | 255.2 | 235.3 | 216.6 | 194.7 | |
| fk+i | −0.235 | −0.141 | −0.120 | −0.266 | −0.888 | |
| fk−j | −0.329 | −0.703 | −0.489 | 0.065 | −0.147 | |
| μk (eV) | 1.356 | 1.481 | 2.532 | 2.884 | 3.002 | |
| ηl (eV) | 2.775 | 2.940 | 3.190 | 3.073 | 3.230 | |
| ωm (eV) | 2.551 | 3.224 | 10.23 | 12.78 | 14.54 | |

To the best of our knowledge, no well-separated six-membered NHC monomer containing a pivotal boron element has been experimentally reported yet. Nevertheless, the experimental data concerning the G13–N bond distances in the six-membered NHCs with various substituents were reported as follows: Al–N (1.957 and 1.963 Å),40,41 Ga–N (2.034–2.063 Å),42,43 In–N (2.268–2.364 Å),44,45 and Tl–N (2.401–2.471 Å).46,47 The corresponding BP86-D3(BJ)/TZVP data of G13-6-Rea provided in Table 1 agree reasonably well with the available experimental values.40–47 Additionally, the experimental data for the central ∠N–G13–N bond angle were reported as follows: ∠N–Al–N (89.86° and 91.85°),40,41 ∠N–Ga–N (87.53°–89.42°),42,43 ∠N–In–N (78.23°–81.12°),44,45 and ∠N–Tl–N (76.20°–78.00°).46,47 Again, these attainable experimental values are in reasonably quantitative agreement with our BP86-D3(BJ)/TZVP results for the G13-6-Rea molecules. It must be noted that the computational and experimental results demonstrate that the central ∠N–G13–N bond angle in G13-6-Rea becomes more acute proceeding down the group 13 family from boron to thallium. This phenomenon can be attributed to the “inert s-pair effect” and “orbital non-hybridization effect,” as discussed elsewhere.82–85
The singlet–triplet splitting ΔEst (= Etriplet − Esinglet; kcal mol−1)86 of G13-6-Rea (Table 1) follows the order B-6-Rea (3.9) < Al-6-Rea (28.2) < Ga-6-Rea (47.0) < In-6-Rea (51.0) < Tl-6-Rea (53.1). That is, ΔEst increases monotonically from B-6-Rea to Tl-6-Rea. Fig. 1 shows that proceeding from B-6-Rea to Tl-6-Rea, a change of the group 13 element (G13) in G13-6-Rea decreases the energy of the nonbonded p2-σ lone pair orbital, which is located at the pivotal G13 atom. Similarly, this G13 replacement down the group 13 family decreases the energy of the vertical p-π orbital at the G13 element. Furthermore, the theoretical data shown in Table 1 anticipates the small ΔEst of B-6-Rea (4.2 kcal mol−1), implying that this molecule could be kinetically unstable and readily undergo chemical reactions with other substrates. In fact, the previous studies have provided similarly small values of ΔEst for borylenes.87 The above theoretical finding could explain why the six-membered-ring boron NHC analogue has not been synthesized and stabilized in experiments.
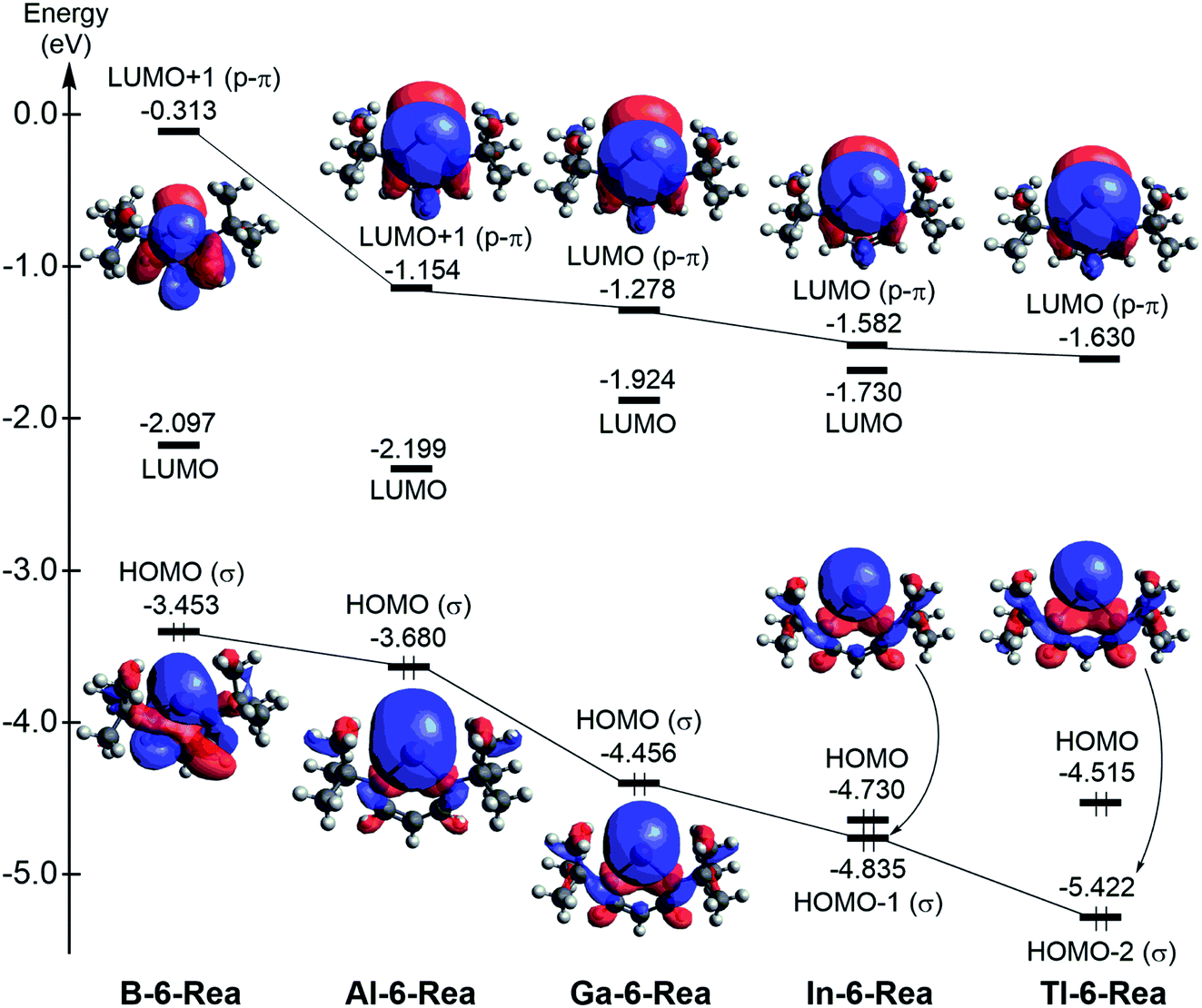 | ||
| Fig. 1 Calculated frontier molecular orbitals of the G13-6-Rea (G13 = B, Al, Ga, In, and Tl) molecules at the BP86-D3(BJ)/def2-TZVP level of theory. For more information see the text. | ||
However, it is noted that the BP86 method cannot provide correct orbital energies due to the reason that the BP86 method is composed of semi-local exchange, and, therefore, decays too rapidly instead of the correct −1.00/R dependence. It has been shown that semi-local functional will collapse to the wrong HOMO–LUMO gap due to an incorrect description of the excitation binding energy in organic systems. For this reason, one single benchmark calculation using a range-separated functional such as LC-BLYP88,89 has been utilized to draw the frontier molecular orbitals of G13-6-Rea, which is given in Fig. S2.† Fortunately, the energy order of the frontier molecular orbitals in Fig. S2† is quite similar to that in Fig. 1. We thus feel confidence for further studies about some properties of G13-6-Rea using the BP86 approach. For more details, one may find some useful references in the literatures.90–92
It is worth noting that the six-membered G13-6-Rea molecules studied in this work, the G13 atom and the molecular backbone (N–C–C–C–N) form an essentially planar six-membered ring. As a result, six π electrons are located in this molecular ring, implying that these cyclic G13-6-Rea molecules should have aromatic character. In fact, from the isolobal analogy viewpoint,93 the six-membered-ring G13-6-Rea species is isolobal with benzene (C6H6), which owns the well-known high aromaticity (Scheme 2). Over the years, the nucleus-independent chemical shift (NICS) approach, developed by Schleyer and et al.,77–79 has become one of the most popular aromaticity probes. We have thus applied the NICS index to investigate the aromatic nature of G13-6-Rea, whose computational NICS(0), NICS(1), and NICS(1)zz values are collected in Table 1. Table 1 shows that the NICS value of G13-6-Rea becomes smaller and less negative as its central G13 atom becomes heavier. These results can be ascribed to the nature of the pivotal G13 elements. In fact, both the orbital size and the orbital energy level of the valence np orbitals of the G13 atom play decisive roles in determining the aromatic character of G13-6-Rea. For instance, the six-membered ring skeleton of B-6-Rea contains only second row elements ( ) and their valence 2p orbitals are similar in size.94 Moreover, these valence 2p orbitals on the cyclic atoms of B-6-Rea are all close in orbital energy. As a result, these phenomena can easily form the p-π resonance on the six-membered ring and make B-6-Rea to possess the highest degree of aromaticity in the G13-6-Rea family. However, the frontier np orbitals of the heavy group 13 elements (such as Al, Ga, In and Tl) bear the higher principal quantum number n (≥3), suggesting that both sizes and energy levels of the frontier np orbitals of a heavy G13 atom are quite larger than those of the valence 2p orbitals of a second row element. Therefore, unlike the significant delocalization over the
) and their valence 2p orbitals are similar in size.94 Moreover, these valence 2p orbitals on the cyclic atoms of B-6-Rea are all close in orbital energy. As a result, these phenomena can easily form the p-π resonance on the six-membered ring and make B-6-Rea to possess the highest degree of aromaticity in the G13-6-Rea family. However, the frontier np orbitals of the heavy group 13 elements (such as Al, Ga, In and Tl) bear the higher principal quantum number n (≥3), suggesting that both sizes and energy levels of the frontier np orbitals of a heavy G13 atom are quite larger than those of the valence 2p orbitals of a second row element. Therefore, unlike the significant delocalization over the  ring of B-6-Rea, there is effectively less overlap of the valence p-π orbital on the heavy G13 center with the two proximal N p-π orbitals. Accordingly, our theoretical analysis demonstrates that the aromatic character of the six-membered ring of G13-6-Rea decreases down the group 13 family from B-6-Rea to Tl-6-Rea (also see their ACID plots in Fig. S3†).
ring of B-6-Rea, there is effectively less overlap of the valence p-π orbital on the heavy G13 center with the two proximal N p-π orbitals. Accordingly, our theoretical analysis demonstrates that the aromatic character of the six-membered ring of G13-6-Rea decreases down the group 13 family from B-6-Rea to Tl-6-Rea (also see their ACID plots in Fig. S3†).
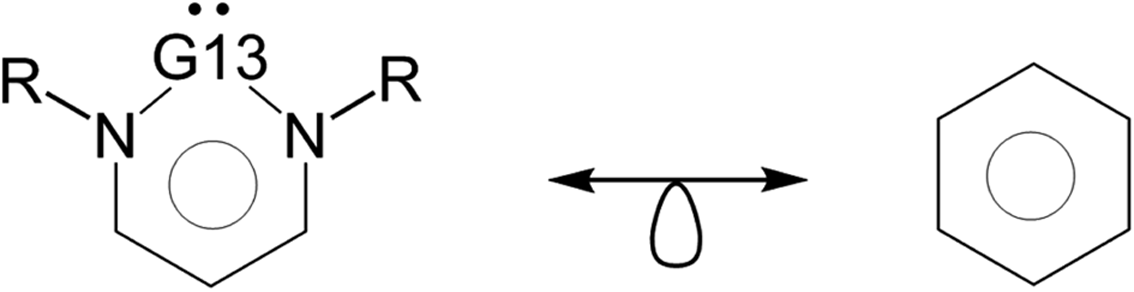 | ||
| Scheme 2 G13-6-Rea is isolobal to benzene. | ||
We are interested in the basicity of G13-6-Rea,95 from which one may understand the nature of its chemical bonding with other substrates. We thus used the chemical equation ((G13-6-Rea)H+(g) → G13-6-Rea(g) + H+) to evaluate the proton affinity (PA) and gas-phase basicity (GPB) of G13-6-Rea, whose BP86 results are provided in Table 1. PA and GPB show a monotonically decreasing trend from B-6-Rea to Tl-6-Rea. The explanation for this phenomenon can be obtained from Fig. 1, in which the HOMO (highest occupied molecular orbital) of B-6-Rea, Al-6-Rea, and Ga-6-Rea is the nonbonding lone pair sp2-σ orbital, which is located on the pivotal boron, aluminum, and gallium center of the corresponding heterocycle plane, respectively. Consequently, this theoretical evidence strongly suggests that B-6-Rea, Al-6-Rea, and Ga-6-Rea will display significant Lewis base chemistry. However, Fig. 1 shows that for In-6-Rea and Tl-6-Rea, the G13 p-π orbital, which is orthogonal to the six-membered ring, is largely associated with the LUMO. Therefore, our theoretical evidence indicates that In-6-Rea and Tl-6-Rea will exhibit Lewis acid chemical behavior.
An intriguing question is what role does G13-6-Rea play when it reacts with other substrates, i.e., is it a nucleophile or an electrophile? The answer was obtained by applying the Fukui function,96 the central site reactivity index of DFT, to the central G13 element of G13-6-Rea. The BP86 results summarized in Table 1 show that the calculated value of the Fukui function of nucleophilic attack (fk+) is obvious larger than that of electrophilic attack (fk−) for B-6-Rea, Al-6-Rea and Ga-6-Rea. This theoretical information strongly suggests that the above three molecules are nucleophilic and thus prefer to undergo nucleophilic attack with other substrates. The supporting evidence comes from the valence orbitals of B-6-Rea, Al-6-Rea and Ga-6-Rea, which are schematically illustrated in Fig. 1. That is, the HOMO (the nonbonding lone pair sp2-σ orbital) of B-6-Rea, Al-6-Rea and Ga-6-Rea would play a dominant role in their chemical reactions. However, Table 1 shows that the Fukui function values of electrophilic attack (fk−) for In-6-Rea and Tl-6-Rea are apparently larger than those of the corresponding nucleophilic attack (fk+), indicating that the LUMO (the unoccupied vertical p-π orbital) of In-6-Rea and Tl-6-Rea plays a vital role in their chemical reactions. As a result, our theoretical evidence suggests that the In-6-Rea and Tl-6-Rea molecules should behave as strong electrophiles.
We also investigated the electrophilicity97 of G13-6-Rea. As suggested by Koopmans,98 Parr and coworkers,99 the electronic chemical potential (μ), chemical hardness (η), and electrophilicity index (ω), respectively, were defined as follows:
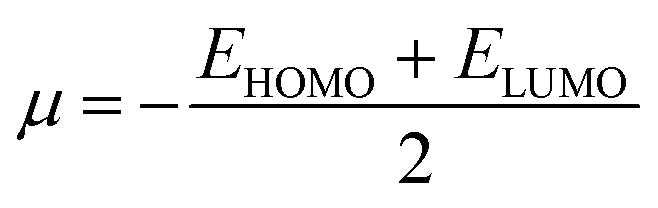 | (3) |
| η = ELUMO − EHOMO | (4) |
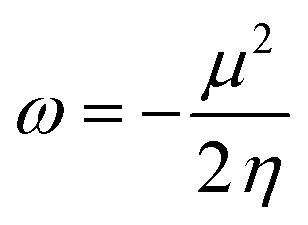 | (5) |
In principle, as with the traditional carbene species, the nature of the chemical reactivity of the six-membered NHC analogues G13-6-Rea depends on two competing frontier orbitals, i.e., the nonbonding lone pair sp2-σ orbital and the unoccupied vertical p-π orbital.1 The same situations also apply to the G13-6-Rea species. Consequently, if the sp2-σ lone pair orbital prevails, then the G13-6-Rea molecule manifests the nucleophilicity itself. Conversely, if the empty p-π orbital predominates, then the G13-6-Rea compound presents the electrophilicity itself. Table 1 clearly shows that the calculated values of the electrophilicity (ω) of G13-6-Rea increase in the order B-6-Rea (2.538) < Al-6-Rea (3.095) < Ga-6-Rea (1.180) ≪ In-6-Rea (11.78) < Tl-6-Rea (14.18). In other words, the larger the atomic number of the pivotal group 13 element in the six-membered ring is and the higher the electrophilicity of G13-6-Rea will be in chemical reactions. Again, the above theoretical observations can be verified from the frontier orbitals of G13-6-Rea depicted in Fig. 1. For instance, B-6-Rea has the sp2-σ lone pair orbital as its HOMO, while its unoccupied vertical p-π orbital is much higher in energy than its LUMO. Therefore, this nonbonding lone pair sp2-σ orbital (HOMO) plays an important role in governing the chemical reactivity of B-6-Rea. This result, in turn, makes B-6-Rea less electrophilic than the other G13-6-Rea molecules. In contrast, Tl-6-Rea has the vertical p-π orbital on Tl as its LUMO, whereas its lone pair sp2-σ orbital is much lower in energy than its HOMO. Consequently, one may easily foresee a crucial role for this empty vertical p-π orbital (LUMO) in the chemical behavior of Tl-6-Rea.100
Bearing the above theoretical findings in mind, we will use the concepts of aromaticity, basicity, and electrophilicity to explore the origin of barrier heights and the chemical reactivity of the G13-6-Rea molecules in the next section.
(2) The insertion reaction of G13-6-Rea (G13 = group 13 elements) with methane
We firstly examine the C–H bond insertion reaction of G13-6-Rea with methane (eqn (1)), whose calculated BP86-D3(BJ)/def2-TZVP results are schematically shown in Fig. 2. The computational Gibbs free activation energy (ΔGACT,CH4) of the present study indicates that the larger the atomic number of the pivotal G13 atom is, the higher the activation energy of G13-6-Rea with methane will be, and its C–H bond insertion reaction will proceed with greater difficulty. For instance, the BP86 ΔGACT,CH4 data (kcal mol−1) given in Fig. 2 show that B-6-CH4-TS (31.9) < Al-6-CH4-TS (48.7) < Ga-6-CH4-TS (64.1) < In-6-CH4-TS (80.4) < Tl-6-CH4-TS (100.4). This theoretical evidence strongly suggests that the C–H bond activation reactions of the six-membered group 13 NHC analogues with methane are kinetically unfavorable. The supporting evidences come from the experimental facts55–59 that the six-membered ring Al analogue only breaks the C–C bond rather than the C–H bond of the hydrocarbons. Moreover, we investigated the theoretical results of their final insertion products (G13-6-CH4-Prod). Again, from Fig. 2, the calculated Gibbs free energy of reaction (ΔGREA,CH4) follows the same trend as the atomic weight of the central G13 element, that is, B-6-CH4-Prod (−32.2) < Al-6-CH4-Prod (−17.9) < Ga-6-CH4-Prod (5.6) < In-6-CH4-Prod (27.8) < Tl-6-CH4-Prod (53.4). However, our above theoretical observations are based on the gas-phase calculations without involving the solvents. Additionally, the B-6-Rea molecule was theoretically proved to form thermodynamically stable product (Fig. 2). As a result, our present theoretical results cannot rule out C–H activation with the B analogue at high temperature in solution.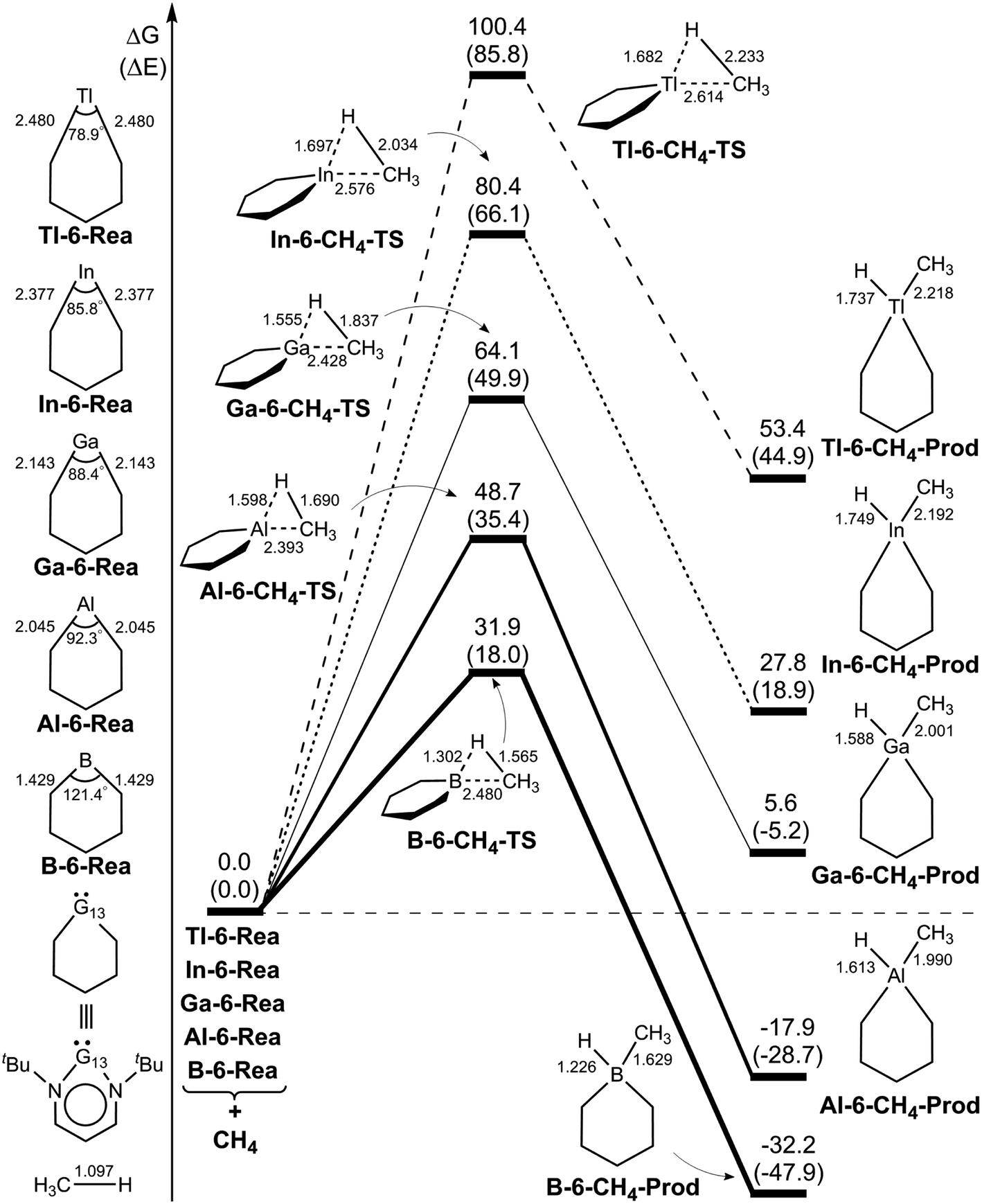 | ||
| Fig. 2 BP86-D3(BJ)/def2-TZVP energy profiles (energy in kcal mol−1 and bond distances in Å) for the insertion reaction of G13-6-Rea (G13 = B, Al, Ga, In, and Tl) with CH4. Also, see Fig. S4 and S5.† | ||
In addition, we found that using the valence bond state correlation diagram (VBSCD) model, which was originally developed by Shaik,61–63 obtain a good linear relationship between the singlet–triplet energy splitting (ΔEst) of G13-6-Rea and Gibbs free activation energy (ΔGACT,CH4) as well as Gibbs free reaction enthalpy (ΔGREA,CH4) for its C–H bond insertion reaction with methane (Fig. 2). That is, ΔGACT,CH4 = 1.17ΔEst + 22.4 (the correction coefficient r = 0.91) (Fig. S6†) and ΔGREA,CH4 = 1.47ΔEst − 46.6 (r = 0.89) (Fig. S7†). That is to say, the smaller the ΔEst of G13-6-Rea, the lower the barrier height and, in turn, the faster the C–H bond activation reaction, and the greater the exothermicity. In other words, our theoretical results based on the VBSCD model demonstrate that ΔEst can be a guideline for predicting the activation energy and reaction enthalpy of the C–H bond activation reaction of G13-6-Rea with hydrocarbons.
To gain a deeper understanding of the origin of the barrier heights for the C–H bond insertion reaction of G13-6-Rea, the activation strain model (ASM)64–67 method was applied in this work. Table 2Table 3 shows that the activation energy (ΔEACT,CH4) is separated into three terms, i.e., the methane deformation energy (ΔEDEF,CH4), the G13-6-Rea deformation energy (ΔEDEF,G13-6-Rea), and the internal energy (ΔEINT). Thus, they are schematically represented in Fig. 3 using the computational data collected in Table 2. Obviously, in the four curves, only ΔEDEF,CH4 increases monotonically down the group 13 family from B-6-CH4-TS to Tl-6-CH4-TS, following a trend identical to that of ΔEACT,CH4. This result strongly suggests that the ΔEDEF,CH4 term is the dominant factor in determining the trend in the activation energy (ΔEACT,CH4). Examining the BP86 geometrical structures of G13-6-CH4-TS in Fig. 2 shows that the H…C stretching bond length (Å) in G13-6-CH4-TS increases in the order 1.565 (B-6-CH4-TS) < 1.690 (Al-6-CH4-TS) < 1.837 (Ga-6-CH4-TS) < 2.034 (In-6-CH4-TS) < 2.233 (Tl-6-CH4-TS). Note that the original H–CH3 bond distance of methane was calculated to be 1.097 Å at the same level of theory. These calculated data can be explained as follows. In the transition state (G13-6-CH4-TS), the H–CH3 bond is breaking and the pivotal G13 element of G13-6-Rea is forming new bonds with the hydrogen and carbon atoms of methane. As a result, the increased elongation of the C–H bond length in the transition state for C–H activation moving down the G13 series is simply due to the Hammond postulate.101 That is to say, as the G13-6-CH4-Prod becomes higher in energy moving down G13, G13-6-CH4-TS will more closely resemble products. This, in turn, makes the C–H bond length of methane in the G13-6-CH4-TS will be longer as well as more product-like. Accordingly, our theoretical examination concludes that the trend in activation barriers of G13-6-CH4-TS species is simply a consequence of both weaker G13–C and G13–H bonds in the product.

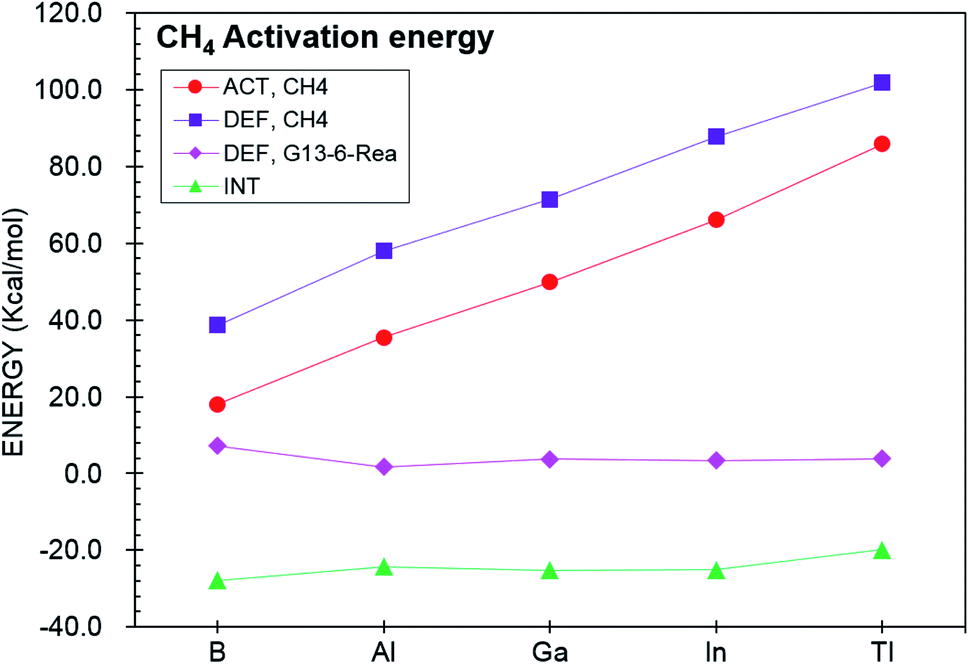 | ||
| Fig. 3 Energy decompositions of the activation energies (ΔEACT,CH4) of the transition states (G13-6-CH4-TS) of the insertion reactions of G13-6-Rea (G13 = group 13 element) with CH4. The data are taken from Table 2. | ||
(3) The [1 + 2] cycloaddition reaction of G13-6-Rea (G13 = group 13 elements) with ethene
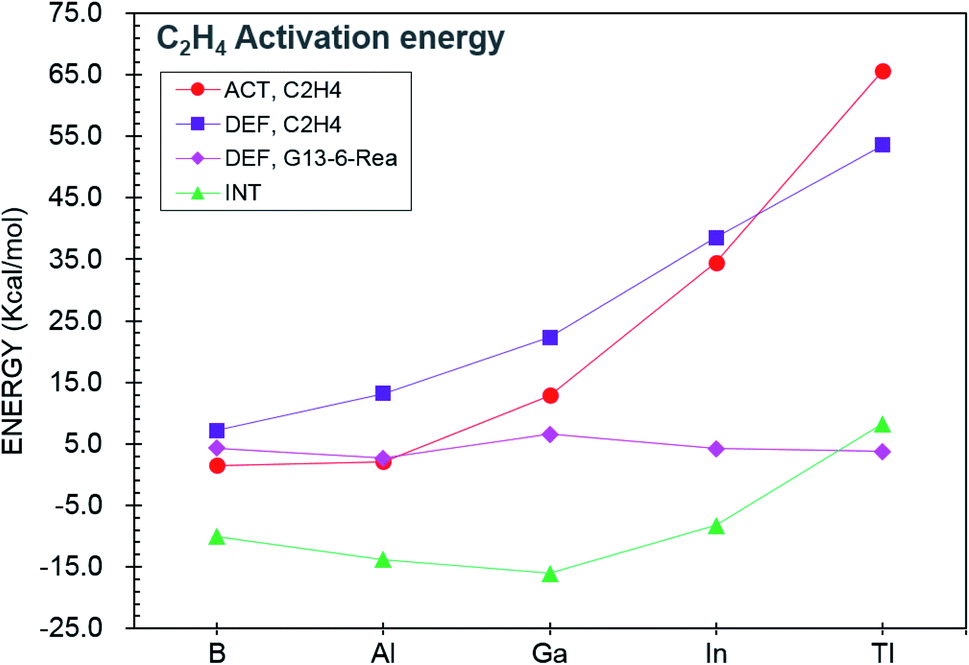
We next investigate the reactivity of G13-6-Rea toward the [1 + 2] cycloaddition to ethene (eqn (2)) at BP86-D3(BJ)/def2-TZVP. Fig. 4, which schematically represents the calculated results, demonstrates that the Gibbs free energy of activation (ΔGACT,C2H4; kcal mol−1) of G13-6-C2H4-TS increases monotonically down the group 13 family from boron to thallium. Specifically, B-6-C2H4-TS (12.0) < Al-6-C2H4-TS (16.2) < Ga-6-C2H4-TS (31.5) < In-6-C2H4-TS (54.5) < B-6-C2H4-TS (85.4). Moreover, the BP86 evidence in Fig. 4 indicates that the Gibbs free energy of reaction (ΔGREA,C2H4; kcal mol−1) also increases in the order B-6-C2H4-Prod (−30.5) < Al-6-C2H4-Prod (−1.5) < Ga-6-C2H4-Prod (26.3) < In-6-C2H4-Prod (52.8) < B-6-C2H4-Prod (84.6). Notably, the relative energy of the cycloaddition product B-6-C2H4-Prod is below those of the corresponding reactants. However, the C–H activation reaction of Al-6-Rea with methane was theoretically predicted to be thermal-neutral. Our theoretical evidence therefore suggests that the [1 + 2] cycloaddition reaction between the six-membered-ring NHC analogue containing one central B element and ethene has favorable kinetics and thermodynamics.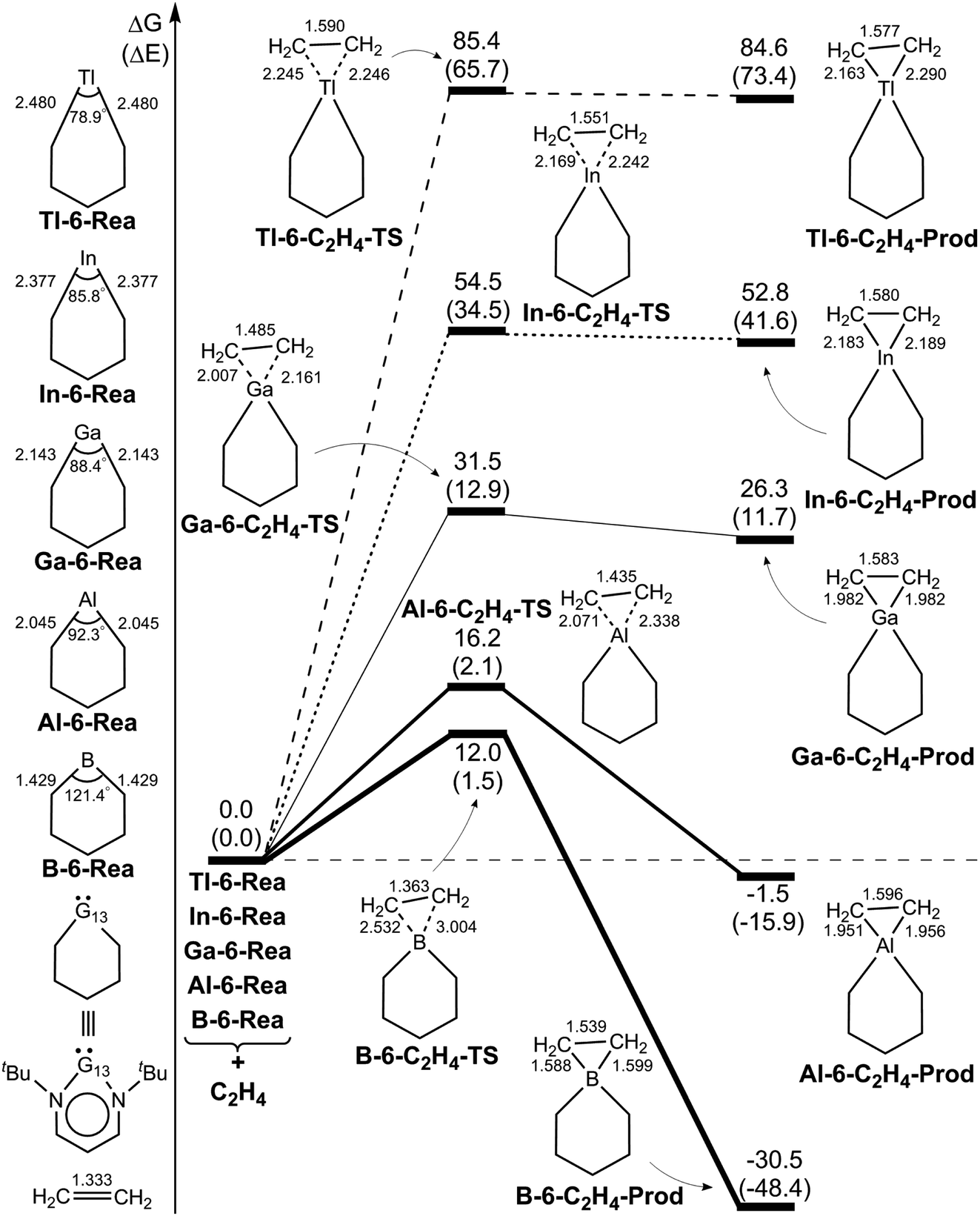 | ||
Fig. 4 BP86-D3(BJ)/def2-TZVP energy profiles (energy in kcal mol−1 and bond distances in Å) for the insertion reaction of G13-6-Rea (G13 = B, Al, Ga, In, and Tl) with H4C2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2H4. Hydrogens are omitted in this picture for clarity. Also see Fig. S8 and S9.† C2H4. Hydrogens are omitted in this picture for clarity. Also see Fig. S8 and S9.† | ||
On the basis of the VBSCD model61–63 mentioned earlier, it was concluded that the G13-6-Rea molecule possessing a smaller value of ΔEst (= Etriplet − Esinglet) should lead to a lower activation energy and a larger exothermicity. Indeed, the present computational results for the [1 + 2] cycloaddition reaction of G13-6-Rea with ethene confirm the accuracy of this prediction. Our BP86 data shown in Table 1 and Fig. 4 demonstrate a linear correlation between ΔEst and the activation energy (ΔGACT,C2H4, kcal mol−1): ΔGACT,C2H4 = 1.15ΔEst − 2.18 (r = 0.84) (Fig. S10†). Likewise, a linear relationship is found between ΔEst and the reaction free energy (ΔGREA,C2H4, kcal mol−1): ΔGREA,C2H4 = 1.99ΔEst − 46.5 (r = 0.92) (Fig. S11†). Accordingly, our theoretical findings suggest that the theoretical singlet–triplet energy splitting (ΔEst) of the six-membered ring group 13 NHC analogue can be a measured criterion for predicting the reactivity.
Again, like the case of the C–H bond activation discussed earlier, the ASM64–67 approach was used to explore the origin of the barrier heights of the cycloaddition reactions of G13-6-Rea. In Fig. 5, comparing the trend in the activation energy (ΔEACT,C2H4) with those of its three components (ΔEDEF,C2H4, ΔEDEF,G13-6-Rea, and ΔEINT), one finds that the ethene deformation energy (ΔEDEF,C2H4) is the key factor determining the trend in ΔEACT,C2H4. This result can be explained in terms of the geometrical structures of G13-6-C2H4-TS given in Fig. 4. The BP86 data indicate that the stretching distance (Å) of the C…C bond in the ethene fragment of G13-6-C2H4-TS increases in the order 1.363 (B-6-C2H4-TS) < 1.435 (Al-6-C2H4-TS) < 1.485 (Ga-6-C2H4-TS) < 1.551 (In-6-C2H4-TS) < 1.590 (Tl-6-C2H4-TS). In comparison, the CC double bond distance in the parent H2CCH2 is 1.333 Å at the same level of theory. The above trend can be attributed to the atomic radius of the central G13 element in G13-6-Rea. During the attacking process between G13-6-Rea and ethene, both carbon atoms in the latter fragment must be separated for better orbital overlaps between the carbons and G13. As noted, the atomic radius of G13 increases with the atomic number.102 As a result, the C–C bond distance of ethene in the transition state should increase with the atomic radius of G13. This phenomenon increases the bonding dissociation energy of ethene, which, in turn, increases the activation barrier, as demonstrated in Fig. 4. In brief, our theoretical observations indicate that the smaller the atomic radius of the pivotal group 13 element on the G13-6-Rea molecule is, the smaller ΔEst of G13-6-Rea, the lower the activation barrier height, the faster the G13-6-Rea cycloaddition reaction with olefins, and the higher the reaction exothermicity of the cycloaddition product.
 | ||
| Fig. 5 Energy decompositions of the activation energies (ΔEACT,C2H4) of the transition states (G13-6-C2H4-TS) of the [1 + 2] cycloaddition reactions of G13-6-Rea (G13 = group 13 element) with C2H4. The data are taken from Table 3. | ||
IV. Conclusion
In this work, DFT accompanied by several well-established concepts (aromaticity/basicity/electrophilicity) and some sophisticated models (VBSCD61–63 and ASM64–67) was used to explore the chemical reactivity of the six-membered-ring G13 NHC analogues (G13-6-Rea).Interestingly, to date, no six-membered-ring boron NHC analogue has been prepared and synthesized in experiments. This can be explained in terms of the small ΔEst of B-6-Rea (∼4 kcal mol−1 according to our DFT data), which implies that this species is kinetically unstable and thus readily reacts with other substrates.
The present theoretical examinations show that the pivotal group 13 element in the six-membered ring of G13-6-Rea plays a crucial role in determining the chemical reactivity of this molecule. Our theoretical analysis suggests that the HOMOs (the sp2-σ lone pair orbital on the G13 element) are the key factors influencing the chemical behavior of B-6-Rea, Al-6-Rea, and Ga-6-Rea, and thus these molecules can be considered nucleophiles. In contrast, the LUMOs (the vacant p-π orbital on the G13 atom) are decisive factors controlling the chemical nature of In-6-Rea and Tl-6-Rea, and thus these molecules can be considered electrophiles. Moreover, the present study predicts that no G13-6-Rea molecule can undergo a C–H bond insertion reaction with hydrocarbons from kinetic and thermodynamic viewpoints. Indeed, recent experimental observations55–59 indicate that the six-membered ring Al analogue only breaks the C–C bond rather than the C–H bond of the hydrocarbons. Nevertheless, our present theoretical observations cannot rule out B-6-Rea undergoing the C–H activation with hydrocarbons at high temperature in solution.
In addition, only B-6-Rea is a good candidate for [1 + 2] cycloaddition reactions with olefins. Furthermore, our theoretical predictions strongly suggest that the lower the atomic number of the group 13 element is, the smaller the atomic radius of the G13 atom, the higher the aromaticity of the six-membered ring of G13-6-Rea, and the smaller ΔEst of this NHC analogue will be. This relationship, in turn, will pertain for all of its insertion reactions with hydrocarbons or cycloaddition reactions with olefins.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Center for High-Performance Computing of Taiwan in providing huge computing resources to facilitate this research. They also thank the Ministry of Science and Technology of Taiwan for the financial support. Special thanks are also due to reviewers 1 and 2 for very helpful suggestions and comments.References
- W. Kirmse, in Carbene Chemistry, Academic Press, New York, 1st edn, 1964, ch. 1 Search PubMed
.
- A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef
- For annual reviews: Coord. Chem. Rev., 2007, 251, 595–895 Search PubMed.
- For annual reviews: Chem. Rev., 2009, 109, 3209–3884 Search PubMed.
- For annual reviews: Eur. J. Inorg. Chem., 2009, 1663–2007 Search PubMed.
- For annual reviews: Dalton Trans., 2009, 6873–7316 Search PubMed.
- D. Martin, M. Melaimi, M. Soleihavoup and G. Bertrand, Organometallics, 2011, 30, 5304–5313 CrossRef CAS PubMed
- See for example: M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed
- See for example: D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed
- See for example: R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS PubMed
- See for example: H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed
- See for example: V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed
- See for example: E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed
- See for example: A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed
- See for example: Á. Vivancos, C. Segarra and M. Albrech, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed
- See for example: S. Kuwata and F. E. Hahn, Chem. Rev., 2018, 118, 9642–9677 CrossRef CAS PubMed
- See for example: A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed
- See for example: C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed
- See for example: Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed
- See for example: Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed
- See for example: R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed
- See for example: C. Boehme and G. Frenking, J. Am. Chem. Soc., 1996, 118, 2039–2046 CrossRef CAS
- V. Nair, S. Bindu and V. Streekumar, Angew. Chem., Int. Ed., 2004, 43, 5130–5135 CrossRef CAS PubMed
- D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed
- G. A. Grasa, R. M. Kissling and S. P. Nolan, Org. Lett., 2002, 4, 3583–3586 CrossRef CAS PubMed
- G. A. Grasa, T. Guveli, R. Singh and S. P. Nolan, J. Org. Chem., 2003, 68, 2812–2819 CrossRef CAS PubMed
- R. Singh, R. M. Kissling, M.-A. Letellier and S. P. Nolan, J. Org. Chem., 2004, 69, 209–212 CrossRef CAS PubMed
- G. A. Grasa, R. S. Singh and P. Nolan, Synthesis, 2004, 971–985 CAS
- F. K. Zinn, M. S. Viciu and S. P. Nolan, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2004, 100, 231–249 RSC
- R. Singh and S. P. Nolan, Chem. Commun., 2005, 5456–5458 RSC
- T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem., 1998, 110, 2631–2633 CrossRef
- T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem., Int. Ed., 1998, 37, 2490–2493 CrossRef CAS PubMed
- L. Ackermann, A. Furstner, T. Weskamp, F. J. Kohl and W. A. Herrmann, Tetrahedron Lett., 1999, 40, 4787–4790 CrossRef CAS
- M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS
- J. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5375–5380 CrossRef CAS
- W. A. Herrmann, Synthetic Methods of Organometallic and Inorganic Chemistry, Georg Thieme Verlag, Stuttgart, NY, 2000, vol. 9 Search PubMed
- E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS
- N. M. Scott and S. P. Nolan, Eur. J. Inorg. Chem., 2005, 1815–1828 CrossRef CAS
- C. Cui, H. W. Roseky and H.-G. Schmidt, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed
- X. Li, X. Cheng, H. Song and C. Cui, Organometallics, 2007, 26, 1039–1043 CrossRef CAS
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC
- D. Dange, S. L. Choong, C. Schenk, A. Stasch and C. Jones, Dalton Trans., 2012, 41, 9304–9315 RSC
- M. S. Hill and P. B. Hitchcock, Chem. Commun., 2004, 1818–1819 RSC
- M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Dalton Trans., 2005, 273–277 RSC
- Y. Cheng, P. B. Hitchcock, M. F. Lappert and M. Zhou, Chem. Commun., 2005, 752–754 RSC
- C.-H. Chen, M.-L. Tsai and M.-D. Su, Organometallics, 2006, 25, 2766–2773 CrossRef CAS
- M. S. Hill, R. Pongtavornpinyo and P. B. Hitchcock, Chem. Commun., 2006, 3720–3722 RSC
- H. Zhu, J. Chai, V. Chandrasekhar, H. W. Roesky, J. Magull, D. Vidovic, H.-G. Schmidt, M. Noltemeyer, P. P. Power and W. A. Merrill, J. Am. Chem. Soc., 2004, 126, 9472–9473 CrossRef CAS PubMed
- T. Chu, S. F. Vyboishchikov, B. M. Gabidullin and G. I. Nikonov, J. Am. Chem. Soc., 2017, 139, 8804–8807 CrossRef CAS PubMed
- C. Helling, C. Wölper and S. Schulz, J. Am. Chem. Soc., 2018, 140, 5053–5056 CrossRef CAS PubMed
- R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed
- L. Song, J. Schoening, C. W. lper, S. Schulz and P. R. Schreiner, Organometallics, 2019, 38, 1640–1647 CrossRef CAS
- J. Krüger, C. Wölper, L. John, L. Song, P. R. Schreiner and S. Schulz, Eur. J. Inorg. Chem., 2019, 11–12, 1669–1678 CrossRef
- P. Bharadwaz and A. K. Phukan, Inorg. Chem., 2019, 58, 5428–5432 CrossRef CAS PubMed
- C. Helling, C. Wölper, Y. Schulte, G. E. Cutsail III and S. Schulz, Inorg. Chem., 2019, 58, 10323–10332 CrossRef CAS PubMed
- C. Helling, C. Wölper and S. Schulz, Eur. J. Inorg. Chem., 2020, 4225–4235 CrossRef CAS
- C. Bakewell, M. Garçon, R. Y. Kong, L. O'Hare, A. J. P. White and M. R. Crimmin, Inorg. Chem., 2020, 59, 4608–4616 CrossRef CAS PubMed
- O. Kysliak, H. Görlsa and R. Kretschmer, Dalton Trans., 2020, 49, 6377–6383 RSC
- C. Helling, C. Wölper and S. Schulz, Dalton Trans., 2020, 49, 11835–11842 RSC
- S. Shaik, J. Am. Chem. Soc., 1981, 103, 3692–3701 CrossRef CAS
- A. Pross, in Theoretical and Physical principles of Organic Reactivity, John Wiley & Sons Inc., USA, 1995 Search PubMed
- S. Shaik and A. Shurki, Angew. Chem., Int. Ed., 1999, 38, 586–625 CrossRef PubMed
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS
- W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed
- I. Fernándeza and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision E.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154122 CrossRef PubMed
- F. Weigenda and R. Ahlrichsb, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed
- B. T. Psciuk, R. L. Lord, C. H. Winter and H. B. Schlegel, J. Chem. Theory Comput., 2012, 8, 4950–4959 CrossRef CAS PubMed
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS
- D. Geuenich, K. Hess, F. Kohler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed
- P. Pyykkö and J.-P. Desclaux, Acc. Chem. Res., 1979, 12, 276–281 CrossRef
- W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1984, 23, 272–295 CrossRef
- P. Pyykkö, Chem. Rev., 1988, 88, 563–594 CrossRef
- P. Pyykkö, Chem. Rev., 1997, 97, 597–636 CrossRef PubMed
- In this work, the unpaired spins in the triplet configuration reside on the HOMO and the LUMO of the G13-6-Rea species rather than the sp2-σ lone pair orbital and the vacant p-π orbital on the pivotal group 13 element..
- Y. Wang and C.-G. Liu, Phys. Chem. Chem. Phys., 2020, 22, 28423–28433 RSC
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109–234117 CrossRef PubMed
- O. A. Vydrov, J. Heyd, A. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106–074114 CrossRef PubMed
- M. Hagemann, R. J. F. Berger, S. A. Hayes, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2008, 14, 11027–11038 CrossRef CAS PubMed
- K. R. Leopold, M. Canagaratna and J. A. Phillips, Acc. Chem. Res., 1997, 30, 57–64 CrossRef CAS
- J. J. C. Trujillo and I. Fernández, Chem.–Eur. J., 2018, 24, 17823–17831 CrossRef PubMed
- R. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 711–724 CrossRef
- A. Liberles, J. Chem. Educ., 1977, 54, 479–482 CrossRef CAS
- L. D. Betowski, J. J. Solomon and R. F. Porter, Inorg. Chem., 1972, 11, 424–427 CrossRef CAS
- P. W. Ayers and R. G. Parr, J. Am. Chem. Soc., 2000, 122, 2010–2018 CrossRef CAS
- P. K. Chattaraj, U. Sarkar and D. R. Roy, Chem. Rev., 2006, 106, 2065–2091 CrossRef CAS PubMed
- T. A. Koopmans, Physica, 1933, 1, 104–113 CrossRef CAS
- R. G. Parr, L. v. Szentpály and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS
- In this work, we have discussed whether the G13-6-Rea molecules would behave as electrophiles or nucleophiles in interactions with a substrate. For instance, we have utilized the concepts of Fukui functions to anticipate that B-6-Rea, Al-6-Rea, and Ga-6-Rea would react as nucleophiles, while In-6-Rea and Tl-6-Rea would act as electrophiles. Nevertheless, it is accepted that many real examples have shown that their chemical reactivity is not governed by the HOMO/LUMO of the reacting molecules. It could be the other adjoining frontier molecular orbitals (such as, HOMO−1 or LUMO−1) that can control the chemical reactivity due to their molecular orbital patterns, symmetries, and the relative energies compared with those of the reacting substrates. As a result, according to the frontier molecular orbitals shown in Fig. 1, it is likely all G13-6-Rea act as ambiphilicity. This, in turn, would make the reacting substrates interact with G13-6-Rea, which should be reexamined to determine the natures of these reactions. Such researches, however, are beyond the scope of the present work..
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 37, 2832–2838 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02703d |
| This journal is © The Royal Society of Chemistry 2021 |