
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving highly efficient and selective cesium extraction using 1,3-di-octyloxycalix[4]arene-crown-6 in n-octanol based solvent system: experimental and DFT investigation†
Kankan Patra*a,
Biswajit Sadhub,
Arijit Sengupta *cd,
C. B. Patila,
R. K. Mishrade and
C. P. Kaushikde
*cd,
C. B. Patila,
R. K. Mishrade and
C. P. Kaushikde
aNuclear Recycle Board, Bhabha Atomic Research Centre, Tarapur 401504, India. E-mail: kankan.patra2010@gmail.com
bHealth Physics Division, Bhabha Atomic Research Centre, Mumbai 400 085, India
cRadiochemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: arijita@barc.gov.in
dHomi Bhabha National Institute, Anushaktinagar, Mumbai 400 094, India
eNuclear Recycle Group, Bhabha Atomic Research Centre, Mumbai 400 085, India
First published on 16th June 2021
Abstract
Due to the long half-life of 137Cs (t1/2 ∼ 30 years), the selective extraction of cesium (Cs) from high level liquid waste is of paramount importance in the back end of the nuclear fuel cycle to avoid long term surveillance of high radiotoxic waste. As 1,3-di-octyloxycalix[4]arene-crown-6 (CC6) is suggested to be a promising candidate for selective Cs extraction, the improvement in the Cs extraction efficiency by CC6 has been investigated through the optimization of the effect of dielectric media on the extraction process. The effects of the feed acid (HNO3, HCl, and HClO4) and the composition of the diluents for the ligand in the organic phase on the extraction efficiency of Cs have been investigated systematically. In 100% n-octanol medium, Cs is found to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ion-pair complex with CC6 (0.03 M) providing a very high distribution ratio of DCs ∼ 22, suggesting n-octanol as the most suitable diluent for Cs extraction. No significant interference of other relevant cations such as Na, Mg and Sr was observed on the DCs value in the optimized solvent system. Density functional theory (DFT) based calculations have been carried out to elucidate the reason of ionic selectivity and enhanced Cs extraction efficiency of CC6 in the studied diluent systems. In addition to the ionic size-based selectivity of the crown-6 cavity, the polarity of the organic solvent system, the hydration energy of the ion, and the relative reorganization of CC6 upon complexation with Cs are understood to have roles in achieving the enhanced efficiency for the extraction of Cs by the CC6 extractant in nitrobenzene medium.
:1 ion-pair complex with CC6 (0.03 M) providing a very high distribution ratio of DCs ∼ 22, suggesting n-octanol as the most suitable diluent for Cs extraction. No significant interference of other relevant cations such as Na, Mg and Sr was observed on the DCs value in the optimized solvent system. Density functional theory (DFT) based calculations have been carried out to elucidate the reason of ionic selectivity and enhanced Cs extraction efficiency of CC6 in the studied diluent systems. In addition to the ionic size-based selectivity of the crown-6 cavity, the polarity of the organic solvent system, the hydration energy of the ion, and the relative reorganization of CC6 upon complexation with Cs are understood to have roles in achieving the enhanced efficiency for the extraction of Cs by the CC6 extractant in nitrobenzene medium.
1. Introduction
A significant amount of radioactivity in the high-level liquid waste obtained from the nuclear fuel recycling process is due to the presence of the 137Cs radionuclide in the solution. Secure management of these nuclear waste solutions is one of the most significant technological and industrial challenges faced by the nuclear industry. The success and public acceptance of the nuclear power program are highly dependent on the successful removal of the harmful radionuclides from the radioactive nuclear waste. After separation of minor actinides, collectively, ∼99% of the remaining radioactivity of the high-level waste (HLW) solution is mainly attributed to 137Cs and 90Sr present in the solution.1 It is well established that these two major radio nuclides, i.e. 137Cs (half-life, t1/2 = 30.1 years) and 90Sr (half-life, t1/2 = 28.9 years), are mainly responsible for the heat and radiation load of the HLW. Furthermore, due to high mobility of Cs, the radionuclide might pose a long-term unpleasant impact on the ecology.2–10The main concept behind the separation of a metal ion into an organic phase through solvent extraction is the screening of the charge on the metal ion through suitable complexation followed by the mass transfer into the nonpolar organic phase. The lower ionic potential of Cs+ makes it difficult to undergo complexation with conventional ligands and hence leaves us with the only choice of complexation based on the size selectivity of the macrocyclic cavities of the extractant molecules.
A variety of solid phase sorbents have been reported in literature for the effective separation of heavy metals from different aqueous streams.11 Multiwall carbon nanotubes functionalized with thorium oxide (MWCNTs/ThO2) was reported for effective adsorption of Pb2+ ions (sorption 93.8%) from aqueous system due to the removal of H+ ions.12 Naushad et al. reported magnetic metal–organic framework for the removal of UO22+ and Th4+ with adsorption capacity of 227.3 and 285.7 mg g−1, respectively.13 Iron based nanocomposite was reported to remove heavy metals including Cd2+, Cr3+ and Co2+ effectively.14 Alqadami et al. reported Pb2+ sorption on nanocomposite using carboxylic and amino functionalities.15 Naushad et al.16 also reported curcumin based magnetic nanocomposite as highly efficient solid phase extractant for Hg2+ with a capacity of 144.9 mg g−1. Khan et al. reported the utilization of hydrochar for the removal of heavy metals including Pb2+ and Cd2+.17,18. Nanomagnetic copper ferrite/drumstick pod biomass (CuFe2O4/DC) composite has also been employed for removal of Pb2+ predominantly following Langmuir isotherm19 Penicillium chrysogenum and Aspergillus ustus were reported to separate Cd2+, Cu2+ and Pb2+ from aqueous medium.20
The Cs-selective extractants, especially those in the calixarene-crown-6 and the calixarene-bis-(crown-6) families, have been known for decade now.21–30 Different extraction methods for the recovery of 137Cs have been reported in the literature based on these crown based systems.31–33 Calix crown ether have been introduced as a strong extracting agent for the alkali/alkaline earth metal cations, because of the presence of suitable pre-organised cavity in the structural frameworks of these ligands.34–37 The complexation selectivity for the metal ions by these ligands depends largely on the conformation adopted by the rigidified calix[4]arene moieties of these molecules. The partial cone isomer of 1-3-dimethoxy-p-terbutylcalix[4]arene-crown-5 has been established for the selective separation of potassium ion.38 Similarly, Casnati et al.25 synthesized and reported another calix-crown based ligand, namely 1,3-di-octyloxycalix[4]arene-crown-6 (CC6) which was found to be highly Cs selective. Further, Sharma et al.39 optimized the composition of the diluent system for the organic phase based on dodecane and iso-decyl alcohol to efficiently extract Cs+ using 0.03 M CC6 and obtained the DCs value as ∼6.6.
Understanding the mechanistic aspects of the metal extraction processes is very essential in the nuclear waste management program. In this respect, the density functional theory (DFT) method is currently widely used40,41 which is an appropriate level of calculation can comprehensively provide molecular level insights of the experimentally obtained results on the selective extraction processes by different ligand systems. For instance, Ali and coworkers42 have reported an unusually high selectivity of Cs+ in a calix-crown based ligand using DFT calculations. Similarly, Boda et al. studied the selective binding of Cs+ over Na+ with a macrocyclic calix-bis-crown ether using DFT studies, whereby the calculated results correlate well with the experimental findings.43 Further, along with counter-ion influence, strong effect of different dielectric media on the solvent extraction process has also been reported in the literature based on the DFT calculations and experimental studies.
Apparently, besides the intrinsic metal binding property of the ligands, solvent polarity of the organic phase also plays a key role in the efficient extraction of the radionuclides from a solution. Thus, choice of proper diluent system for the ligand of consideration in the organic phase is of paramount importance to achieve the maximum efficiency in the extraction process. Moreover, from the environmental point of view, the chosen solvent/diluent system is required to be nontoxic and also should not produce any undesirable bi-products in the overall process. For instance, taking note to their toxicity, use of nitrobenzene and 1,2-dichloroethane is usually avoided in the back end nuclear fuel cycle.
Based on these understandings in the reported literature,25,38,39 in the present study we have explored the experimental conditions, especially through the use of varying dielectric media as the diluents for the ligand in the organic phase, aiming to achieve significantly enhanced distribution ratio (DCs) for the Cs extraction, employing the well-known CC6 ligand as the complexing agent. We have analysed the influences of several solvent systems of varying dielectric properties on the DCs values for Cs extraction using CC6 as the extractant. Based on our study, n-octanol is projected to be an appropriate dielectric medium for CC6 to provide maximum Cs extraction efficiency. Effects of different mineral acids and interference of relevant alkali and alkaline earth metal ions (Na+, Mg2+, Sr2+) on the recovery of Cs+ have also been systematically investigated. Furthermore, to decipher the structures, binding motifs and energetics of the metal–CC6 complexes formed under different experimental conditions, the DFT based calculations have also been carried out to get a better insight of the studied systems.
2. Experimental
2.1 Materials
CC6 ligand was synthesized following the procedure reported by Casnati et al.,25 which was further purified and characterized in-house. Casnati et al. reported25 1,3-dialkoxycalix[4]arene-crown-6 ionophores are obtained in the fixed 1,3-alternate conformation in 63–85% yield, hence further purifications were carried out to avoid the side product interference to the reaction medium. At the same time to avoid the acidic impurity and to reduce the background noise. Chemical formula and geometry optimized structure of CC6 ligand are shown Fig. 1. The radiotracer 137Cs was used in the present investigation and procured from Board of Radiation and Isotope Technology (BRIT), India. 137Cs radiotracer was used throughout the experiment for evaluating extraction efficiency. During the experiment proper personal protective equipment (PPE) and personal radiation monitoring instruments were used. Here, experiments were carried out with a suitable quantity of 137Cs spiked into HNO3 medium. We kept the solution activity less than 1 mCi L−1 as well as gamma radiation field (as 137Cs is a gamma emitter) less than 1 mR h−1. The solvent n-octanol (99.9%) was purchased from S.D. Fine Chemicals, India. Supra-pure HNO3 and quartz double distilled water were used throughout the present investigation.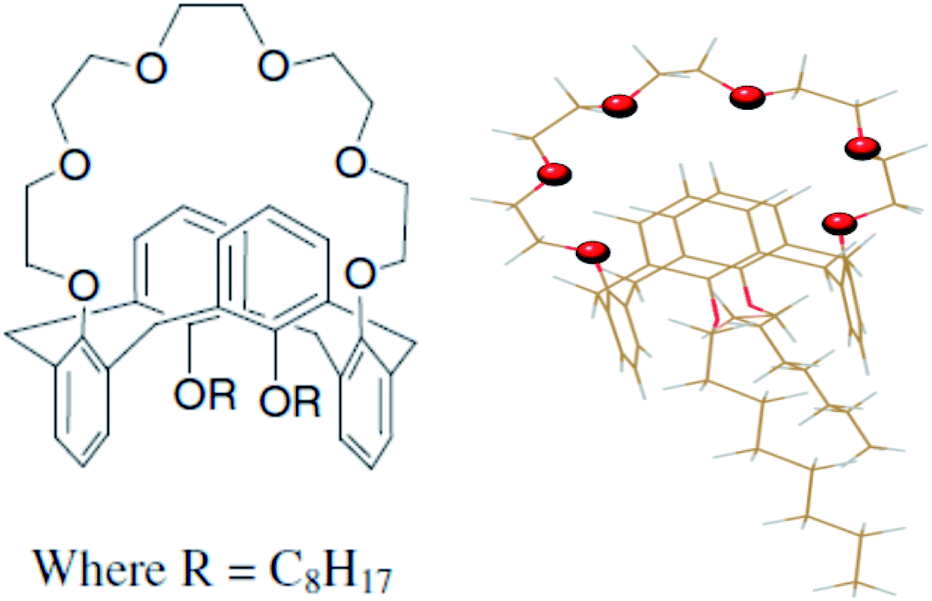 | ||
| Fig. 1 Chemical formula (left panel) and geometry optimized structure (right panel) of 1,3-dioctyloxycalix[4]arene-crown-6 (CC6). In the optimized structure, the oxygen atoms of the crown cavity are presented in ball and stick model, other atoms are presented in wire frame (color code: C = tan; O = red; H = light grey). | ||
2.2 Distribution ratio measurement
To obtain the distribution ratio (DM), equal volume of organic phase containing CC6 in an appropriate solvent system (diluent) and aqueous phase containing 137Cs tracer was equilibrated for 20 min. Then both the phases were separated by centrifugation. DM of the radioactive metal ions was calculated as the ratio of the concentration (or radioactivity) of the radioactive metal ions in organic phase to that in aqueous phase. All the extraction experiments were carried out at the temperature of 25 ± 1 °C using the thermostated water bath. The activity of 137Cs was determined by the gamma counting having an intrinsic Ge detector connected to 4 K MCA at 604.7 keV.2.3 Computational protocol
All structures were geometrically optimized in the gas phase using a medium sized def2-SV(P) basis set44 with pure GGA functional BP86.45,46 Tight convergence criteria (10−8 hartree) were applied in all the calculations. Further, analytical frequency calculations were performed using the AOFORCE module as implemented in TURBOMOLE v6.6 (ref. 47) to verify the obtained structures at the corresponding minima of the potential energy surfaces. Most of the optimized structures were found to be free from any imaginary frequencies. In case of any imaginary frequency, structures were distorted using screwer utility and further structural optimizations were carried out. Finally, single point calculations were performed with hybrid B3LYP functional48,49 using large triple ζ-quality (TZVP) basis set50,51 in the presence of different solvents (dodecane, n-octanol and water) using ORCA 3.0.3 software package.52 Core electrons of Cs+ (replacing 46 electrons in core) and Sr2+ (replacing 28 electrons in core) were modelled using SD-ECP pseudo potential. Solvent effects were incorporated into the single point calculations via implicit COSMO solvation model.53 Dielectric constant values (ε) of 2, 10.3 and 78 were taken for dodecane, n-octanol and water, respectively. In all optimizations as well as in single point calculations, Resolution of Identity (RI/RIJCOSX) approximation was invoked in conjugation with auxiliary basis set, in order to improve the speed of the calculation process. The above methodology was earlier reported to produce good correlation with the experimental results.54–56Finally, binding free energy of the metal binding reactions was calculated using following reaction scheme:
| CC6 (org) + M(H2O)nm+ (aq) = [CC6·M]m+ (org) + (H2O)n (aq) |
Furthermore, the effect of nitrate coordination on the Cs+ binding affinity of CC6 ligand was calculated using the following reaction scheme.
| CC6 (org) + Cs(H2O)6+ (aq) + NO3(H2O)6− (aq) = [Cs·NO3·CC6] (org) + 2(H2O)6 (aq) |
Mülliken population analysis (MPA)61 was also performed to quantify the ligand to metal charge transfer occurred in the complexes formed.
3. Results and discussion
The key idea for the effective separation of a metal ion into an organic phase through solvent extraction is the screening of the charge on the metal ion through suitable complexation followed by the mass transfer into the nonpolar organic phase. This complexation ability of a metal ion highly depends on its chemical potential. The chemical potential is nothing but charge to size ratio. More the charge of the metal ion, higher would be the chemical potential and higher would be the chance of getting coordinated by ligating atoms. Similarly, larger the size of metal ion, smaller would be the chemical potential and hence lesser would be the complexation ability. Cs+ is a mono positive charge metal ion with large size and hence lower is the chemical potential and consequently lower is the chance of getting coordinated. Therefore, for the mass transfer of Cs+ (and also Na+, K+ etc.) an alternative approach has been followed. A size selective pocket was created, where the Cs+ can comfortably sit. In that molecular cavity the metal ions fit by size selectivity and then get coordinated. In crown ether, the ‘O’ donors were the main source of coordination. The substitution in crown ether was utilized to fine tune the cavity size so that the metal ion binding selectivity can improve.3.1 Effect of diluent phase composition on DCs
To assess the role of different diluents for CC6 in the organic phase on the extraction of Cs+ ion,62 corresponding DCs values were determined using different solvent systems. The choice of solvents was made based on prior reports.63 To start with, DCs values were determined in different iso-decyl alcohol and dodecane mixtures by varying their volume fractions but keeping the CC6 concentration constant at 0.030 M. The typical DCs value as obtained in the 50:50 mixture of iso-decyl alcohol and dodecane (DCs = 7.43) was interestingly found to be very similar to that reported by Sharma et al.39 Noticeably, with the increasing polarity of the mixed solvent systems (i.e. with higher concentration of iso-decayl alcohol), the DCs value was noted to be increased systematically (Table 1). This observation prompted us to use a more polar environmentally safe n-octanol medium as the other diluent for the extraction of Cs by CC6 ligand. Indeed, we observed very high DCs value (DCs = 21.79) with n-octanol as the solvent using 0.03 M CC6 ligand in the organic phase. Accordingly, all our subsequent studies were carried out in 100% n-octanol medium only, to understand the effect of varying concentration of CC6 or the effect of changing other experimental conditions.
| Serial no. | Solvent composition | DCs |
|---|---|---|
| 1 | 50% Iso-decyl alcohol + 50% dodecane | 7.43 |
| 2 | 75% Iso-decyl alcohol + 25% dodecane | 12.31 |
| 3 | 100% Iso-decyl alcohol | 19.69 |
| 4 | 100% n-Octanol | 21.79 |
3.2 Dependence of DCs on the nature of different acids (HNO3, HCl, HCIO4)
The extraction profiles for Cs+ as a function of the concentration of different acids (HNO3, HCl, HClO4) were established by determining the DCs values using 0.015 M CC6 + 100% n-octanol in the solution [Fig. 2]. The obtained DCs values followed the trend HNO3 > HCIO4 > HCl for the 3–4 M acidity range, which primarily suggest that the ‘ion-pair’ binding mechanism might be involved in the extraction process at this acidity range. In the presence of HCl, the extraction efficiency of Cs+ for CC6 ligand is found to be the least, possibly because of the lower proficiency of the Cs+–Cl− ion-pair formation, understandably due to relatively higher hydration energy for the Cl− ion.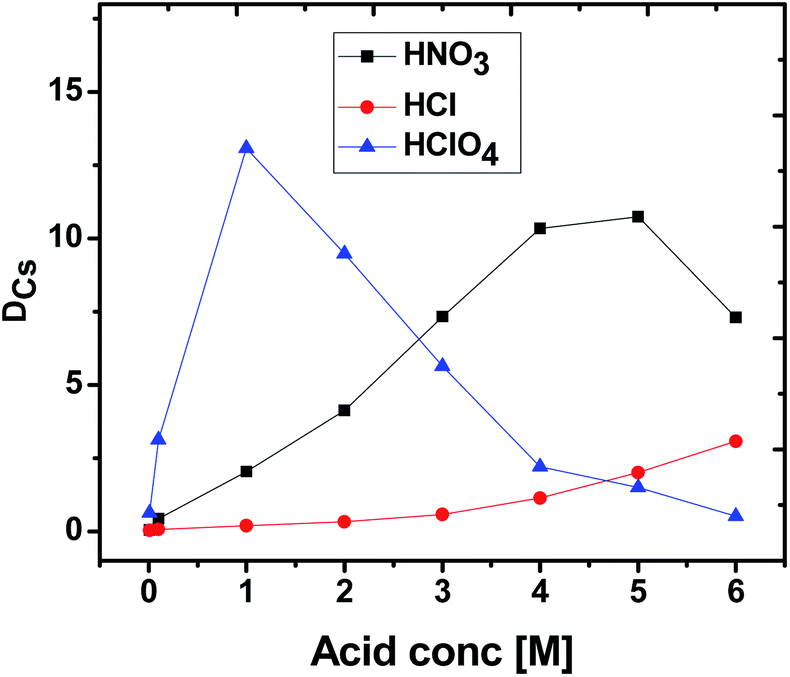 | ||
| Fig. 2 Extraction dependency of cesium on the different acids (HNO3, HCl, HCIO4) concentration. Organic phase: 0.015 M CC6 in 100% n-octanol; aqueous phase: different concentration of acids (HNO3, HCl, HCIO4) spiked with 137Cs metal ions (triplicate measurements were done and the error is within ±5%). | ||
In case of HNO3, with an increase in the feed acidity, the DCs value was found to increase gradually, attending a kind of maximum in the concentration region of 4–5 M and subsequently reducing to a small extent. The initial increase in DCs values with feed HNO3 acidity possibly revealed the gradual requirement of the NO3− ions for the formation of the Cs+–NO3− ion-pair complexes. At very high concentration of HNO3, i.e. beyond about 5 M, the DCs value was decreased marginally, suggesting the competing reactions of H+ and Cs+ for the ion-pair formations with NO3− ions, which would become more prevalent at such high HNO3 concentrations. It is thus suggested that at very high feed HNO3 acidity (>5 M), the availability of large concentration of H+ ions compete largely with the Cs+ ions causing a decrease in the observed DCs value at the higher acid concentrations.
3.3 Determination of metal–ligand binding stoichiometry
In the presence of nitrate, reaction of Cs+ with CC6 can be expressed via following reaction scheme.| Csaq+ + nCC6org + NO3aq− = [Cs·(CC6)n]+[NO3]aq− | (1) |
Thus, the equilibrium constant or the extraction constant (kex) for Cs+ by CC6 can be expressed as,
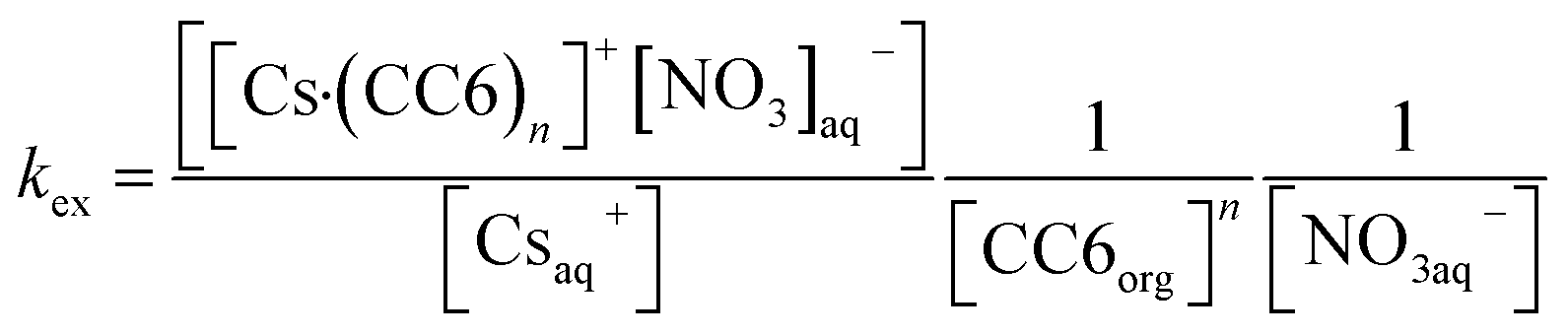 | (2) |
At a particular temperature and at a given HNO3 concentration, nitrate ion concentration in the aqueous phase is constant. Therefore, eqn (2) can be simplified as
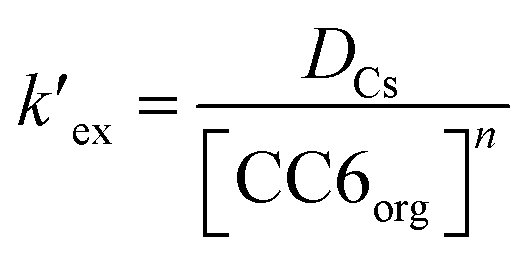 | (3) |
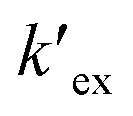 is the conditional extraction constant given as “kex[NO3−]” and DCs is the distribution ratio of the Cs+ ion, i.e. DCs = [Cs+]org/[Cs+]aq. By taking logarithm on both the sides of eqn (3), as given by eqn (4), we can estimate the
is the conditional extraction constant given as “kex[NO3−]” and DCs is the distribution ratio of the Cs+ ion, i.e. DCs = [Cs+]org/[Cs+]aq. By taking logarithm on both the sides of eqn (3), as given by eqn (4), we can estimate the 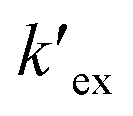 value form the intercept of logDCs versus log[CC6]org plot and subsequently we can evaluate the corresponding free energy change of the reaction (ΔG) using eqn (5).
value form the intercept of logDCs versus log[CC6]org plot and subsequently we can evaluate the corresponding free energy change of the reaction (ΔG) using eqn (5).
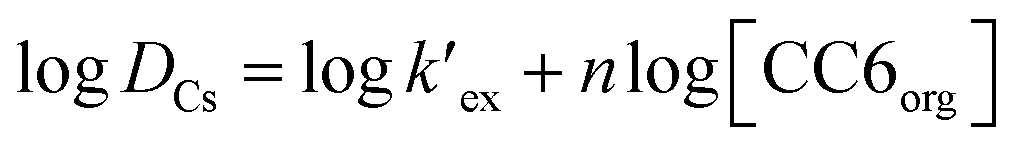 | (4) |
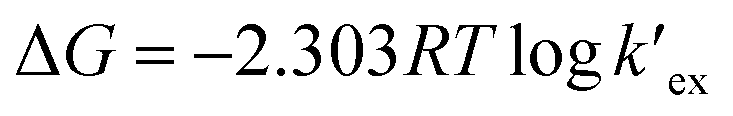 | (5) |
Plot of logDCs against logCC6 (shown in Fig. 3) gave a straight line with a slope of 0.998 ± 0.014 which indicate the formation of 1:1 metal–ligand complex. From the intercept, the conditional extraction constant 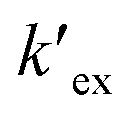 and the change in Gibb's free energy change ΔG have been evaluated as 7.5 × 102 and −3.93 kcal mol−1, respectively. The negative ΔG value revealed that the extraction process is a thermodynamically favourable spontaneous process.
and the change in Gibb's free energy change ΔG have been evaluated as 7.5 × 102 and −3.93 kcal mol−1, respectively. The negative ΔG value revealed that the extraction process is a thermodynamically favourable spontaneous process.
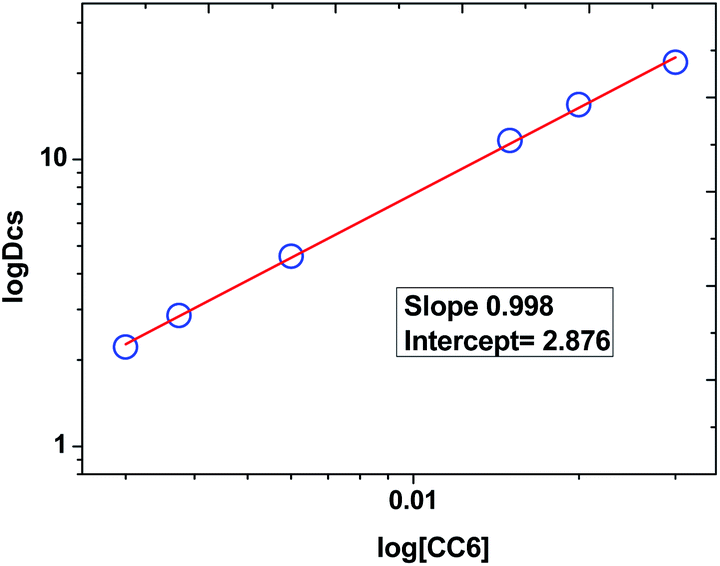 | ||
| Fig. 3 Variation of DCs as a function of CC6 concentration. Organic phase: (0.003–0.03) M·CC6 + 100% n-octanol; aqueous phase: 5 M HNO3 spiked with 137Cs metal ions. | ||
3.4 Interference of Na+, Mg2+ and Sr2+ ions on the extraction efficiency of Cs+
While processing a radioactive waste by an extractant in a solvent system, it is expected to encounter large amounts of other metal ions, especially, Na+, Mg2+ and Sr2+. Therefore, it is required to have a systematic study on the interfering effects of these metal ions in the aqueous phase on the extraction property of Cs+ ions by the extractant used. Fig. 4 demonstrates the results on the interferences of Na+, Mg2+ and Sr2+ ions on the DCs values of Cs+ extraction by using CC6 ligand in 100% n-octanol medium. Experimental data revealed that there was very minor interference of these ions on the extraction behaviour of CC6 for Cs+ ions. Insignificant interferences of Na+, Mg2+ and Sr2+ concentrations on the extraction property of CC6 for Cs+ was also observed in the corresponding extraction profiles.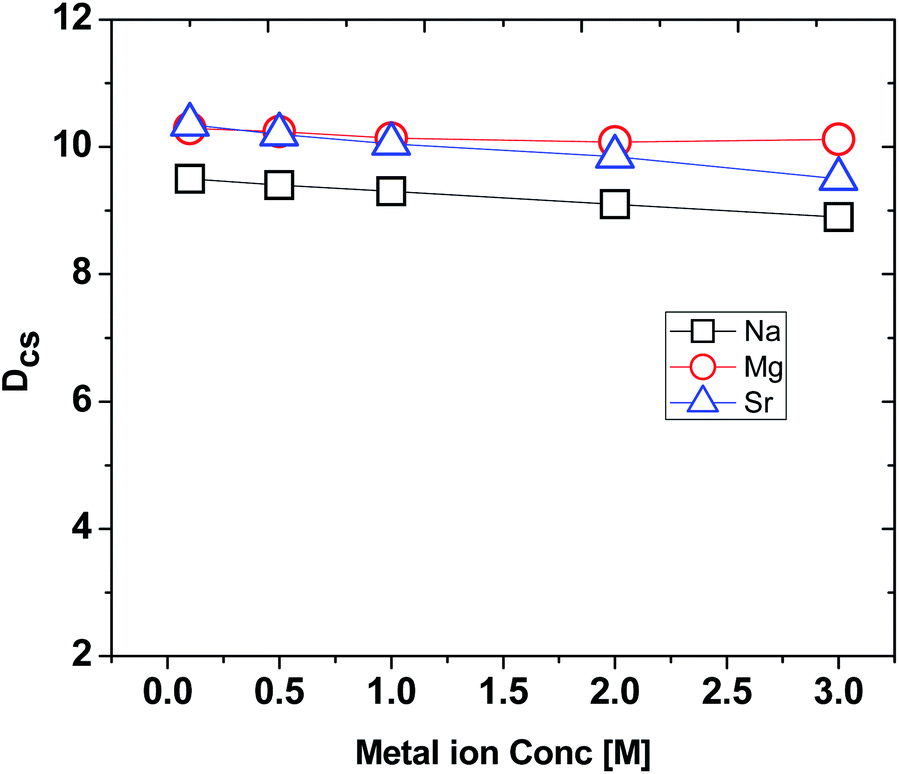 | ||
| Fig. 4 Dependence of the distribution ratio of Cs+ ion (DCs) on the interfering metal ion concentrations. Organic phase: 0.015 M CC6 in 100% n-octanol; aqueous phase: different concentration of metal ions (Na, Mg, Sr) from (0.1–3.0) M in 5 M HNO3 spiked with 137Cs metal ions. (Triplicate measurements were done and the error is within ±5%.) | ||
3.5 Computational results
In order to investigate the reason behind the superior selectivity of Cs+ ion by CC6, DFT calculations were performed. Structural and thermodynamic parameters for the complexes formed by CC6 with various metal ions (M·CC6) are listed in Table 2. According to these results, we find the highest binding free energy for Cs+ (ΔG = −15.11 kcal mol−1). The Cs+ ion is seen to perfectly fit into the crown cavity to interact with all the six oxygen atoms of the crown ring through strong electrostatic interaction (Cs–OCC6: 3.225–3.381 Å). Further, the Cs–Carene bond distance (ortho carbon of benzene) of 3.443 Å and 3.470 Å probably indicate the presence of weak cation–π interaction between the Cs+ ion and the aromatic ring of the calix[4]arene moiety.| Complex | M–OCC6 (Å) | M–Careneb (Å) | ΔH | TΔS | ΔG |
|---|---|---|---|---|---|
| a Values in bracket correspond to dodecane solvent (dielectric constant = 2).b Distance between Cs+ and ortho-carbon of aromatic moiety. | |||||
| Na+·CC6 | 2.430, 2.447, 3.537, 3.613, 4.331, 4.346 | 2.770, 2.914 | −4.62 (+45.76) | −6.08 | +1.46 (+12.46) |
| Mg2+·CC6 | 2.083, 2.114, 2.163, 2.215, 3.679, 4.062 | 2.408, 5.604 | +31.24 (+86.57) | −3.58 | +34.83 (+90.16) |
| Sr2+·CC6 | 2.719, 2.628, 2.650, 2.698, 2.857, 4.869 | 3.119, 3.262 | −5.34 (+6.38) | −12.71 | +7.36 (+58.47) |
| Cs+·CC6 | 3.225, 3.244, 3.283, 3.315, 3.325, 3.381 | 3.443, 3.470 | −17.56 (−6.53) | −2.45 | −15.11 (−4.07) |
| Cs+·NO3−·CC6 | 3.151, 3.185, 3.332, 3.335, 3.502, 3.585 | 3.366, 3.479 | −22.21 (−18.44) | +0.75 | −22.97 (−19.19) |
On the other hand, due to its smaller size, Na+ is seen to interact only with two oxygen atoms of the crown ring (2.430 Å, 2.447 Å), which subsequently resulted in a positive value for the free energy change. Owing to the high ionic potentials, Mg2+ and Sr2+ ions would have attracted stronger electrostatic interaction with the crown cavity as compared to Cs+ ions (see ESI, Fig. S1†). However, smaller ionic size allowed them to interact only with 3–4 oxygen atoms of the crown moiety, resulting in positive values for the free energy changes. Notably, formations of the metal·CC6 complexes require removal of the primary hydration shells of the metal ions. Thus, least hydration energy of Cs+ facilitates its easy migration from aqueous phase to the organic phase. On the contrary, higher hydration free energy values of the divalent metal ions made their transfer from the aqueous to organic phase thermodynamically unfavorable. Partial charges derived from the Mülliken population analysis (MPA) depicted more ligand to metal charge transfer for divalent metal ions as compared to the monovalent metal ions (see ESI, Table S1†). Hence the MPA results failed to produce the actual ionic selectivity trend observed experimentally, suggesting that the metal–ligand charge transfer does not contribute much in the selectivity displayed by the host for the metal·CC6 complex formations (Fig. 5).
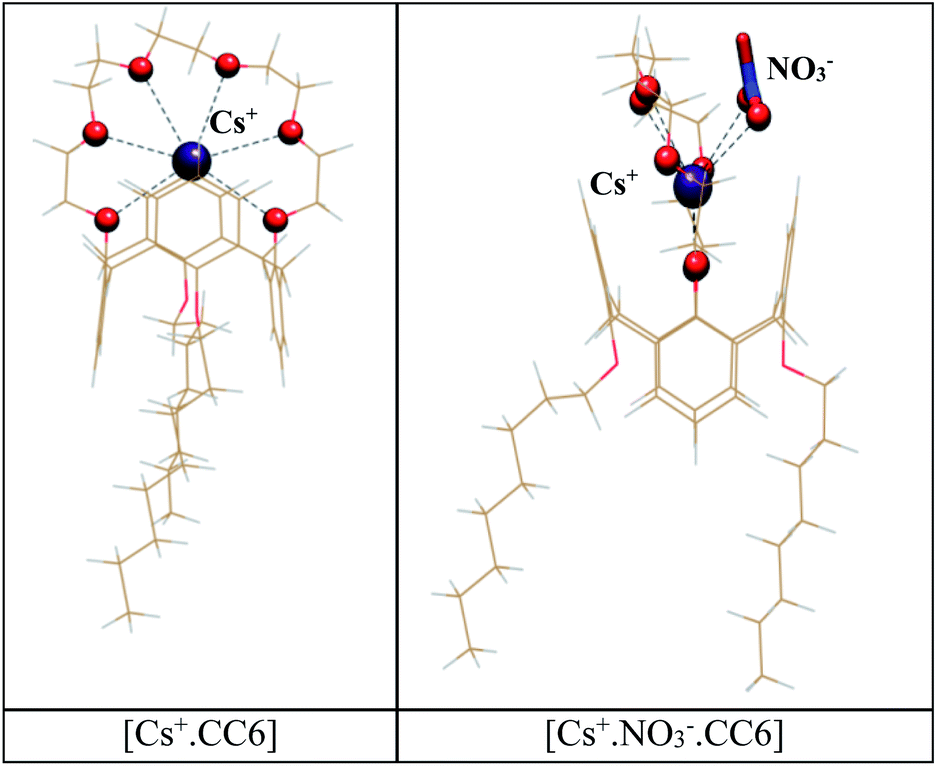 | ||
| Fig. 5 Optimized structures of [Cs+·CC6] complex in the absence and presence of nitrate counter ion. Cs+ coordinated oxygen atoms are presented in ball and stick model; other atoms are presented in wire frame. | ||
Upon complexation, the ligand is also required to reorganize itself in order to capture the metal ion of interest. The energy involved in this reorganize process is known as the re-organization energy of the ligand. In this regard, we also calculated the reorganization energy associated with all the metal·CC6 complexes, as listed in Table S2.†
Expectedly, the highest reorganization energy is noted for Mg2+ (>+34 kcal mol−1), which is followed by Sr2+ (>+22 kcal mol−1) and Na+ (>+6 kcal mol−1) ions. In line with the computed free energy trend, least reorganization energy is found for Cs+ ion. These results reiterate the importance of the size-fit concept of this ligand, suggesting that the divalent metal ions do not fit properly into the binding site of CC6 and hence led to a large deformation of the host molecule and accordingly less efficient binding of these ions as compared to Cs+ ion. Molecular descriptor like HOMO–LUMO gap (HOMO = highest occupied molecular orbital; LUMO = lowest unoccupied molecular orbital) provide information on the stability of the formed metal·CC6 complexes. In line with the experimental selectivity trend, we have also noted the highest HOMO–LUMO gap for Cs+ (−5.54 eV), which confirms the kinetic stability of the Cs+·CC6 complex.
Our experimentally obtained metal–ligand stoichiometry for Cs+·CC6 complex suggests the presence of NO3− in the periphery of Cs+ion to form 1:1 ion-pair complex. As reported earlier,64,65 the presence of counter anion near metal ion in host–guest complexes is expected to provide additional assistance for the metal binding through cooperative effect. In this regard, a set of calculations were also carried out in the presence of NO3− ion at the outer sphere of crown cavity. Here, apart from the coordination with the oxygen atoms of crown ring (Cs–Ocrown: 3.151–3.585 Å), the Cs+ ion is also noticed to coordinate with the oxygen atoms of NO3− ion using a bidentate binding motif (Cs–Onitrate: 3.077–3.147 Å). This resulted in the more negative binding free energy for the Cs+·NO3−·CC6 complex (ΔG = −22.97 kcal mol−1), confirming the formation of the Cs+–NO3− ion-pair complex in the studied system which is transferred favourably from the aqueous phase to the organic phase. To probe the effect of low dielectric media as a solvent in the concerned metal extraction process, we also performed a set of calculations with dodecane solvent (ε = 2). As evident from Table 2, the binding free energy values for all the metal·CC6 complexes are noted to decrease significantly upon decreasing the dielectric constant of the solvent. This is in excellent correlation with our experimental finding (Table 1). Possibly, changing the diluent with a more polar solvent, such as n-octanol, as used in our present study, helps to satisfy the primary coordination shell of Cs+ in the process of aqueous to organic phase transfer, facilitating the extraction of the metal ion by the CC6 ligand used in the present study.
3.6 Comparative evaluation of Cs+ extraction with crown ether
Table 3 is summarizing the results for Cs+ extraction by different crown ether in different solvents that were reported in literature and their comparison with the present investigation. In all the cases they were reported to form 1:1 metal–ligand complex and the size selectivity was the main driving force for the mass transfer of metal ions to the organic phase. The maximum D value achieved in the present case was found to be significantly higher for all the solvent systems reported in the Table 3. However, for p-tertiary-butylsulfonylcalix[4]arene in chloroform the D value was reported to be 25, which was almost similar to that obtained in present investigation. The crown 6 ethers and its derivatives were found to form an appropriate size for Cs+ ion cooperation and hence were explored extensively. The effect of diluent on Cs+ extraction using calix[4]arene-bis(crown 6) ether and calix[4]arene-bis(naphthocrown-6) ether revealed that the extraction efficiency followed the order: nitrobenzene > 1,2-dichloromethane > 1-octanol > 2-nitrophenyl octyl ether > chloroform > toluene. Such diluent dependent Cs+ extraction was also reported using bis-(octyloxy)calix[4]arene-mono-crown-6 ether. They have reported that, the D values followed the trend: NPOE > PTMS > nitrobenzene > MIBK > 1-octanol. Almost no extraction was reported for using chloroform as diluent. The di-tertiary-butylbicylo-18-crown-6 ether in nitrobenzene was reported to achieve the maximum D value of 5.2 for Cs+ ion. This comparative evaluation definitely indicated that, 1,3-dioctyloxycalix[4]arene-crown-6 ether in n-octanol (present case) was a more efficient solvent system for extraction of Cs+.
| Ligand | Solvent composition | Metal:ligand ratio |
DCs | Ref. |
|---|---|---|---|---|
| 1,3-Dioctyloxycalix[4]arene-crown-6 (CC6) | n-Octanol | 1:1 |
21.67 | Present work |
| 1,3-Dioctyloxycalix[4]arene-crown-6 (CC6) | Isodecyl alcohol/n-dodecane | 1:1 |
6.6 | 36 |
| Calix[4]arene-bis(crown-6) | Nitrobenzene | 1:1 |
5.20 | 63 |
| Calix[4]arene-bis(crown-6) | 1,2-Dichloroethane | 1:1 |
3.90 | |
| Calix[4]arene-bis(crown-6) | 1-Octanol | 1:1 |
2.9 | |
| Calix[4]arene-bis(crown-6) | Chloroform | 1:1 |
0.62 | |
| Calix[4]arene-bis(crown-6) | Toluene | 1:1 |
0.30 | |
| Calix[4]arene-bis(crown-6) | 2-Nitrophenyl octyl ether | 1:1 |
1.01 | |
| Calix[4]arene-bis(napthocrown-6) | Nitrobenzene | 1:1 |
7.71 | |
| Calix[4]arene-bis(napthocrown-6) | 1,2-Dichloroethane | 1:1 |
2.62 | |
| Calix[4]arene-bis(napthocrown-6) | 1-Octanol | 1:1 |
0.15 | |
| Calix[4]arene-bis(napthocrown-6) | Chloroform | 1:1 |
0.09 | |
| Calix[4]arene-bis(napthocrown-6) | Toluene | 1:1 |
0.02 | |
| Calix[4]arene-bis(napthocrown-6) | 2-Nitrophenyl octyl ether | 1:1 |
3.78 | |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | Nitrobenzene | 1:1 |
0.91 | 66 |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | NPOE | 1:1 |
1.42 | |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | 1-Octanol | 1:1 |
0.37 | |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | MIBK | 1:1 |
0.74 | |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | Chloroform | 1:1 |
0.05 | |
| Bis-(octyloxy)calix[4]arene-mono-crown-6 | PTMS | 1:1 |
1.04 | |
| p-tert-Butylsulfonylcalix[4]arene (SC4A) | Chloroform | 1:1 |
25 | 67 |
| DTBuB18C6 | Nitrobenzene | 1:1 |
5.2 | 68 |
4. Conclusions
In summary, we have investigated here the effect of different dielectric media on the selective cesium extraction behaviour of CC6 ligand. We report that 100% n-octanol can be used to enhance the distribution ratio (DCs) of Cs+ significantly as compared to iso-decyl alcohol–dodecane based mixed solvent systems used previously. Highly polar solvents tend to satisfy the primary coordination shell of Cs+ ion and thus facilitate efficient migration of the ion from aqueous to the organic phase. The effect of interfering cations (Na+, Mg2+ and Sr2+) was noted to be insignificant in Cs+ extraction by CC6. Metal–ligand stoichiometry analysis predicts the formation of the ion-pair complex with the involvement of the nitrate counter anion, which is further supported by the results from DFT calculation. Theoretical investigations on the structures and binding energies further confirm the thermodynamic and kinetic stability of the Cs+·CC6 complex and also point out the importance of the metal hydration energy, the reorganization energy of the ligand and the size fit concept to operate in a synergistic manner to provide the Cs+ selectivity inside the cavity of the CC6 host molecule.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr H. Pal, former AD, Chemistry Group, BARC, for his constant encouragement and critically reviewing the present manuscript. We also would like to acknowledge Dr. M. S. Kulkarni. Head HPD; Dr. P. K. Pujari, Director RC & IG and Head RCD; Dr R. Acharya, Head, Actinide Spectroscopy Section, RCD, BARC; U. Dani, GM, INRPO (R&WM), BARC.References
- D. H. Meikrantz, T. A. Todd, C. L. Riddle, J. D. Law, D. R. Peterman, B. J. Mincher, C. A. McGrath, J. D. Baker, US Pat., 10/808039, 2004 Search PubMed.
- Radioactive Waste Management and Disposal, ed. L. Cecille, Elsevier Science Publisher, London, New York, 1991 Search PubMed.
- X. Chao, J. Wang and J. Chen, Solvent Extr. Ion Exch., 2012, 30, 623–650 CrossRef.
- Actinides and fission products partitioning and transmutation, in Proceedings of the Fifth International Information Exchange Meeting, Mol, Belgium, 2527 November. 1998, NEA/OECD, 1999 Search PubMed.
- H. Zhou, K. E. Connery, R. A. Bartsch, B. A. Moyer, T. J. Haverlock and L. H. Delmau, Solvent Extr. Ion Exch., 2013, 31, 683–696 CrossRef CAS.
- V. Kumar, J. N. Sharma, P. V. Achuthan and R. C. Hubli, RSC Adv., 2014, 4, 805–810 RSC.
- V. Kumar, J. N. Sharma, P. V. Achuthan and R. C. Hubli, J. Radioanal. Nucl. Chem., 2014, 299, 1547–1553 CrossRef CAS.
- W. R. Wilmarth, G. J. Lumetta, M. E. Johnson, M. R. Poirier, M. C. Thompson, P. C. Suggs and N. P. Machara, Solvent Extr. Ion Exch., 2011, 29, 1–4 CrossRef CAS.
- W. W. Schulz and L. A. Bray, Sep. Sci. Technol., 1987, 22, 191–214 CrossRef CAS.
- J. F. Dozol, M. Dozol and R. M. Macias, J. Inclusion Phenom. Macrocyclic Chem., 2000, 38, 1–22 CrossRef CAS.
- J. Wang and C. Chen, Biotechnol. Adv., 2009, 27, 195–226 CrossRef CAS PubMed.
- A. Mittal, M. Naushad, G. Sharma, Z. A. ALothman, S. M. Wabaidur and M. Alam, Desalin. Water Treat., 2016, 57, 21863–21869 CrossRef CAS.
- A. A. Alqadami, M. Naushad, Z. A. Alothman and A. A. Ghfar, ACS Appl. Mater. Interfaces, 2017, 9(41), 36026–36037 CrossRef CAS PubMed.
- A. A. Alqadami, M. Naushad, M. A. Abdalla, T. Ahamad, Z. A. ALOthman, S. M. Alsehri and A. A. Ghfar, J. Cleaner Prod., 2017, 156, 426–436 CrossRef.
- A. A. Alqadami, M. Naushad, Z. A. ALOthman, M. Alsuhybani and M. Algamdi, J. Hazard. Mater., 2020, 389, 121896–121909 CrossRef CAS PubMed.
- M. Naushad, T. Ahamad, Z. A. AlOthman and A. H. Al-Muhtaseb, J. Mol. Liq., 2019, 279, 1–8 CrossRef CAS.
- M. A. Khan, A. A. Alqadami, M. Otero, M. R. Siddiqui, Z. A. Alothman, I. Alsohaimi, M. Rafatullah and A. E. Hamedelniel, Chemosphere, 2019, 218, 1089–1099 CrossRef CAS PubMed.
- M. A. Khan, A. A. Alqadamia, S. M. Wabaidur, M. R. Siddiqui, B. Jeon, S. A. Alshareef, Z. A. Alothman and A. E. Hamedelniel, J. Hazard. Mater., 2020, 400, 123247–123259 CrossRef CAS PubMed.
- M. A. Khan, M. Otero, M. Kazi, A. A. Alqadami, S. M. Wabaidur, M. R. Siddiqui, Z. A. Alothman and S. Sumbul, J. Hazard. Mater., 2019, 365, 759–770 CrossRef CAS PubMed.
- Z. A. Alothman, A. H. Bahkali, M. A. Khiyami, S. M. Alfadul, S. M. Wabaidur, M. Alam and B. Z. Alfarhan, Sep. Sci. Technol., 2020, 55(10), 1766–1775 CrossRef CAS.
- V. Bohmer, Angew. Chem., Int. Ed., 1995, 34, 713 CrossRef.
- C. D. Gutsche, Calixarenes: An Introduction, The Royal Society of Chemistry, Cambridge, 2nd edn, 2008 Search PubMed.
- R. Ungaro, A. Casnati, F. Ugozzoli, A. Pochini, J. F. Dozol, C. Hill and H. Rouquette, Angew. Chem., Int. Ed. Engl., 1994, 33, 1506–1509 CrossRef.
- A. Casnati, S. Barboso, H. Rouquette, M. J. Schwing-Weill, F. Arnaud-Neu, J. F. Dozol and R. Ungaro, J. Am. Chem. Soc., 2001, 123, 12182–12190 CrossRef CAS PubMed.
- A. Casnati, A. Pochini, R. Ungaro, F. Ugozzoli, F. Arnaud, S. Fanni, M. J. Schwing, R. J. M. Egberink, F. de Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 2767–2777 CrossRef CAS.
- P. Thuéry, M. Nierlich, J. C. Bryan, V. Lamare, J. F. Dozol, Z. Asfari and J. Vicens, Dalton Trans., 1997, 4191–4202 RSC.
- Y. Deng, R. A. Sachleben and B. A. Moyer, J. Chem. Soc., Faraday Trans., 1995, 91, 4215–4222 RSC.
- R. A. Sachleben, P. V. Bonnesen, T. Descazeaud, T. J. Haverlock, A. Urvoas and B. A. Moyer, Solvent Extr. Ion Exch., 1999, 17, 1445–1459 CrossRef CAS.
- T. J. Haverlock, P. V. Bonnesen, R. A. Sachleben and B. A. Moyer, J. Inclusion Phenom. Macrocyclic Chem., 2000, 36, 21–37 CrossRef CAS.
- M. P. Wintergerst, T. G. Levitskaia, B. A. Moyer, J. L. Sessler and L. H. Delmau, J. Am. Chem. Soc., 2008, 130, 4129–4139 CrossRef CAS PubMed.
- A. Casnati, A. Pochini, R. Ungaro, F. Ugozzoli, F. Arnaud, S. Fanni, M. J. Schwing, R. J. M. Egberink, F. de Jong and D. N. Reinhoudt, J. Am. Chem. Soc., 1995, 117, 2767–2777 CrossRef CAS.
- W. W. Schulz and L. A. Bray, Sep. Sci. Technol., 1987, 22, 191–214 CrossRef CAS.
- R. L. Miller, A. B. Pinkerton, P. K. Hurlburt and K. D. Abney, Solvent Extr. Ion Exch., 1995, 13, 813–827 CrossRef CAS.
- L. H. Gerow, J. E. Smith and M. W. Davis, Sep. Sci. Technol., 1981, 16, 519 CrossRef.
- J. Vicens and V. Bhomer, Calixarenes, a versatile class of macrocyclic compounds, Kluwer Academic Publishers, Dordrecht, The Nerherlands, 1991 Search PubMed.
- Z. Asfari, C. Bressot, J. Vicens, C. Hill, J. F. Dozol, H. Rouquette, S. Eymard, V. Lamare and B. Tou-mois, Anal. Chem., 1995, 67, 3133–3139 CrossRef CAS.
- P. Thuery, M. Nierlich, V. Lamare, J. F. Dozol, Z. Asfari and J. Vicens, J. Inclusion Phenom., 2000, 36, 375–408 CrossRef CAS.
- R. Ungaro, A. Casnati, F. Ugozzoli, A. Pochini, J. F. Dozol, C. Hill and H. Rouquette, Angew. Chem., Int. Ed. Engl., 1994, 33, 1506–1509 CrossRef.
- J. N. Sharma, A. Kumar, V. Kumar, S. Pathan, C. Janardahan, V. Tessi and P. K. Wattal, Sep. Sci. Technol., 2014, 135, 176–182 CAS.
- B. Sadhu, M. Sundararajan, G. V. Elmurugan and P. Venuvanalingam, Dalton Trans., 2015, 44, 15450–15462 RSC.
- M. Sundararajan, V. Sinha, T. Bandyopadhyay and S. K. Ghosh, J. Phys. Chem. A, 2012, 116(17), 4388–4395 CrossRef CAS PubMed.
- V. Kumar, J. N. Sharma, P. V. Achuthan, D. K. Singh and S. M. Ali, RSC Adv., 2016, 6(52), 47120–47129 RSC.
- A. Boda and M. A. Sheikh, J. Phys. Chem. A, 2012, 116, 8615 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- R. Ahlrichs, M. Bar, H. P. Baron, R. Bauernschmitt, S. Bocker, M. Ehrig, K. Eichkorn, S. Elliot, F. Furche, F. Haase, M. Haser, H. Horn, C. Huber, U. Huniar, M. Kattannek, C. Kolmel, M. Koolwitz, K. May, C. Ochsenfeld, H. Ohm, A. Schafer, U. Schneider, O. Treutler, M. von Arnim, F. Weigend, P. Weis, and H. Weiss, TURBOMOLE V6.3.1 2011, A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from, http://www.turbomole.com Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, ORCA Version 3.0, an ab initio density functional and semiempirical program package, 2013 Search PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- B. Sadhu, M. Sundararajan, G. Velmurugan and P. Venuvanalingam, Dalton Trans., 2015, 44, 15450–15462 RSC.
- B. Sadhu, M. Sundararajan and T. Bandyopadhyay, Inorg. Chem., 2016, 55, 598–609 CrossRef CAS PubMed.
- B. G. Vats, D. Das, B. Sadhu, I. C. Pius, D. Noronha, S. Kannan and M. Sundararajan, Dalton Trans., 2016, 45, 10319–10325 RSC.
- A. Boda and M. A. Sheikh, J. Phys. Chem. A, 2012, 116, 8615 CrossRef CAS PubMed.
- B. Sadhu, M. Sundararajan and T. Bandyopadhyay, J. Phys. Chem. B, 2015, 119, 12783–12797 CrossRef CAS PubMed.
- B. Sadhu, M. Sundararajan and T. Bandyopadhyay, J. Phys. Chem. B, 2015, 119, 10989–10997 CrossRef CAS PubMed.
- M. Peschke, A. T. Blades and P. Kebarle, J. Phys. Chem. A, 1998, 102, 9978–9985 CrossRef CAS.
- R. S. Mülliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef.
- E. Makrlik, P. Selucký and P. Vaňura, Acta Chim. Slov., 2011, 58, 850 CAS.
- D. R. Raut, P. K. Mohapatra, S. A. Ansari, A. Sarkar and V. K. Manchandda, Desalination, 2008, 232, 262–271 CrossRef CAS.
- B. Sadhu, M. Sundararajan, G. Velmurugan and P. Venuvanalingam, Dalton Trans., 2015, 44, 15450–15462 RSC.
- S. K. Kim, G. I. Vargas-Zúñiga, B. P. Hay, N. J. Young, L. H. Delmau, C. Masselin, C. H. Lee, J. S. Kim, V. M. Lynch, B. A. Moyer and J. L. Sessler, J. Am. Chem. Soc., 2012, 134, 1782–1792 CrossRef CAS PubMed.
- D. R. Raut, P. K. Mohapatra, M. K. Choudhary and S. K. Nayak, J. Membr. Sci., 2013, 429, 197–205 CrossRef CAS.
- C. Liu, D. Zhang, L. Zhao, P. Zhang, X. Lu and S. He, Appl. Sci., 2016, 6, 212–219 CrossRef.
- P. K. Mohapatra, S. A. Ansari, A. Sarkar, A. Bhattacharyya and V. K. Manchanda, Anal. Chim. Acta, 2006, 571, 308–314 CrossRef CAS PubMed.
- B. Sadhu, M. Sundararajan, A. Pillai, R. Singh and T. Bandyopadhyay, Int. J. Quantum Chem., 2017, 117, e25418 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02661e |
| This journal is © The Royal Society of Chemistry 2021 |