
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemoval of per- and polyfluoroalkyl substances (PFAS) from water by ceric(IV) ammonium nitrate†
Jun Sun‡
a,
Sreenu Jennepalli‡a,
Matthew Leec,
Denis M. O'Carrollc,
Björn Åkermark b,
Michael J. Manefieldc,
Biswanath Das‡*ab and
Naresh Kumar*a
b,
Michael J. Manefieldc,
Biswanath Das‡*ab and
Naresh Kumar*a
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: n.kumar@unsw.edu.au; das.biswanath85@gmail.com
bDepartment of Organic Chemistry, Stockholm University, Stockholm 10691, Sweden
cSchool of Civil and Environmental Engineering, Water Research Centre, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 13th May 2021
Abstract
Ceric(IV) ammonium nitrate (CAN) in aqueous medium acts as an excellent precipitating agent for perfluorooctanesulfonic acid (PFOS). The Ce(IV) center plays a crucial role. Interestingly, Ce(III) chloride showed much less effectiveness under similar conditions. The efficacy of CAN was reduced upon changing the substrate to perfluorooctanoic acid (PFOA).
Per- and polyfluoroalkyl substances (PFAS) are toxic and xenobiotic compounds that have been widely used in fire extinguishers, carpet guards, paper, and non-stick cookware.1 However, these groups of organofluorines are highly resistant to degradation and are not easily separable by the usual water purification techniques.2–4 The environmental and health hazards caused by PFAS contamination in drinking water and ground water have stimulated research for innovative strategies to remove PFASs.5 An additional risk while developing these removal techniques is the toxicity of PFASs even at very low concentrations.6,7 Easily available chemicals that can efficiently degrade/precipitate out low concentrations of PFAS from water can provide a futuristic design to develop valuable water purification materials.
The large C–F bond dissociation energy (∼485 kJ mol−1) makes it resistant to oxidation and reduction, and together with simultaneous hydro- and oleophobicity of PFAS, make the overall degradation/separation process challenging.8,9 Methods to degrade/remove PFAS from water include in situ generated radicals, heat treatment, photocatalysis, or strong reducing or oxidizing agents.10–12 There are also important reports on PFAS remediation techniques using carbonaceous nanomaterials that are high surface area sorbents or In2O3 type, which possess oxygen vacancies in the monocrystalline structure.13–15
There are also some reports on the removal of PFASs directly by powdered-activated carbon (PAC) and granular-activated carbon (GAC) adsorption processes.16,17 The conventional coagulation processes (e.g., by iron chloride, alum etc.) are usually inefficient at low concentrations of PFAS. They are slow, produce a lot of sludge, and are usually expensive.18 A natural coagulant Moringa oleifera seeds has been reported to be a better alternative and produces less sludge.19 Nevertheless, the effectiveness at low concentration of PFAS remains as a challenge.
Herein, we report a new approach for precipitation of the commercially available PFOS (as potassium salt) from water (<20 μM) using ceric(IV) ammonium nitrate (CAN). This study was initiated from the perspective of generating in situ high valent/mixed valent oxide species/nanoparticles from FeCl2 and CoCl2 that can oxidatively degrade PFAS. In preliminary studies, CAN was used as an oxidant and resulted in significant reduction in the concentration of PFAS of the supernatant aqueous solution whenever CAN was used. Thereafter a range of concentrations of CAN (from 0.094 mM to 0.75 mM) were tested in consecutive experiments keeping PFOS concentration unchanged at 15 μM (Fig. S3†). After the treatment with 0.38 mM concentration CAN, the supernatant shows ∼80% removal of PFOS (Fig. 1, column 1 and 2). Very slow formation of an off-white precipitate was seen from the solution after seven days of undisturbed standing.
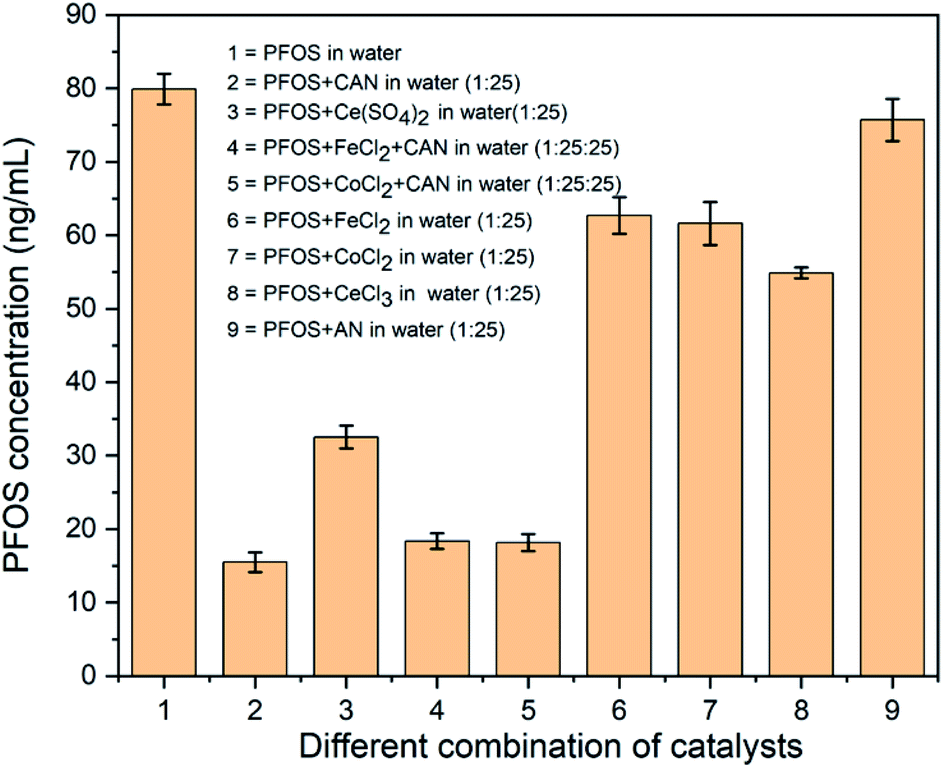 | ||
| Fig. 1 (1) The LC-MS detected concentrations of 15 μM of PFOS in water before treatment, (2) after treatment with 0.38 mM CAN ([(NH4)2CeIV(NO3)6]), (3) 0.38 mM CeIV(SO4)2, (4) 0.38 mM CAN + 0.38 mM FeCl2, (5) 0.38 mM CAN + 0.38 mM CoCl2, (6) 0.38 mM FeCl2, (7) 0.38 mM CoCl2, (8) 0.38 mM CeCl3, and (9) 0.38 mM AN (NH4NO3) (all experiments were repeated in triplicate and the dilution details are provided in the ESI†). | ||
Two 1st row transition metal salts [CoCl2 and FeCl2] and also a combination of these salts with CAN [CAN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CoCl2 (1:1 equivalent); CAN: FeCl2 (1:1 equivalent)] were tested under identical conditions. In all these cases, very little (∼15–20%) diminution of PFOS concentration (in LC-MS) was observed in the absence of CAN (Fig. 1, column 4–7). Since CAN itself was very reactive and either pure Fe and Co salts or a 1:1 mixture appeared to be much less effective, higher ratios of CAN:CoCl2 or CAN:FeCl2 were not tested further.§¶ The lower efficacy of these 1:1 combinations can be explained by possible partial use of CAN to oxidize Fe(II) and Co(II).20
:CoCl2 (1:1 equivalent); CAN: FeCl2 (1:1 equivalent)] were tested under identical conditions. In all these cases, very little (∼15–20%) diminution of PFOS concentration (in LC-MS) was observed in the absence of CAN (Fig. 1, column 4–7). Since CAN itself was very reactive and either pure Fe and Co salts or a 1:1 mixture appeared to be much less effective, higher ratios of CAN:CoCl2 or CAN:FeCl2 were not tested further.§¶ The lower efficacy of these 1:1 combinations can be explained by possible partial use of CAN to oxidize Fe(II) and Co(II).20
To check whether CeIV is playing a crucial role, another CeIV containing commercially available and strong oxidant, CeIV(SO4)2 (0.38 mM) was tested and showed ∼60% disappearance of PFOS under identical conditions (Fig. 1, column 1 and 3). To find if this is a general trend for any oxidation state of cerium, the impact of CeIIICl3 (commercially available) was also investigated. It clearly showed significantly lower efficiency in removing PFOS (at best ∼33%) (Fig. 1 and column 8) from the aqueous medium. No precipitate was found even after two weeks of undisturbed standing. Observing the noteworthy reactivity difference between CAN and CeCl3, another set of experiments was performed using 0.38 mM of ammonium nitrate (AN) (Fig. 1, column 9) and 15 μM of PFOS. Almost no change (<5%) in PFOS concentration indicates strongly towards the significance of the Ce(IV) center.
In order to obtain clarity on the possible species that are precipitating out from the CAN–PFOS interaction, higher concentrations were used. LC-MS analysis indicated that CAN is more effective at higher concentration and complete removal of PFOS (1.5 mM) is possible at around 18.75 mM concentration of CAN (by LC-MS) (Fig. 2) within 3 minutes of continuous stirring after mixing. An off-white precipitate formed over couple of days after mixing CAN and PFOS.
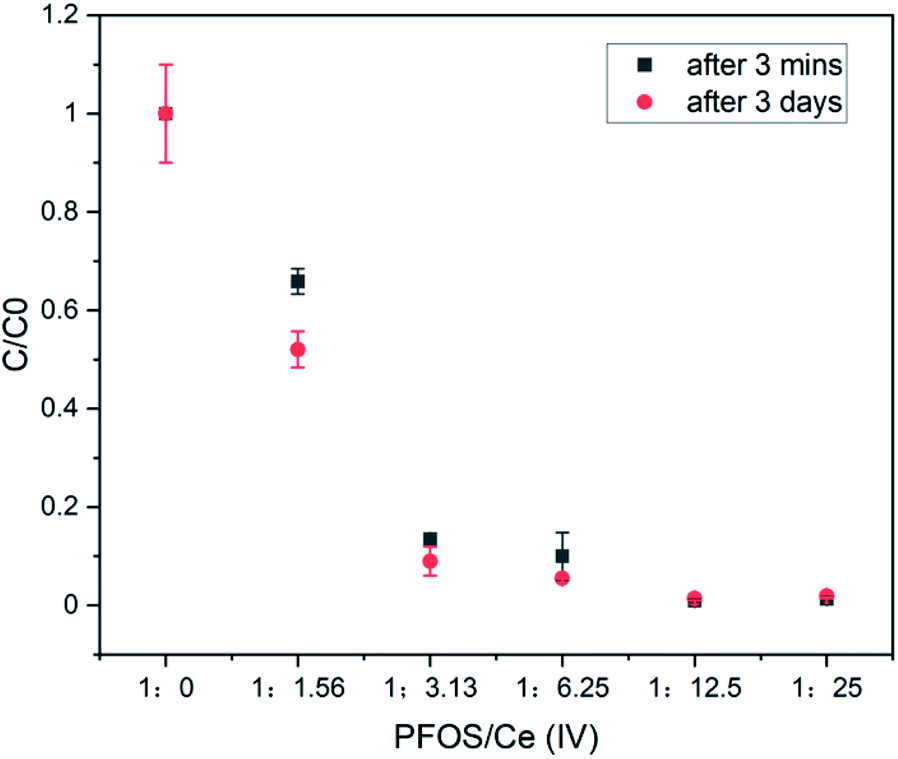 | ||
| Fig. 2 The LC-MS detected 1.5 mM of PFOS in water before ([PFOS:Ce(IV)] = 1:0) and after the treatment (3 minute) with 2.34 mM, 4.69 mM, 9.38 mM, 18.75 mM and 37.5 mM of CAN. | ||
The solutions before and after the treatment with CAN and the precipitate formed during the experiment were also investigated by 400 MHz 1H and 376 MHz 19F NMRs (Fig. 3, S6–S9).† These NMR studies (Fig. 3) of the supernatant liquid showed complete removal (in the NMR detection limit) of PFOS from water. The slight change in the chemical shift values and the splitting pattern of the NMR signals indicate CAN-PFOS interaction or possible complex formation.
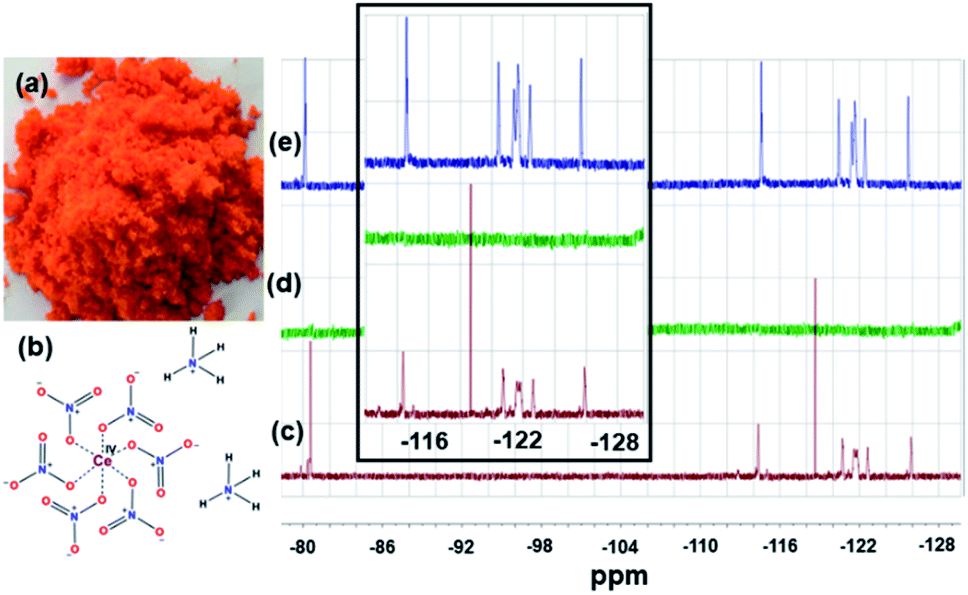 | ||
| Fig. 3 (a) The colour and texture of CAN that was used for this study, (b) pictorial representation of CAN's chemical structure, (c) and (d) are the 376 MHz 19F NMR of aqueous solutions (1.5 mM) of PFOS in DMSO-d6, before and after the treatment with 18.75 mM CAN [Ce(IV)] respectively showing complete disappearance of PFOS from water, and (e) 376 MHz 19F NMR of the precipitate in DMSO-d6. Inset shows magnified picture of c, d and e from −129 ppm to −114 ppm, indicating complexation or strong interaction between CAN [Ce(IV)] and PFOS. | ||
To generalize our findings, we also tested another commercially available and highly regulated PFAS, perfluorooctanoic acid (PFOA, Fig. 6) as a substrate.21 At 15 μM starting concentration, at best 65% of the PFOA could be removed with 0.75 mM of CAN (Fig. S4†). At 1.5 mM PFOA, 78% removal was possible, with 18.75 mM CAN. However, complete removal of PFOA (1.5 mM) was detected (by LC-MS) at a much higher CAN concentration (0.15 M), after 3 minutes of continuous stirring (Fig. 4).
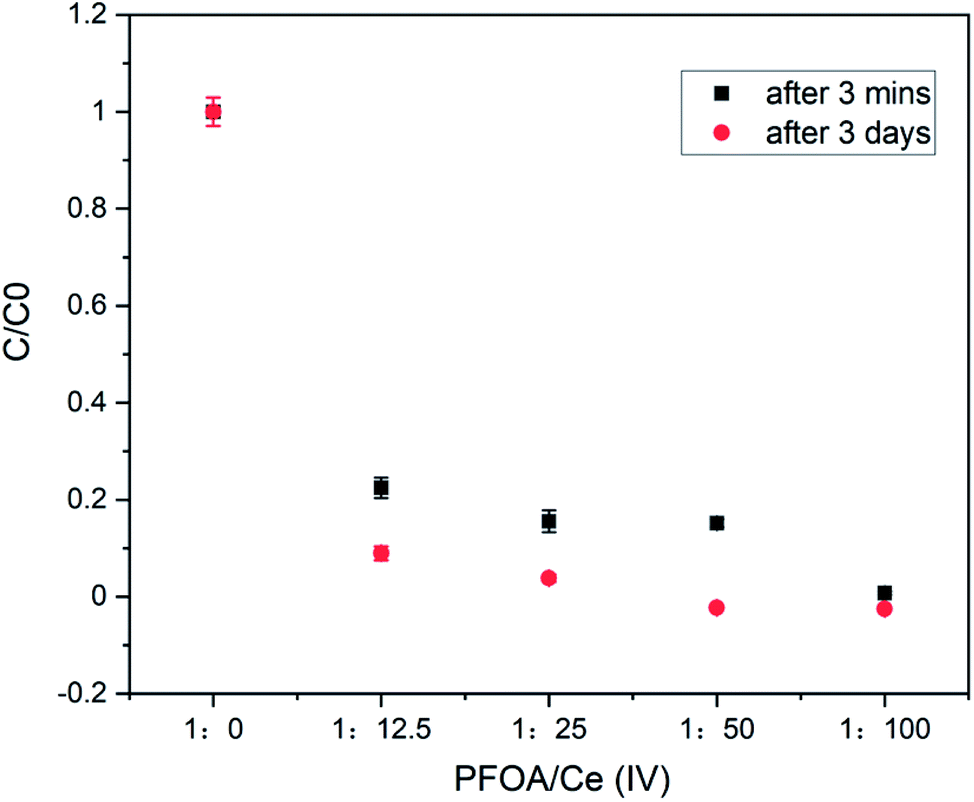 | ||
| Fig. 4 The LC-MS detected concentrations of 1.5 mM of PFOA in water before ([PFOA:Ce(IV)] = 1:0) and after the treatment (3 minute) with 18.75 mM, 37.5 mM, 75 mM and 150 mM of CAN. | ||
In this case, an off-white precipitate was observed after a week of undisturbed standing. Similar NMR investigations were performed by 400 MHz 1H and 376 MHz 19F NMR (Fig. 5, and S10–S13†) spectroscopy. The complete disappearance of the fluoride signal from water (in the NMR detection limit) and the change in the chemical shift values together with the splitting pattern of the precipitate also suggest complexation between Ce and PFOA.22 It is interesting to see more prominent changes in the chemical shift values in the case of the Ce–PFOA complex than that of the Ce–PFOS complex. In the case of Ce–PFOA complex, initial deprotonation of PFOA is expected prior to complexation/interaction with Ce(IV).
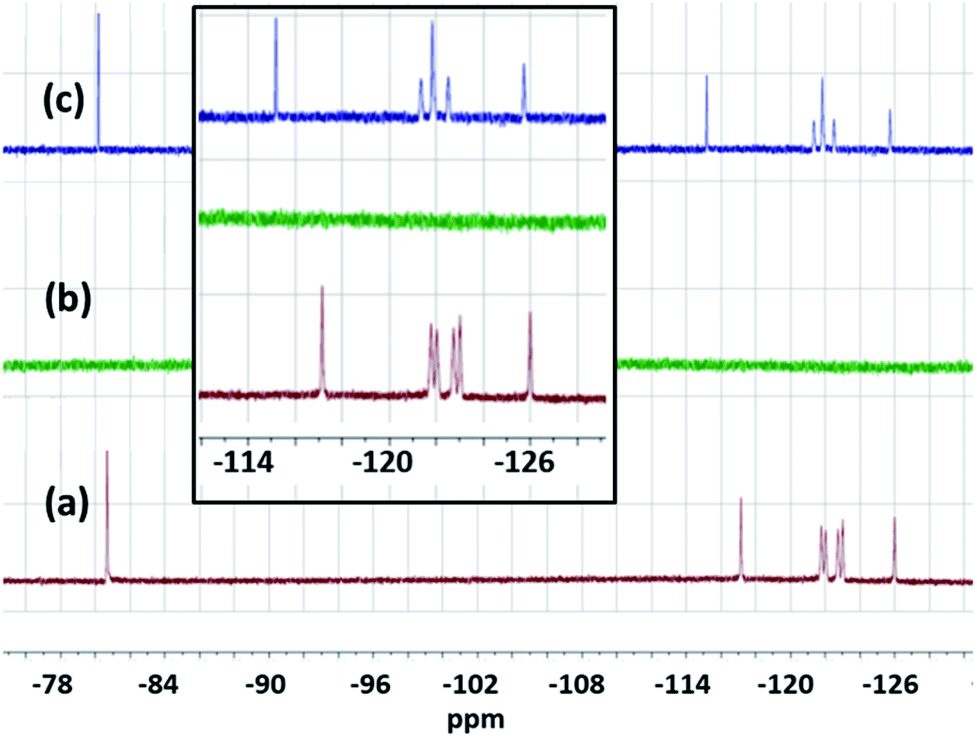 | ||
| Fig. 5 (a) and (b) are the 376 MHz 19F NMR of an aqueous solution (1.5 mM) of PFOA in DMSO-d6, before and after the treatment with 0.15 M CAN [Ce(IV)] respectively showing complete disappearance of PFOA from water. (c) Represents the 376 MHz 19F NMR of the precipitate in DMSO-d6. Inset shows magnified picture of a, b and c from −128 ppm to −113 ppm, indicating complexation between Ce(IV) and PFOA. | ||
 | ||
| Fig. 6 Chemical structures of the two commercially available PFASs used in this study. | ||
Very similar precipitation was observed with CeIV(SO4)2 at the higher concentration study. PFOS (1.5 mM) precipitated out much faster (within a few hours) and complete removal (in the NMR detection limit) (Fig. S16†) was possible at 18.75 mM of CeIV(SO4)2. In comparison, faint white precipitation from PFOA (1.5 mM) and 18.75 mM of CeIV(SO4)2 was observed over a period of two days. NMR of the supernatant aqueous solution shows incomplete removal of PFOA under identical condition (Fig. S17†).
It is clear that PFOS reacts with CAN and CeIV(SO4)2 more efficiently (Fig. 2 and 4) and precipitates out faster than PFOA. This difference in reactivity can be due to the better donor ability (nucleophilicity) of the sulfonate group in PFOS than the carboxylic acid group in PFOA. Due to the higher acidity of the sulfonic acid, the conjugate base sulfonate (nucleophile) remains as the major species in solution in the presence of CAN in aqueous medium.23 In the case of PFOA, to be equally effective, it needs higher concentration of CAN. Moreover, being shorter than PFOS, PFOA has been reported to show a higher tendency to stay in the aqueous phase, and therefore is difficult to precipitate out.24 With AN (NH4NO3), no such precipitation was observed, and NMR results also confirm no detectable interaction between AN and PFOS (Fig. S14 and S15†).
All these observations suggest a possible complexation between Ce(IV) of CAN and these PFASs, followed by precipitation where PFOS and PFOA act as a nucleophile (ligand). No distinct new proton signal in 400 MHz 1H NMR spectra of the aqueous solution and the precipitate after the CAN treatment, confirms the absence of any newly formed hydrocarbons.
HRMS analysis of the diluted (1:20 DMSO:water) solutions of the precipitates (from PFOA + CAN and PFOS + CAN) were quite complicated (Fig. S18 and S19†). It is reported that on the way to complexation with Ce(IV)/Ce(III), PFAS can trigger radical induced pathways to undergo C–C and C–F bond cleavages.25,26 In this regard, the formation of the polymeric compounds [e.g. (Ce)m(PFOA)n(NO3)x(NH4)y(H2O)z, where m, n, x, y and z can be any arbitrary numbers from 0, 1, 2 to n] cannot be completely ruled out. We were unable to identify any new species with reasonable precision from the HRMS experiments. The precise identification of these species needs further detailed investigation with various other spectroscopic techniques.
In conclusion, CAN effectively precipitates out both PFOS and PFOA even at low concentration (<20 μM) and clearly reacts more efficiently with the former. A clear trend of CAN > Ce(SO4)2 > CeCl3 > CoCl2 ≥ FeCl2 > NH4NO3 was observed for precipitating out PFOS and PFOA from the aqueous medium. These PFASs are reported among the most difficult ones to degrade.27 The CeIV center plays an important part in this, as reflected by the comparative studies using CAN, Ce(SO4)2, CeCl3 and AN. The oxidation state of the cerium center, as well as the size of the PFASs play crucial roles in the successful removal/precipitation from the aqueous medium. Thus, a similar simple strategy can potentially be used in future for the design of highly effective filter beds (e.g., using a combination of GAC/PAC and CAN) for purifying PFAS contaminated water at larger scale.
Conflicts of interest
Authors declare no conflict of interests.Acknowledgements
We will like to acknowledge Australian Research Council's (ARC) special research initiatives (Project ID: SR180100030) for providing the research funding. NK and BD will like to thank emeritus Prof. David Black, at School of Chemistry, UNSW for useful discussion.Notes and references
- J. Gluge, M. Scheringer, I. T. Cousins, J. C. DeWitt, G. Goldenman, D. Herzke, R. Lohmann, C. A. Ng, X. Trier and Z. Wang, Environ. Sci.: Processes Impacts, 2020, 22, 2345–2373 RSC.
- M. Haukas, U. Berger, H. Hop, B. Gulliksen and G. W. Gabrielsen, Environ. Pollut., 2007, 148, 360–371 CrossRef CAS PubMed.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, Front. Environ. Sci. Eng. China, 2009, 3, 129–151 CrossRef CAS.
- A. Moller, L. Ahrens, R. Surm, J. Westerveld, F. van der Wielen, R. Ebinghaus and P. de Voogt, Environ. Pollut., 2010, 158, 3243–3250 CrossRef PubMed.
- K. H. Kucharzyk, R. Darlington, M. Benotti, R. Deeb and E. Hawley, J. Environ. Manage., 2017, 204, 757–764 CrossRef CAS PubMed.
- E. P. Hines, S. S. White, J. P. Stanko, E. A. Gibbs-Flournoy, C. Lau and S. E. Fenton, Mol. Cell. Endocrinol., 2009, 304, 97–105 CrossRef CAS PubMed.
- E. M. Quist, A. J. Filgo, C. A. Cummings, G. E. Kissling, M. J. Hoenerhoff and S. E. Fenton, Toxicol. Pathol., 2015, 43, 546–557 CrossRef CAS PubMed.
- D. Lu, S. Sha, J. Luo, Z. Huang and X. Zhang Jackie, J. Hazard. Mater., 2020, 386, 121963 CrossRef CAS PubMed.
- M. F. Rahman, S. Peldszus and W. B. Anderson, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed.
- J. Huang, X. Wang, Z. Pan, X. Li, Y. Ling and L. Li, Chem. Eng. J., 2016, 296, 329–334 CrossRef CAS.
- N. Merino, Y. Qu, R. A. Deeb, E. L. Hawley, M. R. Hoffmann and S. Mahendra, Environ. Eng. Sci., 2016, 33, 615–649 CrossRef CAS.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866–915 CrossRef.
- P. McCleaf, S. Englund, A. Ostlund, K. Lindegren, K. Wiberg and L. Ahrens, Water Res., 2017, 120, 77–87 CrossRef CAS PubMed.
- N. B. Saleh, A. Khalid, Y. Tian, C. Ayres, I. V. Sabaraya, J. Pietari, D. Hanigan, I. Chowdhury and O. G. Apul, Environ. Sci.: Water Res. Technol., 2019, 5, 198–208 RSC.
- Y. Wu, Y. Li, C. Fang and C. Li, ChemCatChem, 2019, 11, 2297–2303 CrossRef CAS.
- V. Ochoa-Herrera and R. Sierra-Alvarez, Chemosphere, 2008, 72, 1588–1593 CrossRef CAS PubMed.
- Y. Qu, C. Zhang, F. Li, X. Bo, G. Liu and Q. Zhou, J. Hazard. Mater., 2009, 169, 146–152 CrossRef CAS PubMed.
- B. K. Pramanik, S. K. Pramanik and F. Suja, Environ. Technol., 2015, 36, 2610–2617 CrossRef CAS PubMed.
- M. Asrafuzzaman, A. N. Fakhruddin and M. A. Hossain, ISRN Microbiol., 2011, 2011, 632189 Search PubMed.
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178–1186 CrossRef CAS PubMed.
- S. Huang and P. R. Jaffe, Environ. Sci. Technol., 2019, 53, 11410–11419 CrossRef CAS PubMed.
- M. Shamsipur, M. Sarkouhi, J. Hassan and S. Haghgoo, Afr. J. Pharm. Pharmacol., 2011, 5, 1573–1579 CrossRef CAS.
- S. Rayne and K. Forest, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2009, 44, 1145–1199 CrossRef CAS PubMed.
- S. T. Le, T. C. G. Kibbey, K. P. Weber, W. C. Glamore and D. M. O'Carroll, Sci. Total Environ., 2021, 764, 142882 CrossRef CAS PubMed.
- D. W. Brixius, Doctor of Philosophy, Iowa State University, 1972.
- Q. Zhuo, S. Deng, B. Yang, J. Huang and G. Yu, Environ. Sci. Technol., 2011, 45, 2973–2979 CrossRef CAS PubMed.
- J. Liu, D. J. Van Hoomissen, T. Liu, A. Maizel, X. Huo, S. R. Fernández, C. Ren, X. Xiao, Y. Fang, C. E. Schaefer, C. P. Higgins, S. Vyas and T. J. Strathmann, Environ. Sci. Technol. Lett., 2018, 5, 289–294 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02635f |
| ‡ Contributed equally to the experimental work. |
| § ICP-MS measurement was not successful due to the presence of the excess nitrate (from ammonium cerium(IV) nitrate) in the medium. |
| ¶ The higher oxidative activity of Ce(IV) is well known at high concentration and in acidic medium. |
| This journal is © The Royal Society of Chemistry 2021 |