
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Competing HB acceptors: an extensive NMR investigations corroborated by single crystal XRD and DFT calculations†
Surbhi Tiwaria,
Neeru Aryaa,
Sandeep Kumar Mishrab and
N. Suryaprakash *a
*a
aNMR Research Centre and Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore 560012, India. E-mail: nsp@iisc.ac.in; suryaprakash1703@gmail.com; Fax: +91 80 23601550; Tel: +91 80 23607344 Tel: +91 80 22933300 Tel: +91 98 45124802
bDepartment of Physics and NMR Research Centre, Indian Institute of Science Education and Research, Pune 411008, India
First published on 22nd April 2021
Abstract
A series of N-benzoylanthranilamide derivatives have been synthesized with the substitution of competitive HB acceptors and investigated by NMR spectroscopy and single crystal XRD. The interesting rivalry for HB acceptance between ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and X (F or OMe) is observed in the investigated molecules which leads to an unusual increase in the electron density at the site of one of the NH protons, reflecting in the high field resonance in the 1H NMR spectrum. The NMR experimental findings and single crystal XRD are further reinforced by the DFT studies.
O and X (F or OMe) is observed in the investigated molecules which leads to an unusual increase in the electron density at the site of one of the NH protons, reflecting in the high field resonance in the 1H NMR spectrum. The NMR experimental findings and single crystal XRD are further reinforced by the DFT studies.
Introduction
The hydrogen bond (HB) plays a key role in stabilizing the three-dimensional structures of many organic and biomolecules and has tremendous influence in chemistry, biology, drug design, etc.1–3 The HB can exist between an H atom covalently bonded to a donor atom (D) and acceptor atom(s) (A), where both D and A should be more electronegative than H.4 Based on the number of acceptor atoms the HB could be two-centered, or three-centered (bifurcated). The bifurcated HB can either be of (A⋯H⋯A) or (H⋯A⋯H) type.5–9 These HBs can be inter- or intra-molecular or a mixture of both types. The existence of inter- and/or intra-molecular HB may administer the architecture of various natural and synthetic compounds. The selective introduction of HBs may lead to the desired conformation of a molecule.10 The strength of any HB is directly related to the electronegativity of acceptor atom(s)10 and also depends on geometrical parameters, such as, the angle and the distance between H and acceptor atom. Owing to the electronegativity of N and O atoms, the strong HBs of N–H⋯O, O–H⋯O, and O–H⋯N motifs are usually encountered.11 The substantial fraction of the commercially available pharmaceutical drugs possess the fluorine atom(s) which alter their physical properties and improves the binding affinity with the target molecules through HB(s).12–16 Despite being the most electronegative atom,17 the participation of organic fluorine in the HB has been extensively debated. Earlier it was believed that organic fluorine hardly participates in the intramolecular HB.18–22 Nonetheless, a number of recent reports established the existence of intramolecular HB with the participation of fluorine attached to the carbon atom. The recent report also states that, “it is now difficult to doubt the existence of hydrogen bonds involving organic fluorine”.19Among many available analytical techniques, the NMR spectroscopy has been proved to be the most valuable one in the study of HB. The change in chemical shift upon dilution with the solvents of different polarities, variable temperature studies, 2D HOESY and 2D HSQC experiments, clearly ascertain the presence or the absence of HB.23,24 The participation of fluorine in the HB is also evidenced by the detection of 1hJFH, mediated through non-covalent bond.25 It has been reported that the couplings between F and H separated by 5 covalent bonds (5JFH) is always less than 1 Hz. The detection of the significantly large coupling strength between 19F and 1H has been attributed to be HB mediated.26–28 There are several examples of detection of direct through space couplings between many homo- and hetero-nuclear spins, such as JHF, JFF JPF, and JPP, and between many other NMR active nuclei, and many reports have discussed the mechanism and strengths of such through-space interactions.29 The detection of JFH has also been extensively debated as, whether it arises because of hydrogen bond or due to the overlap of electronic clouds.29–33 Some studies also attributed the term “through-space” and evidenced that the spin polarization could be transferred between the two nuclei, H and F, via hydrogen bonds.34,35
The anthranilamide derivatives are pharmacologically important and known for their applications as antibacterial,36,37 antiviral,38 anticoagulants agents39 and also serve as a potent inhibitor of human factor Xa.40,41 Consequently, a series of N-benzoylanthranilamide were synthesized with ortho substitution at the benzoyl ring where one H, one donor (N) and two H acceptors (O and X = F, OMe) are present which satisfies the requirement of bifurcated (three-centered) intramolecular HB (Scheme 1). All these molecules were characterized and subjected to investigations by the utility of NMR experiments to ascertain the presence or absence of HBs. The basic structural frameworks of the molecules are reported in Scheme 1.
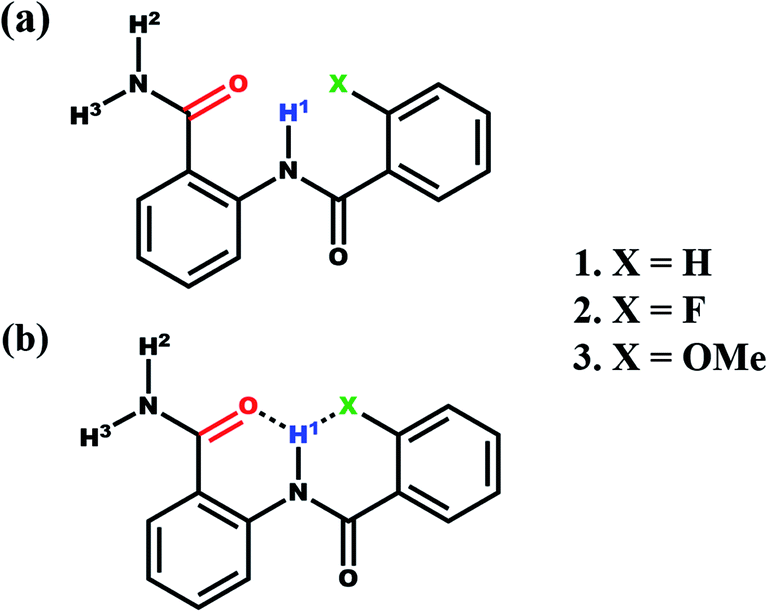 | ||
| Scheme 1 (a) The structural frameworks of N-benzoylanthranilamide and its derivatives along with the numbering of H atoms involved in HB; (b) illustration of the possible formation of a bifurcated HB. | ||
Results and discussions
Generally, the amide protons resonate between 5–9 ppm in the 1H NMR spectrum, and the formation of hydrogen bond leads to significant downfield shift. In the present study, the NH1 proton of molecule 1, resonated at 12.22 ppm. The extensive deshielding of this proton may be attributed to the intramolecular HB between NH1 proton and the carbonyl oxygen (CO) of amide group. The 2D 1H–1H NOESY establishes the spatial proximity between two spins and aids in arriving at the favorable conformation of the molecule. The detection of correlation peak in the NOESY spectrum of molecule 1 (Fig. 1), corroborates the spatial proximity between the proton of NH1 and H17.
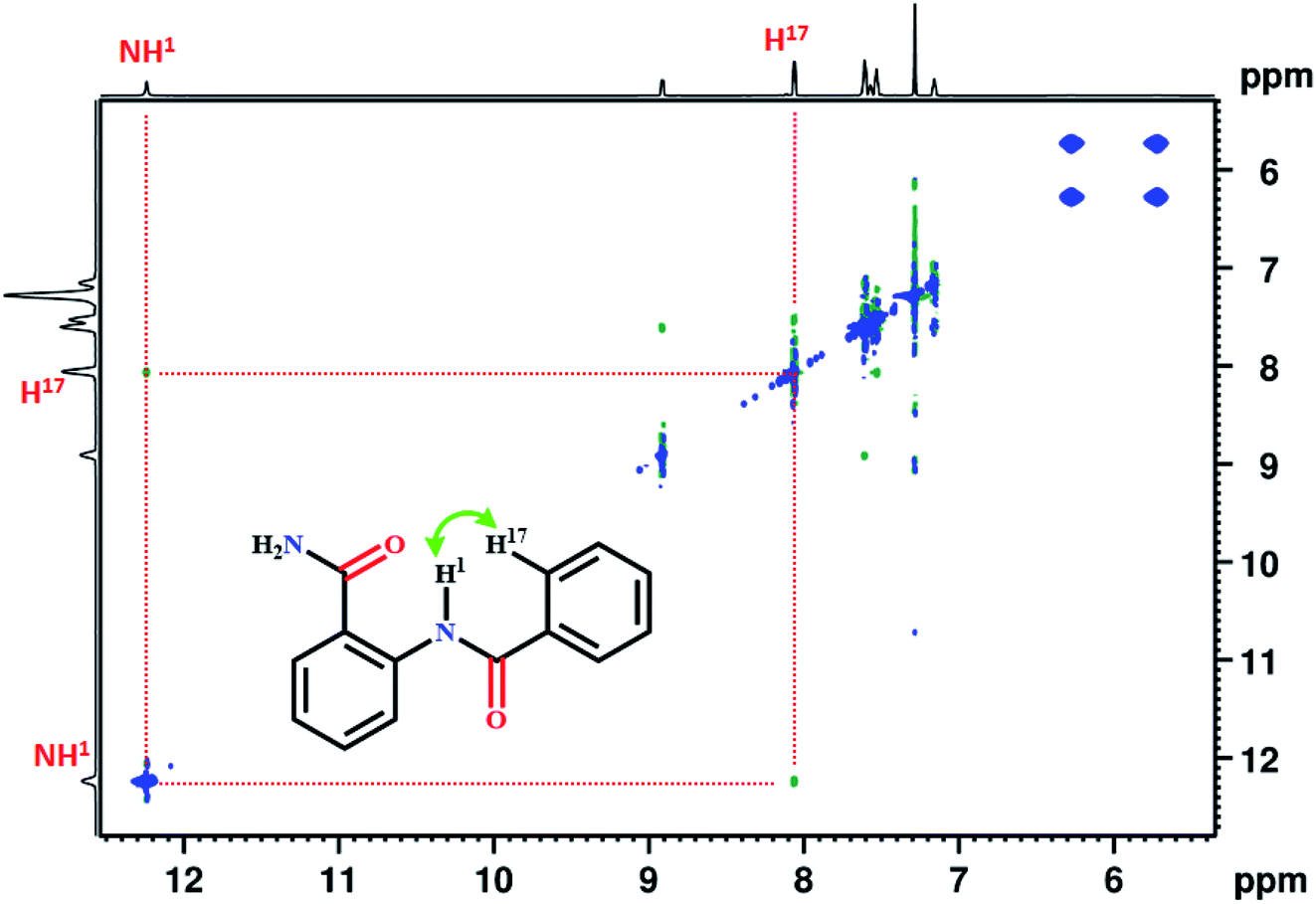 | ||
| Fig. 1 800 MHz 2D 1H–1H NOESY spectrum of molecule 1 in the solvent CDCl3 at 298 K. | ||
In molecule 2, the fluorine atom is presumed to be involved in an additional intramolecular HB with NH1, rendering the bifurcation. This assumption is strengthened by the detection of strong correlation peak between 19F and NH1 proton (Fig. S14†) in the 2D 19F–1H HOESY (Heteronuclear Overhauser Effect SpectroscopY) spectrum. Hence these results direct towards the existence of bifurcated HB in the investigated systems.
Differentiating inter- and intra-molecular HB
To validate the intermolecular or intramolecular HB and to ascertain the effect of monomeric water on HB42 if any, the dilution study23 using a non-polar solvent CDCl3 was carried out. The dilution results in the dispersion of the molecules and consequently a substantial change in the chemical shift of protons when the interactions are of intermolecular type. However, the chemical shift remains invariant when the interaction is intramolecular. The plot of NH1 chemical shifts as a function of dilution with solvent CDCl3, for all the molecules, is given in Fig. 2. Although the solute concentration was not diluted to a large extent, the invariance of chemical shifts of NH1 proton when diluted to half its value, safely discards the possibility of any intermolecular interactions and confirming the existence of intramolecular HBs. However, the NH2 and NH3 protons exhibited a slight shift towards the shielded region on dilution. The negligible change in the chemical shift of residual water peak (1.54 ppm) ascertains the trifling effect of monomeric water8,23,42 on the intramolecular HB.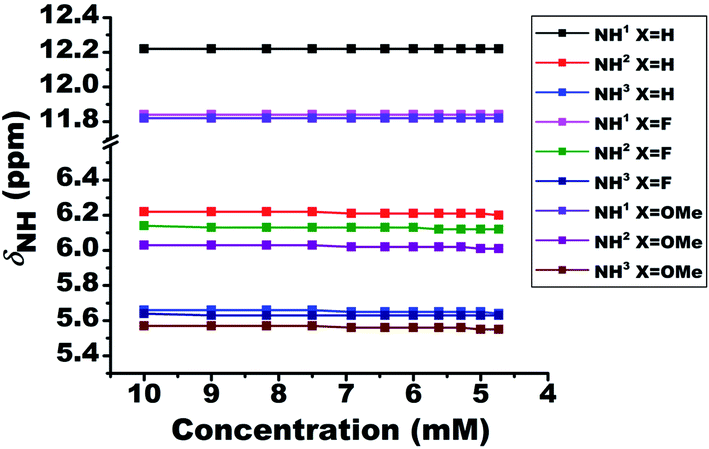 | ||
| Fig. 2 Variation in the chemical shifts of NH proton as a function of the concentration of solvent CDCl3, for the molecules 1–3. The initial concentration was taken as 10 mM in 450 ml of solvent at 298 K. The NH protons and the molecules are identified by numbers and different colour codes given in the inset. | ||
Relative strengths of HB
The relative strengths of intramolecular HB interactions can be estimated by the titration study with a highly polar solvent dimethyl sulfoxide (DMSO).43 Due to the high affinity towards HB acceptance, the solvent DMSO is capable of rupturing a variety of inter- or intra-molecular HBs. Hence the titration with systematic addition of DMSO-d6 to the 10 mM solutions of the molecules 1–3 in CDCl3 were carried out and the variation in chemical shift of NH1 peak was monitored (Fig. 3). The severe overlap of the NH2 and NH3 resonances with the aromatic protons hindered the determination of the effect of DMSO on these peaks. On incremental addition of DMSO-d6, the chemical shift of NH1 proton was shifted to higher frequency region for all the molecules, which is attributed to the engagement of NH1 proton in the intermolecular HB with DMSO (Fig. 3).8,44,46 The deshielding in the NH1 proton chemical shift is inversely proportional to the strength of intramolecular HB in such systems44,46 because the intramolecular HB minimizes the accessibility of sites for DMSO around the acidic proton. The small change in chemical shifts upon DMSO-d6 addition could possibly be attributed to the favorable near planar geometry of these molecules which also limits the accessibility of the sites for the association of DMSO by creating hindrance. The change in the NH1 chemical shifts for molecules 1–3 on addition of 0.65 mole fraction of DMSO are reported in Table 1.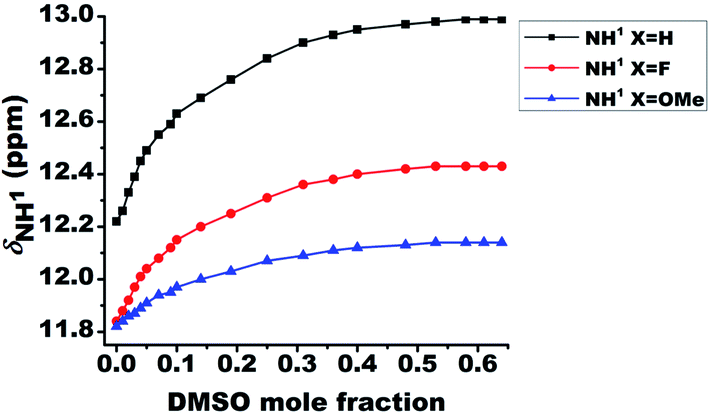 | ||
| Fig. 3 Change in the chemical shift of NH1 protons for the molecules 1–3 on addition of 0.65 mole fraction of DMSO-d6 at 298 K. The initial concentration taken was 10 mM in 450 ml of CDCl3 and DMSO-d6 was systematically added to it. | ||
| Molecule | Substituent (X) | δNH1 (ppm) | Difference in chemical shift δNH1 (ppm) | |
|---|---|---|---|---|
| DMSO-d6 (in mole fraction = 0) | DMSO-d6 (in mole fraction = 0.65) | |||
| 1 | H | 12.22 | 12.99 | 0.77 |
| 2 | F | 11.84 | 12.43 | 0.59 |
| 3 | OMe | 11.83 | 12.14 | 0.31 |
Effect of temperature
The lowering of temperature leads to the strengthening of HB, which results in the deshielding of the proton involved in the HB. On systematically varying the temperature from 300 K to 230 K the chemical shift of the NH1 proton moved towards the higher resonance frequency as a consequence of the strengthening of the intramolecular HB (Fig. 4). However, in the case of NH2 and NH3 protons, the shift towards the deshielded region (Fig. 4) is attributed to the decrease in the electron density on amide (>CO–NH2) group. Additionally, the deshielding in NH2 and NH3 proton chemical shifts also point towards the existence of HB. However, from the close inspection of the chemical structures, these deshielding can be attributed to intermolecular HBs between carbonyl oxygen and these protons at low temperature (Fig. 4), rather than intramolecular HB. This possibility was also inferred from the dilution studies with CDCl3 solvent (Fig. 2), which is now confirmed. The calculated amide temperature coefficient values (ΔδNH1/ΔT) also reveal the small variation in the chemical shift of NH1 protons with temperature, viz., from −0.3 to −1.3 ppb K−1 and are assimilated in Table S1.† These values, which is more positive than −0.4 ppb K−1, the HB predictivity is more than 85%.47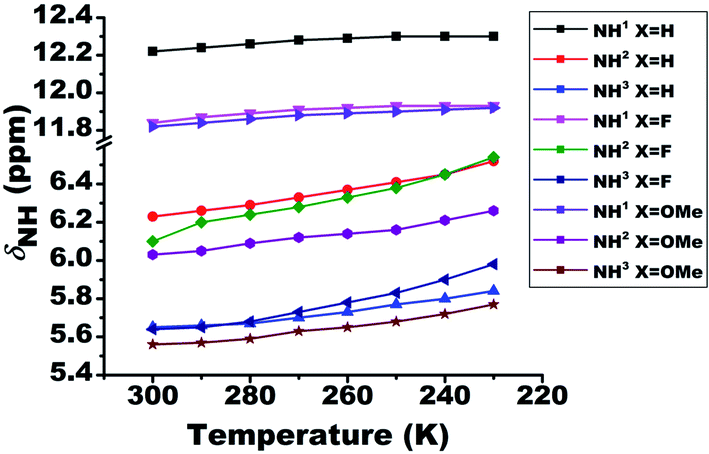 | ||
| Fig. 4 Effect of temperature on the chemical shifts of NH protons (δNH) of molecules 1–3. The temperature was varied from 300 K to 230 K. The solution concentration in CDCl3, was maintained at 10 mM. | ||
Detection of 1hJXH
The interaction of NH1 proton with the acceptor atom X can be reflected as J coupling in the 1D 1H NMR spectrum, if X is also an NMR active nucleus. The NH1 proton in the molecule 2 appeared as a doublet with the frequency separation of 6.1 Hz. The 1D 1H{19F} NMR experiment on molecule 2 confirmed the interaction between 1H and 19F, where NH1 proton appeared as a singlet (Fig. 5b). In the present study, the observed 1hJFH value of 6.1 Hz (Fig. 5a) is small compared to the previous reports,8,44,45 and can be attributed to the presence of another strong HB acceptor (CO group of amide) which pulls the NH1 proton far from the F atom, which resulted in the decreased F⋯HN strength. To extract the hidden couplings, if any, the 1D 1H{14N} and 2D 15N–1H coupled HSQC experiments at the natural abundance of 15N were carried out for molecule 2, and the spectra are reported in Fig. S10 and S17 respectively in the ESI.† These spectra yielded 1hJFH of 7.68 Hz, which implied that the observed doublet of 6.1 Hz for NH1 proton in Fig. 5a, also had the contribution from the unresolved 1J14N–1H and the additional broadening arose due to the 14N quadrupole relaxation.
 | ||
| Fig. 5 400 MHz 1H spectrum corresponding to proton NH1 of molecule 2 in the solvents CDCl3; (a) 19F coupled spectrum and (b) 19F decoupled spectrum. | ||
In the solvent DMSO-d6, the observed 1hJFH was only 3.8 Hz in both 1D 1H and 1D 1H{14N} spectra (Fig. 6(b) and S13†). However, in many earlier reports, this coupling completely vanished45,46,48,49 in DMSO except in molecules where the structural restraint resists the complete breaking of HB by DMSO.26,44 The retention of the 1hJFH for molecule 2 in the DMSO-d6 is due to the favorable cis geometry (Fig. 1), which prevents the complete solvation of HB. The molecules tend to surround the NH1 proton causing the steric hindrance whereby the distance between NH1 proton and fluorine atom increases, leading to the decrease in 1hJFH. However, in the earlier reports, the cis form of the structure stabilizes in a non-polar solvent by the influence of intramolecular HB and the molecules attain the lowest energy structure by rupturing the HB in DMSO which is different from the cis form, resulting in a complete nullification of 1hJFH.
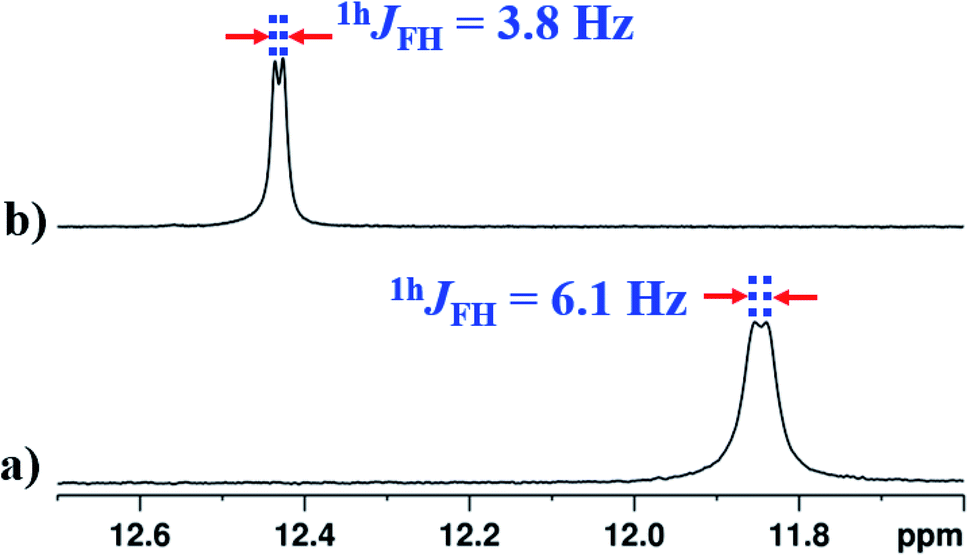 | ||
| Fig. 6 400 MHz of 1H NMR spectrum of molecule 2, at 298 K, corresponding to NH1 proton; (a) in CDCl3 solvent; (b) in DMSO-d6 solvent. | ||
Variation in 1hJFH
On lowering the temperature from 300 K to 230 K, the value of 1hJFH in molecule 2 changed from 6.1 Hz to 8.9 Hz (Fig. 7). The covalent bond mediated scalar coupling remains practically invariant, while the HB mediated coupling varies with changing the distance between H and acceptor atom.23 Therefore, this significant variation in the 1hJFH for molecule 2, is attributed to the coupling mediated through HB.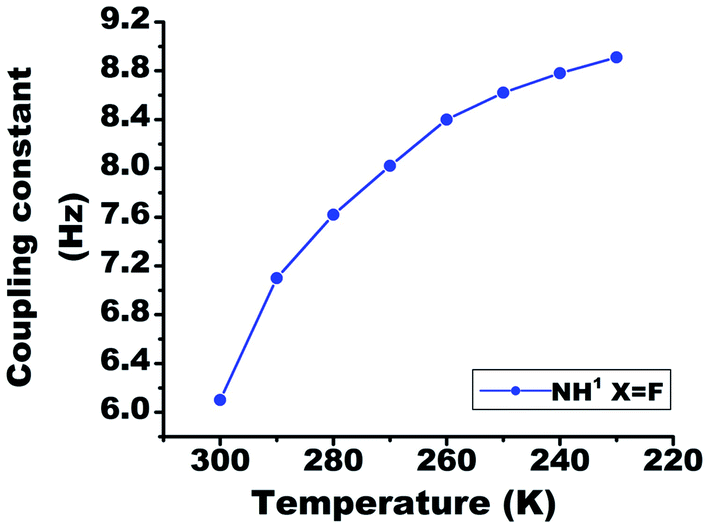 | ||
| Fig. 7 The temperature dependent variation in the 1hJFH in molecule 2, in the range 300 K to 230 K. The solute concentration was taken 10 mM in the solvent CDCl3. | ||
Unusual chemical shift value of NH1 proton
Conventionally, the presence of HB or the introduction of additional HB(s) results in the deshielding of proton in the 1H-NMR spectrum. Contrary to the expectation, the NH1 proton is observed to be more shielded (Fig. 8(b) and (c)) on substitution of X at the ortho position of benzoyl ring (in molecules 2 and 3). This is due to the fact that the NH1 proton is involved in a strong HB with theCO oxygen of amide group. Furthermore, the substitution of F or OMe increases the electron density on the NH1 proton by stabilizing an equilibrium between two hydrogen acceptors resulting in the shielding of proton.49 Another possible reason could be the weakening of strong two-center CO⋯H HB during the competition with another HB acceptor.
 | ||
| Fig. 8 400 MHz 1H NMR spectra in CDCl3 solvent at 298 K, corresponding to the NH1 proton region of; (a) molecule 1; (b) molecule 2; and (c) molecule 3. | ||
To derive more insight, additionally the molecules 4–7 were synthesized (Fig. 9a, S31, 9b and S36). When the F in the molecule 4 was displaced from ortho to meta position on the benzoyl ring with respect to NH1, the value of chemical shift of NH1 was observed to be similar to that of molecule 1 (Fig. 9c). Also considering the electronic effect, when the F was displaced to para position in molecule 5 (ESI, Fig. S31†), the observed chemical shift value was similar to those detected in molecules 1 and 4. Subsequently, when the amide group of the molecule 2 was displaced from ortho to meta position (molecule 6), the NH1 proton exhibited a doublet with a separation of 16.25 Hz (Fig. 9d), whose value is similar to those observed for other reported molecules.45,46,48–52
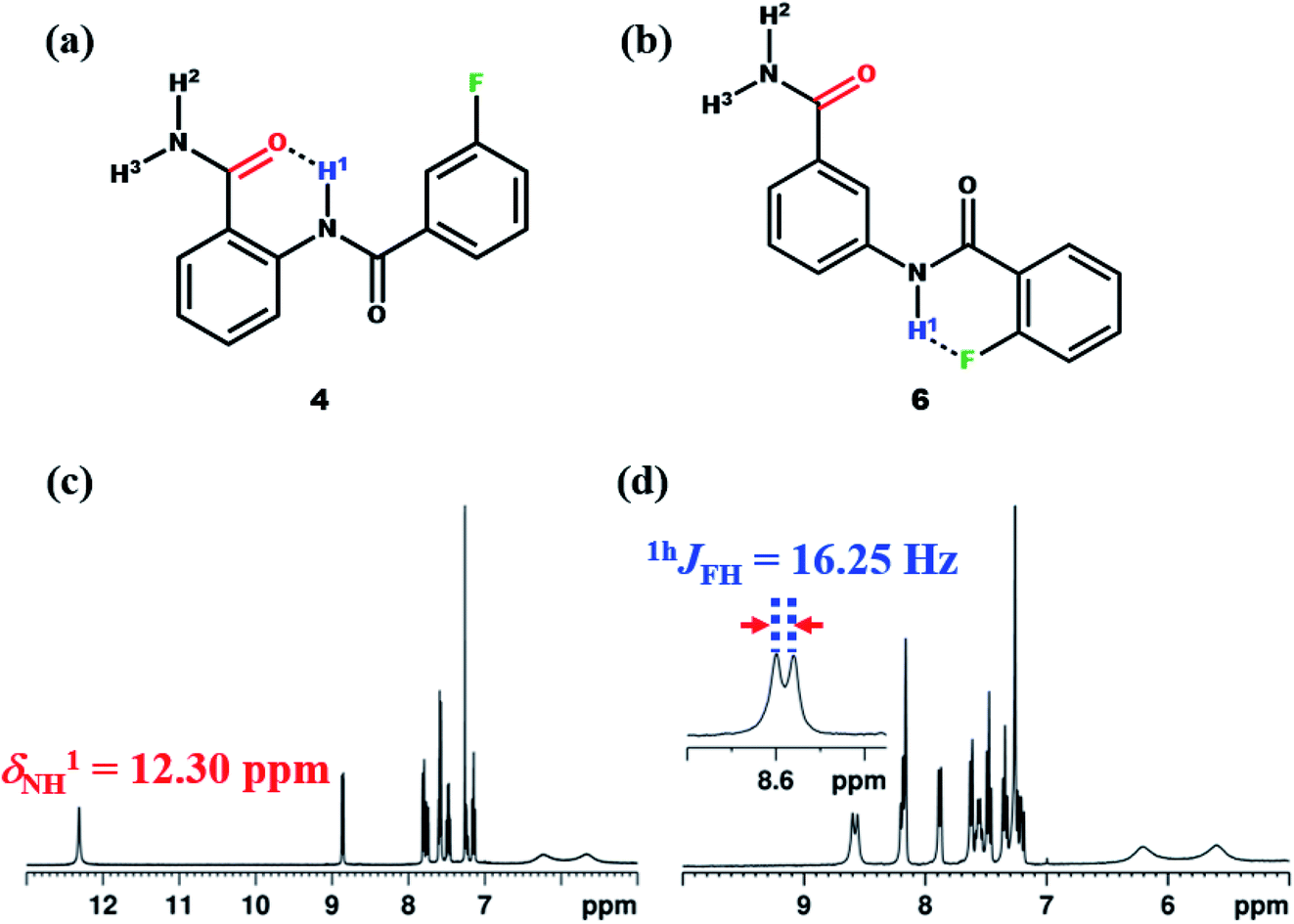 | ||
| Fig. 9 The chemical structures of; (a) molecule 4, and (b) molecule 6; (c and d) the corresponding 400 MHz 1H NMR spectra in the CDCl3 solvent recorded at 298 K. | ||
All these in-depth NMR studies leads to the conclusion that the N–H⋯X interactions are influenced by strong CO⋯H HB, which is also reflected in the shielding of NH1 proton and reduced value of 1hJFH in the molecule 2. These narrations suggest that the competitive equilibrium between N–H⋯X and CO⋯H–N type HBs debilitate with each other and is the possible reason for the shielding of NH1 proton in the molecules 2 and 3 compared to 1. The aforesaid facts are also corroborated by findings from the single crystal XRD and theoretical calculations discussed in the forthcoming part of this manuscript. A good single crystal for molecule 2 was obtained, and unfortunately, we failed in our efforts to crystallize other molecules, and thus XRD study is restricted only to molecule 2.
Single crystal X-ray diffraction (XRD) studies
The XRD is another powerful technique for the investigation of HB, where the linearity in the bond angle i.e. D–H⋯A of 180° and closer to this value validates the presence of stronger HB.53,54 The distance between the H and the acceptor atom between 1.2–1.5 Å also indicates a strong HB, whereas the distance of 1.5–2.2 Å suggests the HB of moderate strength and the value > 2.2 Å establishes a relatively weak HB.53 The XRD structure of the molecule 2 is reported in Fig. 10.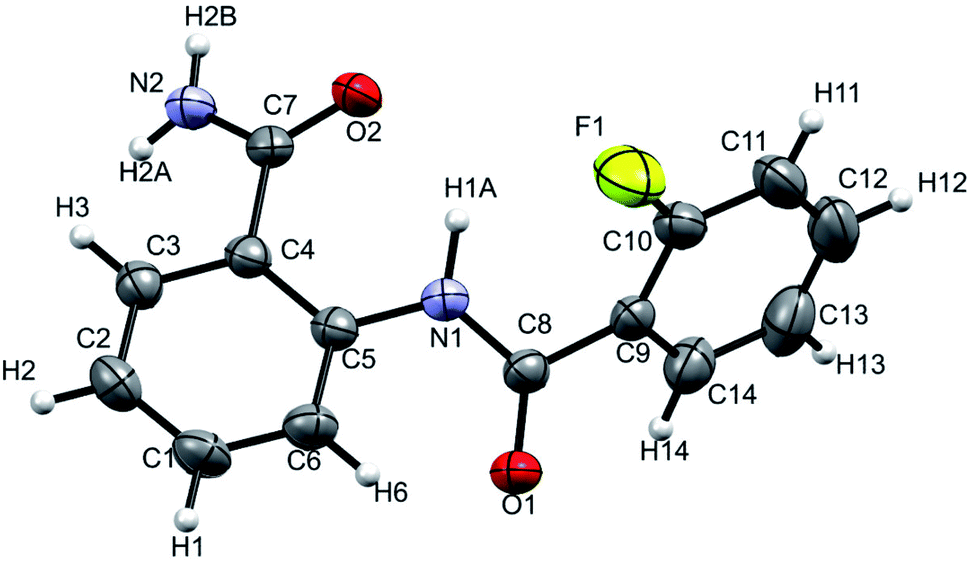 | ||
| Fig. 10 Single-crystal XRD structure of molecule 2 at 298 K with ORTEP view with 50% probability level (CCDC number: 2049223†). | ||
The NH1⋯F distance and the N–H1⋯F angle were determined as 2.310 Å and 110° respectively, which corresponds to a weak HB in the solid state. The results derived from the single crystal XRD are assimilated in Table 2 and the experimental details are provided in ESI (Table S2†).
| Parameter | DFT | XRD | ||||||
|---|---|---|---|---|---|---|---|---|
| Molecule | Molecule | |||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 2 | |
| dN–H1 | 1.018 | 1.019 | 1.018 | 1.019 | 1.018 | 1.009 | 1.018 | 0.965 |
| dO⋯1HN | 1.821 | 1.847 | 2.013 | 1.798 | 1.808 | — | 1.829 | 1.974 |
| dNH1⋯F/OMe | — | 2.310 | 1.938 | — | — | 1.895 | — | 2.322 |
| φNH1O | 139.2 | 138.3 | 130.1 | 139.94 | 139.71 | — | 138.60 | 139.2 |
| φNH1F/NH1OMe | — | 110.0 | 128.4 | — | — | 137.34 | — | 109.3 |
| Φ1N1C5C4C7 | −2.41 | −6.30 | −0.68 | −2.41 | −2.60 | — | −2.43 | −2.4(3) |
| Φ2N1C8C9C10 | −22.82 | 38.24 | −15.025 | 21.86 | 18.31 | 2.24 | −26.96 | 39.8(3) |
| Energy of HB (EHB) (kcal mol−1) | — | −2.823 | 1.283 | — | — | 2.902 | — | — |
DFT computations
The DFT based computations carried out also reinforces the NMR and XRD findings. The lowest energy structures were optimized using Gaussian09 suite with B3LYP/6-311+g(d,p) level of basis set using chloroform as the default solvent.55 The optimized structure of molecule 2 is reported in Fig. 11 and of molecules 1 and 3 in Fig. S40 and S41† respectively, and the computed structural parameters are provided in Table 2. The formation of three-center HB usually increases the D–H bond lengths with decrease in the H⋯X distance and D–H⋯A bond angle4 tending towards the planar geometry. Thus, the theoretical computations highlight the following aspects: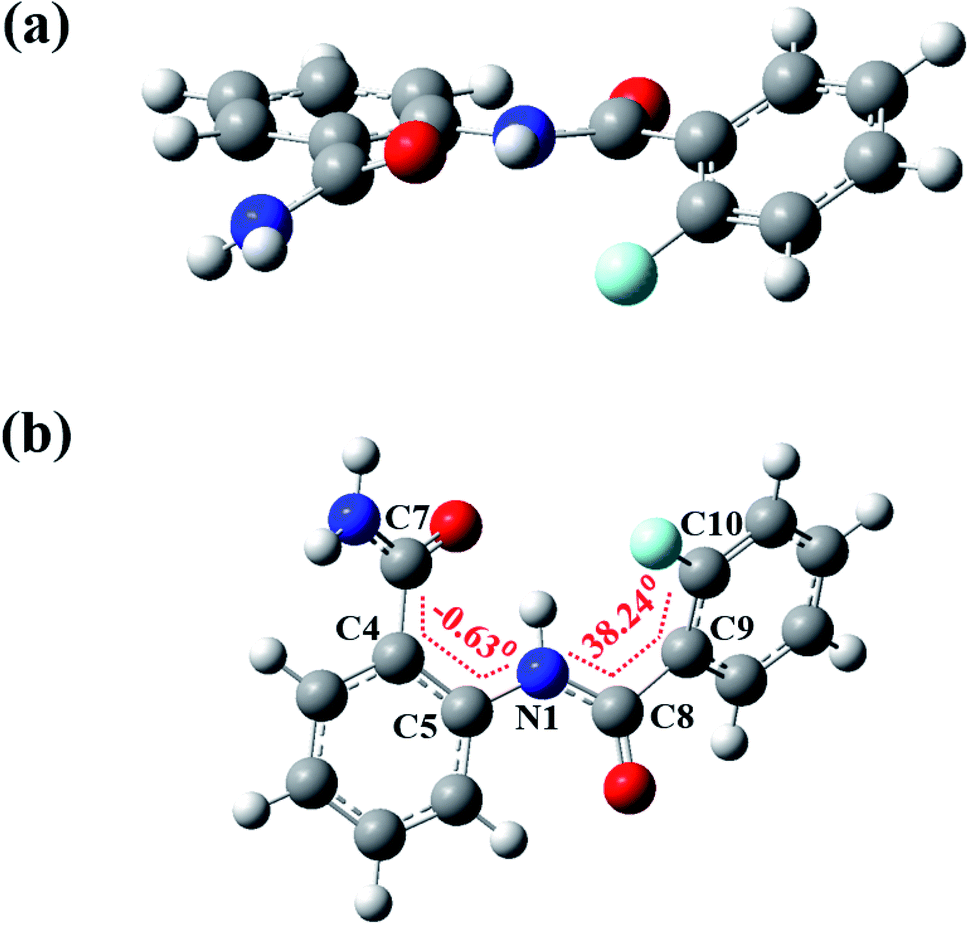 | ||
| Fig. 11 The DFT optimized spatial structure of molecule 2; (a) and (b) are two different projections. | ||
• The bond length (dN–H1) of molecule 2 (1.019 Å) is observed to be longer than the molecule 1 (1.018 Å) whereas the molecule 3 showed no significant change (1.018 Å).
• The distance between H and the X (X = F or OMe) was observed 2.310 Å and 1.938 Å in the molecules 2 and 3 respectively, suggesting the presence of a weak HB.
• The N–H1⋯O bond angles in molecules 1, 2 and 3 were determined to be, 139.2°, 138.3° and 130.1° respectively, whereas the N–H1⋯X (X = F or OMe) bond angles in molecules 2 and 3 were found to be 110° and 128.4°, respectively, suggesting the comparatively strong N–H1⋯OC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) HB than N–H1⋯X.
HB than N–H1⋯X.
• The molecules 1–3 lacks the planarity which is also noticed on comparing the torsion angles (Table 2). In the molecule 2 the torsion angle (Φ2N1C8C9C10 = 38.24°) was found to be higher than in the molecule 1 (Φ2N1C8C9C10 = −22.82°), whereas in the molecule 3 it was observed much close to the planarity (Φ2N1C8C9C10 = −15.02°), which indicates the stronger interaction by methoxy group compared to the fluorine substituent.
• The energies of HBs (EHB) were calculated as −2.823 and −1.283 kcal mol−1 for molecules 2 and 3 respectively, showing agreement with the presence of weak HBs.
• The substitution of X (F or OMe) in the molecules 2 and 3 leads to the increase in the O⋯H distance in O⋯1H–N (dO⋯H1). All the O⋯H distances are reported in the Table 2.
The theoretically computed structural parameters of the molecules 1–3 are also compared with those of molecules 4–7 in the Table 2. The DFT optimized minimum energy structures of molecules 4–7 are reported in Fig. S42–S45 of ESI.† The displacement of X (F or OMe) from ortho to meta position in the molecules 4 and 7 resulted in the decrease in the O⋯H distances (dO⋯H1 = 1.798 Å and 1.829 Å, respectively) compared to the molecules 2 and 3 (dO⋯H1 = 1.847 Å and dO⋯H1 = 2.013 Å, respectively), indicating the strong O⋯H HB in the molecules 4 and 7. However, on shifting the amide group from ortho to meta position in the molecule 6, the strength of observed H⋯F HB increases and displayed the planarity in the structure with a torsion angle of 2.24° which is significantly less compared to that of molecule 2 (Φ2N1C8C9C10 = 38.24°) (ESI, Fig. S44†). The chemical shift of NH1 (δNH1) computed from the DFT optimized minimum energy structures and the experimently observed chemical shifts of molecules 1–7 are reported in Table S3.† The results obtained from DFT and single crystal XRD for molecule 2 are compared in Table 2.
Conclusions
The presence of weak HB in the N-bezoylanthranilamide and its derivatives is substantiated by one- and two-dimensional NMR experimental investigations. The strong correlated peak in the 2D NOESY (molecule 1) and 2D HOESY (molecule 2) spectra ascertain the spatial propinquity between NH1 and X (F or methoxy), leading to the existence of bifurcated HB. The doublet for NH1 proton provided clear evidence for the HB between 19F and NH1 proton. The residual 1hJFH (∼60%) in the high polarity solvent DMSO-d6 and 2D 1H–1H NOESY experiments confirmed the existence of favorable cis conformers for the investigated molecules. The rivalry between N–H⋯X andCO⋯H–N types of HBs is perceived as unusual shielding in NH1 resonance frequency of molecules 2 and 3 and comparatively small 1hJFH coupling in the molecule 2. The NMR experimental findings are strongly supported by the single crystal XRD and DFT computational studies.
Experimental
All the NMR spectra were recorded using 400 MHz and 800 MHz spectrometers at 298 K, except for the variable temperature studies. The TMS was used as an internal reference to measure the proton chemical shifts. The synthesized molecules were characterized by electron spray ionization mass spectrometry (ESI-HRMS) and various one- and two-dimensional NMR techniques. The commercially available chemicals, including deuterated solvents, were purchased and used as received. The XRD data was collected on a diffractometer with Mo Kα radiation. The structure was solved by direct methods using SHELXS97 (ref. 56) and refined in the spherical atom approximation (based on F2) by SHELXL97 (ref. 56) using the WinGX suite (ref. 57).General synthesis of molecules 1 to 7
The 1 equivalent of benzoyl chloride (500 mg, 3.67 mmol) of interest and pyridine (290.29 mg, 3.67 mmol) was added dropwise to the 1.09 equivalent of amino benzamide (4.003 mmol) of interest solution in 15 ml of chloroform at 0 °C. After that, the ice bath is removed, and the reaction mixture was stirred at room temperature for 1 hour. A precipitate obtained was filtered and washed with a copious amount of water. The trace of pyridine was evaporated by adding toluene solvent. The formation of N-benzoylanthranilamide and its derivatives was characterized by electron spray ionization mass spectrometry (ESI-HRMS) and using NMR techniques.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
ST is grateful to CSIR for Senior Research Fellowship. NA is grateful IISc for RA position. NS gratefully acknowledges the generous financial support by the Science and Engineering Research Board (SERB), New Delhi (Grant Number: CRG/2018/002006).References
- B. Kojić-Prodić and K. Molčanov, Acta Chim. Slov., 2008, 55, 692 Search PubMed.
- G. C. Pimentel and A. L. McClellan, Annu. Rev. Phys. Chem., 1971, 22, 347 CrossRef CAS.
- P. Schuster and P. Wolschann, Annu. Rev. Biochem., 1999, 960, 947 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637 CAS.
- R. Taylor, O. Kennard and W. Versichel, J. Am. Chem. Soc., 1984, 106, 244 CrossRef CAS.
- G. A. Jeffrey and H. A. Maluszynska, J. Mol. Struct., 1986, 147, 127 CrossRef CAS.
- C. Z. Gómez-Castro, I. I. Padilla-Martínez, E. V. García-Báez, J. L. Castrejón-Flores, A. L. Peraza-Campos and F. J. Martínez-Martínez, Molecules, 2014, 19, 14446 CrossRef PubMed.
- A. Lakshmipriya and N. Suryaprakash, J. Phys. Chem. A, 2016, 120, 7810 CrossRef CAS PubMed.
- E. S. Feldblum and I. T. Arkin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4085 CrossRef CAS PubMed.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, NY, 3rd edn, 1960, vol. 30, p. 464 Search PubMed.
- L. Pauling and M. Delbrück, Science, 1940, 92, 77 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- A. Tressaud, J. Fluorine Chem., 2011, 132, 651 CrossRef CAS.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 1939 Search PubMed.
- J. D. Dunitz, ChemBioChem, 2004, 5, 614 CrossRef CAS PubMed.
- P. A. Champagne, J. Desroches and J. F. Paquin, Synthesis, 2015, 47, 306 CAS.
- H. J. Schneider, Chem. Sci., 2012, 3, 1381 RSC.
- J. D. Dunitz and R. Taylor, Chem. –Eur. J., 1997, 3, 89 CrossRef CAS.
- J. A. K. Howard, V. J. Hoy, D. O'Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613 CrossRef CAS.
- S. K. Mishra and N. Suryaprakash, Molecules, 2017, 22, 423 CrossRef PubMed.
- F. J. Martínez-Martínez, I. I. Padilla-Martínez, M. A. Brito, E. D. Geniz, R. C. Rojas, J. B. R. Saavedra, H. Höpfl, M. Tlahuextl and R. Contreras, J. Chem. Soc., Perkin Trans., 1998, 2, 401 RSC.
- K. Pervushin, A. Ono, C. Fernández, T. Szyperski, M. Kainosho and K. Wüthrich, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14147 CrossRef CAS PubMed.
- A. K. Patel, S. K. Mishra, K. Krishnamurthy and N. Suryaprakash, RSC Adv., 2019, 9, 32759 RSC.
- T. M. Barbosa, R. V. Viesser, L. G. Martins, R. Rittner and C. F. Tormena, ChemPhysChem, 2018, 19, 1358 CrossRef CAS PubMed.
- R. J. Abraham, S. L. R. Ellison, M. Barfield and W. A. Thomas, J. Chem. Soc. Perkin Trans. 2, 1987, 977 RSC.
- J. C. Hierso, Chem. Rev., 2014, 114, 4838 CrossRef CAS PubMed.
- H. Takemura, M. Kotoku, M. Yasutake and T. Shinmyozu, Eur. J. Org. Chem., 2004, 2019 CrossRef CAS.
- R. A. Cormanich, M. P. Freitas, C. F. Tormena and R. Rittner, RSC Adv., 2012, 2, 4169 RSC.
- S. J. Grabowski, Chem. Rev., 2011, 111, 2597 CrossRef CAS PubMed.
- I. Hyla-Kryspin, G. Haufe and S. Grimme, Chem. –Eur. J., 2004, 10, 3411 CrossRef CAS PubMed.
- J. Hilton and L. H. Sutcliff, Progress in NMR Spectroscopy, 1975, 10, 27 CrossRef.
- Y. A. Cheburkov, N. Mukhamadalier and I. L. Knunyants, Tetrahedron, 1968, 24, 1341 CrossRef CAS.
- V. R. Aldilla, R. Chen, A. D. Martin, C. E. Marjo, A. M. Rich, D. S. Black, P. Thordarson and N. Kumar, Sci. Rep., 2020, 10, 770 CrossRef CAS PubMed.
- R. Kuppusamy, M. Yasir, E. Yee, M. Willcox, D. S. Blacka and N. Kumar, Org. Biomol. Chem., 2018, 16, 5871 RSC.
- S. J. Barraza, P. C. Delektaf, J. A. Sindac, C. J. Dobryf, J. Xiang, R. F. Keep, D. J. Millerf and S. D. Larsen, Bioorg. Med. Chem., 2015, 23, 1569 CrossRef CAS PubMed.
- D. O. Arnaiz, Y. L. Chou, B. D. Griedel, R. E. Karanjawala, M. J. Kochanny, W. Lee, A. M. Liang, M. M. Morrissey, G. B. Phillips, K. L. Sacchi, S. T. Sakata, K. J. Shaw, R. M. Snider, S. C. Wu, B. Ye and Z. Zhao, US. Pat., 6140351, October 31, 2000 Search PubMed.
- B. Ye, D. O. Arnaiz, Y. L. Chou, B. D. Griedel, R. Karanjawala, W. Lee, M. M. Morrissey, K. L. Sacchi, S. T. Sakata, K. J. Shaw, S. C. Wu, Z. Zhao, M. Adler, S. Cheeseman, W. P. Dole, J. Ewing, R. Fitch, D. Lentz, A. Liang, D. Light, J. Morser, J. Post, G. Rumennik, B. Subramanyam, M. E. Sullivan, R. Vergona, J. Walters, Y. X. Wang, K. A. White, M. Whitlow and M. J. Kochanny, J. Med. Chem., 2007, 50, 2967 CrossRef CAS PubMed.
- P. Zhang, L. Bao, J. F. Zuckett, Z. J. Jia, J. Woolfrey, A. Arfsten, S. Edwards, U. Sinha, A. Hutchaleelaha, J. L. Lambing, S. J. Hollenbach, R. M. Scarborough and B. Y. Zhu, Bioorg. Med. Chem. Lett., 2004, 14, 989 CrossRef CAS PubMed.
- N. Masaru and W. Chihiro, Chem. Lett., 1992, 21, 809 CrossRef.
- G. N. Manjunatha Reddy, M. V. Vasantha Kumar, T. N. Guru Row and N. Suryaprakash, Phys. Chem. Chem. Phys., 2010, 12, 13232 RSC.
- P. Dhanishta, P. Sai Siva Kumar, S. K. Mishra and N. Suryaprakash, RSC Adv., 2018, 8, 11230 RSC.
- S. K. Mishra and N. Suryaprakash, RSC Adv., 2015, 5, 86013 RSC.
- N. Arya, S. K. Mishra and N. Suryaprakash, New J. Chem., 2019, 43, 13134 RSC.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249 CrossRef CAS PubMed.
- P. Dhanishta, S. K. Mishra and N. Suryaprakash, J. Phys. Chem. A, 2018, 122, 199 CrossRef CAS PubMed.
- S. K. Mishra and N. Suryaprakash, Phys. Chem. Chem. Phys., 2015, 17, 15226 RSC.
- S. R. Chaudhari, S. Mogurampelly and N. Suryaprakash, J. Phys. Chem. B, 2013, 117, 1123 CrossRef CAS PubMed.
- R. Dalterio, X. H. S. Huang and K. L. Yu, Appl. Spectrosc., 2007, 61, 603 CrossRef CAS PubMed.
- L. Hennig, K. Ayala-Leon, J. Angulo-Cornejo, R. Richter and L. Beyer, J. Fluorine Chem., 2009, 130, 453 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2049223. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02538d |
| This journal is © The Royal Society of Chemistry 2021 |