
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed one-pot synthesis of quinoxalines: transfer hydrogenative condensation of 2-nitroanilines with vicinal diols†
Ramachandra Reddy Puttaa,
Simin Chuna,
Seok Beom Leea,
Junhwa Honga,
Dong-Chan Ohb and
Suckchang Hong *a
*a
aResearch Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea. E-mail: schong17@snu.ac.kr
bNatural Products Research Institute, College of Pharmacy, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 08826, Republic of Korea
First published on 20th May 2021
Abstract
Here, we report iron-catalyzed one-pot synthesis of quinoxalines via transfer hydrogenative condensation of 2-nitroanilines with vicinal diols. The tricarbonyl (η4-cyclopentadienone) iron complex, which is well known as the Knölker complex, catalyzed the oxidation of alcohols and the reduction of nitroarenes, and the corresponding carbonyl and 1,2-diaminobenzene intermediates were generated in situ. Trimethylamine N-oxide was used to activate the iron complex. Various unsymmetrical and symmetrical vicinal diols were applied for transfer hydrogenation, resulting in quinoxaline derivatives in 49–98% yields. A plausible mechanism was proposed based on a series of control experiments. The major advantages of this protocol are that no external redox reagents or additional base is needed and that water is liberated as the sole byproduct.
Introduction
Quinoxaline is a nitrogen-containing heterocyclic compound with both benzene and pyrazine rings. Quinoxaline structures are widely found in natural products and biologically active molecules.1 Several commercial drugs, such as quinacillin, sulfaquinoxaline, clofazimine and echinomycin, contain quinoxaline moieties. Due to their immense importance for not only medicinal chemistry but also organic semiconductors2 and fluorescent dyes,3 diverse synthetic routes have been developed so far. Among such methods, the traditional method involves the double condensation of 1,2-diamines with dicarbonyl compounds.4 Alternatively, oxidative annulation of 1,2-diamines with α-hydroxy ketones5 or phenacyl bromides6 and epoxide opening with 1,2-diamine have also been developed.7 However, the use of additional oxidants, harsh reaction conditions, multistep processes and low atom efficiency are the major concerns for these protocols. In the last decade, several research groups have made considerable progress in the synthesis of quinoxalines by employing vicinal diols via an acceptorless dehydrogenative coupling strategy (ADC).8 In this strategy, alcohol is oxidized to carbonyl compounds in situ, and hydrogen gas is liberated as a byproduct. From the viewpoint of atom economy, the transfer hydrogenation (TH) strategy has emerged as a more attractive method for the construction of C–C and C–N bonds.9 In transfer hydrogenative condensation, alcohols can act as hydrogen donors and coupling components at the same time, which are easily handled, renewable and inexpensive. On the other hand, nitroarenes are usually considered precursors of anilines, which can be hydrogenated as hydrogen acceptors in the TH process.10 Nitroarenes are highly versatile and common synthetic blocks in the organic synthesis and chemical industries. Through the TH process, alcohols could serve as electrophilic carbonyl intermediates and nitroarenes as nucleophilic aniline intermediates (Fig. 1). The main advantage of this protocol is that no additional oxidant and reductant are required and water is generated as the only byproduct. Thus, it is considered an environmentally benign process and a green alternative.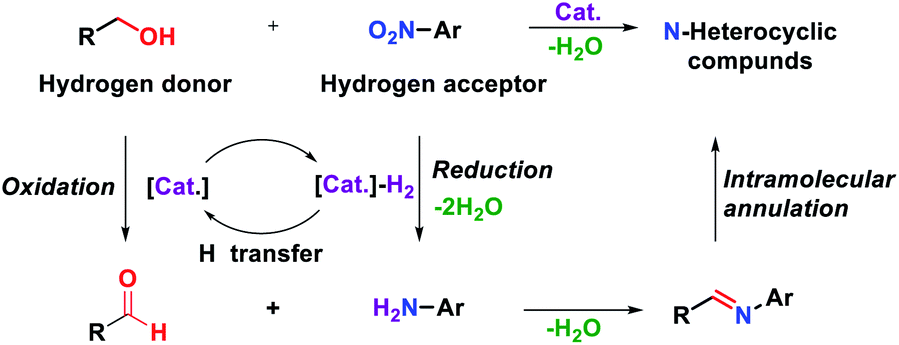 | ||
| Fig. 1 Transfer hydrogenation process between nitroarenes and alcohols. | ||
In 2012, Climent et al. developed the synthesis of quinoxalines by using a Au-heterogeneous catalyst from 1,2-dinitrobenzenes and vicinal diols.11 However, an external high-pressure hydrogen source was used for the reduction of 1,2-dinitrobenzenes. Inspired by that work, Zhang's group reported ruthenium- and dppp-catalyzed transfer hydrogenative condensation to synthesize quinoxalines from 2-nitroanilines and vicinal diols (Scheme 1).12 Later, Kundu's group employed Ir, Co and NiBr2/1,10-phenanthroline catalysts in the transfer hydrogenation between 2-nitroanilines and vicinal diols to afford quinoxalines.13 Nevertheless, these reaction systems required an additional base and excess amount of alcohols to achieve satisfactory yields. Therefore, it is still highly desirable to develop new methods using nonprecious, earth-abundant catalysts without an additional base. Among transition metals, iron is the most abundant metal with a low price and low toxicity. For these reasons, the reduction of nitro arene using an iron-based catalyst and the synthesis of N-heterocycles through redox annulation have been reported.14 More recently, we reported the synthesis of N-heterocycles from nitroarenes and alcohols via a transfer hydrogenative condensation catalyzed by cyclopentadienyl iron complex, which is well known as Knölker complex.15 Based on this work, we reasoned that the transfer hydrogenative condensation of 2-nitroanilines with vicinal diols might be possible, leading to the formation of quinoxalines. During the preparation of this manuscript, transfer hydrogenative synthesis of quinoxaline using same iron catalyst was reported by Darcel's group.16 However, they still used excess amounts of base and alcohol in their method. Herein, we report the iron-catalyzed one-pot synthesis of quinoxalines from 2-nitroanilines with vicinal diols by a transfer hydrogenative condensation strategy. No additional base or external redox agents were required, and water was generated as the sole byproduct (Scheme 1).
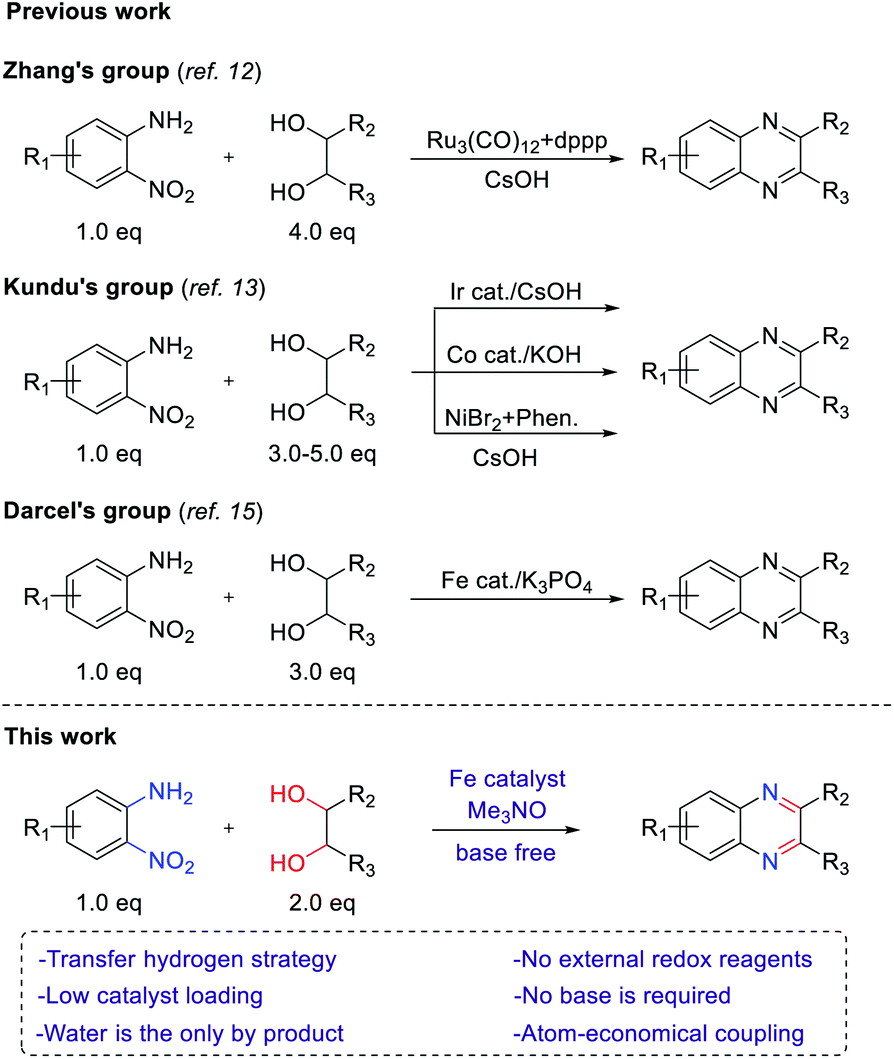 | ||
| Scheme 1 Quinoxaline synthesis from 2-nitroanilines and vicinal diols via transfer hydrogenative condensation. | ||
Results and discussion
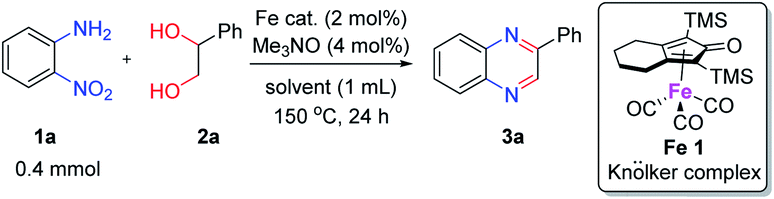
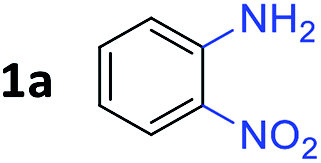
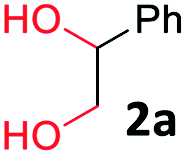
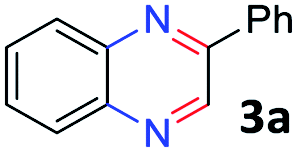
To determine the optimal condition of transfer hydrogenative condensation for the quinoxalines, we chose 2-nitroaniline 1a and 1-phenylethane-1,2-diol 2a as model substrates (Table 1). Initially, 2 mol% FeCl2 as the catalyst and o-xylene (1 mL) as the solvent were employed, and the reaction mixture was heated at 150 °C for 24 h (entry 1). Unfortunately, desired 2-phenyl quinoxaline 3a was observed in low yield. To find more efficient catalysts, various iron complexes were screened (entries 1–4). Cyclopentadienyl iron complex Fe 1 exhibited the highest activity in the formation of desired product 3a. Trimethylamine N-oxide (Me3NO) was used to oxidize CO ligand and liberate a vacant site on iron carbonyl complex. Next, we applied different kinds of solvents, such as toluene, CPME, chlorobenzene and 1,4-dioxane (entries 5–8). Toluene showed the best result, affording 3a in 84% isolated yield (entry 5). Both decreasing and increasing the amount of alcohol (1.0 eq. or 3.0 eq.) showed lower conversion to the desired product (entries 9 and 10). When the catalyst loading was increased from 2 to 3 mol%, an increased yield for the desired product 3a was obtained (87%, entry 11). Hence, we chose the optimal reaction conditions as indicated in Table 1, entry 11. It is worth mentioning that trace amounts of 2-phenyl-benzimidazole were observed as byproducts, which could be generated via annulation of 1,2-diaminobenzene with benzaldehyde. We supposed that some 2a might undergo C–C bond cleavage to form benzaldehyde during the dehydrogenative process.
|
||||
|---|---|---|---|---|
| Entry | 2a (equiv.) | Fe cat. | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.4 mmol), 2a (0.8 mmol), catalyst (2 mol%), Me3NO (4 mol%) and solvent (1 mL) in a sealed tube at 150 °C for 24 h.b Isolated yield.c 3 mol% of Fe 1 and 6 mol% of Me3NO. | ||||
| 1 | 2.0 | FeCl2 | o-Xylene | 13 |
| 2 | 2.0 | FeCl3·6H2O | o-Xylene | <5 |
| 3 | 2.0 | Fe2(CO)9 | o-Xylene | 40 |
| 4 | 2.0 | Fe 1 | o-Xylene | 62 |
| 5 | 2.0 | Fe 1 | Toluene | 84 |
| 6 | 2.0 | Fe 1 | CPME | 41 |
| 7 | 2.0 | Fe 1 | Chlorobenzene | 66 |
| 8 | 2.0 | Fe 1 | 1,4-Dioxane | 30 |
| 9 | 1.0 | Fe 1 | Toluene | 73 |
| 10 | 3.0 | Fe 1 | Toluene | 67 |
| 11c | 2.0 | Fe 1 | Toluene | 87 |
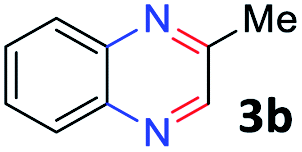
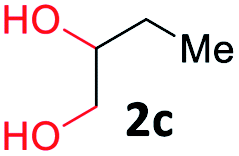
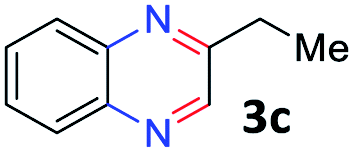
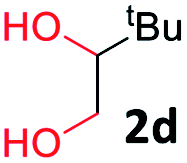
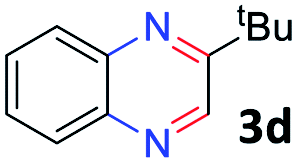
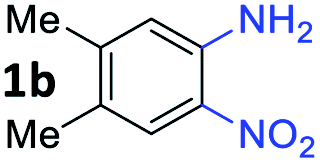
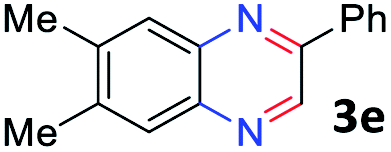
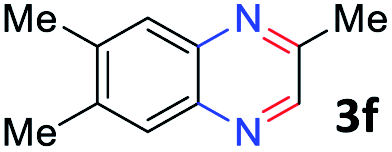
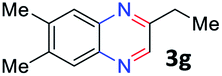
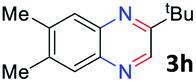
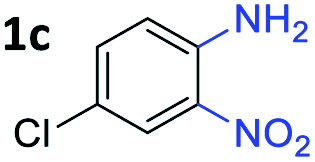
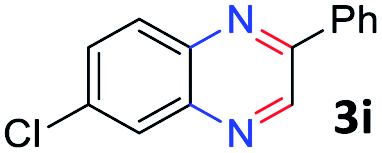
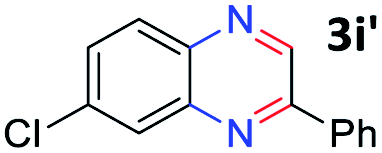
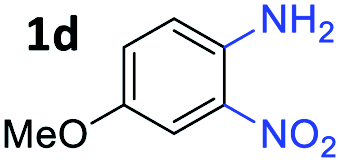
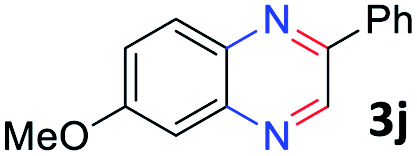
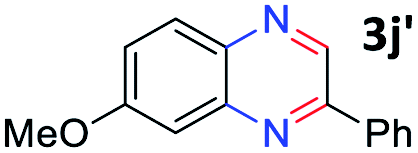
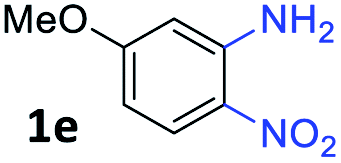
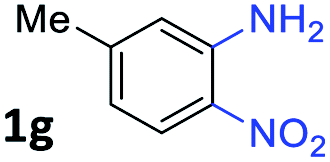
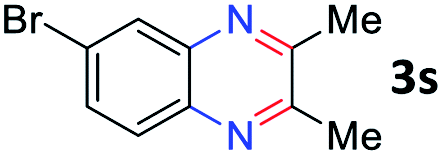
With the optimized reaction conditions, we explored the versatility and limitations of the synthetic method. First, we explored the scope of unsymmetrical vicinal diols 2 with a variety of 2-nitroanilines 1 for transfer hydrogenative condensation (Table 2). Alkyl-substituted unsymmetrical vicinal diols also underwent smooth condensation followed by cyclization with 2-nitroaniline 1a, affording 2-alkyl-substituted quinoxalines 3b–d in good yields. 4,5-Dimethyl-2-nitroaniline 1b reacted well with aryl- and alkyl-substituted unsymmetrical vicinal diols, resulting in 2-substituted quinoxalines 3e–3h with excellent yields (82–94%). Reaction of 4-chloro-2-nitroaniline 1c with 2a gave the two regioisomers 3i and 3i′ in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 ratio. Similarly, 4-methoxy-2-nitroaniline 1d and 5-methoxy-2-nitroaniline 1e were annulated with 2a, resulting in a mixture of regioisomers 3j and 3j′ in 1:1.9 and 1:1.6 ratios, respectively. In addition, the reaction of 3-nitropyridin-4-amine 1f with 2a also gave an inseparable mixture of regioisomers 3k and 3k′. Even if the regioselectivities are not good, it was found that more 2,7-disubstituted quinoxalines are produced than 2,6-disubstituted quinoxalines, regardless of the substituent position on the starting materials. Generally, aryl-substituted vicinal diols 2a showed better yields than alkyl-substituted vicinal diols 2b–2d. Moreover, electron-donating groups (Me and OMe) containing nitroanilines 1b and 1d–1e afforded quinoxalines in higher yields than electron-deficient groups (Cl) containing substrate 1c.
:1.5 ratio. Similarly, 4-methoxy-2-nitroaniline 1d and 5-methoxy-2-nitroaniline 1e were annulated with 2a, resulting in a mixture of regioisomers 3j and 3j′ in 1:1.9 and 1:1.6 ratios, respectively. In addition, the reaction of 3-nitropyridin-4-amine 1f with 2a also gave an inseparable mixture of regioisomers 3k and 3k′. Even if the regioselectivities are not good, it was found that more 2,7-disubstituted quinoxalines are produced than 2,6-disubstituted quinoxalines, regardless of the substituent position on the starting materials. Generally, aryl-substituted vicinal diols 2a showed better yields than alkyl-substituted vicinal diols 2b–2d. Moreover, electron-donating groups (Me and OMe) containing nitroanilines 1b and 1d–1e afforded quinoxalines in higher yields than electron-deficient groups (Cl) containing substrate 1c.
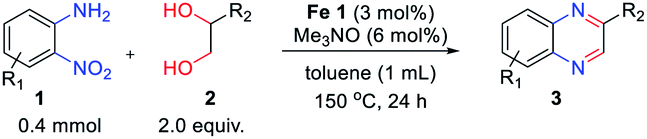
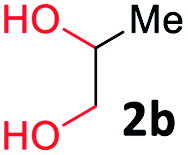
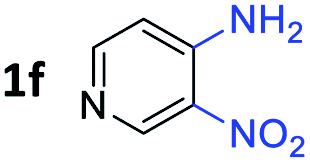
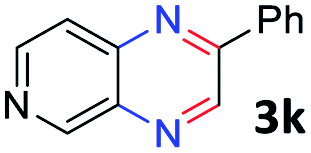
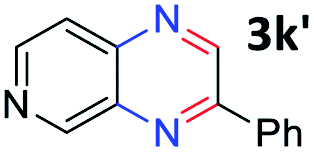
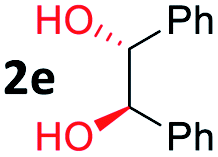
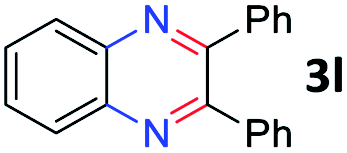
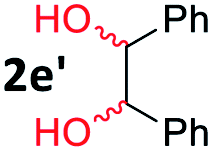
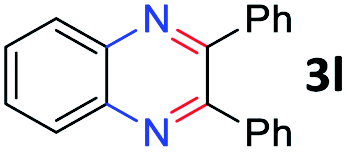
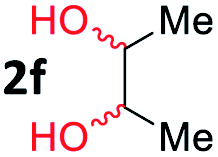
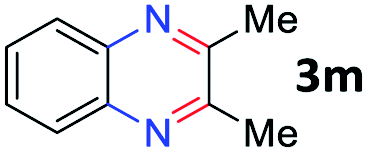
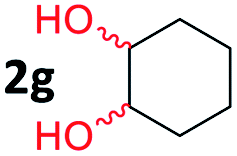
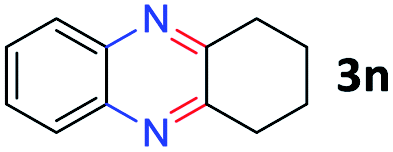
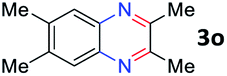
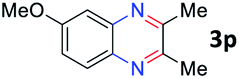
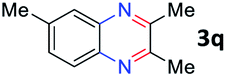
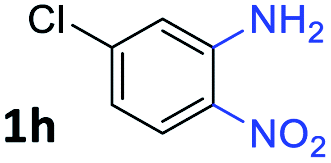
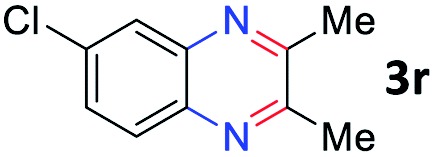
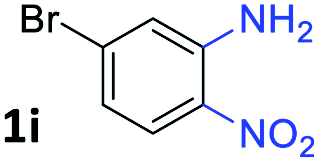
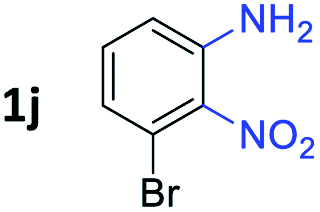
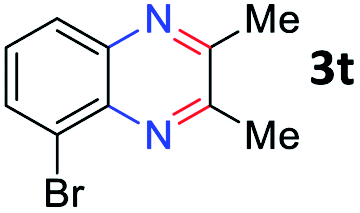
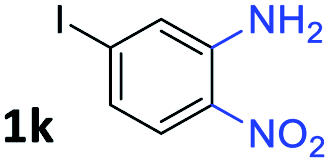
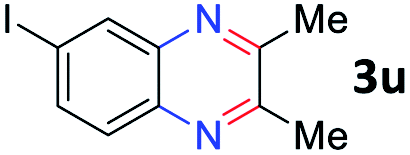
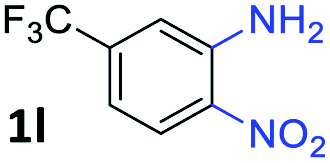
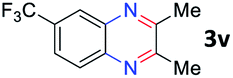
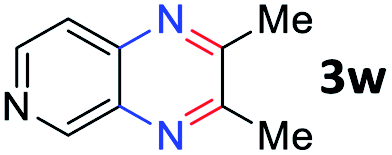
After investigating the scope of the unsymmetrical diols, we turned our attention toward the transfer hydrogenative condensation of symmetrical diols 2 with 2-nitroanilines 1, as illustrated in Table 3. The reaction of (R,R)-1,2-diphenylethane-1,2-diol 2e with 2-nitroaniline 1a afforded a low yield of the desired product 3l along with a significant amount of 2-phenyl benzimidazole as a side product. In addition, when meso 2e' was applied to the reaction, the similar result was obtained. This result implies that the stereocenter of diol does not affect the reaction. In the case of 1,2-dimethylethane-1,2-diol 2f, the corresponding quinoxaline product 3m was obtained in 80% yield, even if a longer reaction time was needed for full conversion. On the other hand, cyclohexane-1,2-diol 2g failed to produce desired product 3n under the reaction conditions. This result was in contrast to previous Darcel's conditions using bases.16 We expected that the base influences the imine condensation or the annulation process rather than transfer hydrogenation. Therefore, we thought the ring strain of fused tricyclic product 3n retards the annulation process. Because 1,2-diaminobenzene and cyclic ketone could be observed in this reaction, that are supposed as intermediates went through transfer hydrogenation. Based on the above results, we chose 2f as a coupling partner to explore the reaction scope of 2-nitroaniline with a symmetrical diol. Various substituted 2-nitroanilines 1 reacted well with 2f, resulting in the corresponding products with moderate to good yields 3o–3v (61–80%). In particular, 2-nitroanilines bearing electron-donating groups (Me and OMe) afforded the respective products 3o–3q in good yields. Although 2-nitroanilines with electron-withdrawing groups (Cl, Br, I and CF3) were less efficient than electron-rich 2-nitroanilines (3r–3v vs. 3o–3q), the halogen groups were tolerated well under the reaction conditions. No dehalogenated products were observed in the reaction with 2-nitroanilines containing halogen substituents (Cl, Br and I), which could be affected in Darcel's conditions.16 The tolerance of halogen groups opens up a pathway for further modification of the quinoxaline products. 3-Nitropyridin-4-amine 1f underwent smooth cyclization with 2e, leading to desired product 3x, whereas no desired product was observed with 2f.
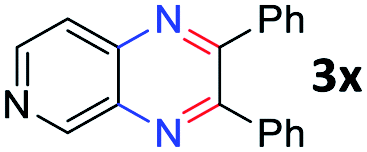
Encouraged by the above results, 1,2,3-triol 4 (glycerol) was applied in this reaction system instead of vicinal diol (Scheme 2). Interestingly, 2-benzimidazoyl quinoxaline product 5a was obtained by the reaction of 2-nitroaniline 1a with glycerol 4, and no other products were observed. We expected that the quinoxaline ring might be formed at first and that the benzimidazole moiety can be formed followed by cyclocondensation of another 2-nitroaniline with a quinoxaline-2-aldehyde intermediate. The reaction of 4,5-dimethyl-2-nitroaniline 1b also affords the desired product 5b in moderate yield. However, in the case of 4-methoxy-2-nitroaniline 1d, a messy products were observed in TLC, which are expected as a mixture of regioisomers. And only trace amount of product was observed in the reaction of 3-nitropyridin-4-amine 1f. Although the scope of the reaction is somewhat limited, this result opens up a new route for the synthesis of 2-benzimidazoyl quinoxaline derivatives via one-pot transfer hydrogenative condensation.
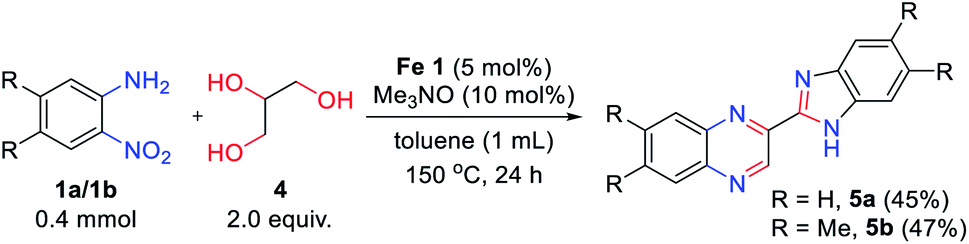 | ||
| Scheme 2 Transfer hydrogenative condensation of 2-nitroaniline with glycerol. | ||
To further extend the scope of the reaction system, we employed 1,2-dinitrobenzene 6 instead of 2-nitroaniline 1 as a coupling partner to explore the possibility of quinoxaline formation (Scheme 3). Under the reaction with twice the amount of hydrogen donor 2a, 1,2-dinitrobenzene 6 resulted in the desired product 3a with high yield (81%).
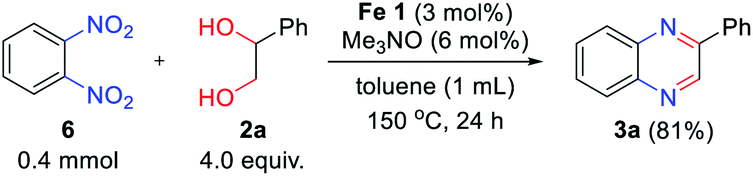 | ||
| Scheme 3 Transfer hydrogenative condensation of 1,2-dinitrobenzene with vicinal diol. | ||
To investigate the mechanism for the developed reaction, few control experiments were performed, as shown in Scheme 4. First, we examined the reaction without a catalyst to investigate the role of the iron complex (Scheme 4a). No reaction was observed in the absence of catalyst, and most of the starting materials remained, which indicates the vital role of iron catalysts in transfer hydrogenation. Next, to determine the intermediates, the reaction of 1b with 2a was interrupted after 12 h (Scheme 4b). Unfortunately, no intermediates were observed; only product 3e was observed in 38% yield, and the starting materials remained. Thus, we can expect that the cyclization step occurs very quickly after transfer hydrogenation. Furthermore, an acceptorless dehydrogenative coupling (ADC) strategy has been applied to determine the feasibility of the formation of quinoxaline. For this purpose, we examined the reaction between 1,2-diaminobenzene 7 and 2a; however, a lower yield of the desired product 3a was observed (31%, Scheme 4c). Based on these results, we speculate that sufficient diol oxidation does not proceed well without a hydrogen acceptor or nitro group. We also suppose that the formation of an imine intermediate from 2-nitroanilines is favored over the formation of a 1,2-diaminobenzene intermediate in the whole reaction process.
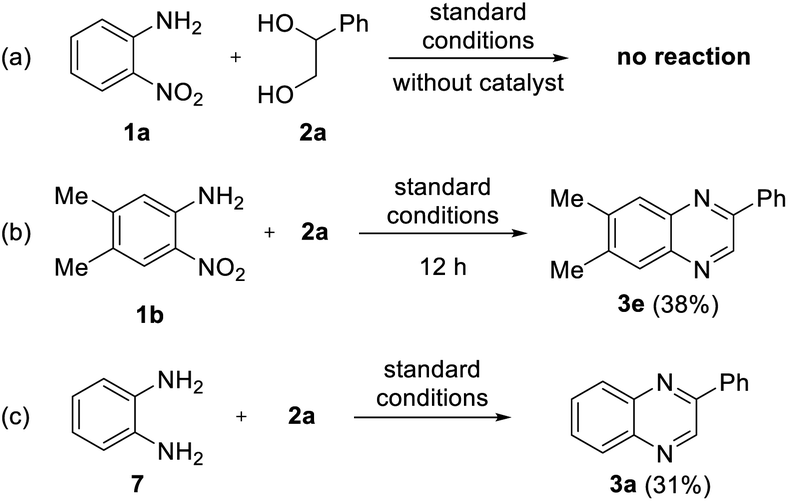 | ||
| Scheme 4 Control experiments. | ||
Based on these results and the related literature,12,13b,15 a plausible mechanism for the transfer hydrogenative condensation of 2-nitroanilines with vicinal diols was proposed, as depicted in Scheme 5. Initially, Me3NO oxidizes a CO ligand on Knölker complex Fe 1 and liberate CO2. The activated catalyst [Fe] is formed and mediates the oxidation of diols 2 to afford the corresponding carbonyl intermediate and [Fe]–H2. Followed by the condensation of carbonyl compounds with 2-nitroanilines 1 produce the α-hydroxyimine intermediate (I or II). Then, α-keto amine intermediates III or IV can be formed by the hydrogenation of nitro groups and oxidation of the remaining alcohol via an iron-mediated transfer hydrogenation process. Finally, the α-keto amine intermediate (III or IV) undergoes intramolecular condensation followed by aromatization and produces the desired products 3 or 3′. Throughout the whole TH process, at least 1.5 equivalents of diol are required as hydrogen donors to reduce one nitro functionality.
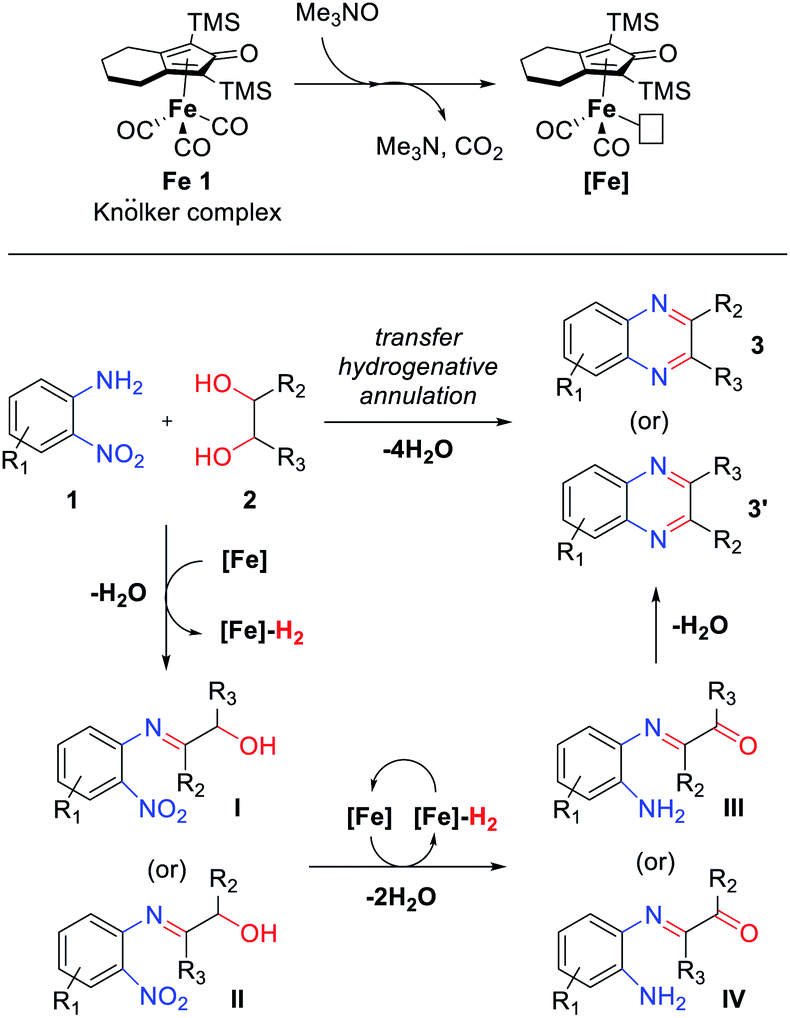 | ||
| Scheme 5 Plausible mechanism for the formation of quinoxalines. | ||
Conclusions
In summary, we have developed an iron-catalyzed transfer hydrogenative condensation process for the one-pot synthesis of quinoxalines from 2-nitroanilines and vicinal diols. A cyclopentadienyl iron complex has been applied to a hydrogen transfer strategy between nitro and alcohol functionalities. In this protocol, alcohols serve as hydrogen donors, and nitroarenes act as hydrogen acceptors. Various unsymmetrical and symmetrical vicinal diols underwent efficient cyclization and produced quinoxaline derivatives in 49–98% yields. A plausible mechanism for quinoxaline formation was proposed based on a series of control experiments. Furthermore, glycerol was successfully employed in the reaction system, leading to a 2-benzimidazoyl quinoxaline product. This result opens up a new route for the synthesis of 2-benzimidazoyl quinoxaline derivatives via the one-pot condensation of 2-nitroaniline with glycerol. Additionally, a hydrogen transfer strategy was applied in the reaction of 1,2-dinitrobenzene with a vicinal diol, resulting in the desired quinoxaline in 81% yield. This hydrogen transfer process can proceed smoothly without the use of external redox reagents and an additional base. Utilization of a nonprecious iron metal catalyst and the generation of water as the sole byproduct make this reaction an eco-friendly method for synthesizing quinoxalines. Further application of this transfer hydrogen protocol in the synthesis of N-heterocycles is ongoing in our laboratory.Author contributions
R. R. Putta – investigation (reaction scope), methodology, writing-original draft. S. Chun – conceptualization, project administration, investigation (preparation of catalyst). S. B. Lee – investigation (reaction scope), resources. J. Hong – investigation (mechanism), formal analysis, validation. D.-C. Oh – data curation, funding acquisition, writing-review & editing. S. Hong – conceptualization, supervision, funding acquisition, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (2021R1C1C1007657 and 2021R1A4A2001251) and Creative-Pioneering Researchers Program through Seoul National University (SNU).Notes and references
-
(a) M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed
; (b) J. A. Pereira, A. M. Pessoa, M. N. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed
-
(a) J. Zhao, H. Li, H. Li, Q. Zhao, H. Ling, J. Li and W. Huang, Dyes Pigm., 2019, 167, 255–261 CrossRef CAS
-
(a) S. Kothavale, K. H. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2019, 11, 17583–17591 CrossRef CAS PubMed
-
(a) M. Ayaz, Z. Xu and C. Hulme, Tetrahedron Lett., 2014, 55, 3406–3409 CrossRef CAS PubMed
-
(a) K. Aghapoor, F. Mohsenzadeh, M. M. Morad and H. R. Darabi, C. R. Chim., 2012, 15, 764–767 CrossRef CAS
-
(a) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Lett., 2015, 145, 1600–1605 CrossRef CAS
-
(a) A. Vidal-Albalat, S. Rodríguez and F. V. González, Org. Lett., 2014, 16, 1752–1755 CrossRef CAS PubMed
-
(a) A. Mondal, M. K. Sahoo, M. Subaramanian and E. Balaraman, J. Org. Chem., 2020, 85, 7181–7191 CrossRef CAS PubMed
-
(a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed
-
(a) V. K. Das, S. Mazhar, L. Gregor, B. D. Stein, D. G. Morgan, N. A. Maciulis, M. Pink, Y. Losovyj and L. M. Bronstein, ACS Appl. Mater. Interfaces, 2018, 10, 21356–21364 CrossRef CAS PubMed
- M. J. Climent, A. Corma, J. C. Hernández, A. B. Hungría, S. Iborra and S. Martínez-Silvestre, J. Catal., 2012, 292, 118–129 CrossRef CAS
- F. Xie, M. Zhang, H. Jiang, M. Chen, W. Lv, A. Zhenga and X. Jiana, Green Chem., 2015, 17, 279–284 RSC
-
(a) S. Shee, K. Ganguli, K. Jana and S. Kundu, Chem. Commun., 2018, 54, 6883–6886 RSC
-
(a) G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed
-
(a) R. R. Putta, S. Chun, S. H. Choi, S. B. Lee, D.-C. Oh and S. Hong, J. Org. Chem., 2020, 85, 15396–15405 CrossRef CAS PubMed
- J. Wu and C. Darcel, J. Org. Chem., 2021, 86, 1023–1036 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02532e |
| This journal is © The Royal Society of Chemistry 2021 |