
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New α-galactosidase-inhibiting aminohydroxycyclopentanes†‡
Patrick Weber *a,
Roland Fischerb,
Seyed A. Nasseric,
Arnold E. Stütza,
Martin Thonhofera,
Stephen G. Withersc,
Andreas Wolfsgrubera and
Tanja M. Wrodnigga
*a,
Roland Fischerb,
Seyed A. Nasseric,
Arnold E. Stütza,
Martin Thonhofera,
Stephen G. Withersc,
Andreas Wolfsgrubera and
Tanja M. Wrodnigga
aGlycogroup, Institute of Chemistry and Technology of Biobased Systems, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria. E-mail: patrick.weber@tugraz.at; Tel: +43 316 873 32078
bInstitute of Inorganic Chemistry, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria
cChemistry Department, University of British Columbia, 2036 Main Mall, Vancouver, BC, V6T 1Z1 Canada
First published on 29th April 2021
Abstract
A set of cyclopentanoid α-galactosidase ligands was prepared from a partially protected ω-eno-aldose via a reliable (2 + 3)-cycloaddition protocol with slightly modified conditions. The obtained N-benzylisoxazolidine ring was selectively opened and the configuration of the hydroxymethylgroup was inverted. Consecutive deprotection provided an aminocyclopentane, which was N-alkylated to furnish a set of potential α-galactosidase inhibitors. Their glycosidase inhibitory activities were screened with a panel of standard glycosidases of biological significance.
1. Introduction
Glycoside hydrolases are a class of abundant and essential enzymes for carbohydrate catabolism, controlled trimming of oligosaccharide motifs in glycoproteins, as well as the release of stored polysaccharides in plants and animals. The Carbohydrate-Active enZYmes database1,2 (CAZY) currently features well over one hundred and sixty sequence-associated families of glycosidases.α-D-Galactosides are, in a quite diverse context, an important class of oligosaccharides and carbohydrate conjugates. Their corresponding hydrolases, α-D-galactosidases, are found in glycoside hydrolase (GH) families 4, 27 (containing the Fabry disease related human lysosomal enzyme), 36, 57, 97 and 110 as well as in GH109. GH 4 and GH109 enzymes follow a mechanism requiring NAD+ and Mn2+ that starts with the oxidation at C-3 followed by β-elimination of the aglycone, with subsequent water addition and reduction,3 while the enzymes of the other five families follow standard Koshland retaining mechanisms.4
α-D-Galactosidases have been employed or modulated in several quite diverse applications. For example, α-galactosidases are frequently employed in combination with proteases to remove oligosaccharides from food to improve their nutritional availability and quality.5,6 Improving the activity of lysosomal α-galactosidase through chaperoning is a new approach to the management of Fabry's disease, a hereditary lysosomal disorder resulting from mutations in the GLA gene that result in catalytically compromised mutants of this essential enzyme.7–11 In a completely different context, α-galactosidases that selectively and efficiently remove the immunodominant α-1,3-galactosyl epitope (Galili antigen) which hampers xenotransplantation from otherwise suitable mammalian sources, have been a focus of organ and tissue transplant medicine.12–14 Similarly, for the purpose of generating red blood cells for readily available universal donor blood, enzymatic conversion of B type red blood cells into type O, by removal of α-1,3-bound galactosides from the cell surface, has been a field of intense and sustained research.15–19
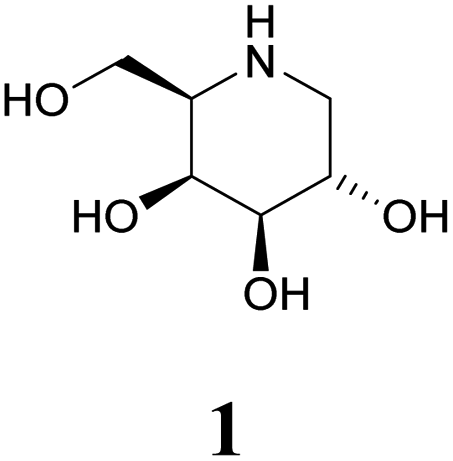
Specific inhibitors of α-galactosidases have been developed as chaperones for treatment of the hereditary lysosomal disorder, Fabry's disease. A prime example is 1,5-dideoxy-1,5-imino-D-galactitol (DGJ, 1), which enhances the residual activity of various α-galactosidase mutants, ameliorating Fabry related symptoms and providing improved quality of life for afflicted patients. α-Galactosidase inhibitors may also help the discovery, purification and characterization of enzymes for managed degradation of pathologically active α-galactosides.
Amongst competitive α-galactosidase inhibitors, compound 1 is the current bench mark (IC50 1.6 nM and 0.24 μM with α-galactosidases from green coffee beans (gcb, GH 27) and E. coli, (GH 36) respectively).20 Homolog 2 inhibits gcb galactosidase with IC50 0.06 μM and E. coli enzyme with IC50 32 μM.21 Contrasting this, structurally related 4-epi-isofagomine 3 exhibits β-galactosidase activity practically exclusively (IC50 200 μM, gcb).22 Conversely, N-alkyl substituted analogues of the corresponding 5-fluoro derivative, such as 4, are sub-micromolar (Ki 0.50 μM) inhibitors of gcb α-galactosidase.23 Pyranoid carbasugar 4-epi-validamine (5) is only a weak inhibitor with IC50 500 μM (gcb) and 890 μM (E. coli), respectively.24 Interestingly, closely related bicyclic analogue 6 is much more potent with Ki 0.54 μM and IC50 80 μM with these enzymes.25
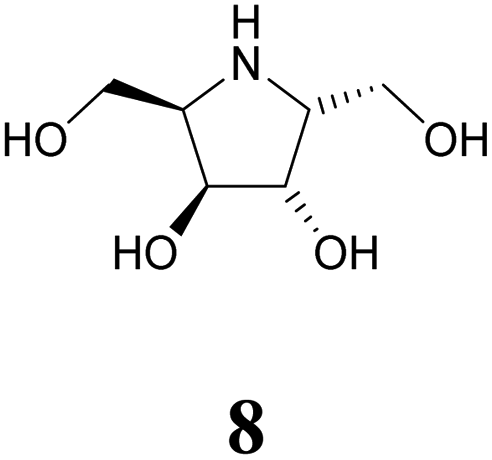
Turning to furanoid structures, 1,4-dideoxy-1,4-imino-D-lyxitol (7) was found to be an inhibitor of gbc α-galactosidase (Ki 0.13 μM).26 Its C-6-homolog, 2,5-dideoxy-2,5-imino-D-altritol (8), exhibited an IC50 of 0.78 μM with α-galactosidase from green coffee beans without any noteworthy inhibition of a panel of other glycosidases.27 Other researchers reported an IC50 of 5.2 μM for compound 8 with the same enzyme.28 These authors also noticed that meso-2,5-dideoxy-2,5-iminogalactitol (10) was an even better inhibitor (IC50 0.19 μM) of gcb α-galactosidase while D-galacto configured aminocyclopentane 12 inhibits it with Ki 0.43 μM.29 In contrast to that, aminocyclopentane 12 does not show any inhibitory activity against bovine liver α-galactosidase.30 Furthermore, a study by Zheng and co-workers31 revealed that non-carbohydrate-based structures such as lansoprazole (IC50 6.4 μM) may also serve as gcb α-galactosidase inhibitors.
Human lysosomal α-galactosidase (Fabrazyme®, GH 27) is strongly inhibited by compounds 1 (Ki 0.23 μM, Fabrazyme®),32 4 (Ki 116 μM, Fabrazyme®),23 8 (IC50 0.69 μM, human lysosomal),27 and moderately by 11 (Ki 110 μM, Fabrazyme®).32 Compound 9, the 1-aminodeoxy derivative of 8, exhibits IC50 0.053 μM (pH 7) and IC50 0.67 μM (pH 4.6) with Fabrazyme®33 (Fig. 1). These data demonstrate that only very few small glycomimetics known thus far exhibit noteworthy activity as well as selectivity for α-galactosidases.
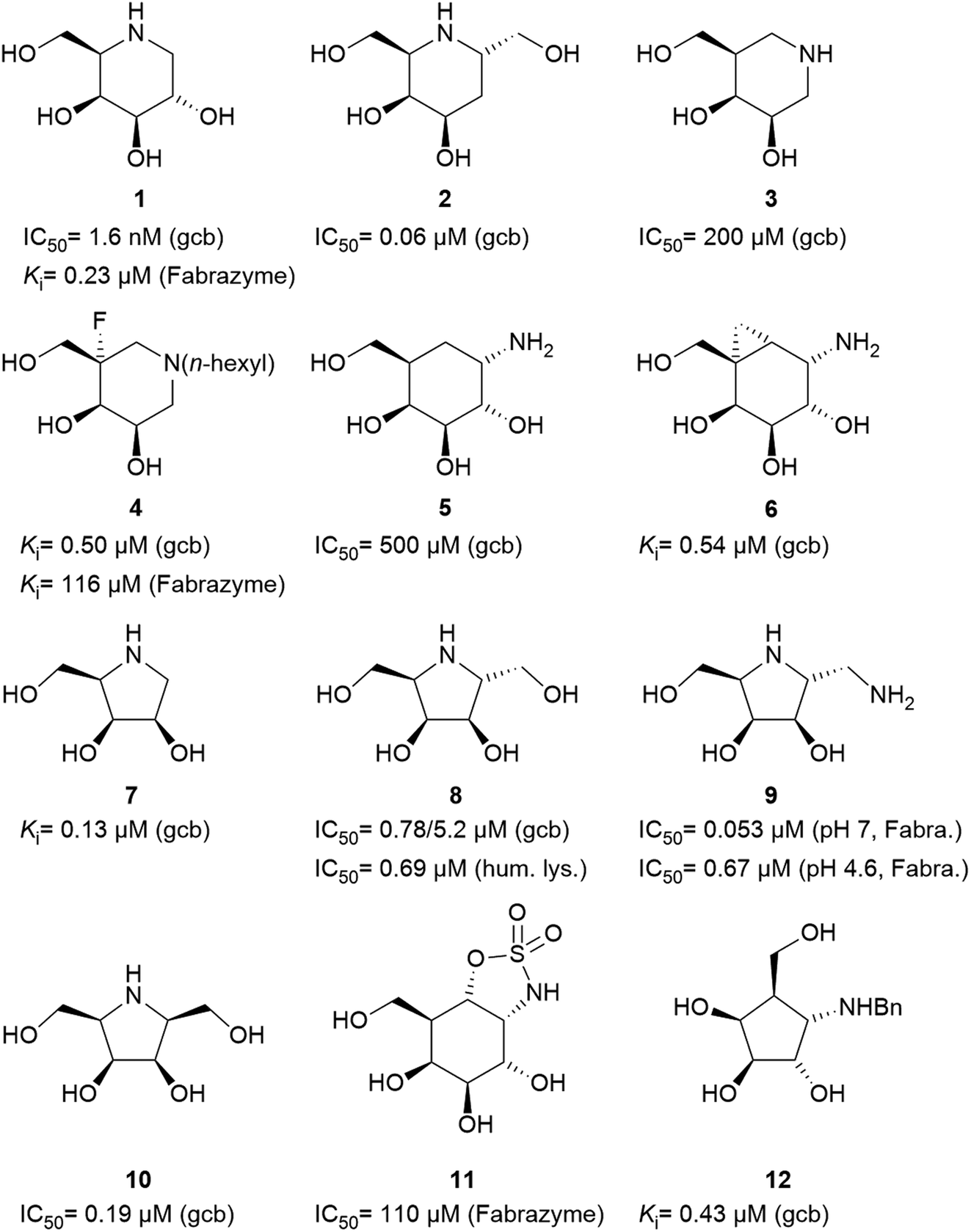 | ||
| Fig. 1 Carbohydrate-related α-galactosidase inhibitors and inhibitory activities. | ||
We have recently been interested in hydroxymethyl-branched di- and trihydroxycyclopentylamines such as compound 12, which may be regarded as resulting from ring contraction by formal homolytic extraction of the sugar ring oxygen and bond formation between C-1 and C-5. The new carbacyclic scaffold maintains the stereochemical information of the corresponding parent free sugar or glycosylamine albeit with the characteristic conformational features of five-membered rings. There are only four leading references in which such sugar analogues are shown as proven D-galactosidase inhibitors.29,30,34,35 As already outlined, there is only one N-alkylated hydroxymethyl-trihydroxycyclopentylamine 12 reported in literature. Yet, this compounds shows sufficient inhibitory activity against α-galactosidase (0.43 μM, gbc) but also inhibitory activity against β-galactosidase (1.5 μM, bovine liver) as well as β-glucosidase (0.51 μM, almonds). Aim of this work, is the synthesis of new aminocyclopentanes, as powerful and selective GH27 α-galactosidase inhibitors. These structures were prepared, relying on Vasella's (2 + 3)-cycloaddition approach.36
2. Results and discussion
2.1. Synthesis
Intermediate 21 was synthesised starting with reductive elimination of known iodosugar 13 (ref. 37) with zinc under slightly acidic conditions to give hemiacetal 14. Its treatment with N-benzylhydroxylamine in CH2Cl2, provided a mixture of syn-product 15 and the desired known38,39 anti-configured N-benzylisoxazolidine 16 (experimental details as well as NMR-data had not been reported for this compound) in a ratio of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (15:16). The stereochemical outcome of this cyclisation is highly dependent on the solvent. Jäger and co-workers39 pointed out that if the reaction is carried out in a polar solvent such as methanol, the main product is compound 15. In contrast, usage of an unpolar solvent such as CH2Cl2 or CHCl3 mainly provides compound 16. Separation of compounds 15 and 16 was easily achieved by silica gel chromatography. Protection of the free hydroxyl function in compound 16 with chloromethyl methyl ether (MOMCl) in the presence of Hünig's base (i-Pr2NEt) yielded fully protected compound 17. “One-pot” hydrogenolytic removal of the N-benzyl group over Pd(OH)2/C (20%) in the presence of Boc2O (di-tert-butyl-dicarbonate) and Na2CO3 resulted in N-Boc-protected primary alcohol 18. Subsequent Swern or Dess–Martin oxidation furnished the corresponding aldehyde 19. Treatment of aldehyde 19 with pyridine or i-Pr2NEt in methanol allowed, via enolization, almost complete isomerisation to epimer 20. This reaction could easily be followed by thin-layer chromatography. The epimerization reaction was then quenched by addition of NaBH4 at 0 °C to obtain desired primary alcohol 21. Unexpectedly, the conditions of this attempted in situ reduction reaction favoured re-isomerization at C-5, now exclusively – via intermediate 19 – providing starting alcohol 18, possibly influenced by unfavourable boronate complexation of aldehyde 19 or by easier access of the reagent trans to the isopropylidene group and consequently higher reactivity of the “wrong” aldehyde epimer, 19, in the equilibrium. Improvement of experimental conditions by isomerisation of aldehyde 19 in CH2Cl2 in the presence of i-Pr2NEt to obtain desired epimer 20, and consecutive in situ reduction with diisobutylaluminium hydride (DIBAL-H) in CH2Cl2 resulted in a mixture of desired alcohol 21 and intermediate 18 in a ratio of about 2:1 (21:18). The diastereomers were separated chromatographically to obtain pure 21 (47%) and recovered starting material 18 (25%) (Scheme 1).
:7 (15:16). The stereochemical outcome of this cyclisation is highly dependent on the solvent. Jäger and co-workers39 pointed out that if the reaction is carried out in a polar solvent such as methanol, the main product is compound 15. In contrast, usage of an unpolar solvent such as CH2Cl2 or CHCl3 mainly provides compound 16. Separation of compounds 15 and 16 was easily achieved by silica gel chromatography. Protection of the free hydroxyl function in compound 16 with chloromethyl methyl ether (MOMCl) in the presence of Hünig's base (i-Pr2NEt) yielded fully protected compound 17. “One-pot” hydrogenolytic removal of the N-benzyl group over Pd(OH)2/C (20%) in the presence of Boc2O (di-tert-butyl-dicarbonate) and Na2CO3 resulted in N-Boc-protected primary alcohol 18. Subsequent Swern or Dess–Martin oxidation furnished the corresponding aldehyde 19. Treatment of aldehyde 19 with pyridine or i-Pr2NEt in methanol allowed, via enolization, almost complete isomerisation to epimer 20. This reaction could easily be followed by thin-layer chromatography. The epimerization reaction was then quenched by addition of NaBH4 at 0 °C to obtain desired primary alcohol 21. Unexpectedly, the conditions of this attempted in situ reduction reaction favoured re-isomerization at C-5, now exclusively – via intermediate 19 – providing starting alcohol 18, possibly influenced by unfavourable boronate complexation of aldehyde 19 or by easier access of the reagent trans to the isopropylidene group and consequently higher reactivity of the “wrong” aldehyde epimer, 19, in the equilibrium. Improvement of experimental conditions by isomerisation of aldehyde 19 in CH2Cl2 in the presence of i-Pr2NEt to obtain desired epimer 20, and consecutive in situ reduction with diisobutylaluminium hydride (DIBAL-H) in CH2Cl2 resulted in a mixture of desired alcohol 21 and intermediate 18 in a ratio of about 2:1 (21:18). The diastereomers were separated chromatographically to obtain pure 21 (47%) and recovered starting material 18 (25%) (Scheme 1).
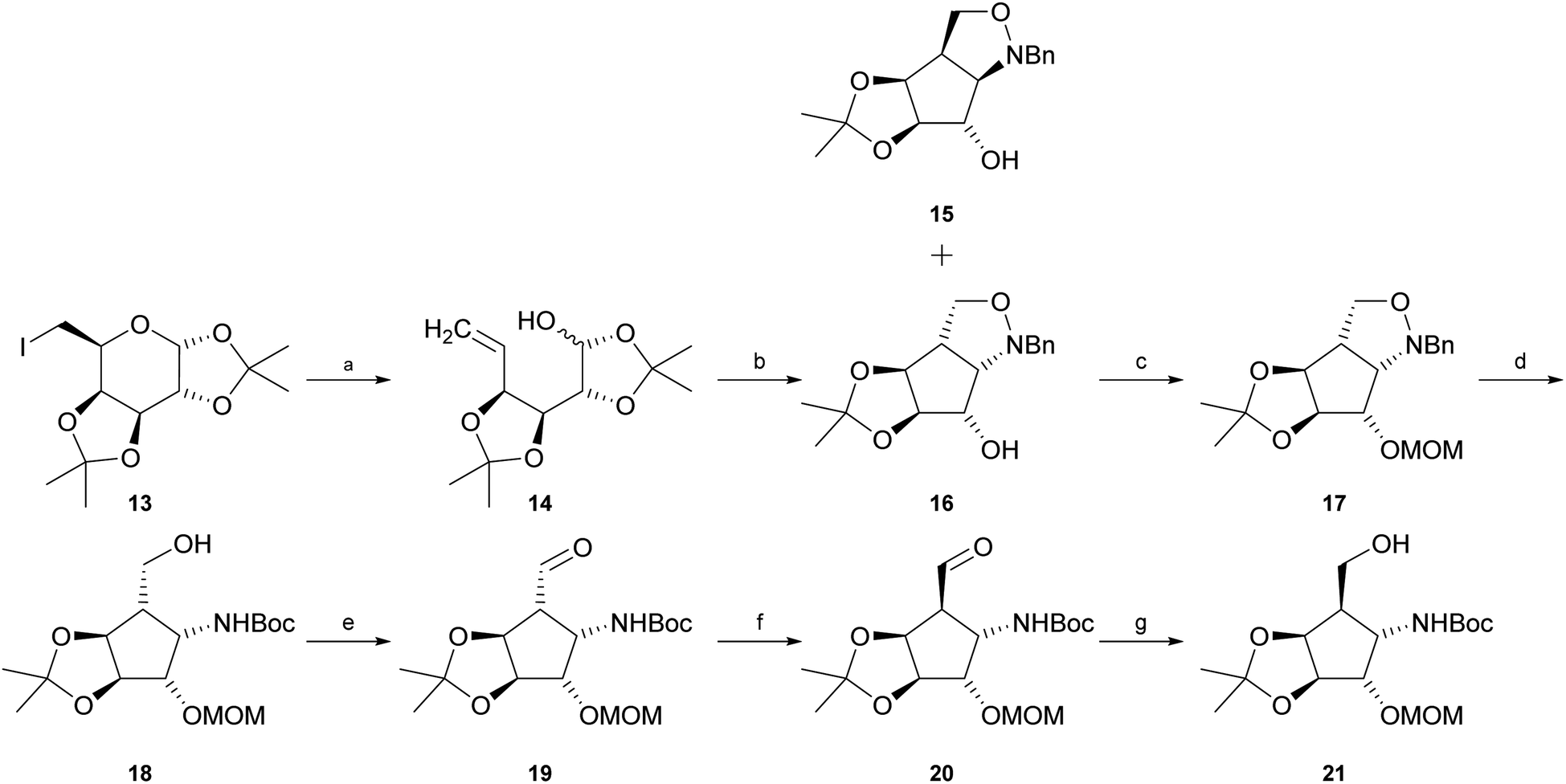 | ||
| Scheme 1 Synthesis of intermediate 21. (a) Zn, NH4Cl, MeOH; (b) BnNHOH·HCl, pyridine, CH2Cl2, 75% over two steps, 86% by conversion; (c) MOMCl, i-Pr2NEt, CH2Cl2, reflux, 91%; (d) H2, 20% Pd(OH)2/C, Boc2O, Na2CO3, MeOH, 90%; (e) Dess–Martin periodinane, CH2Cl2 or oxalyl chloride, DMSO, i-Pr2NEt, CH2Cl2, −60 °C; (f) pyridine or i-Pr2NEt, CH2Cl2; (g) DIBAL-H, CH2Cl2, 0 °C, 47% over three steps, 62% by conversion. | ||
Crystallization from cyclohexane/ethyl acetate afforded crystals of sufficient quality to verify the structural identity of diastereomer 21 by X-ray structure determination (Fig. 2, CCDC 2065318).
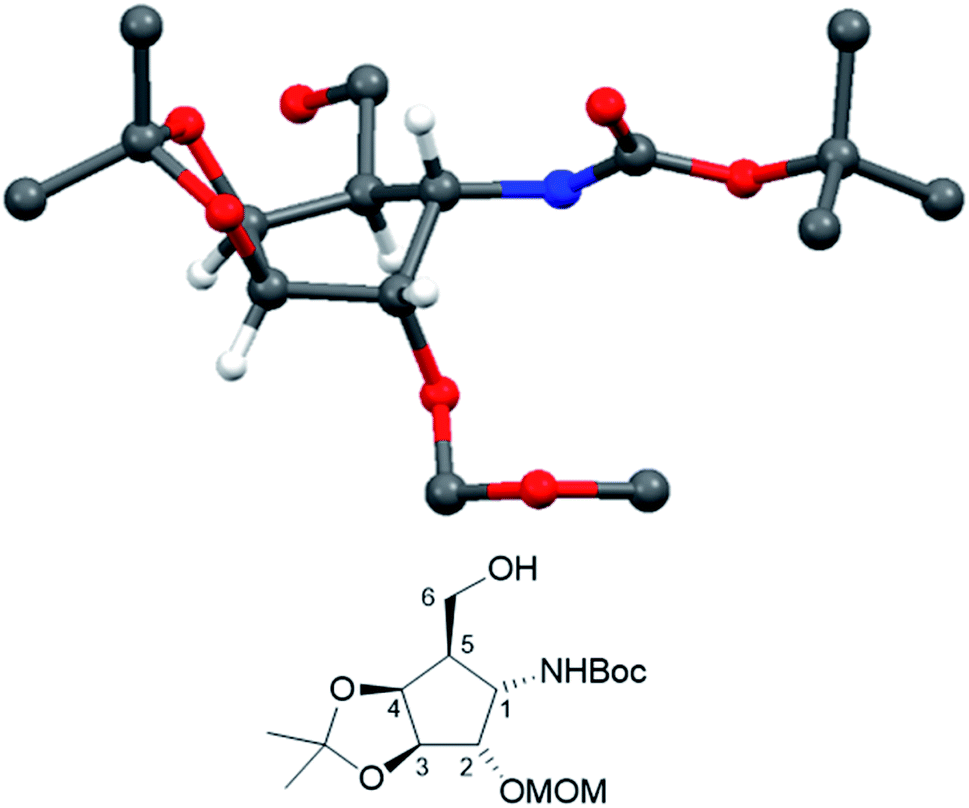 | ||
| Fig. 2 Crystal structure of compound 21. (CCDC: 2065318, for the sake of consistency, numbering follows the monosaccharide pattern starting with the amino substituted carbon (C-1) clockwise along the ring.) | ||
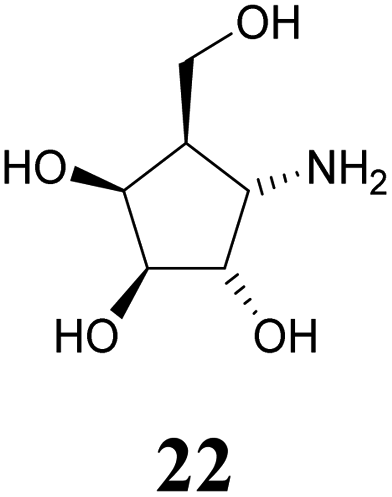
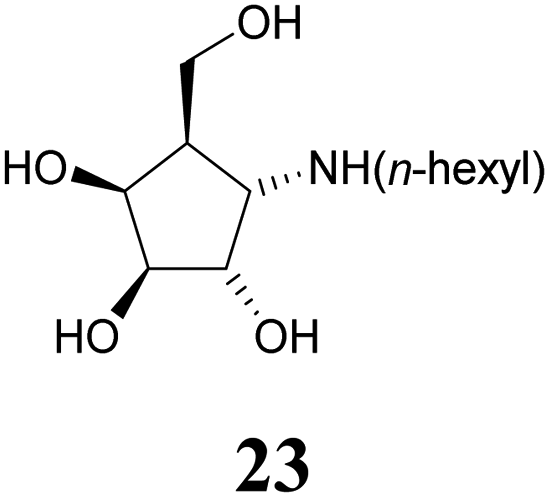
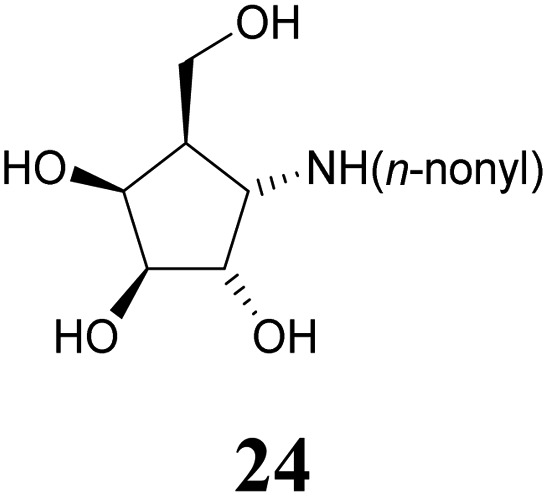
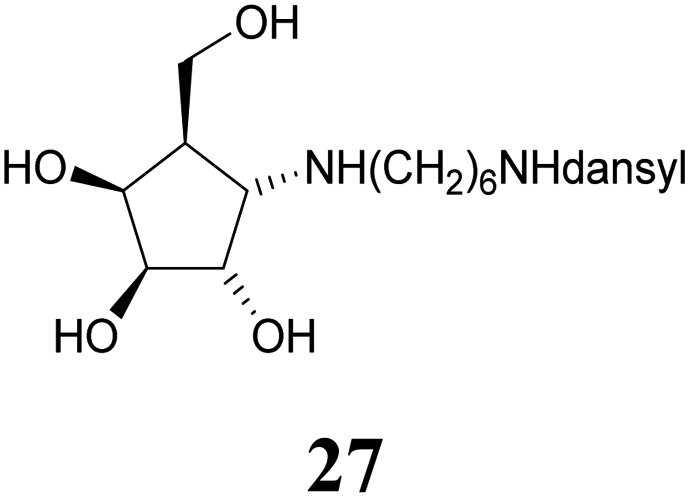
Deprotection of alcohol 21 with trifluoroacetic acid in THF–MeOH–H2O (3:1:1) resulted in known30 aminotetraol 22. Reductive amination with the respective aldehyde in the presence of Pd/C under an atmosphere of H2 gave chain extended inhibitors 23 and 24, respectively. Alkylation of amine 22, by treatment with ω-bromo hexanoic nitrile furnished compound 25, which was subsequently reduced in the presence of RANEY®-Ni under an atmosphere of H2 to obtain primary amine 26. Its conventional N-dansylation yielded inhibitor 27 (Scheme 2).
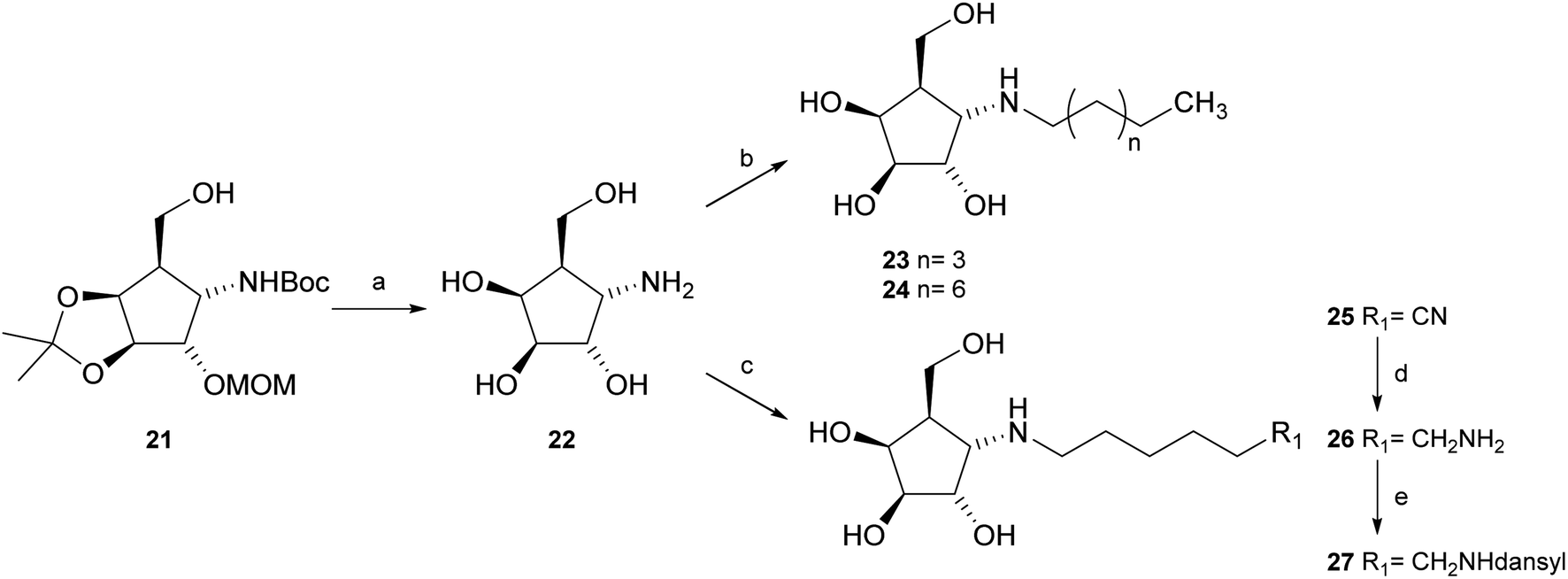 | ||
| Scheme 2 Synthesis of inhibitors 23, 24 and 27. (a) Trifluoroacetic acid, THF–MeOH–H2O (3:1:1), 60 °C, 75%; (b) R–CHO, AcOH, H2, 20% Pd(OH)2/C, MeOH, 57% (23), 64% (24); (c) Br(CH2)5CN, NaHCO3, DMF, 60 °C; (d) H2, RANEY®-Ni, MeOH, 49%, two steps; (e) dansyl chloride, Na2CO3, MeOH, 55%. | ||
2.2. Inhibitory activities
Amines 22–24 and 27 were screened for inhibitory activities with a panel of standard glycosidases (Table 1). As can be seen the compounds act as inhibitors of both α- and β-galactosidases, though overall they perform better on α-galactosidases with inhibition constants down to the high nanomolar/low micromolar range, as discussed in more detail below.| Enzyme (GH family) | Compounds | |||||
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
| a Abg = β-glucosidase/β-galactosidase from Agrobacterium sp.; E. coli = lac Z β-galactosidase from E. coli; bovine liver = β-galactosidase from bovine liver; Fabrazyme® = commercial recombinant lysosomal α-galactosidase; gcb = α-galactosidase from green coffee beans S. cer. = α-glucosidase from S. cerevisiae; GCase = recombinant human lysosomal β-glucocerebrosidase. h. lys. = human lysosomal α-galactosidase. n.d. = not determined, n.i. = no inhibition, with Ki values estimated to be >1 mM. | ||||||
| Abg (β-Glc/β-Gal) (GH 1) | 0.014 ± 0.003 | 0.013 ± 0.003 | 0.0037 ± 0.0016 | 0.005 ± 0.001 | 12 (ref. 40) | n.d. |
| E. coli (β-Gal) (GH 2) | 200 ± 90 | 120 ± 20 | 23 ± 7 | 7.8 ± 4.6 | 13 (ref. 40) | n.d. |
| Bovine liver (β-Gal) (GH 35) | 1.4 ± 1.0 (3.3 (ref. 30)) | 0.093 ± 0.054 | 0.19 ± 0.07 | 0.18 ± 0.08 | n.i.41 | IC50: 190 (ref. 27) |
| Fabrazyme® (α-Gal) (GH 27) | 9.5 ± 1.9 | 4.9 ± 0.9 | 0.52 ± 0.21 | 4.4 ± 1.1 | 0.23 (ref. 32) | IC50: 0.69 (ref. 27) (h. lys.) |
| gcb (α-Gal) (GH 27) | 6.7 ± 0.9 (12 (ref. 30)) | 0.19 ± 0.05 | 0.25 ± 0.05 | 0.14 ± 0.04 | 0.0016 (ref. 40) | IC50: 0.78 (ref. 27) |
| S. cer. (α-Glc) (GH 13) | 160 ± 60 | 130 ± 40 | 73 ± 17 | 16 ± 4 | n.i.41 (yeast) | n.d. |
| GCase (β-Glc) (GH 30) | 18 ± 3 | 40 ± 8 | 1.2 ± 0.5 | 0.63 ± 0.09 | n.d. | n.d. |
While the parent molecule with a free amine proved to be a modest inhibitor with inhibition constants in the low to mid micromolar range, alkylation of this nitrogen improved inhibition in each case (with one exception in the case of GCase). Further extension of the alkyl chain generally improved inhibition yet more indeed for the β-glycosidases, all of which belong to the same, structurally related clan GHA, largely parallel increases in inhibition were seen with each addition of carbon atoms. This is consistent with common interactions with closely related structures.42,43 For the α-galactosidases, the alkylation also generally improved inhibition, but not as predictably as with the β-galactosidases, despite the fact that both, Fabrazyme® and gcb, belong to the same sequence-related family, GH27 (Fig. 3).
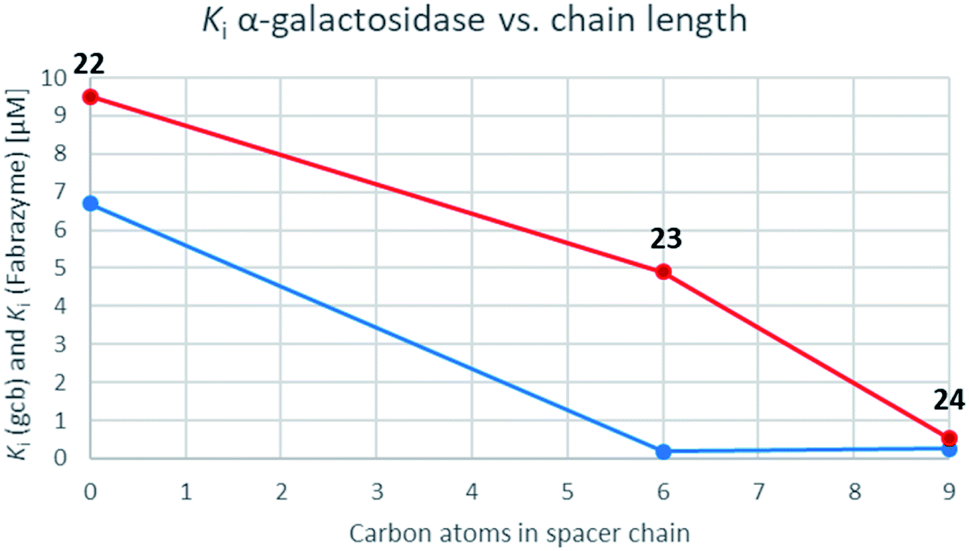 | ||
| Fig. 3 Graphs correlating inhibition constants of gcb α-galactosidase (blue) as well as Fabrazyme® (red) vs. spacer chain length. | ||
Optimal affinity for the human enzyme, Fabrazyme®, was obtained with the n-nonyl substituted derivative 24, with a Ki value comparable to that of galacto-isofagomine. This is the compound in current clinical use under the name Migalastat® or Galafold. The presence of the n-nonyl chain in 24 would likely improve its pharmacokinetic profile by keeping the compound in circulation longer, making this a candidate for testing as a chaperone. However, the fact that this compound is also a good inhibitor of other glycosidases tested here, means that its selectivity needs to be further improved.
3. Conclusions
Following a simple protocol previously outlined by Vasella's and Jäger's groups,35,36 a series of novel derivatives of 12, bearing chain extensions at the nitrogen at position C-1 have been synthesized. Inhibition constants for these compounds with a range of D-galactosidases, including human lysosomal α-galactosidase, were then measured. The results of these inhibitory studies revealed that free amine 22 is the worst inhibitor within this series and the activity increases with the introduction of an alkyl chain as seen with compounds 23, 24 as well as 27.Furthermore, it is both unexpected and interesting that the α-configured compounds developed here are good inhibitors of the β-glycosidases in the first place, since these have the wrong anomeric configuration for these enzymes. Further work will elucidate the reason for this unexpected finding.
4. Experimental
4.1. General methods
Optical rotations were measured at 20 °C on a Perkin Elmer 341 polarimeter at a wavelength of 589 nm and a path length of 10 cm. [α]20D values are given in 10−1 degree cm2 per g. NMR spectra were recorded on a Bruker Ultrashield spectrometer at 300.36 (1H) and 75.53 MHz (13C). CDCl3 was employed for protected compounds and CD3OD or D2O for unprotected inhibitors. Carbon and hydrogen numbering in NMR spectra was conducted in analogy to carbohydrate nomenclature and clockwise, starting with the amino bearing carbon as C-1 (Fig. 2). Chemical shifts are listed in delta employing residual, non-deuterated solvent as the internal standard. Signals were assigned unambiguously by COSY, HSQC as well as APT analysis. Coupling constants ‘J’ for protecting groups, alkyl chains as well as the dansyl group (spacer arms) were found in the expected range and are not listed. For intermediate 21, the structure was confirmed by XRD structural analysis: suitable single crystals of compounds were immersed in silicone oil, mounted using a glass fiber and frozen in the cold nitrogen stream (100 K). X-Ray diffraction data were collected at low temperature on a Bruker Kappa APEX II diffractometer using Mo Kα radiation (l = 0.71073 Å) generated by an INCOATEC micro-focus source. The data reduction and absorption correction was performed with the Bruker SMART and Bruker SADABS, respectively. The structures were solved with SHELXT44 by direct methods and refined with SHELXL45 by least-square minimization against F2 using first isotropic and later anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were added to the structure models on calculated positions using the riding model. The space group assignments and structural solutions were evaluated using PLATON.46,47 MALDI-TOF Mass Spectrometry was performed on a Micromass TofSpec 2E Time-of-Flight Mass Spectrometer. Analytical thin-layer chromatography (TLC) was performed on precoated aluminium plates silica gel 60 F254 (E. Merck 5554) and detected with UV light (254 nm). For staining, a solution of vanillin (9 g) in a mixture of H2O (950 mL)/ethanol (750 mL)/H2SO4 (120 mL) or ceric ammonium molybdate (100 g ammonium molybdate/8 g ceric sulfate in 10% H2SO4 (1 L)) were employed followed by heating on a hotplate. For column chromatography, silica gel 60 (230–400 mesh, E. Merck 9385) or silica gel 60 (Acros Organics, AC 24036) were used. Reaction monitoring was done by TLC.4.2. Kinetic studies
Kinetic studies were performed at room temperature in an appropriate buffer for each enzyme (specific conditions can be found below). All reactions were performed in half-area 96-well-plates (Corning) and monitored with a Synergy H1 plate reader (BioTek). In each experiment, the appropriate concentration of the enzyme was incubated with different concentrations of the inhibitors for 2–5 minutes before initiating the reaction by the addition of the glycoside substrate. The initial rate was then measured by monitoring the increase in, either absorbance at 405 nm (where the aglycone is either p-nitrophenol (pNP) or 2,4-dinitrophenol (dNP)) or fluorescence at 365/455 nm (where the aglycone is 4-methylumbelliferone (MU)) for up to five minutes. Ki determinations were performed using two different substrate concentrations. For each substrate concentration, a range of three to six inhibitor concentrations was used. Dixon plots (1/v vs. [I]) were constructed to validate the use of competitive inhibition model and to assess the fit of the data. The data were then fit to a competitive inhibition model using non-linear regression analysis with Grafit 7.0.0. (Erithacus Software).Specific assay conditions for each enzyme:
Agrobacterium sp. β-glucosidase:48,49 50 mM sodium phosphate buffer (pH 7) substrate: Gal-β-pNP (Km = 4.1 mM).
E. coli lac z β-galactosidase: 50 mM sodium phosphate containing 1.0 mM MgCl2 (pH 7) substrate: Gal-β-pNP (Km = 60 μM).
Bovine liver β-galactosidase: 50 mM sodium phosphate buffer (pH 7) substrate: Gal-β-pNP (Km = 0.65 mM).
Green coffee bean α-galactosidase: 50 mM sodium phosphate buffer (pH 7) substrate: Gal-α-MU, Km = 250 μM.
Fabrazyme® (acid α-galactosidase): 20 mM sodium citrate, 50 mM sodium phosphate, 1.0 mM tetrasodium EDTA, 0.25% v/v Triton X-100® and 0.25% w/v taurocholic acid buffer (pH 5.5) substrate Gal-α-dNP (Km = 0.65 mM).
S. cerevisiae α-glucosidase: 50 mM sodium phosphate buffer (pH 7). Substrate: Glc-α-pNP, Km = 0.75 mM.
GCase (β-glucocerebrosidase): 20 mM sodium citrate, 50 mM sodium phosphate, 1.0 mM tetrasodium EDTA, 0.25% v/v Triton X-100® and 0.25% w/v taurocholic acid buffer (pH 7) susbtrate: Glc-β-dNP (Km = 2.7 mM).
:1) the solids were removed by filtration and the filtrate was concentrated under reduced pressure. The residue containing crude 14 was taken up in CH2Cl2 (100 mL) and this suspension was treated with pyridine (3.45 mL, 43.6 mmol) and N-benzylhydroxylamine hydrochloride (2.78 g, 17.4 mmol). After consumption of the starting material (TLC, 3 h, cyclohexane–EtOAc 2:1) the suspension was consecutively washed with saturated aqueous NaHCO3 solution and brine. The combined organic layers were dried (Na2SO4) and the solvents were removed under reduced pressure to obtain a yellow syrup. Purification on silica gel (cyclohexane–EtOAc 6:1 to 1:1) provided desired anti-configurated N-benzylisoxazolidine 16 (3.19 g, 10.9 mmol, 75% over two steps, 86% by conversion, Rf = 0.6). The melting point as well as the optical rotation [α]20D were in accordance with reported38 data. 1H-NMR (300 MHz, CDCl3) δ = 7.40–7.24 (m, 5H, aromatics), 4.51 (bs, 2H, J2,3 < 1 Hz, J3,4 < 1 Hz, J4,5 < 1 Hz, H-3, H-4), 4.24 (dd, 1H, J5,6a = J6a,6b 8.6 Hz, H-6a), 4.14 (d, 1H, N–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–Ph), 3.98 (dd, 1H, J1,2 5.9 Hz, H-2), 3.94 (dd, 1H, J1,5 6.0 Hz, H-1), 3.86 (d, 1H, N–C2-Ph), 3.68 (dd, 1H, J5,6b 5.3 Hz, H-6b), 3.66 (bs, 1H, OH), 3.22 (dddd, 1H, H-5), 1.45, 1.31 (2 s, 3H each, C(C3)2). 13C-NMR (75.5 MHz, CDCl3): δ = 136.6 (ipso), 129.1, 128.8, 128.0 (aromatics), 111.2 (
2–Ph), 3.98 (dd, 1H, J1,2 5.9 Hz, H-2), 3.94 (dd, 1H, J1,5 6.0 Hz, H-1), 3.86 (d, 1H, N–C2-Ph), 3.68 (dd, 1H, J5,6b 5.3 Hz, H-6b), 3.66 (bs, 1H, OH), 3.22 (dddd, 1H, H-5), 1.45, 1.31 (2 s, 3H each, C(C3)2). 13C-NMR (75.5 MHz, CDCl3): δ = 136.6 (ipso), 129.1, 128.8, 128.0 (aromatics), 111.2 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) (CH3)2), 87.5, 84.0 (C-3, C-4), 75.6 (C-2), 71.3 (C-1), 70.2 (C-6), 61.4 (N–H2–Ph), 54.1 (C-5), 27.6, 25.2 (C(H3)2). MS (MALDI): calculated for [C16H21NO4H]: m/z 292.1549 [M + H]+; found [M + H]+ 292.1547.
(CH3)2), 87.5, 84.0 (C-3, C-4), 75.6 (C-2), 71.3 (C-1), 70.2 (C-6), 61.4 (N–H2–Ph), 54.1 (C-5), 27.6, 25.2 (C(H3)2). MS (MALDI): calculated for [C16H21NO4H]: m/z 292.1549 [M + H]+; found [M + H]+ 292.1547.The already reported35 corresponding syn-configurated N-benzylisoxazolidine 15 (438 mg, 1.50 mmol, 10.3%) was obtained as colourless solid.
:1), methanol (20 mL) was added and the solution was stirred for 20 min. The reaction mixture was allowed to reach ambient temperature and was consecutively washed with HCl (2 M) and saturated aqueous NaHCO3. The combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified on silica gel (cyclohexane–EtOAc 5:1) to provide compound 17 as a colourless oil (3.96 g, 11.8 mmol, 91%). [α]20D: −15.7 (c = 1.1, CHCl3); 1H-NMR (300 MHz, CDCl3) δ = 7.46–7.24 (m, 5H, aromatics), 4.78 (dd, 1H, J2,3 5.1 Hz, J3,4 < 1 Hz, H-3), 4.71 (dd, 2H, O–C2–O–CH3), 4.46 (dd, 1H, J4,5 5.9 Hz, H-4), 4.25 (dd, 1H, J1,2 5.5 Hz, H-2), 4.21 (dd, 1H, J5,6a = J6a,6b 8.6 Hz, H-6a), 4.05 (d, 1H, N–C2-Ph), 3.73 (dd, 1H, J5,6b 4.5 Hz, H-6b), 3.58 (dd, 1H, J1,5 7.0 Hz, H-1), 3.40 (s, 3H, O–CH2–O–C3), 3.16 (dddd, 1H, H-5), 1.53, 1.34 (2s, 3H each, C(C3)2). 13C-NMR (75.5 MHz, CDCl3): δ = 137.7 (ipso), 129.2, 128.4, 127.4 (aromatics), 112.1 ((CH3)2), 95.7 (O–H2–O–CH3), 84.8 (C-3), 83.7 (C-4), 82.0 (C-2), 71.0 (C-1), 70.3 (C-6), 62.1 (N–H2–Ph), 55.6 (O–CH2–O–H3), 50.8 (C-5), 27.9, 25.6 (C(H3)2). MS (MALDI): calculated for [C16H21NO4H]: m/z 336.1811 [M + H]+; found [M + H]+ 336.1818.:1) the solids were removed by filtration. The solvent was removed under reduced pressure and the residue was chromatographed (cyclohexane–EtOAc 3:1) to furnish alcohol 18 (3.71 g, 10.7 mmol, 90%) as a colourless oil. [α]20D: −2.0 (c = 0.87, CHCl3); 1H-NMR (300 MHz, CDCl3) δ = 5.02 (dd, 1H, J1,NH 7.1 Hz, NH), 4.73 (dd, 2H, O–C2–O–CH3), 4.59 (dd, 1H, J3,4 < 1 Hz, J4,5 6.9 Hz, H-4), 4.46 (dd, 1H, J2,3 5.8 Hz, H-3), 4.42 (dd, 1H, J1,2 < 1 Hz, J1,5 7.0 Hz, H-1), 4.05 (dd, 1H, H-2), 3.72–3.59 (m, 2H, H-6), 3.39 (s, 3H, O–CH2–O–C3), 3.09 (bs, 1H, OH), 2.53 (m, 1H, H-5), 1.44 (s, 12H, C(C3)2, NH–CO–O–C(C3)3), 1.27 (s, 3H, C(C3)2). 13C-NMR (75.5 MHz, CDCl3): δ = 156.1 (NH–O–O–(C(CH3)3)), 111.1 ((CH3)2), 96.7 (O–H2–O–CH3), 82.8 (C-3), 82.3 (C-2), 80.6 (C-4), 80.1 (NH–CO–O–((CH3)3)), 59.9 (C-6), 56.4 (O–CH2–O–H3), 52.9 (C-1), 49.4 (C-5), 28.5 (NH–CO–O–(C(H3)3)), 26.6, 24.1 (C(H3)2). MS (MALDI): calculated for [C16H29NO7H]: m/z 348.2022 [M + H]+; found [M + H]+ 348.2021.:1), the reaction mixture was consecutively washed with HCl (2 M) and saturated aqueous NaHCO3 solution. The collected organic layers were combined, dried (Na2SO4), filtered and concentrated under reduced pressure. The remaining syrup was quickly passed through a pad of silica gel (CH2Cl2 to EtOAc) and the solvents were removed under reduced pressure. The mixture of desired aldehyde 20 (cyclohexane–EtOAc 1:1, Rf = 0.6) and precursor 19 (cyclohexane–EtOAc 1:1, Rf = 0.7) was dissolved in CH2Cl2 (20 mL) and treated with i-Pr2NEt (1.64 mL, 9.67 mmol) at 0 °C. The reaction mixture was stirred until only the desired diastereomer 20 was observed (1 h, cyclohexane–EtOAc 1:1). Then DIBAL-H (9.67 mL, 1 M in CH2Cl2) was dropwise added to the solution. After completed reduction (30 min, cyclohexane–EtOAc 1:1), 20 mL of saturated aqueous potassium sodium tartrate solution were added. The mixture was vigorously stirred overnight and was consecutively washed with HCl (2 M) and saturated aqueous NaHCO3 solution, the combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified on silica gel (cyclohexane–EtOAc 4:1) to give alcohol 21 (785 mg, 2.26 mmol, Rf = 0.32, 47%, 62% by conversion) as a colourless oil. Epimer 18 (418 mg, 1.20 mmol, Rf = 0.40, 25%) was recovered. Crystallisation from EtOAc–cyclohexane gave colourless crystals of 21 with sufficient quality for XRD. Mp = 113–115 °C; [α]20D: −56.0 (c = 0.77, CHCl3); 1H-NMR (300 MHz, CDCl3) δ = 5.02 (bs, 1H, NH), 4.71 (dd, 1H, J3,4 = J4,5 6.0 Hz, H-4), 4.70 (dd, 2H, O–C2–O–CH3), 4.46 (dd, 1H, J2,3 < 1 Hz, H-3), 4.19–4.04 (m, 1H, H-1), 3.95 (dd, 1H, J1,2 4.7 Hz, H-2), 3.91–3.80 (m, 2H, H-6), 3.38 (s, 3H, O–CH2–O–C3), 2.33 (bs, 1H, OH), 2.12–2.01 (m, 1H, H-5), 1.44 (s, 12H, C(C3)2, NH–CO–O–C(C3)3), 1.28 (s, 3H, C(C3)2). 13C-NMR (75.5 MHz, CDCl3): δ = 156.1 (NH–O–O–(C(CH3)3)), 110.8 ((CH3)2), 96.5 (O–H2–O–CH3), 81.3, 81.2 (C-2, C-3), 79.9 (NH–CO–O–((CH3)3)), 79.0 (C-4), 60.6 (C-6), 56.0 (O–CH2–O–H3), 52.4 (C-2), 48.2 (C-5), 28.5 (NH–CO–O–(C(H3)3)), 26.0, 23.9 (C(H3)2). MS (MALDI): calculated for [C16H21NO4H]: m/z 348.2022 [M + H]+; found [M + H]+ 348.2022.:1:1, 10 mL) and treated with 5 mL trifluoroacetic acid. The solution was heated to 60 °C until completed deprotection was observed (12 h, CHCl3–MeOH–NH4OH (25%) 4:4:1). The solvents were removed under reduced pressure. The remaining syrup was co-evaporated with toluene (3 times) and was purified by silica gel chromatography (CHCl3–MeOH–NH4OH (25%) 8:4:1) to provide polyol 22 (245 mg, 1.50 mmol, 75%) as colourless solid. The corresponding hydrochloride was obtained by addition of conc. HCl to a methanolic solution of amine 22 (10 mg) adjusting the pH value to 1. Removal of the solvents under reduced pressure yielded known30 22·HCl. The optical rotation was in accordance to the already reported.30 free base: 1H-NMR (300 MHz, D2O) δ = 4.15 (dd, 1H, J3,4 = J4,5 4.6 Hz, H-4), 3.99 (dd, 1H, J1,2 = J2,3 7.4 Hz, H-2), 3.91 (dd, 1H, H-3), 3.83–3.70 (m, 2H, H-6), 3.05 (dd, 1H, J1,5 8.1 Hz, H-1), 2.00–1.89 (m, 1H, H-5). 13C NMR (75.5 MHz, D2O) δ = 78.0 (C-3), 74.8 (C-2), 70.1 (C-4), 59.9 (C-6), 51.4 (C-1), 49.4 (C-5). Hydrochloride: 1H-NMR (300 MHz, D2O) δ = 4.28 (dd, 1H, J1,2 = J2,3 7.8 Hz, H-2), 4.23 (dd, 1H, J3,4 = J4,5 4.7 Hz, H-4), 4.02 (dd, 1H, H-3), 3.87 (dd, 1H, J5,6a 7.3 Hz, J6a,6b 11.0 Hz, H-6a), 3.77 (dd, 1H, J5,6b 7.1 Hz, H-6b), 3.59 (dd, 1H, J1,5 8.3 Hz, H-1), 2.34 (dddd, 1H, H-5). 13C NMR (75.5 MHz, D2O) δ = 77.9 (C-3), 72.3 (C-2), 70.1 (C-4), 59.5 (C-6), 51.9 (C-1), 45.9 (C-5). MS (MALDI): calculated for [C6H13NO4H]: m/z 164.0923 [M + H]+; found [M + H]+ 164.0922.:1 + 1 vol% NH4OH (25%)). The catalyst was filtered off and the solvents were removed under reduced pressure. The residue was quickly passed through a pad of silica gel (CHCl3–MeOH 8:1 + 1 vol% NH4OH (25%)) to obtain inhibitor 23 (21.3 mg, 86.1 μmol, 57%) as a colourless solid. [α]20D: −23.5 (c = 0.82, MeOH); 1H-NMR (300 MHz, CD3OD) δ = 4.25 (dd, 1H, J3,4 4.4 Hz, J4,5 6.8 Hz, H-4), 4.05 (dd, 1H, J1,2 6.5 Hz, J2,3 4.1 Hz, H-2), 3.88 (dd, 1H, H-3), 3.79 (ddd, 2H, J5,6a = J5,6b 5.4 Hz, J6a,6b 12.2 Hz, H-6), 3.30 (dd, 1H, J1,5 7.5 Hz, H-1), 2.82 (t, 2H, H-1′), 2.16–2.04 (m, 1H, H-5), 1.65–1.26 (m, 8H, H-2′, H-3′, H-4′, H-5′), 0.91 (t, 3H, H-6′). 13C NMR (75.5 MHz, CD3OD) δ = 79.7 (C-3), 74.1 (C-2), 72.2 (C-4), 61.8 (C-6), 61.7 (C-1), 48.7 (C-1′), 47.8 (C-5), 32.7, 29.1, 27.7, 23.6 (C-2′, C-3′, C-4′, C-5′), 48.7 (C-6′). MS (MALDI): calculated for [C12H25NO4H]: m/z 248.1862 [M + H]+; found [M + H]+ 248.1862.:1 + 1 vol% NH4OH (25%)), the catalyst was removed by filtration and the filtrate was concentrated under reduced pressure. Silica gel filtration (CHCl3–MeOH 8:1 + 1 vol% NH4OH (25%)) of the residue provided inhibitor 24 (26.8 mg, 92.6 μmol, 64%) as a colourless oil. [α]20D: −22.7 (c = 1.1, MeOH); 1H-NMR (300 MHz, CD3OD) δ = 4.22 (dd, 1H, J3,4 4.8 Hz, J4,5 6.2 Hz, H-4), 4.00 (dd, 1H, J1,2 6.2 Hz, J2,3 4.4 Hz, H-2), 3.86 (dd, 1H, H-3), 3.84–3.72 (m, 2H, H-6), 3.12 (dd, 1H, J1,5 7.6 Hz, H-1), 2.68 (t, 2H, H-1′), 2.05–1.91 (m, 1H, H-5), 1.62–1.19 (m, 14H, H-2′, H-3′, H-4′, H-5′, H-6′, H-7′, H-8′), 0.90 (t, 3H, H-9′). 13C NMR (75.5 MHz, CD3OD) δ = 79.8 (C-3), 74.6 (C-2), 72.2 (C-4), 62.1 (C-6), 61.6 (C-1), 49.2 (C-1′), 48.7 (C-5), 33.0, 30.6, 30.6, 30.4, 30.2, 28.3, 23.7 (C-2′, C-3′, C-4′, C-5′, C-6′, C-7′, C-8′), 14.4 (C-9′). MS (MALDI): calculated for [C15H31NO4H]: m/z 290.2331 [M + H]+; found [M + H]+ 290.2335.:1 + 1 vol% NH4OH (25%)) and thereafter the solvent was removed under reduced pressure. The residue was chromatographically purified (CHCl3–MeOH 14:1 + 1 vol% NH4OH (25%)) to obtain crude nitrile 25 as a colourless oil. 1H-NMR (300 MHz, CD3OD) δ = 4.21 (dd, 1H, J3,4 4.6 Hz, J4,5 6.4 Hz, H-4), 3.98 (dd, 1H, J1,2 6.5 Hz, J2,3 4.2 Hz, H-2), 3.86 (dd, 1H, H-3), 3.84–3.76 (m, 2H, H-6), 3.08 (dd, 1H, J1,5 8.1 Hz, H-1), 2.72–2.61 (m, 2H, H-1′), 2.47 (t, 1H, H-5′), 2.00–1.89 (m, 1H, H-5), 1.75–1.42 (m, 6H, H-2′, H-3′, H-4′). 13C NMR (75.5 MHz, CD3OD) δ = 121.2 (C-6′), 79.9 (C-3), 74.8 (C-2), 72.3 (C-4), 62.2 (C-6), 61.6 (C-1), 49.1 (C-5), 49.0 (C-1′), 29.8 (C-2′), 27.4, 26.3 (C-3′, C-4′), 17.2 (C-5′).A solution of crude nitrile 25 in methanol (2 mL) was stirred in the presence of RANEY®-Ni under an atmosphere of hydrogen gas at ambient pressure. After completed reduction (30 min, CHCl3–MeOH 3:1 + 1 vol% NH4OH (25%)), the catalyst was filtered off and the filtrate was concentrated under reduced pressure. The residual oil was purified on silica gel (CHCl3–MeOH–NH4OH (25%) 8:4:1) to yield amine 26 (40.5 mg, 0.154 mmol, 49%, 2 steps) as a colourless oil. [α]20D: −30.2 (c = 1.2, MeOH); 1H-NMR (300 MHz, D2O) δ = 4.19 (dd, 1H, J3,4 4.8 Hz, J4,5 5.4 Hz, H-4), 4.09 (dd, 1H, J1,2 = J2,3 7.0 Hz, H-2), 3.92 (dd, 1H, H-3), 3.82–3.68 (m, 2H, H-6), 2.89 (dd, 1H, J1,5 7.8 Hz, H-1), 2.85 (t, 2H, H-6′), 2.58 (t, 2H, H-1′), 2.07–1.95 (m, 1H, H-5), 1.65–1.26 (m, 8H, H-2′, H-3′, H-4′, H-5′). 13C NMR (75.5 MHz, D2O) δ = 78.0 (C-3), 73.6 (C-2), 70.1 (C-4), 60.1 (C-6), 58.0 (C-1), 47.5, 47.5 (C-5, C-1′), 39.8 (C-6′), 28.3, 28.2, 26.0, 25.5 (C-2′, C-3′, C-4′, C-5′). MS (MALDI): calculated for [C12H26N2O4H]: m/z 263.1971 [M + H]+; found [M + H]+ 263.1971.
:1 + 1 vol% NH4OH (25%)) the suspension was concentrated under reduced pressure. The residue was purified on silica gel (CHCl3–MeOH 8:1 + 1 vol% NH4OH (25%)) to provide dansyl amide 27 (31.2 mg, 62.9 μmol, 55%) as a yellow syrup. [α]20D: −6.7 (c = 0.92, MeOH); 1H-NMR (300 MHz, CD3OD) δ = 8.56 (d, 1H, dansyl), 8.36 (d, 1H, dansyl), 8.19 (d, 1H, dansyl), 7.63–7.54 (m, 2H, dansyl), 7.28 (d, 1H, dansyl), 4.32 (dd, 1H, J3,4 4.3 Hz, J4,5 7.1 Hz, H-4), 4.16 (dd, 1H, J1,2 = 6.5 Hz, J2,3 3.8 Hz, H-2), 3.94 (dd, 1H, H-3), 3.86 (dd, 1H, J5,6a 5.6 Hz, J6a,6b 10.8 Hz, H-6a), 3.78 (dd, 1H, J5,6a 7.0 Hz, H-6b), 3.57 (dd, 1H, J1,5 7.7 Hz, H-1), 2.98 (t, 2H, H-1′), 2.89 (s, 6H, dansyl), 2.84 (t, 2H, H-6′), 2.32 (dddd, 1H, H-5), 1.65–1.14 (m, 8H, H-2′, H-3′, H-4′, H-5′). 13C NMR (75.5 MHz, CD3OD) δ = 153.2, 137.2 (ipso), 131.3, 131.1, 131.0, 130.1, 129.1, 124.3, 120.6, 119.5 (dansyl), 79.3 (C-3), 73.3 (C-2), 72.1 (C-4), 61.9 (C-1), 61.4 (C-6), 47.8 (C-1′), 46.4 (C-5), 45.8 (dansyl), 43.6 (C-6′), 30.4, 27.0, 26.9, 26.9 (C-2′, C-3′, C-4′, C-5′). MS (MALDI): calculated for [C24H37N3O6SH]: m/z 496.2481 [M + H]+; found [M + H]+ 496.2481.Author contributions
P. W. performed the synthesis, M. T. and A. W. assisted with the development and the syntheses. R. F. provided crystal structure for compound 21. S. A. N. performed biochemical experiments and S. G. W. supervised and evaluated biochemical studies. P. W. and A. E. S. conceived and designed the synthetic experiments. P. W., S. A. N., A. E. S., S. G. W. and T. M. W. prepared and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. G. W. thanks GlycoNet, the Canadian Network of Centres of Excellence in glycoscience, for financial support. S. A. N. is the recipient of a Vanier graduate fellowship. P. W. is recipient of a DOC Fellowship of the Austrian Academy of Sciences at the Institute of Organic Chemistry and the Institute of Chemistry and Biology of Biobased Systems, Graz University of Technology. A. W. and T. M. W. thank the Austrian Science Fund FWF (P30372-B21). Open access funding by Graz University of Technology is appreciated.Notes and references
- B. Henrissat, Biochem. J., 1991, 280(Pt 2), 309–316 CrossRef CAS PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS PubMed.
- V. L. Y. Yip and S. G. Withers, Biochemistry, 2006, 45, 571–580 CrossRef CAS PubMed.
- D. E. Koshland Jr, Biol. Rev., 1953, 28, 416–436 CrossRef.
- S. Ghazi, J. A. Rooke and H. Galbraith, Br. Poult. Sci., 2003, 44, 410–418 CrossRef CAS PubMed.
- L. Weignerová, P. Simerská and V. Křen, Biocatal. Biotransform., 2009, 27, 79–89 CrossRef.
- Y. Yu, T. Mena-Barragán, K. Higaki, J. L. Johnson, J. E. Drury, R. L. Lieberman, N. Nakasone, H. Ninomiya, T. Tsukimura, H. Sakuraba, Y. Suzuki, E. Nanba, C. O. Mellet, J. M. García Fernández and K. Ohno, ACS Chem. Biol., 2014, 9, 1460–1469 CrossRef CAS PubMed.
- A. Markham, Drugs, 2016, 76, 1147–1152 CrossRef PubMed.
- R. J. Desnick, in Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease, ed. R. N. Rosenberg and J. M. Pascual, 6th edn, Academic Press, 2020, pp. 575–587, DOI:10.1016/B978-0-12-813955-4.00042-8.
- M. Martínez-Bailén, A. T. Carmona, F. Cardona, C. Matassini, A. Goti, M. Kubo, A. Kato, I. Robina and A. J. Moreno-Vargas, Eur. J. Med. Chem., 2020, 192, 112173 CrossRef PubMed.
- K. Kok, K. C. Zwiers, R. G. Boot, H. S. Overkleeft, J. M. F. G. Aerts and M. Artola, Biomolecules, 2021, 11(2), 271 CrossRef CAS PubMed.
- U. Galili, E. A. Rachmilewitz, A. Peleg and I. Flechner, J. Exp. Med., 1984, 160, 1519–1531 CrossRef CAS PubMed.
- U. Galili, B. A. Macher, J. Buehler and S. B. Shohet, J. Exp. Med., 1985, 162, 573–582 CrossRef CAS PubMed.
- B. A. Macher and U. Galili, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 75–88 CrossRef CAS PubMed.
- N. Harpaz, H. M. Flowers and N. Sharon, Arch. Biochem. Biophys., 1975, 170, 676–683 CrossRef CAS PubMed.
- J. Goldstein, G. Siviglia, R. Hurst, L. Lenny and L. Reich, Science, 1982, 215, 168 CrossRef CAS PubMed.
- Q. P. Liu, G. Sulzenbacher, H. Yuan, E. P. Bennett, G. Pietz, K. Saunders, J. Spence, E. Nudelman, S. B. Levery, T. White, J. M. Neveu, W. S. Lane, Y. Bourne, M. L. Olsson, B. Henrissat and H. Clausen, Nat. Biotechnol., 2007, 25, 454–464 CrossRef CAS PubMed.
- P. Rahfeld, L. Sim, H. Moon, I. Constantinescu, C. Morgan-Lang, S. J. Hallam, J. N. Kizhakkedathu and S. G. Withers, Nat. Microbiol., 2019, 4, 1475–1485 CrossRef CAS PubMed.
- P. Rahfeld and S. G. Withers, J. Biol. Chem., 2020, 295, 325–334 CrossRef CAS PubMed.
- G. Legler and S. Pohl, Carbohydr. Res., 1986, 155, 119–129 CrossRef CAS PubMed.
- N. Asano, S. Ishii, H. Kizu, K. Ikeda, K. Yasuda, A. Kato, O. R. Martin and J.-Q. Fan, Eur. J. Biochem., 2000, 267, 4179–4186 CrossRef CAS PubMed.
- Y. Ichikawa, Y. Igarashi, M. Ichikawa and Y. Suhara, J. Am. Chem. Soc., 1998, 120, 3007–3018 CrossRef CAS.
- M. Thonhofer, A. Gonzalez Santana, R. Fischer, A. Torvisco Gomez, R. Saf, M. Schalli, A. E. Stütz and S. G. Withers, Carbohydr. Res., 2016, 420, 6–12 CrossRef CAS PubMed.
- Y. Kameda, K. Kawashima, M. Takeuchi, K. Ikeda, N. Asano and K. Matsui, Carbohydr. Res., 1997, 300, 259–264 CrossRef CAS PubMed.
- Y. Wang and A. J. Bennet, Org. Biomol. Chem., 2007, 5, 1731–1738 RSC.
- T. B. Mercer, S. F. Jenkinson, B. Bartholomew, R. J. Nash, S. Miyauchi, A. Kato and G. W. J. Fleet, Tetrahedron: Asymmetry, 2009, 20, 2368–2373 CrossRef CAS.
- A. Kato, Y. Yamashita, S. Nakagawa, Y. Koike, I. Adachi, J. Hollinshead, R. J. Nash, K. Ikeda and N. Asano, Bioorg. Med. Chem., 2010, 18, 3790–3794 CrossRef CAS PubMed.
- B. J. Ayers, N. Ngo, S. F. Jenkinson, R. F. Martínez, Y. Shimada, I. Adachi, A. C. Weymouth-Wilson, A. Kato and G. W. J. Fleet, J. Org. Chem., 2012, 77, 7777–7792 CrossRef CAS PubMed.
- L. Gartenmann Dickson, E. Leroy and J.-L. Reymond, Org. Biomol. Chem., 2004, 2, 1217–1226 RSC.
- M. Bøjstrup and I. Lundt, Org. Biomol. Chem., 2005, 3, 1738–1745 RSC.
- O. Motabar, K. Liu, N. Southall, J. J. Marugan, E. Goldin, E. Sidransky and W. Zheng, Curr. Chem. Genomics, 2010, 4, 67–73 CrossRef CAS PubMed.
- M. Artola, C. Hedberg, R. J. Rowland, L. Raich, K. Kytidou, L. Wu, A. Schaaf, M. J. Ferraz, G. A. van der Marel, J. D. C. Codée, C. Rovira, J. M. F. G. Aerts, G. J. Davies and H. S. Overkleeft, Chem. Sci., 2019, 10, 9233–9243 RSC.
- W.-C. Cheng, J.-H. Wang, H.-Y. Li, S.-J. Lu, J.-M. Hu, W.-Y. Yun, C.-H. Chiu, W.-B. Yang, Y.-H. Chien and W.-L. Hwu, Eur. J. Med. Chem., 2016, 123, 14–20 CrossRef CAS PubMed.
- E. Leroy and J.-L. Reymond, Org. Lett., 1999, 1, 775–777 CrossRef CAS PubMed.
- M. Kleban, P. Hilgers, J. N. Greul, R. D. Kugler, J. Li, S. Picasso, P. Vogel and V. Jäger, ChemBioChem, 2001, 2, 365–368 CrossRef CAS PubMed.
- B. Bernet and A. Vasella, Helv. Chim. Acta, 1979, 62, 1990–2016 CrossRef CAS.
- P. J. Garegg and B. Samuelsson, J. Chem. Soc., Perkin Trans. 1, 1980, 2866–2869, 10.1039/P19800002866.
- S. Henkel, M. Kleban and V. Jäger, Z. Kristallogr. - New Cryst. Struct., 2001, 216, 89–90 CAS.
- J. N. Greul, M. Kleban, B. Schneider, S. Picasso and V. Jäger, ChemBioChem, 2001, 2, 368–370 CrossRef CAS PubMed.
- R. F. G. Fröhlich, R. H. Furneaux, D. J. Mahuran, R. Saf, A. E. Stütz, M. B. Tropak, J. Wicki, S. G. Withers and T. M. Wrodnigg, Carbohydr. Res., 2011, 346, 1592–1598 CrossRef PubMed.
- R. C. Bernotas, M. A. Pezzone and B. Ganem, Carbohydr. Res., 1987, 167, 305–311 CrossRef CAS PubMed.
- M. Schalli, P. Weber, C. Tysoe, B. M. Pabst, M. Thonhofer, E. Paschke, A. E. Stütz, M. Tschernutter, W. Windischhofer and S. G. Withers, Bioorg. Med. Chem. Lett., 2017, 27, 3431–3435 CrossRef CAS PubMed.
- M. Schalli, C. Tysoe, R. Fischer, B. M. Pabst, M. Thonhofer, E. Paschke, T. Rappitsch, A. E. Stütz, M. Tschernutter, W. Windischhofer and S. G. Withers, Carbohydr. Res., 2017, 443–444, 15–22 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- J. B. Kempton and S. G. Withers, Biochemistry, 1992, 31, 9961–9969 CrossRef CAS PubMed.
- H. Prade, L. F. Mackenzie and S. G. Withers, Carbohydr. Res., 1997, 305, 371–381 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Dr Inge Lundt to the happy occasion of her 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2065318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02507d |
| This journal is © The Royal Society of Chemistry 2021 |