
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymerization of new aniline derivatives: synthesis, characterization and application as sensors†
Akhat G. Mustafin *a,
Lyaysan R. Latypova*a,
Anastasia N. Andriianova*a,
Ilnur N. Mullagalievb,
Shamil M. Salikhova,
Renat B. Salikhovb and
Gulsum S. Usmanovaa
*a,
Lyaysan R. Latypova*a,
Anastasia N. Andriianova*a,
Ilnur N. Mullagalievb,
Shamil M. Salikhova,
Renat B. Salikhovb and
Gulsum S. Usmanovaa
aUfa Institute of Chemistry of the Russian Academy of Sciences, Republic of Bashkortostan, pr. Oktyabrya 71, Ufa 450054, Russia. E-mail: an.chem17@gmail.com
bBashkir State University, Republic of Bashkortostan, Z. Validi St 32, Ufa, 450076, Russia
First published on 14th June 2021
Abstract
This work is focused on modifying aniline monomers with various characteristics that allows one to study the effect of the substituent on the respective polymer. A series of new polyaniline (PANI) derivatives based on an ortho-substituted aniline derivative, 2-(1-methylbut-2-en-1-yl)aniline, were synthesized and characterized. The structures and composition of the polymers that we synthesized were confirmed by elemental analysis, proton nuclear magnetic resonance (1H NMR) spectroscopy, carbon nuclear magnetic resonance (13C NMR) spectroscopy, Fourier-transform infrared spectroscopy (FT-IR), and ultraviolet-visible spectroscopy (UV). Characterization by FT-IR and UV-visible spectroscopy techniques indicated that the polymers exist in protonated emeraldine forms. Scanning electron microscope (SEM) results revealed that the surface morphology of the resulting polymers changed from a heterogeneous hierarchical to spherical structure upon changing the substituent in the aniline monomers. The polymers are soluble in common organic solvents, so they can be used to make films. The electrical properties of the polymers were studied and their high sensitivity to moisture and ammonia was demonstrated. The results of the studies showed the prospects of using thin polymer films in the design of chemical sensors. The impact of the substituent on the polymer characteristics is rationalized in terms of steric and electronic effects.
Introduction
Polyaniline (PANI) is among the most famous representatives of the class of electrically conductive polymers. It has a unique set of properties: redox activity, acid–base properties, electronic and ionic conductivity, and high general stability and thermal stability.1–4 For this reason, the polymer finds use in energy-saving devices, for screening electromagnetic radiation, to obtain antistatic and electrically conductive coatings, and as a corrosion inhibitor.5–8 There is a significant potential of PANI application in medicine and heterogeneous catalysis.9–11 A considerable drawback of PANI, similarly to all conductive polymers, lies in its extremely low solubility in the majority of typical solvents, which significantly narrows the scope of its application.12–14 One of the ways to overcome these problems involves the functionalization of PANI that makes it possible to increase the solubility while retaining many of its important properties. A review of literature data has shown that incorporation of substituents such as alkyl,15–17 alkoxy,18,19 and hydroxy groups20,21 at the ortho position of the aromatic ring has a beneficial effect on the solubility of polymers. As a result, polymers can be soluble not only in high polarity but also in medium polarity solvents. If sulfo groups are incorporated into the ring, the polymers become soluble in aqueous solutions.22 The use of this method of PANI modification not only improves its solubility but also favors the formation of uniform thin polymer films.Recently, films of electrically conductive polyconjugated polymers in resistive sensors, which are used to determine various toxic gases due to high sensitivity, relatively low cost, and simple design, were intensely studied.23–25
One of the main advantages of sensors based on electrically conductive polyconjugated polymer films is that they can operate at room temperature.26
The most important feature that makes PANI a promising material for sensors is that its chemical and optical properties change due to protonation (doping) and deprotonation (dedoping) processes that occur when it interacts with components that can change the oxidation state of the polymer. PANI derivatives are efficient materials for application in potentiometric and amperometric sensors for various gases (NH3, HCl, H2S, etc.) and in pH sensors.27–29 Therefore, the development of a technology for producing electrically conductive films based on PANI derivatives and for creating chemical sensors based thereon is a problem of current interest.
Previously, we performed a study on the synthesis of a new functionalized monomer, ortho-(1-methylbut-2-en-1-yl)aniline, followed by conversion into a polymer.30 The resulting polymer is soluble in NMP, DMF, DMSO and moderately soluble in CHCl3, acetone and THF. The presence of a double bond makes it possible to involve this monomer in various chemical reactions followed by conversion to new polymeric products with physicochemical properties that differ from those of the original polymer.
The purpose of this work was to synthesize new functional monomers based on ortho-(1-methylbut-2-en-1-yl)aniline and study how the structure of the initial aniline derivatives affects the properties of the resulting polymeric products.
Materials and methods
Materials
Aniline was distilled under reduced pressure prior to use. Dimethyl sulfoxide (DMSO), N-methylpyrrolidone (NMP), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), toluene, methanol (MeOH), chloroform (CHCl3), ethyl alcohol (EtOH), ethyl acetate (EtOAc), benzene, acetonitrile (MeCN), methylene chloride (CH2Cl2), petroleum ether, hexane, as well as penta-1,3-diene were distilled prior to use. All the other chemicals were analytical grade reagents and were used as procured. 4-Chloropent-2-ene31 and 2-(1-methylbut-2-en-1-yl)aniline32 were obtained by known procedures.Synthesis of the monomers
2-(1-Methylbut-1-en-1-yl)aniline (2). Yield: 1.9 g (95%). 1H NMR (CDCl3, 500 MHz) δ Z[E]: 1.04 [1.18] (3H, t, 3J = 7.5 Hz, H-4′), 1.92–1.98 [2.29–2.35] (2H, m, H-3′a, H-3′b), 2.07 [2.06] (3H, d, 4J5′–2′ = 1.0 Hz, H-5′), 3.78 (2H, br.s, NH2); 5.69 [5.61] (1H, dt, 3J2′–3′a = 7.5 Hz, 3J2′–3′b = 7.5 Hz, 4J2′–5′ = 1.0 Hz, H-2′), 6.79 [6.77] (1H, dd, 3J6–5 = 8.1 Hz, 4J6–4 = 1.5 Hz, H-6), 6.86 [6.84] (1H, dt, 3J4–5 = 7.5, 3J4–3 = 7.5 Hz, 3J4–6 = 1.5 Hz, H-4), 7.05 [7.11] (1H, dd, 3J3–4 = 7.5 Hz, 3J3–5 = 1.3 Hz, H-3), 7.18 [7.15] (1H, ddd, 3J5–6 = 8.1 Hz, 3J5–4 = 7.5 Hz, 3J5–3 = 1.3 Hz, H-5). 13C NMR (CDCl3, 500 MHz) δ: 14.37 [14.28] (C-4′), 22.62 [21.77] (C-3′), 24.58 [24.59] (C-5′), 115.05 [115.51] (C-6), 118.30 [118.35] (C-4), 127.74 [127.56] (C-5), 128.85 [128.90] (C-3), 131.36 [132.37] (C-2′), 131.63 [131.35] (C-1′), 133.16 [133.11] (C-2), 142.91 [143.16] (C-1).
2-(1-Methylbutyl)aniline (3). Yield: 2.8 g (93%). 1H NMR (CDCl3, 500 MHz) δ: 1.02 (3H, t, 3J = 7.4 Hz, H-4′), 1.34 (3H, d, 3J = 6.8 Hz, H-5′), 1.38–1.49 (2H, m, H-3′a, H-3′b), 1.60–1.67 (1H, m, H-2′b), 1.71–1.79 (1H, m, H-2′a), 2.84 (1H, m, H-1′), 6.76 (1H, d, 3J6–5 = 7.6 Hz, H-6), 6.89 (1H, t, 3J4–3 = 7.6 Hz, H-4), 7.11 (1H, t, 3J5–6 = 7.6 Hz, H-5), 7.20 (1H, d, 3J3–4 = 7.6 Hz, H-3). 13C NMR (CDCl3, 500 MHz) δ: 14.36 (C-4′), 20.66 (C-5′), 20.93 (C-3′), 32.71 (C-1′), 39.28 (C-2′), 116.08 (C-6), 119.13 (C-4), 126.21 (C-3), 126.45 (C-5), 132.07 (C-2), 143.63 (C-1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
N-[2-(1-Hydroxy-1-methylbutyl)phenyl]acetamide (4) and N-{2-[1-methylbut-1-en-1-yl]phenyl}acetamide (5). Yields: 2.9 g (70%) of 4 and 0.6 g (17%) of 5. For 4, 1H NMR (CDCl3, 500 MHz) δ: 0.79 (3H, t, 3J = 7.4 Hz, H-4′), 1.17–1.32 (2H, m, H-3′a, H-3′b), 1.49 (3H, s, H-5′), 1.66–1.78 (2H, m, H-2′b, H-2′a), 2.02 (3H, s, Ac), 6.90 (1H, d, 3J6–5 = 7.5 Hz, H-6), 7.01–7.04 (2H, m, H-3, H-5), 7.12 (1H, t, 3J4–5 = 7.5 Hz, 3J4–3 = 7.5 Hz, H-4). For 5, 1H NMR (CDCl3, 500 MHz) δ: 0.87 (3H, t, 3J = 7.5 Hz, H-4′), 1.70–1.76 (2H, m, H-3′a, H-3′b), 1.93 (3H, s, H-5′), 2.10 (3H,s, Ac), 5.65 (1H, t, 3J2′–3′a = 7.3 Hz, 3J2′–3′b = 7.3 Hz, H-2′), 6.59–7.06 (2H, m, H-3, H-4), 7.21 (1H, t, 3J5–4 = 8.0 Hz, 3J5–6 = 8.0 Hz, H-5), 8.27 (1H, d, 3J6–5 = 8.0 Hz, H-6). For 4, 13C NMR (CDCl3, 500 MHz) δ: 14.12 (C-4′), 16.92 (C-3′), 21.74 (Ac), 27.97 (C-5′), 44.28 (C-2′), 80.70 (C-1′), 122.80 (C-6), 124.02 (C-3), 126.06 (C-5), 128.17 (C-4), 128.91 (C-2), 138.30 (C-1), 160.08 (Ac). For 5, 13C NMR (CDCl3, 500 MHz) δ: 14.03 (C-4′), 22.62 (C-3′), 24.79 (Ac), 25.07 (C-5′), 120.09 (C-6), 123.81 (C-3), 127.60 (C-5), 128.12 (C-4), 131.33 (C-2), 131.94 (C-1′), 132.69 (C-2′), 134.29 (C-1), 168.08 (Ac).
:1).
2-(2-Aminophenyl)pentan-2-ol (6). Yield: 1.7 g (75%). 1H NMR (CDCl3, 500 MHz) δ: 0.91 (3H, t, 3J4′–3′ = 7.4 Hz, H-4′), 1.16–1.24 (1H, m, H-3′a), 1.32–1.41 (1H, m, H-3′b), 1.62 (3H, s, H-5′), 1.84–1.89 (1H, m, H-2′a), 1.95–2.0 (1H, m, H-2′b), 6.64 (1H, d, 3J6–5 = 7.5 Hz, H-6), 6.70 (1H, t, 3J5–6 = 7.5 Hz, 3J5–4 = 7.5 Hz, H-5), 7.04–7.07 (2H, m, H-4, H-5). 13C NMR (CDCl3, 500 MHz) δ: 14.47 (C-4′), 17.80 (C-3′), 27.97 (C-5′), 42.81 (C-2′), 76.76 (C-1′), 117.75 (C-6), 117.77 (C-5), 126.69 (C-3), 128.08 (C-4), 129.99 (C-2), 145.41 (C-1).
Synthesis of polymers
Poly[2-(1-methylbut-1-en-1-yl)aniline] (P-PA). Dark brown powder, yield: 0.9 g (88%). 1H NMR (CDCl3, 500 MHz) δ: 0.80–1.03 (m, H-4′), 1.67–1.92 (m, H-3′), 1.98–2.08 (m, H-5′), 5.60–5.74 (m, H-2′), 7.01–7.35 (m, Ph). 13C NMR (CDCl3, 500 MHz) δ: 14.01 (C-4′), 22.75 (C-3′), 25.32 (C-5′), 127.65–130.23 (C-3, C-5, C-6, C-1′), 134.20 (C-2′), 135.92 (C-2), 140.07 (C-4), 151.41 (C-1).
Poly[2-(1-methylbutyl)aniline] (P-MB). Dark brown powder, yield: 0.8 g (85%). 1H NMR (CDCl3, 500 MHz) δ: 0.73–0.94 (m, H-4′), 1.10–1.34 (m, H-3′, H-5′), 1.43–1.64 (m, H-2′), 2.88–3.05 (m, H-1′), 6.95–7.51 (m, Ph). 13C NMR (CDCl3, 500 MHz) δ: 14.19 (C-4′), 20.71 (C-3′), 21.60 (C-5′), 33.48 (C-1′), 39.75 (C-2′), 125.68–127.68 (C-3, C-5, C-6), 130.88 (C-2), 143.11 (C-4), 150.45 (C-1).
Poly[2-(2-aminophenyl)pentan-2-ol] (P-AP). Dark brown powder, yield: 0.6 g (60%).1H NMR (CDCl3, 500 MHz) δ: 0.72–0.92 (m, H-4′), 1.14–1.42 (m, H-3′), 1.47–1.68 (m, H-5′), 1.75–1.95 (m, H-2′), 7.05–7.12 (m, H-6), 7.24–7.32 (m, H-5), 7.36–7.41 (m, H-6). 13C NMR (CDCl3, 500 MHz) δ: 14.21 (C-4′), 17.50 (C-3′), 28.32 (C-5′), 43.72 (C-2′), 76.55 (C-1′), 123.27–130.17 (C-3, C-5, C-6) 137.16 (C-2), 139.54 (C-1).
Apparatus and characterization
Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were measured in CDCl3 using a Bruker Avance III spectrometer (500 MHz). Chemical shifts are represented in parts per million downfield from tetramethylsilane as the internal standard. FT-IR spectra were recorded between 200 and 4000 cm−1 in KBr pellets using a Specord M80 spectrometer. Elemental analysis was performed on a Euro-2000 CHNS(O) analyzer. UV-vis spectra were recorded on a Shimadzu Spectrophotometer 2600 in DMSO solution at 298 K, at wavelengths ranging from 190 to 900 nm (slit width – 2.0 nm, scanning speed – fast), using a 1 cm thick quartz tube. Fluorescence spectral analysis was conducted using an RF-5301 PC Shimadzu spectrofluorophotometer in DMSO solution at 298 K. The microstructure of nanoparticles was studied with a TESCAN MIRA 3 LMH high-resolution scanning electron microscope (SEM) equipped with a detector of scattered and backward scattered electrons. The survey was conducted at 10 kV accelerating voltage and under 10−3 Pa vacuum. For SEM analysis a drop of the polymer solution was applied onto a substrate by spin coating. For sensing study the sitall substrate was cleaned in an ethanol solution and dried for 10 min in an oven at 50 °C. A polymer film was applied on top of substrate by spin-coating from a 0.1 M DMF solution. The spin time was 2 minutes, and the spin rates for three samples were 700 rpm. The following equipment was used in the experiments to measure sensor properties: MASTECH DC POWER SUPPLY HY3005D-2 power supplies, a DMM 4020 multimeter as the ammeter, an Arduino Uno programmable controller, a display, as well as DHT-11 and MQ135 sensors in Fig. 1.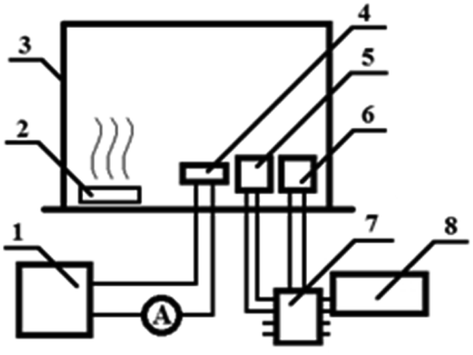 | ||
| Fig. 1 Calibration of humidity and ammonia sensors: (1) power supply unit, (2) container with water or ammonia, (3) sealed cabinet, (4) polymer film humidity sensor, (5) electronic hygrometer, (6) ammonia sensor, (7) Arduino UNO, (8) display. | ||
Results and discussion
Synthesis of samples
2-(1-Methylbut-2-en-1-yl)aniline (1) was used as the starting compound. To obtain 2-(1-methylbut-1-en-1-yl)aniline (2), double bond isomerization was performed in the starting compound by treatment with KOH at 300 °C to give product 2 in 98% yield. Catalytic hydrogenation of compound 1 on Pd/C (10%) in ethyl acetate at room temperature gave 2-(1-methylbutyl)aniline (3) in 92% yield. 2-(2-Aminophenyl)pentan-2-ol (6) was obtained due to an unusual reaction in the course of compound 2 acylation with acetyl chloride. In this synthesis, we observed that yet another product, N-[2-(1-hydroxy-2-methylbutyl)phenyl]acetamide (4), was formed along with N-{2-[1-methylbut-1-en-1-yl]phenyl}acetamide (5). The content of product 4 in the product mixture was 70%. The N-acetate group was readily hydrolyzed upon refluxing compound 4 with KOH in methanol to give tertiary alcohol 6 in 75% yield (Scheme 1). The structures of compounds 2, 3 and 6 were identified by 1H NMR and 13C spectra.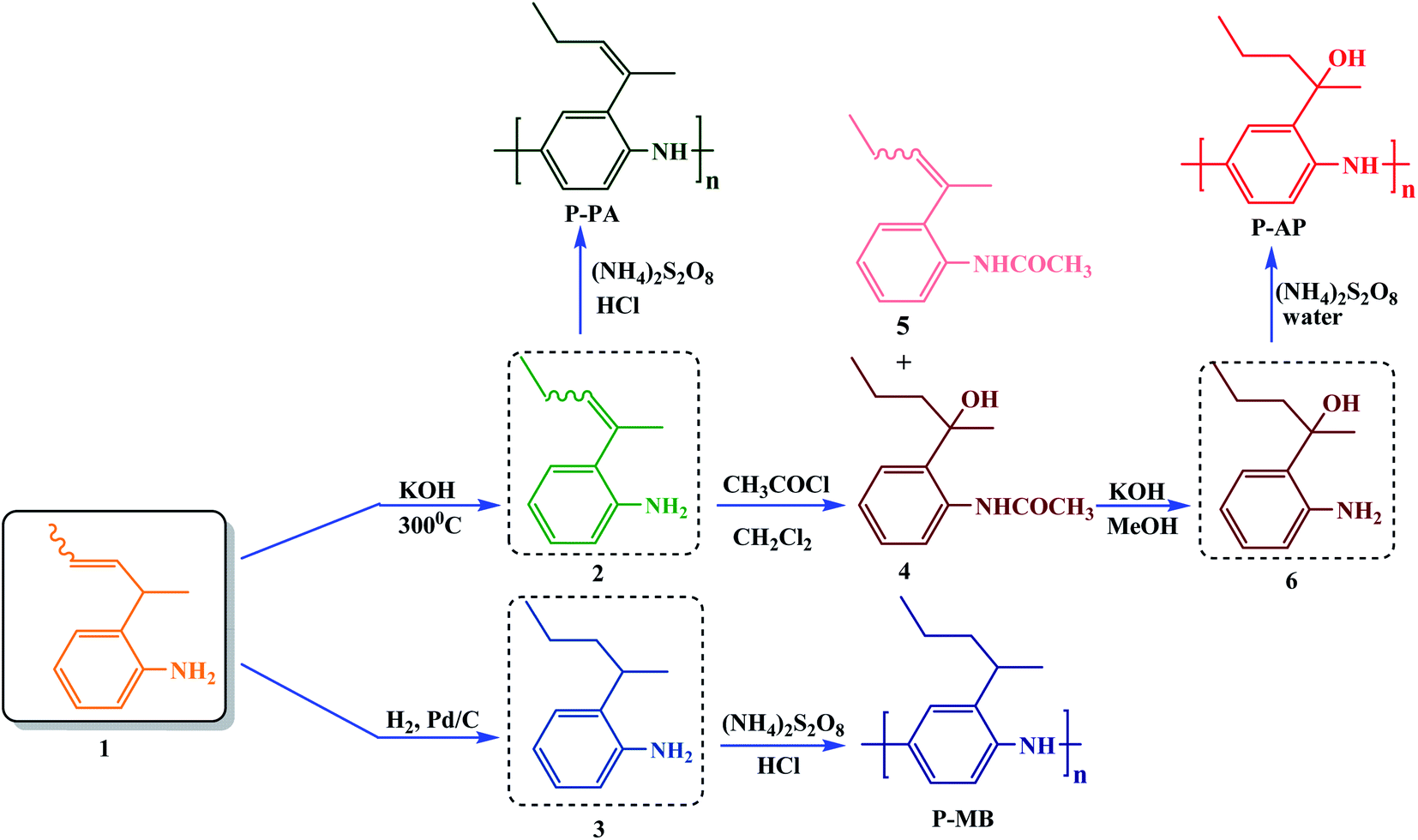 | ||
| Scheme 1 Synthesis of 2-(1-methylbut-2-en-1-yl)aniline derivatives followed by the production of PANIs. | ||
Chemical oxidative polymerization was carried out to synthesize PANI derivatives; monomers 2 (1 g, 6.2 mmol) and 3 (1 g, 6.1 mmol) were added to 0.2 mol L−1 aqueous solution of HCl (50 mL), and then the polymerization was conducted by dropwise addition of an ammonium persulfate solution (monomer/PSA = 1.00/1.25) in 0.2 mol L−1 aqueous solution of HCl (50 mL) as the oxidant at 20 °C for 24 h. Then the PANIs was filtered and the precipitate was washed with petroleum ether and 0.2 mol L−1 HCl to remove aniline oligomers. After washing and filtration, the solid was dried in vacuum to evaporate the solvent from the sample completely.
Monomer 6 (1 g, 5.6 mmol) was dissolved in 15 mL of distilled water and the solution was oxidized with a solution of ammonium persulfate (1.28 g, 5.6 mmol) in water. The polymerization was continued for 24 h without disturbance at room temperature (20 °C). The product was filtered off and washed with a large amount of water followed by petroleum ether until the filtrate became colorless. For the doping procedure, the polymer was treated with 1 M HCl solution for 24 h. The powder was then filtered off, washed with petroleum ether, and dried under vacuum (Scheme 1).
FT-IR spectra
The structures of the polymers that we synthesized were confirmed by FT-IR spectroscopy. One can see in Fig. 2 that the spectra of poly[2-(1-methylbut-1-en-1-yl)aniline] (P-PA), poly[2-(1-methylbutyl)aniline] (P-MB) and poly[2-(2-aminophenyl)pentan-2-ol] (P-AP) samples exhibit peaks characteristic of PANI, in particular, the bands at 3032 cm−1, 3245 cm−1 and 3038 cm−1 corresponding to the stretching vibrations of the![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H bond. The absorption maxima in the regions of 3350 cm−1, 3420 cm−1 and 3390 cm−1 correspond to the vibrations of the –N–H bond. The presence of alkyl substituents in P-PA, P-MB and P-AP is confirmed by the absorption of C–H bonds in the ranges of 2773–2968 cm−1, 2870–2957 cm−1, and 2829–2961 cm−1. The absorption maxima in the regions of 1616 cm−1, 1608 cm−1 and 1613 cm−1 correspond to the stretching vibrations of the CN bond in the conjugated system. These absorption bands are consistent with the presence of quinonediimine moieties in the polyaniline chain. The intense narrow absorption maxima in the samples at 1510 cm−1, 1557 cm−1 and 1504 cm−1 are consistent with the presence of a benzene ring and correspond to its pulsation vibrations. Moreover, the presence of a benzene ring is consistent with the absorption bands at 798 cm−1, 755 cm−1 and 759 cm−1 corresponding to the out-of-plane deformation vibrations of the C–H bonds. The peaks at 1461 cm−1, 1496 cm−1 and 1450 cm−1 correspond to the NN bonds. The peaks at 1373 cm−1, 1368 cm−1 and 1393 cm−1 correspond to the stretching vibrations of the C–N bonds. The absorption peaks at 1124 cm−1, 1138 cm−1 and 1117 cm−1 are due to the vibrations of the –NH+ groups formed upon protonation of the PANI's molecular chain (Table 1). This is evidence of the degree of electron delocalization and the nature of its electrical conductivity. The spectra of the P-AP polymer contain bands in the region of 1368–1358 cm−1 indicating the formation of a tertiary alcohol. The peaks at 594–462 cm−1, 536 cm−1 and 541 cm−1 correspond to various vibrations of the substituted rings. Thus, it follows from IR spectroscopy data that the structures of the polymers that we obtained are close to the emeraldine structure.
C–H bond. The absorption maxima in the regions of 3350 cm−1, 3420 cm−1 and 3390 cm−1 correspond to the vibrations of the –N–H bond. The presence of alkyl substituents in P-PA, P-MB and P-AP is confirmed by the absorption of C–H bonds in the ranges of 2773–2968 cm−1, 2870–2957 cm−1, and 2829–2961 cm−1. The absorption maxima in the regions of 1616 cm−1, 1608 cm−1 and 1613 cm−1 correspond to the stretching vibrations of the CN bond in the conjugated system. These absorption bands are consistent with the presence of quinonediimine moieties in the polyaniline chain. The intense narrow absorption maxima in the samples at 1510 cm−1, 1557 cm−1 and 1504 cm−1 are consistent with the presence of a benzene ring and correspond to its pulsation vibrations. Moreover, the presence of a benzene ring is consistent with the absorption bands at 798 cm−1, 755 cm−1 and 759 cm−1 corresponding to the out-of-plane deformation vibrations of the C–H bonds. The peaks at 1461 cm−1, 1496 cm−1 and 1450 cm−1 correspond to the NN bonds. The peaks at 1373 cm−1, 1368 cm−1 and 1393 cm−1 correspond to the stretching vibrations of the C–N bonds. The absorption peaks at 1124 cm−1, 1138 cm−1 and 1117 cm−1 are due to the vibrations of the –NH+ groups formed upon protonation of the PANI's molecular chain (Table 1). This is evidence of the degree of electron delocalization and the nature of its electrical conductivity. The spectra of the P-AP polymer contain bands in the region of 1368–1358 cm−1 indicating the formation of a tertiary alcohol. The peaks at 594–462 cm−1, 536 cm−1 and 541 cm−1 correspond to various vibrations of the substituted rings. Thus, it follows from IR spectroscopy data that the structures of the polymers that we obtained are close to the emeraldine structure.
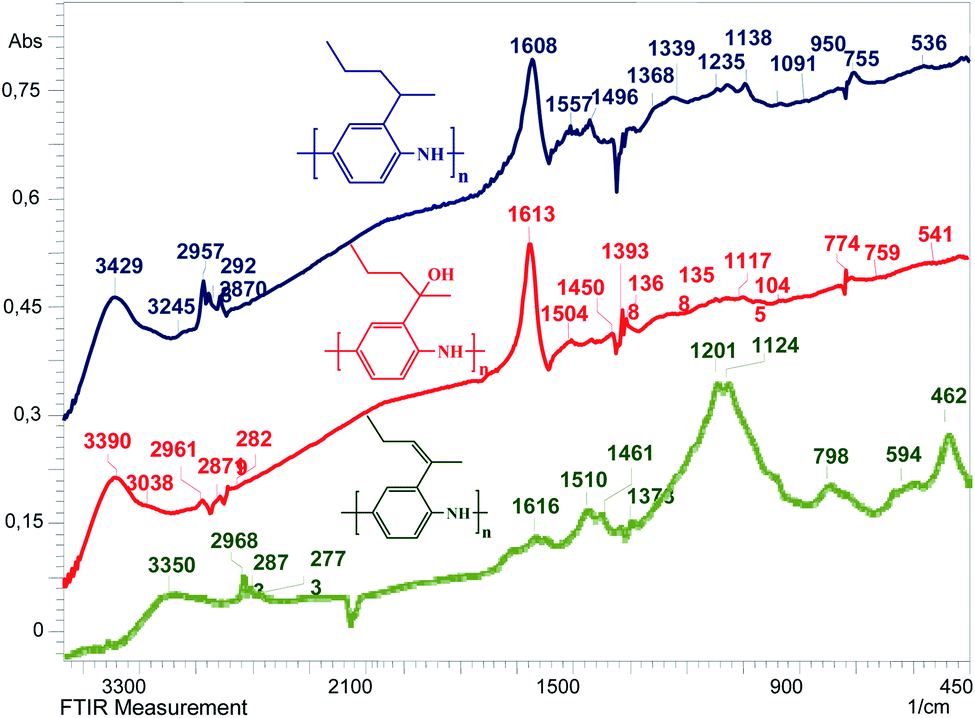 | ||
| Fig. 2 FT-IR spectra of P-PA, P-MB and P-AP. | ||
| Bands and peaks (cm−1) | Assignments |
|---|---|
| 3350–3429 cm−1 | H-Bonded N–H groups |
| 2773–2968 cm−1 | C–H stretching of alkyl structures |
| 1608–1616 cm−1 | Quinone-ring stretching |
| 1504–1557 cm−1 | Aromatic CC |
| 1368–1393 cm−1 | C–N stretching |
| 1117–1138 cm−1 | N+–/–N+˙– |
Elemental analysis
The results of elemental analysis are shown in Table 2. The total of carbon, hydrogen, and nitrogen amounts in the polymers synthesized was over 90% in all cases. This is consistent with a pure polymer in the emeraldine oxidation state, as demonstrated by the comparison with the calculated values.
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polymera | C (%) | H (%) | N (%) | Cl (%) | O (%) | Total (%) | ||||||
| Calc. | Found | Calc. | Found | Calc. | Found | Calc. | Found | Calc. | Found | Calc. | Found | |
| a Repeat unit. | ||||||||||||
| P-PA | 74.68 | 71.21 | 7.36 | 6.78 | 7.92 | 7.57 | 10.04 | 3.33 | — | — | 100.00 | 88.89 |
| P-MB | 73.85 | 71.89 | 8.39 | 8.19 | 7.83 | 7.35 | 9.93 | 4.87 | — | — | 100.00 | 92.30 |
| P-AP | 67.78 | 65.44 | 7.70 | 7.37 | 7.19 | 6.87 | 9.11 | 1.19 | 8.22 | 8.05 | 100.00 | 88.92 |
Solubility
As pointed out in the Introduction, the main purpose for attaching alkyl chains to the polyaniline backbone was to increase the poor solubility of unsubstituted polyaniline, especially in common organic solvents. Typically, when PANI doped with mineral or organic acids is added to bipolar aprotic solvents such as DMSO or NMP, a fraction of the polymer material (presumably oligomers or low molecular mass materials) dissolves, but a residual mass is always present in the mixture. After the successful synthesis of P-PA, P-MB and P-AP, the solubility was determined in various organic solvents. P-PA and P-MB (dark-brown solution) readily dissolved in polar solvents such as DMSO, DMF, NMP and THF, as well as in less polar solvents such as chloroform, acetone, and acetonitrile, but were insoluble in benzene and toluene. It is noteworthy that P-AP (dark-brown solution) was soluble in all the organic solvents tested (Table 3).| Form | Film color | Solubility |
|---|---|---|
| a The solubility of polymers was measured using 100 mg of a sample in 5 mL of a solvent. | ||
| P-PA | Dark-brown | DMSO, DMF, NMP, CHCl3, CH2Cl2, acetone |
| P-MB | Dark-brown | DMSO, DMF, NMP, CHCl3, CH2Cl2, acetone, EtOH, MeCN, THF |
| P-AP | Dark-brown | DMSO, DMF, NMP, CHCl3, CH2Cl2, acetone, EtOH, MeCN, THF, EtOAc, benzene, toluene |
Thus, modifying the alkyl in the PANI side chain is an efficient method for enhancing the solubility of the polymer in various types of solvents. Such a versatile solubility should be advantageous in the preparation of polymer films for chemical sensors.
1H NMR spectroscopy studies
Additional information about the structures of monomers and polymers was obtained from NMR spectra. Interestingly, displacement and hydrogenation of the double bond, as well as the presence of a hydroxy group in the polymers, made these samples soluble in polar solvent such as CHCl3, which enabled us to record their NMR spectra. The NMR spectra of monomers and polymers recorded in CDCl3 are shown in Fig. 3.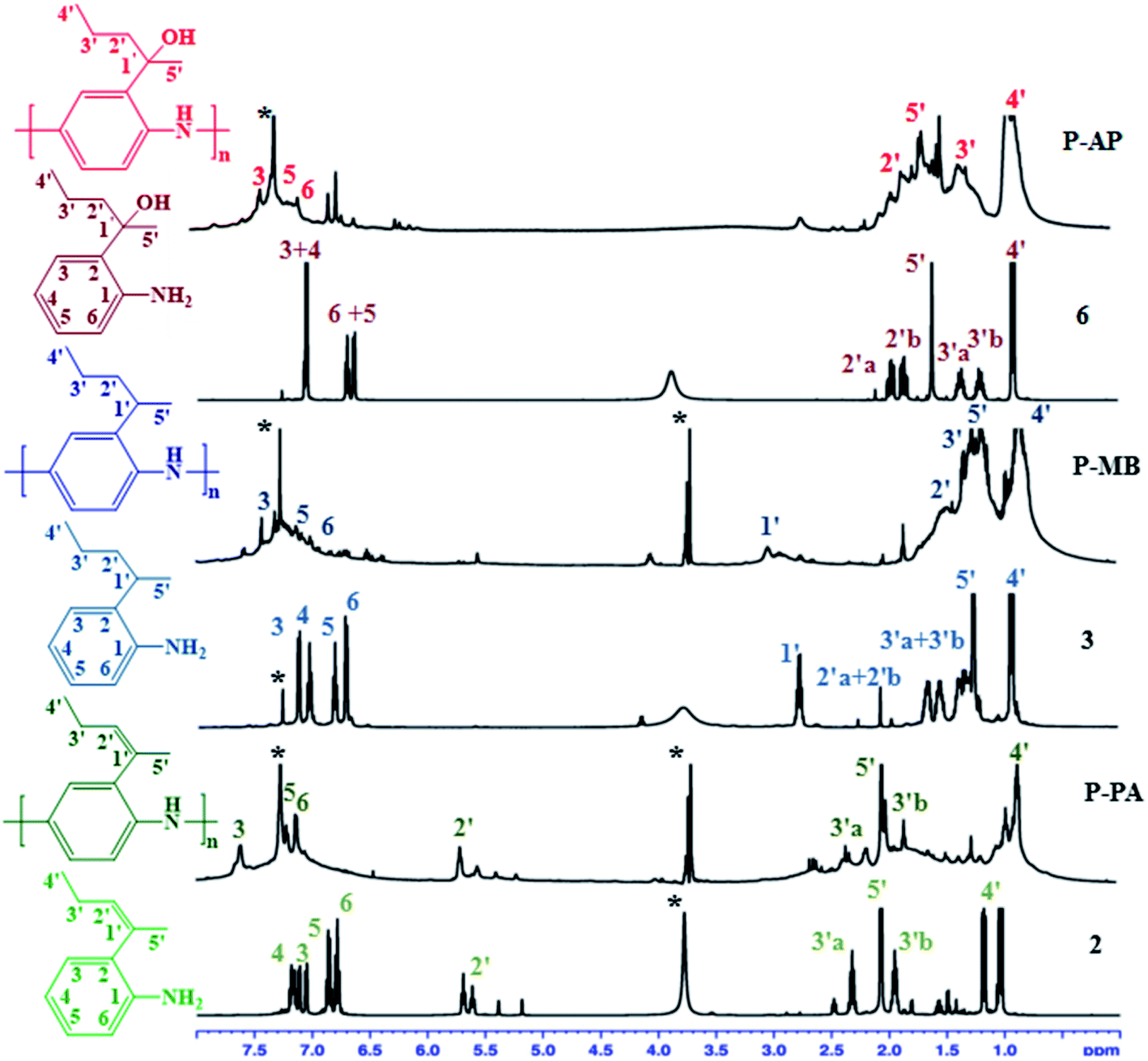 | ||
| Fig. 3 1H NMR spectra of the monomers and the corresponding polyaniline in CDCl3. | ||
Analysis of the 1H NMR spectra recorded at 500 MHz showed that aromatic protons in the starting monomers appeared in the δ 6.71–7.17 range as sharp peaks with clearly visible coupling constants, while the corresponding signals in the polymers were shifted downfield to the region of δ 7.06–7.62 and appeared as broadened lines. The signals of the H-4 proton at 7.0 ppm and of –NH2 at 3.80 ppm from the monomers disappear in 1H NMR after polymerization. The protons of the methylene groups in the side chains are expected to appear at 0.72–5.74 ppm.
Optical properties
Studying the absorption spectra is the best way to perform a comparative analysis of the effect of various structural factors on the change in the molecular geometry of the polymer chain.33 It is known that in most cases, PANI and its derivatives manifest two absorption maxima in the regions of 300 and 600 nm corresponding to the π–π* electron transition in the benzoid moiety and the n–π* electron transition in the quinoid moiety.33 Similarly to literature data, the PANI derivatives synthesized exhibit two absorption peaks in the electronic spectra (Fig. 4a). The presence of a substituent at the aromatic ring of the polymer resulted in a hypsochromic shift of the absorption maxima (by 10–20 nm) in the electronic spectra relative to those of unsubstituted PANI. The observed changes are due to the steric effect of bulky substituents, the presence of which favors an increase in the torsion angle between the aromatic rings and a decrease in the degree of conjugation (Table 4).16 It was found that in the series of PANI derivatives in question, the presence of a hydroxy group in the substituent of P-AP favors the largest shift in the absorption maximum due to a greater steric effect compared to Alk-substituted PANI derivatives (P-MB, P-PA) that manifest smaller hypsochromic shifts. Comparative analysis of literature data on other alkyl-substituted PANI derivatives confirms the above regularities.16,30,33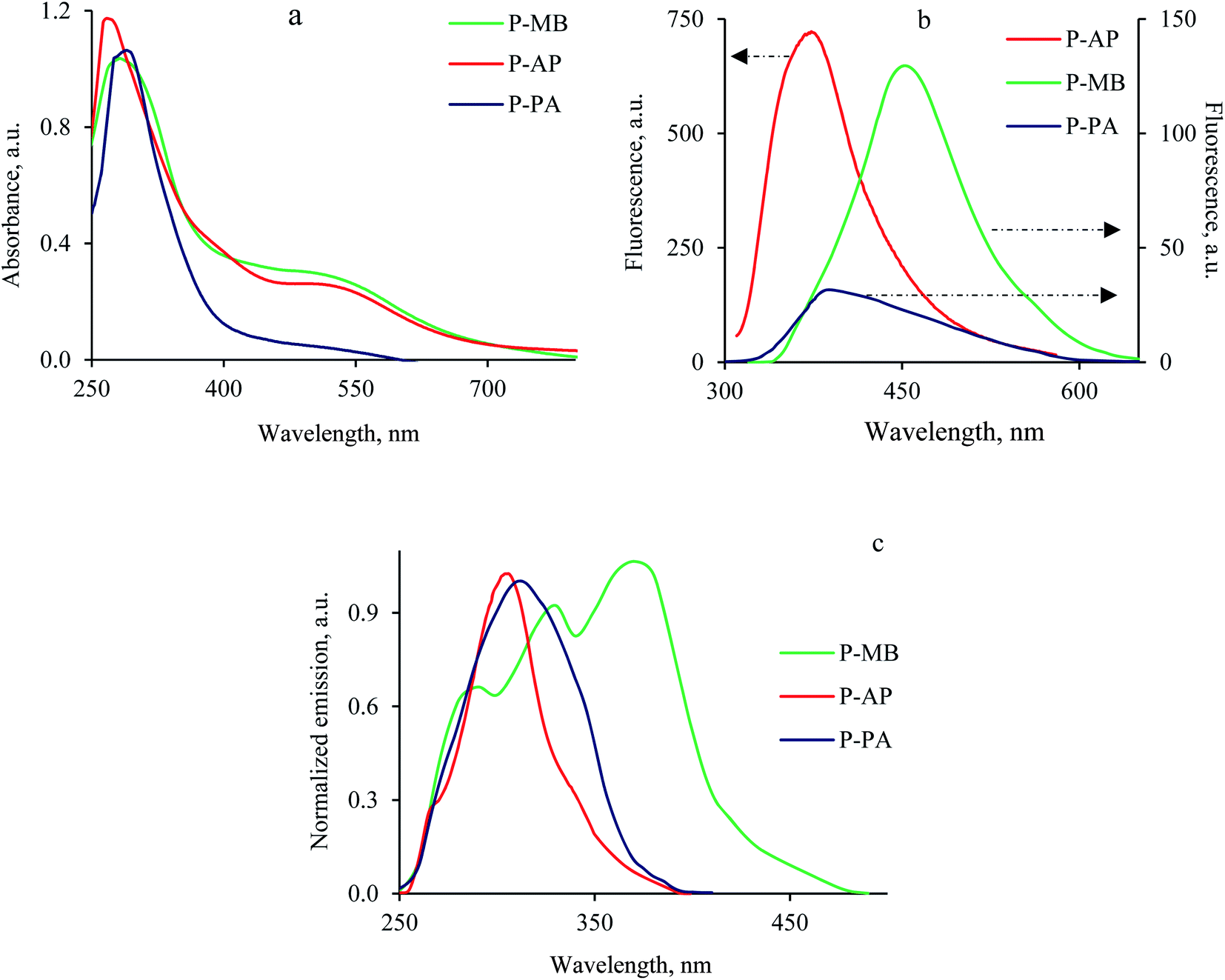 | ||
| Fig. 4 UV spectra (a), emission (b) and normalized excitation (c) photoluminescence spectra of the polymers in DMSO (10−2 g L−1). | ||
Previously, unsubstituted PANI was found to have luminescent properties with a quantum yield of 0.1 only NMP solution,34 whereas the luminescence quantum yields of substituted PANI derivatives are much higher.35 In addition, the luminescent properties of the resulting material can be controlled by changing the synthesis conditions and by varying the doping agent. In view of this, fluorescence studies of new PANI derivatives are very important. At the same time, analysis of changes in the excitation and emission spectra would make it possible to estimate the effect of the substituent on the structure of the polymer chain and on the optical properties of the compounds being studied.
Fig. 4b shows the excitation and emission spectra of P-AP, P-MB and P-PA recorded in DMSO solutions at room temperature. The fluorescent properties of conjugated polymers can be explained in terms of the band theory of semiconductors.36 On exposure to radiation, electrons pass from the valence band of a conjugated polymer to the conduction band and then migrate back to the polymer chain. An excited electron and an oppositely charged hole attract each other and undergo recombination to emit a photon.37 According to the general concept of the photoluminescence of compounds comprising a system of conjugated bonds, incorporation of a substituent into the aromatic system should have resulted in a decrease in the luminescence intensity due to a decrease in the structure rigidity.36 However, the opposite picture is observed for all the PANI derivatives studied, namely, incorporation of a bulky alkyl substituent increases the luminescence intensity. It was found previously that a PANI polymer should contain more than two sequentially linked benzenoid moieties to make photoluminescence observable.38 Thus, the highest luminescence intensity is demonstrated by undoped PANI in fully reduced form. Analysis of the set of the PANI derivatives presented in this work allows one to make the assumption that incorporation of an alkyl substituent complicates the oxidation of the amino group to an imino group.33 This assumption is confirmed by the increase in the photoluminescence intensity with an increase in the substituent volume, that is, in accordance with the steric hindrance that it exerts.
The photoluminescence excitation–emission maps (PLE) were studied for the series of PANI derivatives obtained in Fig. 5. One can see from the PLE maps that the position of the photoluminescence maximum does not depend on the excitation wavelength and shifts with an increase in the substituent volume, which also allows one to make the assumption that the steric effect has an influence on the position of the photoemission maximum.
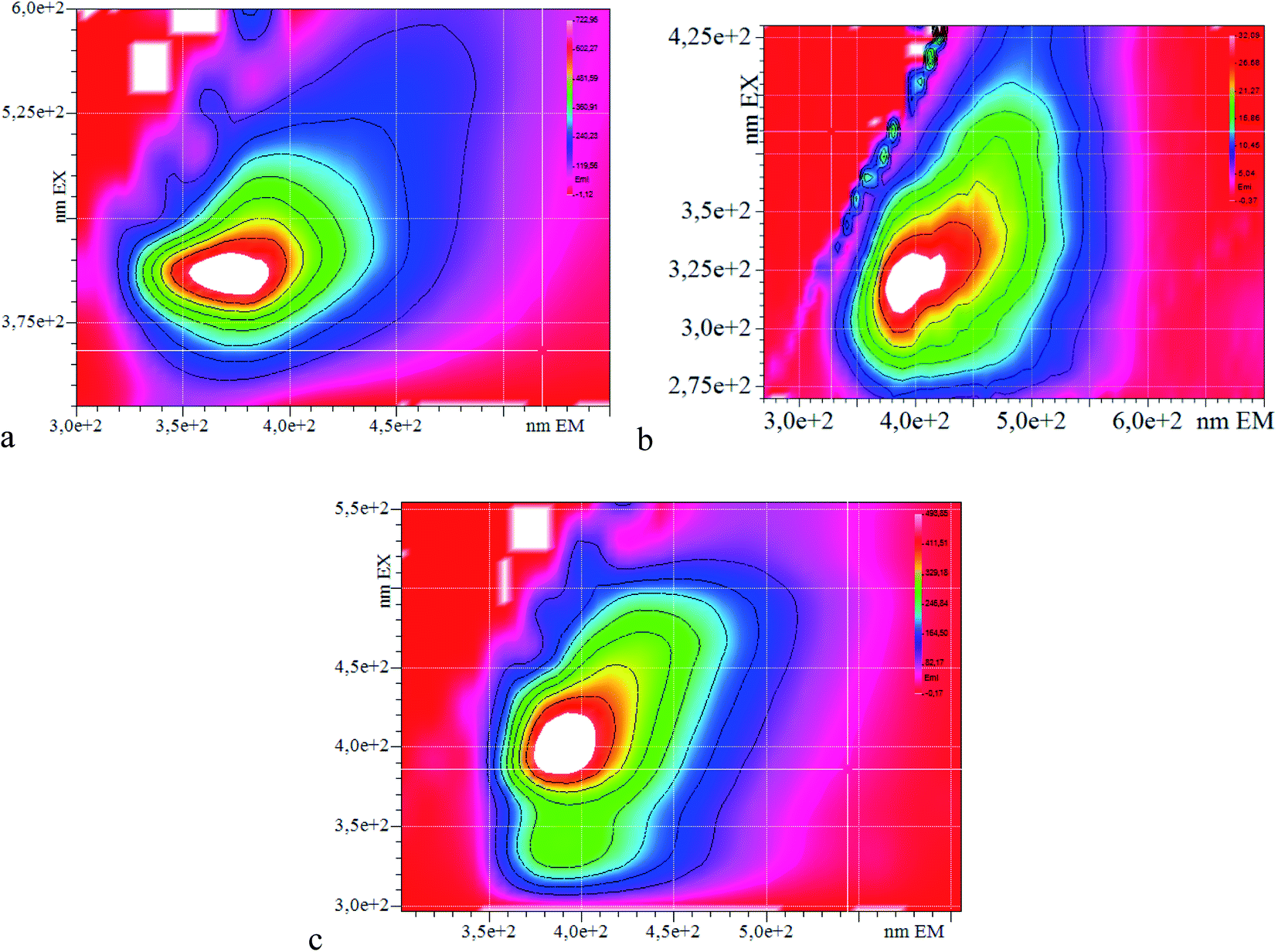 | ||
| Fig. 5 PLE map of the solutions of P-AP (a), P-PA (b) and P-MB (c). | ||
Scanning electron microscopy
The structural flexibility and the dependence of physicochemical properties on the synthesis conditions are considered to be among the most important PANI properties. They make it possible to identify the structure–property relationship for nanosized materials and develop potential applications adapted for various types of morphologies, depending on the properties required.39Fig. 6 shows the results of scanning electron microscopy. The supramolecular structure of the samples obtained differs significantly from the structure of PANI and its alkyl derivatives studied previously.39 Unsubstituted PANI obtained in the classical way has a fibrillar structure, whereas its ortho-substituted derivatives are most commonly characterized by a globular morphology comprising distinct spheres.39 Similarly to the samples studied previously, the P-AP and P-PA polymers obtained have a globular morphology, in contrast to P-MB which has a heterogeneous hierarchical structure.
 | ||
| Fig. 6 SEM images of the polymers. | ||
The observed difference between the supramolecular structures formed by the polymers may be due to the nature of the reacting monomer and the effect of the substituent at the aromatic ring in the polymer chain.
Mw determination of polymers
Molecular weights of the polymers P-MB and P-AP were obtained by gel permeation chromatography analyses. The samples were dissolved in THF. Weight average MW (Mw) varies from 59796 to 67268 g mol−1. Table 5 summarizes the Mw, number average MW (Mn), and polydispersity index (PDI) of these samples.
| Sample | Mn (g mol−1) | Mw (g mol−1) | PDI |
|---|---|---|---|
| P-MB | 31454 |
67268 |
2.1 |
| P-AP | 22353 |
59796 |
2.7 |
The sensing properties of polymers
An important advantage of PANI and its derivatives that allows one to use them in various fields of technology is that their physicochemical parameters depend on the chemical composition of the environment.40 Gas sensors are considered to be one the promising applications of PANI-based polymers.41 In view of this, the P-MB, P-AP and P-PA polymers obtained were used as sensitive materials in resistive sensors. The results are presented in Fig. 7 and 8.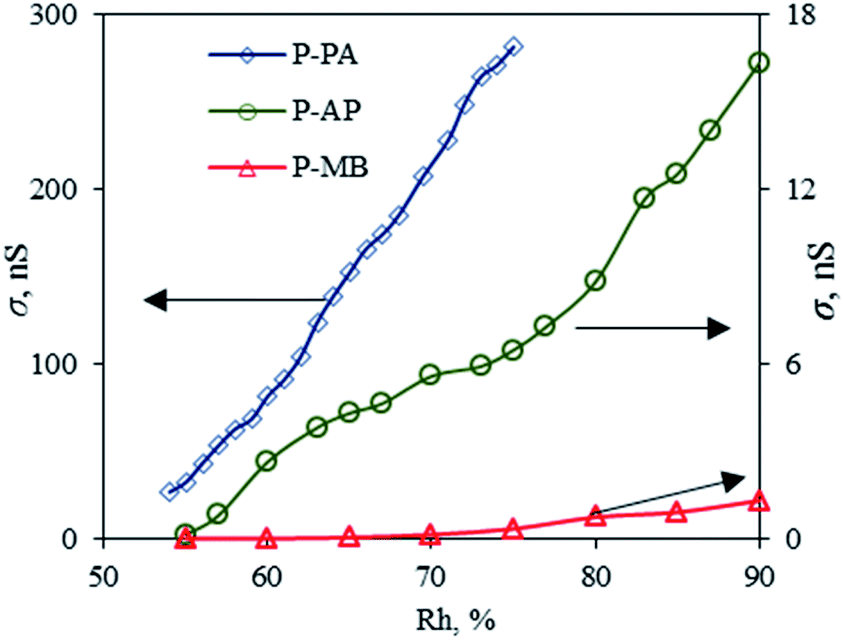 | ||
| Fig. 7 Plots of the conductivity of P-PA, P-AP and P-MB films on the air humidity. | ||
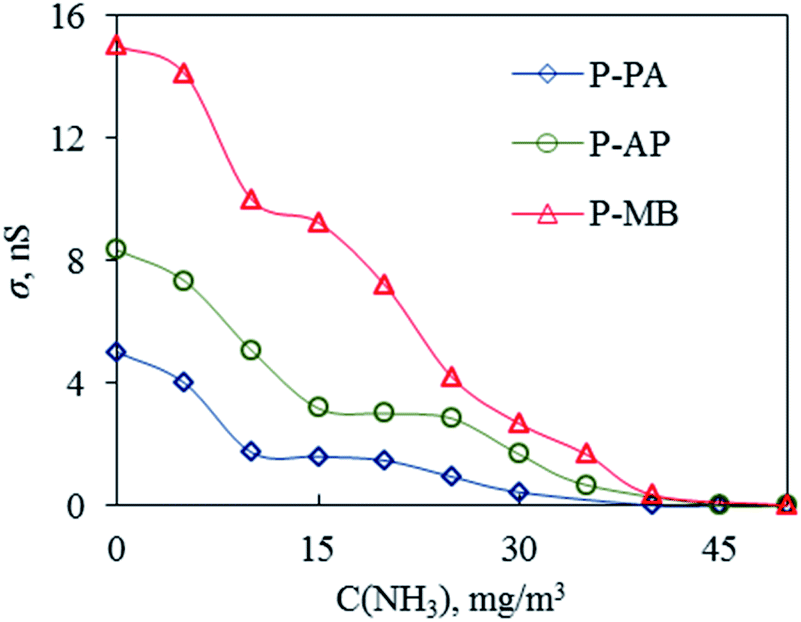 | ||
| Fig. 8 Plots of the conductivity of P-PA, P-AP and P-MB films on the concentration of ammonia vapor. | ||
Based on SEM results, it is assumed that P-PA (Fig. 7) should have the best response to chemicals in air because it has a more developed surface. P-PA showed the highest film conductivity in humid air (Fig. 8). Presumably, an increase in the electrical conductivity of a polymer film with an increase in the ambient humidity is due to an increase in the mobility of the doping ion that is weakly bound to the polymer chain through van der Waals forces.42 As the environment humidity increases, the polymer film absorbs moisture, which results in polymer swelling followed by unfolding of the compact spiral-shaped polymer chain. The formation of a more aligned structure of the PANI derivative favors the charge transfer along the polymer chain.43 It is also assumed that saturation of the polymer with moisture is accompanied by the formation of a complex due to the transfer of an unshared electron pair from the nitrogen atom in the polymer to water molecules.43 The results of measuring the current–voltage characteristics confirm the assumption that the mobility of charge carriers along individual polymer chains and across adjacent chains is high due to the geometrically ordered, closely packed structure of the polymer chains.44
On the other hand, the polymer chains within the micro-aggregated P-MB polymer film were much less ordered and less densely packed, which resulted in a greater number of structural defects that apparently improve the sensitivity to ammonia vapor (Fig. 7 and 8). The P-MB film had the largest roughness (Table 6).
| Compound | P-PA | P-MB | P-AP |
|---|---|---|---|
| Average roughness Ra, nm | 1.7 | 2.5 | 3.7 |
| Root-mean-square roughness Rq, nm | 2.4 | 3 | 4.7 |
It is well known that PANI doping with HCl results in an increase in electrical conductivity. Upon protonation, neutral PANI molecules acquire protons to form positively charged local centers located on the nitrogen atoms, which facilitate the motion of valence electrons from one such center to another, thus causing the hopping conductivity. When PANI reacts with ammonia, the molecules of the latter absorb protons from PANI to form energetically favorable ammonium (NH4+). As a result, PANI dedoping occurs.
Considering that the carrier mobility depends on the ordering of film molecules on the surface45,46 and to explain the reasons of the strong difference in the conductivity of films, we analyzed the film surfaces on a scanning probe microscope in atomic force microscopy (AFM) mode in Fig. 9. As one can see from Fig. 9, the P-PA film has a more uniform and smoother surface than the P-AP and P-MB films. In addition, P-PA films have lower roughness.
 | ||
| Fig. 9 Atomic force microscopy of the surface of polymer films. | ||
Thus, the presented results of studying the surface of films with a scanning probe microscope in atomic force microscopy mode show that the transport layers with higher conductivity and moisture sensitivity have lower surface roughnesses.
Conclusions
Thus, a series of hitherto unknown poly(o-alkylanilines) have been synthesized in good yields, namely: poly[2-(1-methylbut-1-en-1-yl)aniline], poly[2-(1-methylbutyl)aniline] and poly[2-(2-aminophenyl)pentan-2-ol]. The polymers vary in solubility, depending on the chemical structure of the substituents. The PANI derivatives presented here demonstrated a high photoluminescence intensity. The changes in the emission spectra made it possible to estimate the effect of structural factors on the molecular geometry of the polymers obtained. However, the electrical conductivity of modified polyanilines is lower than that of pure PANI due to the smaller conjugation length. The considerable increase in the solubility of the presented series of polymers, i.e., P-MB, P-AP and P-PA, made it possible to form homogeneous thin films and use them as active layers in resistive gas sensors. Varying the substituents in the polymer structure resulted in sensors with various sensitivity to the ambient humidity and ammonia concentration. The greatest response to the moisture variation was obtained for the sensor based on P-PA. The quality of the film surface plays an important role in the sensor properties of polymers; therefore, the PANI derivatives were studied by AFM and SEM. It was found that the polymer with the smallest roughness was most sensitive to the environment quality.Thus, a detailed study of a number of PANI derivatives with different substituents showed that the physicochemical properties of the resulting polymers can be tuned, which makes it possible to use them in various fields of organic electronics.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation as part of the state task no. AAAA-A19-119020890014-7.Notes and references
- M. A. Shishov, V. A. Moshnikov and I. Y. Sapurina, Glass Phys. Chem., 2011, 37, 106–110 CrossRef CAS.
- S. Trey, S. Jafarzadeh and M. Johansson, ACS Appl. Mater. Interfaces, 2012, 4, 1760–1769 CrossRef CAS PubMed.
- V. G. Kulkarni, L. D. Campbell and W. R. Mathew, Synth. Met., 1989, 30, 321–325 CrossRef CAS.
- Z. A. Boeva and V. G. Sergeyev, Polym. Sci., Ser. C, 2014, 56, 144–153 CrossRef CAS.
- S. Bhadra, D. Khastgir, N. K. Singha and J. H. Lee, Prog. Polym. Sci., 2009, 34, 783–810 CrossRef CAS.
- Z. Li and L. Gong, Materials, 2020, 13, 548 CrossRef CAS PubMed.
- E. Akbarinezhad, M. Ebrahimi and H. R. Faridi, Prog. Org. Coat., 2009, 64, 361–364 CrossRef CAS.
- E. Song and J. W. Choi, Nanomaterials, 2013, 3, 498–523 CrossRef CAS PubMed.
- M. R. Gizdavic-Nikolaidis, J. R. Bennett, S. Swift, A. J. Easteal and M. Ambrose, Acta Biomater., 2011, 17, 4204–4209 CrossRef PubMed.
- L. A. Gallarato, L. E. Mulko, M. S. Dardanelli, C. A. Barbero, D. F. Acevedo and E. I. Yslas, Colloids Surf., B, 2017, 150, 1–7 CrossRef CAS PubMed.
- M. R. Gizdavic-Nikolaidis, J. C. Pagnon, N. Ali, R. Sum, N. Davies, L. F. Roddam and M. Ambrose, Colloids Surf., B, 2015, 136, 666–673 CrossRef CAS PubMed.
- S. Shreepathi and R. Holze, Chem. Mater., 2005, 17, 4078–4085 CrossRef CAS.
- H. Swaruparani, S. Basavaraja, C. Basavaraja, D. S. Huh and A. Venkataraman, J. Appl. Polym. Sci., 2010, 117, 1350–1360 CAS.
- J. Kim, S. Kwon and D. W. Ihm, Curr. Appl. Phys., 2007, 7, 205–210 CrossRef.
- R. Bodalia, R. Stern, C. Batich and R. Duran, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2123–2127 CrossRef CAS.
- L. R. Latypova, A. N. Andriianova, S. M. Salikhov, I. N. Mullagaliev, R. B. Salikhov, I. B. Abdrakhmanov and A. G. Mustafin, Polym. Int., 2020, 69, 804–812 CrossRef CAS.
- A. R. Modarresi-Alam, H. A. Amirazizi, F. Movahedifar, A. Farrokhzadeh, G. R. Asli and H. Nahavandi, J. Mol. Struct., 2015, 1083, 17–26 CrossRef CAS.
- W. F. Alves, E. C. Venancio, F. L. Leite, D. H. Kanda, L. F. Malmonge, J. A. Malmonge and L. H. Mattoso, Thermochim. Acta, 2010, 502, 43–46 CrossRef CAS.
- G. D'Aprano, M. Leclerc, G. Zotti and G. Schiavon, Chem. Mater., 1995, 7, 33–42 CrossRef.
- P. Anilkumar and M. Jayakannan, Langmuir, 2008, 24, 9754–9762 CrossRef CAS PubMed.
- S. M. Sayyah, A. A. Bahgat and H. M. A. El-Salam, Int. J. Polym. Mater., 2002, 51, 291–314 CrossRef CAS.
- J. Huang and M. Wan, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1277–1284 CrossRef CAS.
- R. B. Salikhov, A. N. Lachinov, R. G. Rakhmeev, R. M. Gadiev, A. R. Yusupov and S. N. Salazkin, Meas. Tech., 2009, 52, 427–431 CrossRef CAS.
- J. Janata and M. Josowicz, Nat. Mater., 2003, 2, 19–24 CrossRef CAS PubMed.
- G. Harsányi, Sens. Rev., 2000, 20, 98–105 CrossRef.
- A. Piqué, R. C. Y. Auyeung, J. L. Stepnowski, D. W. Weir, C. B. Arnold, R. A. McGill and D. B. Chrisey, Surf. Coat. Technol., 2003, 163, 293–299 CrossRef.
- F. W. Zeng, X. X. Liu, D. Diamond and K. T. Lau, Sens. Actuators, B, 2010, 143, 530–534 CrossRef CAS.
- L. Kumar, I. Rawal, A. Kaur and S. Annapoorni, Sens. Actuators, B, 2017, 240, 408–416 CrossRef CAS.
- T. F. Wu and J. D. Hong, RSC Adv., 2016, 6, 96935–96941 RSC.
- A. Andriianova, A. Shigapova, Y. Biglova, R. Salikhov, I. Abdrakhmanov and A. Mustafin, Chin. J. Polym. Sci., 2019, 37, 774–782 CrossRef CAS.
- V. I. Levashova and T. P. Mudrik, Pet. Chem., 2008, 48, 314–317 CrossRef.
- R. R. Gataullin, I. S. Afon'kin, A. A. Fatykhov, L. V. Spirikhin and I. B. Abdrakhmanov, Russ. J. Org. Chem., 2001, 37, 834–840 CrossRef CAS.
- A. N. Andriianova, Y. N. Biglova and A. G. Mustafin, RSC Adv., 2020, 10, 7468–7491 RSC.
- P. S. Antonel, E. M. Andrade and F. V. Molina, J. Electroanal. Chem., 2009, 632, 72–79 CrossRef.
- A. N. Andriyanova, D. E. Gribko, T. T. Sadykov, I. N. Mullagaliev, R. B. Salikhov and A. G. Mustafin, Polym. Sci., Ser. B, 2021, 63, 135–141 CrossRef.
- C. A. Parker, Photoluminescence of Solutions with Applications to Photochemistry and Analytical Chemistry, ed. C. A. Parker,New York, 1969 Search PubMed.
- F. A. Rafiqi and K. Majid, Synth. Met., 2015, 202, 147–156 CrossRef CAS.
- J. Y. Shimano and A. G. MacDiarmid, Synth. Met., 2001, 123, 251–262 CrossRef CAS.
- H. D. Tran, D. Li and R. B. Kaner, Adv. Mater., 2009, 21, 1487–1499 CrossRef CAS.
- O. S. Ahmad, T. S. Bedwell, C. Esen, A. Garcia-Cruz and S. A. Piletsky, Trends Biotechnol., 2019, 37, 294–309 CrossRef CAS PubMed.
- D. Nicolas-Debarnot and F. Poncin-Epaillard, Anal. Chim. Acta, 2003, 475, 1–15 CrossRef CAS.
- M. V. Kulkarni, S. K. Apte, S. D. Naik, J. D. Ambekar and B. B. Kale, Sens. Actuators, B, 2013, 178, 140–143 CrossRef CAS.
- M. V. Kulkarni and A. K. Viswanath, Polym. Eng. Sci., 2007, 47, 1621–1629 CrossRef CAS.
- F. W. Zeng, X. X. Liu, D. Diamond and K. T. Lau, Sens. Actuators, B, 2010, 143, 530–534 CrossRef CAS.
- C. D. Dimitrakopoulos and D. J. Mascaro, IBM J. Res. Dev., 2001, 45, 11–27 CAS.
- A. R. Tameev, R. G. Rakhmeev, V. R. Nikitenko, R. B. Salikhov, A. A. Bunakov, A. N. Lachinov and A. V. Vannikov, Phys. Solid State, 2011, 53, 195–200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02474d |
| This journal is © The Royal Society of Chemistry 2021 |