
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insight into B(C6F5)3 catalyzed imine reduction with PhSiH3 under stoichiometric water conditions†
Yunqing He‡
 *a,
Wanli Nie‡*b,
Ying Xuec and
Qishan Hud
*a,
Wanli Nie‡*b,
Ying Xuec and
Qishan Hud
aFaculty of Materials and Chemical Engineering, Computational Physics Key Laboratory of Sichuan Province, Yibin University, Yibin, Sichuan 64400, China. E-mail: yunqing0228@163.com
bNatural Products and Small Molecule Catalysis Key Laboratory of Sichuan Province, Leshan Normal University, Leshan, Sichuan 614000, China. E-mail: niewl126@126.com
cCollege of Chemistry, Sichuan University, Chengdu 610064, China
dSchool of Chemistry and Chemical Engineering, Sichuan University of Arts and Science, Dazhou, Sichuan 635000, China
First published on 14th June 2021
Abstract
A DFT and experimental study on the mechanism of B(C6F5)3 catalyzed imine reduction is performed using PhSiH3 as reductant under stoichiometric water conditions. Ingleson’s path B is reconfirmed here. And four novel (C6F5)3B–OH2 induced pathways (paths C2, C3, D2 and D3) entirely different from all the previous mechanisms were determined for the first time. They are all B(C6F5)3 and water/amine catalyzed cycles, in which the nucleophilic water or amine catalyzed addition step between PhSiH3 and the N-silicon amine cation is the rate-determining step of paths C2/D2 and C3/D3 with activation Gibbs free energy barriers of 23.9 and 18.3 kcal mol−1 in chloroform, respectively, while the final desilylation of the N-silicon amine cation depends on an important intermediate, (C6F5)3B–OH−. The competitive behavior of the 5 paths can explain the experimental facts perfectly; if all the reactants and catalysts are added into the system simultaneously, water amount and nucleophiles (excess water and produced/added amine) provide on–off selectivity of the pathways and products. 1 eq. water leads to quick formation of (C6F5)3B–OH−, leading to B-II being turned off, and nucleophiles like excess water and produced/added amine switch on CD-II, leading to production of the amine. B-I′ of Ingleson’s path B is the only mechanism for anhydrous systems, giving N-silicon amine production only; B-I and C-I are competitive paths for systems with no more than 1 eq. water, producing the N-silicon amine and the [PhHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NHPh]+[(C6F5)3B–OH]− ion pair; and paths C2, C3, D2 and D3 are competitive for systems with 1 eq. water and nucleophiles like excess water or added/produced amine, directly giving amination products.
NHPh]+[(C6F5)3B–OH]− ion pair; and paths C2, C3, D2 and D3 are competitive for systems with 1 eq. water and nucleophiles like excess water or added/produced amine, directly giving amination products.
1 Introduction
Reductive amination of carbonyl compounds (Scheme 1, which includes two steps: the condensation step with water generation first, and then the imine reduction) represents an efficient one-step approach to a variety of amines and is of significant importance in the chemical, biochemical and polymer industries.1 In recent years, frustrated Lewis pairs (FLPs), especially B(C6F5)3-based ones, have been widely considered as prospective non-metallic catalysts capable of effectively activating H2 or Si–H as hydride sources in the catalytic reduction of imines and enamines.2,3 It is a priori assumed that any reaction system involving B(C6F5)3 requires strictly anhydrous conditions,4 so an imine (or enamine) must be synthesized and dehydrated in advance.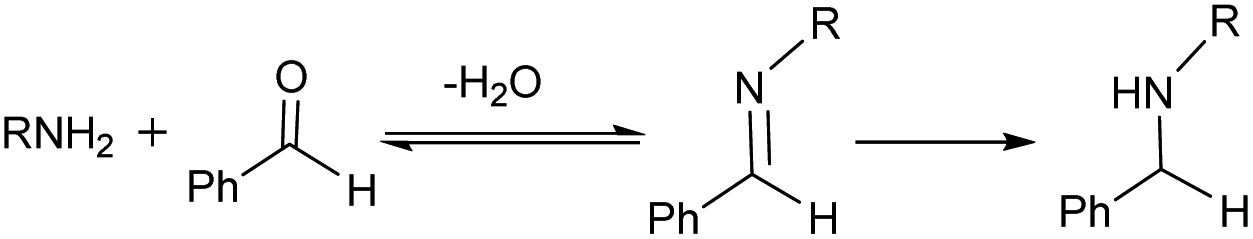 | ||
| Scheme 1 Reductive amination. | ||
Recently, Ashley and Stephan’s research groups reported the hydrogenation of aldehydes catalyzed by B(C6F5)3 in reagent grade polar solvents under high pressure of H2, in which the authors suggested that B(C6F5)3 has sufficient water tolerance that a small amount of water in the solvent does not affect the activity of the catalyst in polar solvents; Soós and coworkers developed a boron/nitrogen-centered FLP with remarkably high water tolerance; and Chang and coworkers reported a one-pot borane catalyzed reductive amination/hydrosilylation of α,β-unsaturated aldehydes, producing β-silylated secondary amines.5 In 2016, Ingleson’s research team reported a successful one-pot reductive amination of aldehydes catalyzed by BAr3 in the presence of hydridosilanes. Based on the viewpoint of dissociative borane catalysis, and its poor tolerance to water/amine combinations, Ingleson suggested that B(C6F5)3 is only a viable catalyst for arylamines but not alkylamines due to the irreversible deprotonation of (C6F5)3B–OH2 by the latter (Scheme 2);6 that is to say irreversible formation of (C6F5)3B–OH− was disadvantageous to the reaction process. On the other hand, the acidity of (C6F5)3B–OH2 is comparable to that of hydrochloric acid.7 Broadbelt et al. proposed a transition state in which (C6F5)3B–OH2 donates a proton in ring-opening reactions of epoxyoctane.8 Our group recently confirmed that B(C6F5)3 catalyzed reductive amination of aldehydes with alkoxyamines can proceed under moist conditions with hydridosilanes as reducing agents.9
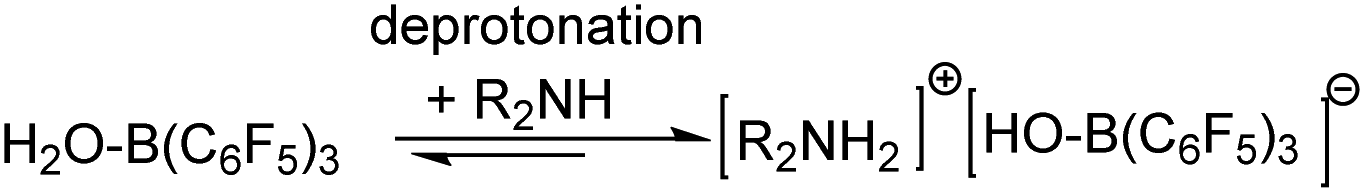 | ||
| Scheme 2 Deprotonation of (C6F5)B–OH2 with amines. | ||
The mechanistic study of FLP chemistry under “water or moist” conditions has attracted great attention. Imine reduction is a key process in reductive amination reactions, and the mechanism of borane catalyzed imine reduction under water conditions is a burning question. Ingleson et al. proposed two alternative catalytic cycles to explain the reduction of the imine intermediate in the borane catalyzed reductive amination.6 Based on Ingleson’s work, a density functional theory (DFT) study was performed by Wei et al. on the mechanism of B(C6F5)3 catalyzed reductive amination under wet conditions, in which Ingleson’s cycles were believed to be disfavored for high barriers and another path with very low barriers was suggested.10 But we noted that a four-molecule elementary reaction step is involved in Wei’s pathway which in fact makes it barely possible kinetically.11 In all the paths proposed by Ingleson et al. and Wei et al., imine cations PhHCN+RPh (R = H or {Si}) and (C6F5)3B–H− are considered as key intermediates (Scheme 3), and amine should certainly be produced under water conditions. However, this is not consistent with our experiments; we have found that water amount and nucleophiles are very crucial for producing different hydrosilylation or amination products (see Table 1). The interaction between the Lewis base (LB), water and B(C6F5)3 may play an important role in the reductive process, and perhaps some Si–H bond activation processes which do not produce (C6F5)3B–H− should be considered.
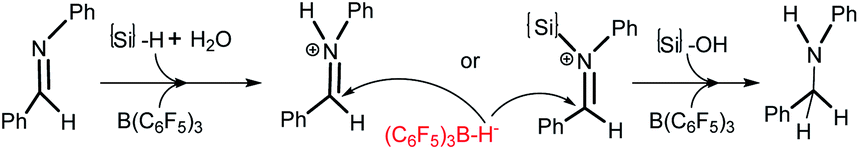 | ||
| Scheme 3 The mechanisms previously proposed for imine reduction by Ingleson et al. and Wei et al. | ||
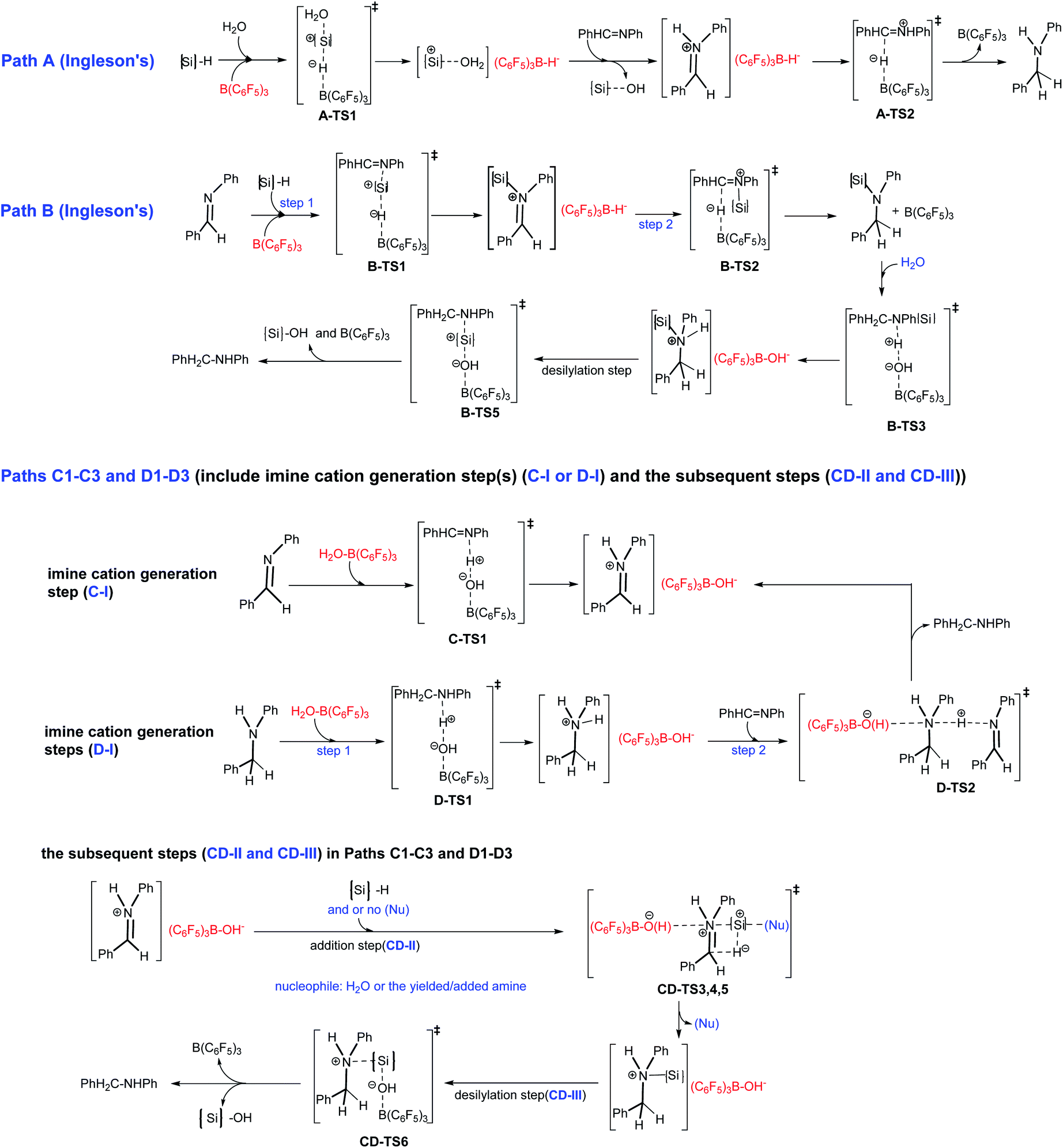
In this work, we present a DFT computational and experimental investigation into the mechanism of B(C6F5)3 catalyzed imine reduction with PhSiH3 under water conditions, and eight reaction pathways are reported (see Scheme 4). Paths A and B are Ingleson’s catalytic cycles which were determined to be disfavored for high barriers by Wei et al. using PhSiMe2H; here we reconfirmed them using PhSiH3. The (C6F5)3B–OH2 induced paths C1–C3 and D1–D3 are newly proposed and completely different from the above cycles, in that (C6F5)3B–OH− is the key intermediate, and nucleophilic reagents like water or amines play an important role. As shown in Scheme 4, the imine cation generation step(s) (C-I or D-I), addition step (CD-II) and desilylation step (CD-III) are involved in paths Cn and Dn; C-I involves a direct proton transfer from (C6F5)3B–OH2 to the imine, giving an imine cation, and direct proton transfer from (C6F5)3B–OH2 to the amine occurs first in D-I. The pathways involving C-I and D-I are denoted as paths Cn and Dn, respectively. And then the pathways involving CD-II without nucleophile assistance are denoted as paths C1/D1, and those that involve water assisted and amine assisted CD-II are denoted as paths C2/D2 and paths C3/D3, respectively.
 | ||
| Scheme 4 Possible pathways of the title reaction. | ||
2 Computational details
The computational study was performed using the Gaussian 16 program package.12 All geometry optimizations and corresponding frequency analyses were performed at the B3LYP/6-311G(d,p) level in the gas phase, and the relationships between the transition states and the intermediates were verified with intrinsic reaction coordinate (IRC)13 calculations. Solvent effect correction was done at the B3LYP/6-311G(d,p) level with Grimme’s D3 BJ dispersion correction14 using the SMD model15–17 in chloroform. Single point energies in the solvent were combined with the thermodynamic corrections in the gas phase to obtain the Gibbs free energies in chloroform. Gibbs free energies are used in the following discussion and dissociated B(C6F5)3 is selected as zero for comparison of different mechanisms. Transition states and intermediates are denoted as TS and IM in the following discussion, respectively. The optimized Cartesian coordinates of the stationary points are given in the ESI.†3 Results and discussion
3.1 Experimental data
The influence of water and amine on B(C6F5)3 catalyzed imine reduction using PhSiH3 under water conditions is studied experimentally. As shown in Table 1, entries 1–4 give the corresponding hydrosilylation products in imine (PhCHN–Ph/–tBu) reduction systems containing 0 or 1 equiv. water with respect to the imine, while amination products were formed for entries 5 and 6 upon simply increasing the amount of water to 2 equiv. with respect to the imine, and entries 7 and 8 give the corresponding amination products, with just a slight difference (0.2 equiv. Et2NH is used) compared to entries 3 and 4.
|
|||||
|---|---|---|---|---|---|
| Entry | Imines | H2O equiv. | Et2NH equiv. | Temp. (°C)/time | Main products (% yield) |
| a Reaction conditions: imine 0.1 mmol; hydrosilane 0.15 mmol; B(C6F5)3 10 mol%; solvent: CDCl3; products were determined by 1H NMR spectroscopy. | |||||
| 1 | PhCHN–Ph |
0 | 0 | 30/12 h | PhCH2NPhSiH2Ph (100) |
| 2 | PhCHN–tBu |
0 | 0 | 50/12 h | PhCH2NtBuSiH2Ph (100) |
| 3 | PhCHN–Ph |
1 | 0 | 30/12 h | PhCH2NPhSiH2Ph (100) |
| 4 | PhCHN–tBu |
1 | 0 | 50/12 h | PhCH2NtBuSiH2Ph (100) |
| 5 | PhCHN–Ph |
2 | 0 | 30/12 h | PhCH2NHPh (100) |
| 6 | PhCHN–tBu |
2 | 0 | 50/12 h | PhCH2NHtBu (100) |
| 7 | PhCHN–Ph |
1 | 0.2 | 30/12 h | PhCH2NHPh (60) |
| 8 | PhCHN–tBu |
1 | 0.2 | 50/12 h | PhCH2NHtBu (83) |
3.2 Computational data
PhN![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) B(C6F5)3 (IM0) in anhydrous systems, and (C6F5)3BO(H)–HNPhCHPh (IM1) and (C6F5)3BO(H)–HNH(Ph)–CH2Ph (IM2) under water conditions, and the optimized structures of the complexes are shown in Fig. 1. So, IM0 is the starting point in anhydrous systems, and IM1 and IM2 are the starting points for paths Bn and Cn under water conditions.
B(C6F5)3 (IM0) in anhydrous systems, and (C6F5)3BO(H)–HNPhCHPh (IM1) and (C6F5)3BO(H)–HNH(Ph)–CH2Ph (IM2) under water conditions, and the optimized structures of the complexes are shown in Fig. 1. So, IM0 is the starting point in anhydrous systems, and IM1 and IM2 are the starting points for paths Bn and Cn under water conditions.
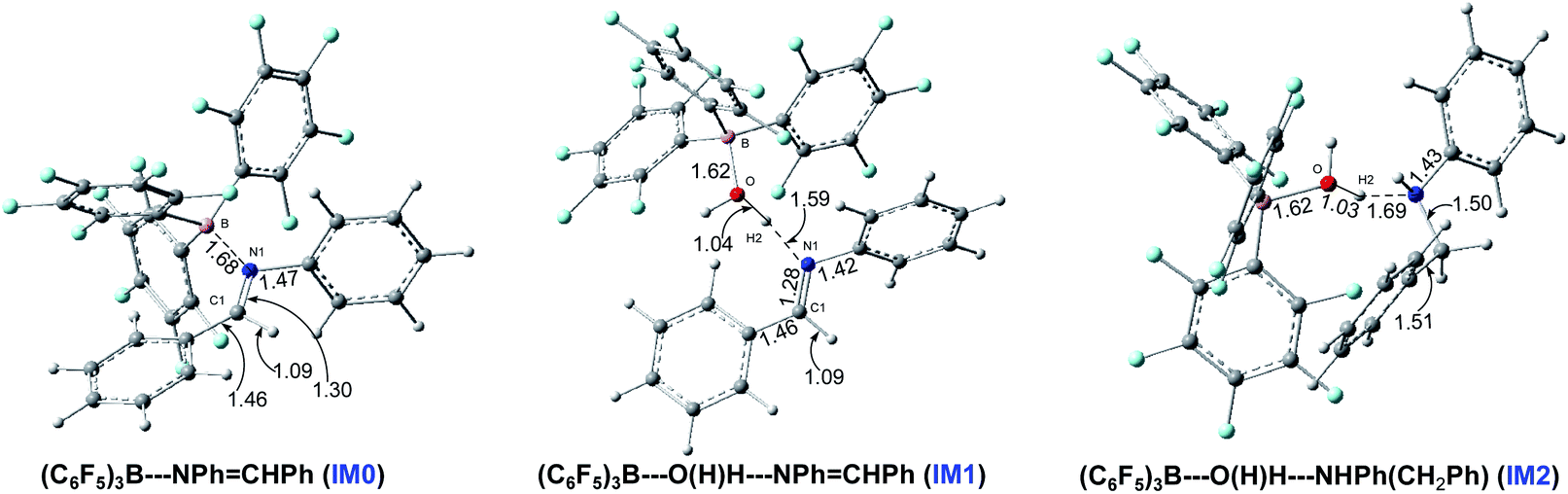 | ||
| Fig. 1 The optimized structures of the initial complexes at the B3LYP/6-311G(d,p) level in the gas phase with corresponding interatomic distances in angstroms. | ||
Here we give an abbreviated presentation of path B for necessity of discussion. The energy profile of path B is shown in Fig. 2, and the optimized structures of the reactants and products, the stationary points in the N-silicon amine forming process (B-I′ and B-I in Fig. 2 for anhydrous and water containing conditions, respectively) and the stationary points in the subsequent steps (B-II) are shown in Fig. S2–S4,† respectively. Path B (IM1 → PhH2C–NHPh) is a possible pathway under water conditions due to its low Gibbs free energy barriers (19.9, 4.3, 4.8 and 15.9 kcal mol−1 for B-TS1, B-TS2, B-TS4 and B-TS5, respectively, and no barrier for B-TS3). B-TS4 is a transition state for N–Si rotation. While IM0 → N-silicon amine (B-I′) may be the mechanism of imine reduction in anhydrous conditions, without subsequent hydrolysis and desilylation.
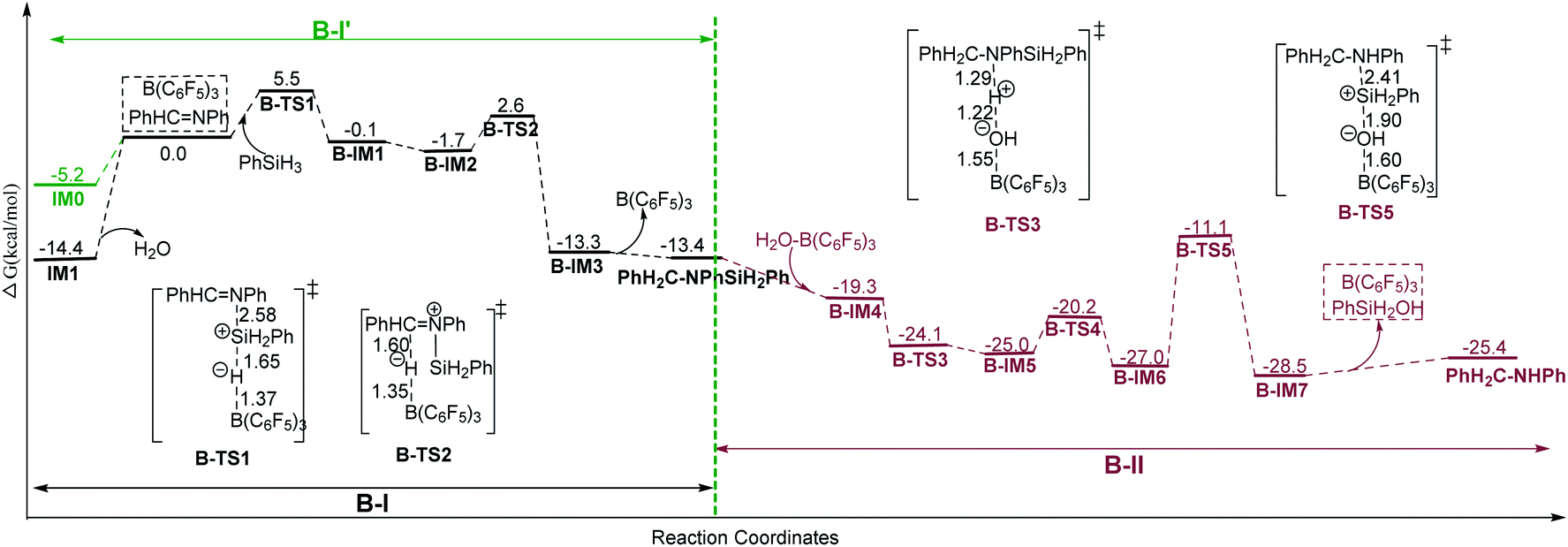 | ||
| Fig. 2 The Gibbs free energy profile of path B in chloroform with corresponding distances in angstroms (unit: kcal mol−1). | ||
3.2.3.1 Imine cation generation steps (C-I and D-I).
3.2.3.1.1 Imine cation generation step C-I. As shown in Fig. 3, IM1 is turned into the imine cation PhHC
N+HPh and (C6F5)3B–OH− anion via C-TS1 with a Gibbs free energy barrier of 1.8 kcal mol−1. The only imaginary frequency of C-TS1 is 355.82i cm−1 and its vibrational mode corresponds to the transfer of H2 from O1 to N1; the B–O1, O1–H2 and N1–H2 interatomic distances in C-TS1 are 1.58, 1.13 and 1.40 Å , respectively. In CD-IM3, the B–O1, O1–H2 and N1–H2 interatomic distances are 1.52, 1.60 and 1.07 Å, respectively, showing that the imine cation PhHCN+HPh and (C6F5)3B–OH− have been generated. Water acts as a protonic acid in this step.
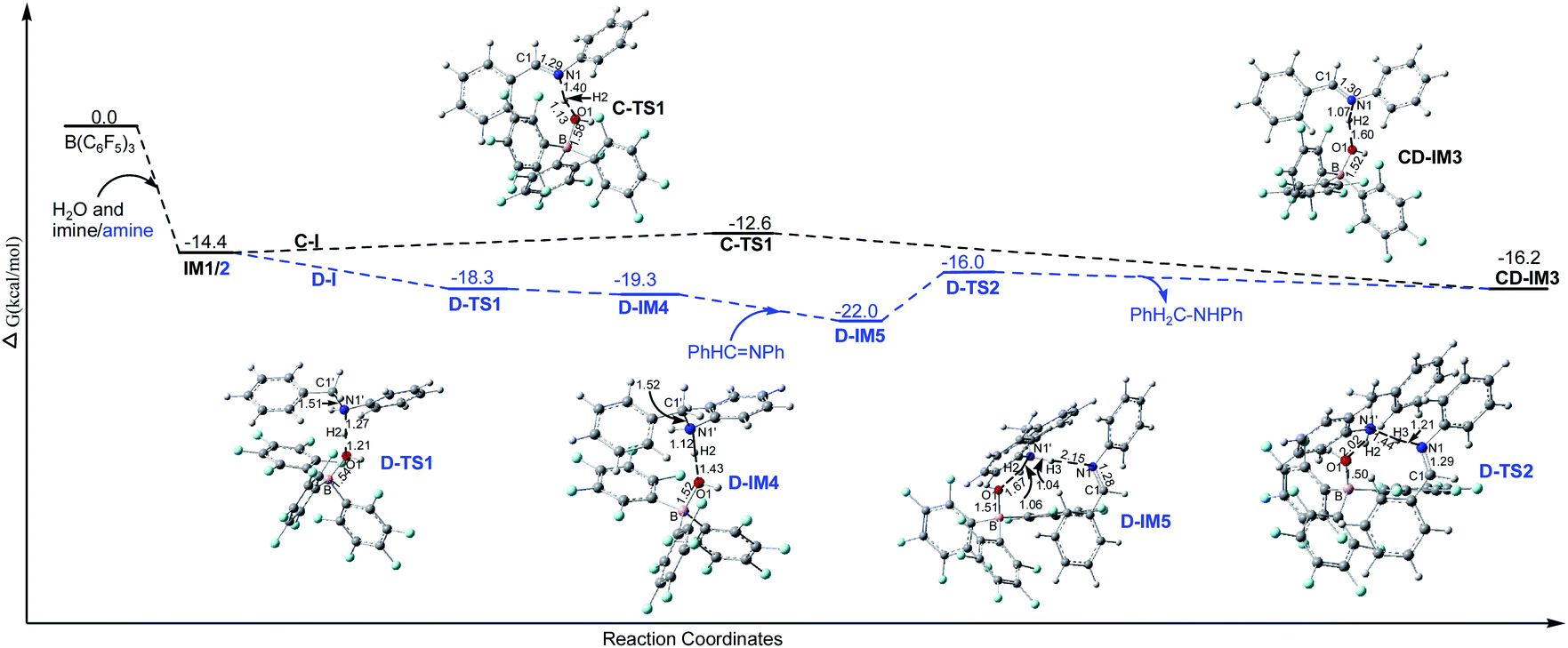 | ||
| Fig. 3 The Gibbs free energy profiles of the imine cation generation steps C-I and D-I in chloroform with the corresponding interatomic distances in the optimized structures in angstroms (unit: kcal mol−1, IM1 and IM2 share the same ΔG). | ||
3.2.3.1.2 Imine cation generation steps D-I. As shown in Fig. 3, IM2 is turned into the amine cation PhH2C–N+H2Ph and (C6F5)3B–OH− anion via D-TS1 with no barrier. The only imaginary frequency of D-TS1 is 583.10i cm−1 and its vibrational mode corresponds to the transfer of H2 from O1 to N1′; the B–O1, O1–H2 and N1′–H2 interatomic distances in D-TS1 are 1.54, 1.21 and 1.27 Å, respectively. In D-IM4, the B–O1, O1–H2 and N1′–H2 interatomic distances are 1.52, 1.43 and 1.12 Å, respectively, implying that the amine cation PhH2C–N+H2Ph and (C6F5)3B–OH− have been produced. And then the amine cation gives a proton to N1 in the imine by D-TS2 with a Gibbs free energy barrier of 6.0 kcal mol−1 to produce the imine cation PhHC
N+HPh with the amine PhH2C–NHPh being regenerated simultaneously. The only imaginary frequency of D-TS2 is 836.56i cm−1 and its vibrational mode corresponds to the transfer of H3 from N1′ in the amine cation to N1 in the imine; the N1′–H3 and N1–H3 interatomic distances in D-TS2 are 1.44 and 1.21 Å, respectively. CD-IM3 is obtained after releasing the amine PhH2C–NHPh.
3.2.3.2 The subsequent addition steps giving the N-silicon amine cation (CD-II).
3.2.3.2.1 Direct addition step. As shown in Fig. 4, H1− transfers from PhSiH3 to C1 in PhHC
N+HPh, and the Si of PhH2Si+ approaches N1 by CD-TS3 with a Gibbs free energy barrier of 41.4 kcal mol−1, giving the N-silicon amine cation PhH2C–N+HPh(SiH2Ph). The only imaginary frequency of CD-TS3 is 415.94i cm−1 and its vibrational mode corresponds to the transfer of H1 from Si to C1 and Si approaching N1; the C1–H1, Si–H1, N1–Si and C1–N1 interatomic distances in CD-TS3 are 1.20, 1.82, 2.53 and 1.43 Å, respectively. In CD-IM4, the C1–H1, N1–Si and C1–N1 distances are 1.09, 1.90 and 1.53 Å, respectively, implying that C1–H1 and N1–Si bonds have formed and C1N1 has turned into a C1–N1 single bond. CD-IM5 is the most stable complex of the N-silicon amine cation and (C6F5)3B–OH−.
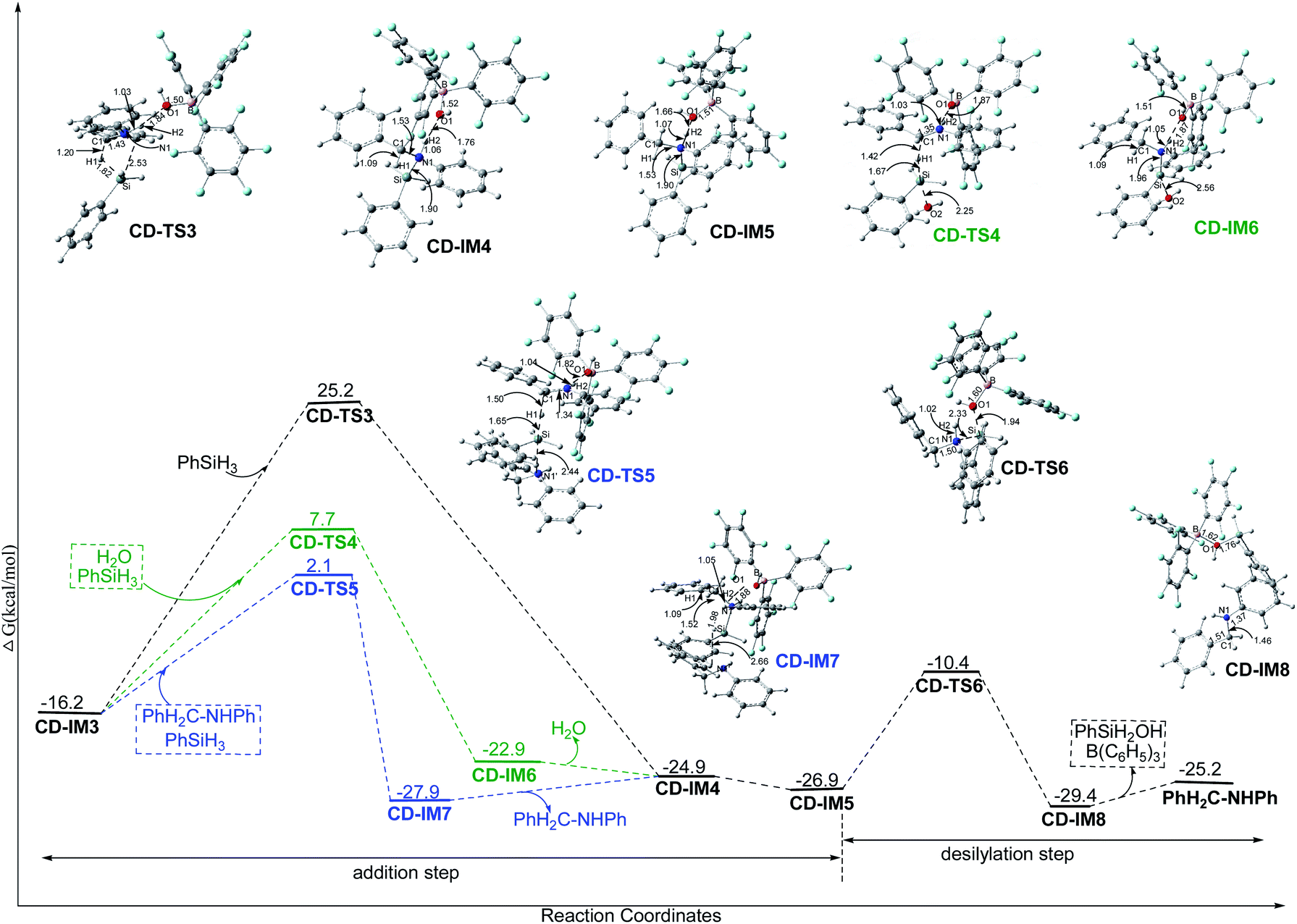 | ||
| Fig. 4 The Gibbs free energy profiles of the addition and desilylation steps (CD-II and CD-III) in chloroform with the corresponding interatomic distances in the optimized structures in angstroms (unit: kcal mol−1). | ||
3.2.3.2.2 Water assisted addition step. As shown in Fig. 4, with the assistance of nucleophilic H2O, H1− transfers from PhSiH3 to C1 in PhHC
N+HPh by CD-TS4 with a Gibbs free energy barrier of 23.9 kcal mol−1. The only imaginary frequency of CD-TS4 is 377.91i cm−1 and its vibrational mode corresponds to the transfer of H1 from Si to C1 and O2 of H2O approaching Si; the C1–H1, Si–H1, O2–Si and C1–N1 interatomic distances in CD-TS4 are 1.42, 1.67, 2.25 and 1.35 Å, respectively. When H1− is transferred to C1, PhH2Si+ is simultaneously attracted by O2 and the neighboring N1 with lone pair electrons. In CD-IM6, the C1–H1, O2–Si and N1–Si interatomic distances are 1.09, 2.56 and 1.96 Å, respectively, suggesting that the attraction from N1 is stronger than that from O2, an N1–Si bond is formed and the N-silicon amine cation PhH2C–N+HPh(SiH2Ph) is produced; there is no bond formed between O2 and Si. CD-IM5 is obtained when CD-IM6 releases H2O. Water acts as a catalyst to decrease the barrier of this step (23.9 kcal mol−1 for CD-TS4 vs. 41.4 kcal mol−1 for CD-TS3); the weak nucleophilicity of H2O decreases the barrier of H1− transfer from Si to C1, and the (C6F5)3B–OH− location and the O1H2N1 hydrogen bond increase the attraction of N1 to PhH2Si+.
3.2.3.2.3 Amine assisted addition step. When amine is produced in the system, it may assist the addition step just like water. As shown in Fig. 4, with the assistance of the amine PhH2C–NHPh, H1− transfers from PhSiH3 to C1 in PhHC
N+HPh by CD-TS5 with a Gibbs free energy barrier of 18.3 kcal mol−1. The only imaginary frequency of CD-TS5 is 425.87i cm−1 and its vibrational mode corresponds to the transfer of H1 from Si to C1 and N1′ of the amine approaching Si; the C1–H1, Si–H1, N1′–Si and C1–N1 interatomic distances in CD-TS5 are 1.50, 1.65, 2.44 and 1.34 Å, respectively. Similar to the water assisted addition step, PhH2Si+ is attracted by N1′ and N1 simultaneously. In CD-IM7, the C1–H1, N1–Si and N1′–Si distances are 1.09, 1.98 and 2.66 Å, respectively, and an N-silicon amine cation is produced with N1–Si bond formation. The amine (PhH2C–NHPh) acts as a catalyst to decrease the barrier of this step (18.3 kcal mol−1 for CD-TS5 vs. 41.4 kcal mol−1 for CD-TS3) even more; the nucleophilicity of the amine decreases the barrier of H1− transfer from Si to C1 tremendously, while the (C6F5)3B–OH− location and the O1H2N1 hydrogen bond increase the attraction of N1 to PhH2Si+. CD-IM5 is obtained when CD-IM7 releases PhH2C–NHPh.
3.2.3.3 The common final desilylation step. As shown in Fig. 4, the final step is that (C6F5)3B–OH− attacks Si in the N-silicon amine cation by CD-TS6 with a Gibbs free energy barrier of 16.5 (for paths Cn) or 17.5 (for paths Dn) kcal mol−1, producing PhSiH2OH and amine PhH2C–NHPh with B(C6F5)3 regeneration. The only imaginary frequency of CD-TS6 is 130.10i cm−1 and its vibrational mode corresponds to the breaking of the N1–Si and B–O1 bonds and the formation of the Si–O1 bond. The N1–Si, Si–O1 and B–O1 interatomic distances in CD-TS6 are 2.33, 1.94 and 1.60 Å, respectively. CD-TS6 is similar to B-TS5 with the only difference being the orientation of the substituent groups on N1.
| Path A | Imine protonation and (C6F5)3B–H− formation | H− transfer to imine cation, giving amine | |||
| A-TS1/31.6 | A-TS2/4.6 | ||||
| Path B | B-I: N-silicon amine generation steps | B-II: hydrolysis for desilylation steps | |||
| B-TS1/19.9 | B-TS2/4.3 | B-TS3/— | B-TS4/1.8 | B-TS5/15.9 | |
| Paths Cn/Dn | Imine cation generation step(s) | CD-II: addition step | CD-III: common desilylation step |
|---|---|---|---|
| C1 | C–I: C-TS1/1.8 | CD-TS3/41.4 | CD-TS6/16.5 for paths Cn; 17.5 for paths Dn |
| C2 | CD-TS4/23.9 | ||
| C3 | CD-TS5/18.3 | ||
| D1 | D-I: D-TS1/— | CD-TS3/41.4 | |
| D2 | D-TS2/6.0 | CD-TS4/23.9 | |
| D3 | CD-TS5/18.3 |
| Paths Cn/Dn | Imine cation generation step(s) | CD-II: addition step | CD-III: common desilylation step |
|---|---|---|---|
| C1 | C–I: C-TS1/1.8 | CD-TS3/41.4 | CD-TS6/16.5 for paths Cn; 17.5 for paths Dn |
| C2 | CD-TS4/23.9 | ||
| C3 | CD-TS5/18.3 | ||
| D1 | D-I: D-TS1/; D-TS2/6.0 | CD-TS3/41.4 | |
| D2 | CD-TS4/23.9 | ||
| D3 | CD-TS5/18.3 |
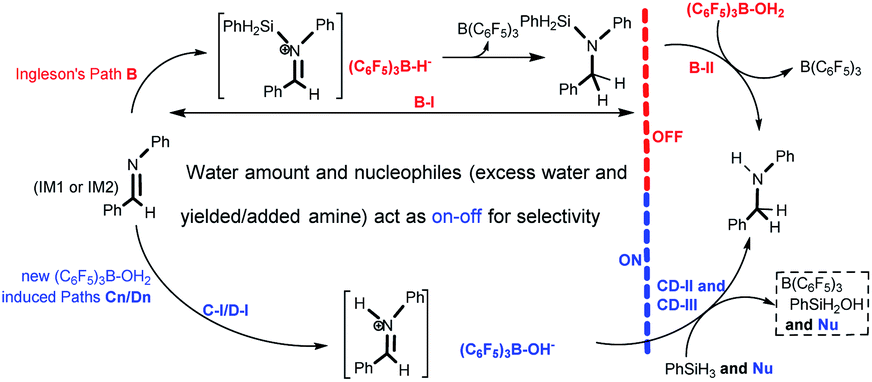 | ||
| Fig. 5 The competitive behavior of paths B, C2, C3, D2 and D3 results in water and nucleophiles (excess water and produced/added amine) providing on–off selectivity for imine reduction with PhSiH3 under water conditions. | ||
3.2.4.1 For stepwise imine reduction. Ingleson’s path B is the only mechanism. The N-silicon amine is formed in anhydrous systems first along B-I′ (IM0 → N-silicon amine, see Fig. 2), and then water is added into the system to produce the amine along B-II. So only hydrosilylation products can be obtained in anhydrous conditions; this is consistent with entries 1 and 2 in Table 1.
3.2.4.2 For the one-time feeding method. For the one-time feeding method, all the reactants and catalysts were added into the system simultaneously. As illustrated in Fig. 2–5 and Table 2, water and nucleophiles (excess water and produced/added amine) provide on–off selectivity of the pathways and products.
(1) When the water amount is no more than 1 eq. without amine in the system, if only the barriers of their rate-determining steps are considered, one can say that paths B and C2 can proceed competitively, leading to amination products; this is inconsistent with entries 3 and 4 in Table 1. The competitive behavior of paths B and C2 is discussed as follows: The initial dominant presence of IM1 is favorable for C-I instead of B-I; the much lower barrier of C-TS1 (1.8 kcal mol−1) than B-TS1 (19.9 kcal mol−1) gives C-I a competitive advantage over B-I, so C-I dominates dynamically in comparison with B-I, suggesting that B(C6F5)3 is turned into (C6F5)3B–OH− quickly. This means that hydrolysis for desilylation (B-II) of the N-silicon amine can’t proceed due to the lack of dissociated B(C6F5)3 (i.e. B-II is turned off by C-I), and there is no remaining free water for the water assisted addition step (CD-IM3 → CD-TS4, CD-II) of path C2. Hence, in fact, just B-I and C-I proceed competitively, delivering the N-silicon amine and the [PhHCNHPh]+[(C6F5)3B–OH]− ion pair, which is consistent with entries 3 and 4 in Table 1.
(2) When the water amount is more than 1 eq. without amine in the system, B-II still can’t proceed due to the lack of free B(C6F5)3, while the remaining free water after C-I means that the subsequent water assisted addition step (CD-IM3 → CD-TS4) in path C2 can proceed, and amine is finally yielded by the desilylation step; the yielded amine makes paths C3, D2, and D3 join the competition. These four pathways lead to amination products which is consistent with entries 5 and 6 in Table 1. Similarly, when 1 eq. water combined with a catalytic amount of amine is added into the system, amine will be produced by paths C3, C2, D3 and D2 competitively; this is consistent with entries 7 and 8 in Table 1. Obviously, nucleophilic excess water and produced/added amine open up the CD-II process and finally lead to amine production. Accordingly, the imine intermediate in reductive amination with PhSiH3 should be reduced to amine through paths C3, C2, D3 and D2.
(3) Paths C2, C3, D2 and D3 can proceed successfully only when the water amount is above a critical amount (more than 1 eq. water is suggested for it is a key reactant in C-I and D-I). In brief, 1 eq. water leads to quick formation of (C6F5)3B–OH−, leading to B-II being turned off, and then nucleophiles like excess water and produced/added amine switch on CD-II, leading to the production of amine.
(4) When using the alkylamine Me2NH instead of the produced arylamine, the Gibbs free energy of the ion pair [Me2NH2]+(C6F5)3B–OH− is located at −24.4 kcal mol−1 (lower than −19.3 kcal mol−1 for D-IM4, Fig. 3); this is disfavorable for producing the amine with high selectivity, and the arylamine is suggested to catalyze the addition step, which is consistent with entries 7 and 8 in Table 1 and Ingleson’s result.
4 Conclusions
In systems of imine reduction with PhSiH3 under stoichiometric water conditions, the most stable initial complex of B(C6F5)3 is the ternary complex (C6F5)3BOH(H)LB (LB is PhNCHPh or PhH2C–NHPh), which is convenient for the (C6F5)3B–OH2 induced reaction. Ingleson’s path B is reconfirmed as one possible competitive pathway. And four novel B(C6F5)3 and water/amine catalyzed competitive pathways (paths C2, C3, D2 and D3) induced by (C6F5)3B–OH2 were determined for the first time, in which the nucleophilic water or amine catalyzed addition step between PhSiH3 and the N-silicon amine cation is the rate-determining step of paths C2/D2 and C3/D3 with Gibbs free energy barriers of 23.9 and 18.3 kcal mol−1 in chloroform, respectively, while (C6F5)3B–OH− is an important intermediate and the final desilylation of the N-silicon amine cation depends on it. Water acts as a reactant in the imine cation generation step(s); water/amine (providing an appropriate nucleophilic electric field) acts as a catalyst in the addition step CD-II. The competitive behavior of paths B, C2, C3, D2 and D3 can explain the experimental facts perfectly. For the one-time feeding method of imine reduction under stoichiometric water conditions with PhSiH3, water amount and nucleophiles (excess water and produced/added amine) provide on–off selectivity of the pathways and products. 1 eq. water leads to quick formation of (C6F5)3B–OH−, leading to the B-II path being closed, and nucleophiles like excess water and produced/added amine open up CD-II, leading to production of amine: (1) when the water amount is no more than 1 eq., the N-silicon amine and the ion pair [PhHCNHPh]+[(C6F5)3B–OH]− are produced through competitive B-I and C-I; (2) when 1 eq. water is combined with a catalytic amount of arylamine or if more than 1 eq. water is added initially, amine will be produced by the competitive paths C2, C3, D2 and D3. In brief, B-I′ of Ingleson’s path B is the only mechanism for anhydrous systems, giving N-silicon amine production only, B-I and C-I are competitive paths for systems with no more than 1 eq. water, producing the N-silicon amine and the ion pair [PhHCNHPh]+[(C6F5)3B–OH]−, and paths C2, C3, D2 and D3 are competitive for systems with 1 eq. water and nucleophiles like excess water or added/produced amine, directly giving amination products.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (21542011), the Startup Project supported by Yibin University (2021QH03), the Scientific Research Fund of Sichuan Provincial Education Department (17ZA0196), the Science and Technology Planning Project of Leshan Science and Technology Bureau (18JZD118), and the Startup Project supported by Leshan Normal University (Z1601).References
- (a) S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, U.K., 2005, Vol. 9, pp. 1015–1025 Search PubMed; (b) T. C. Nugent and M. El-Shazly, Adv. Synth. Catal., 2010, 352, 753–819 CrossRef CAS; (c) J. P. Wolfe, S. Wagaw, J. F. Marcoux and S. Buchwald, Acc. Chem. Res., 1998, 31, 805–818 CrossRef; (d) J. Zheng, T. Roisnel, C. Darcel and J.-B. Sortais, ChemCatChem, 2013, 5, 2861–2864 CrossRef CAS.
- (a) D. J. Parks and W. E. Piers, J. Am. Chem. Soc., 1996, 118, 9440–9441 CrossRef CAS; (b) J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed; (c) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed; (d) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880–1881 CrossRef CAS PubMed; (e) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fraeohlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072–5074 RSC; (f) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed; (g) S. Tussing, L. Greb, S. Tamke, B. Schirmer, C. Muhle-Goll, B. Luy and J. Paradies, Chem.–Eur. J., 2015, 21, 8056–8059 CrossRef CAS PubMed; (h) S. Tussing, K. Kaupmees and J. Paradies, Chem.–Eur. J., 2016, 22, 7422–7426 CrossRef CAS PubMed.
- (a) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997–6000 CrossRef CAS PubMed; (b) J. Hermeke, M. Mewald and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 17537–17546 CrossRef CAS PubMed; (c) A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed; (d) W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed; (e) J. Zhang, S. Park and S. Chang, J. Am. Chem. Soc., 2018, 140, 13209–13213 CrossRef CAS PubMed; (f) J. Zhang and S. Chang, J. Am. Chem. Soc., 2020, 142, 12585–12590 CrossRef CAS PubMed.
- (a) J. M. Blackwell, E. R. Sonmor, T. Scoccitti and W. E. Piers, Org. Lett., 2000, 2, 3921–3923 CrossRef CAS PubMed; (b) P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC; (c) Y.-B. Liu and H.-F. Du, Acta Chim. Sin., 2014, 72, 771–777 CrossRef CAS.
- (a) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed; (b) D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed; (c) T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed; (d) T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed; (e) Á. Gyömöre, M. Bakos, T. Földes, I. Pápai, A. Domján and T. Soós, ACS Catal., 2015, 5, 5366–5372 CrossRef; (f) E. Dorkó, M. Szabó, B. Kótai, I. Pápai, A. Domján and T. Soós, Angew. Chem., Int. Ed., 2017, 56, 9512–9516 CrossRef PubMed; (g) E. Kim, S. Park and S. Chang, Chem.–Eur. J., 2018, 24, 5765–5769 CrossRef CAS PubMed.
- (a) V. Fasano, J. E. Radcliffe and M. J. Ingleson, ACS Catal., 2016, 6, 1793–1798 CrossRef CAS; (b) V. Fasano and M. J. Ingleson, Chem.–Eur. J., 2017, 23, 2217–2224 CrossRef CAS PubMed.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef CAS.
- Y. Yu, Y. Zhu, M. N. Bhagat, A. Raghuraman, K. F. Hirsekorn, J. M. Notestein, S. T. Nguyen and L. J. Broadbelt, ACS Catal., 2018, 8, 11119–11133 CrossRef CAS.
- (a) C. Tian, Y. Jiang, M. Borzov and W.-L. Nie, Acta Chim. Sin., 2015, 73, 1203–1206 CrossRef CAS; (b) X. Hu, C. Tian, Y. Jiang, M. Borzov and W.-L. Nie, Acta Chim. Sin., 2015, 73, 1025–1030 Search PubMed; (c) Z.-G. Wen, C. Tian, Y. Jiang, M. Borzov and W.-L. Nie, Acta Chim. Sin., 2016, 74, 498–502 CrossRef CAS; (d) W.-L. Nie, G.-F. Sun, C. Tian and M. Borzov, Z. Naturforsch., B: J. Chem. Sci., 2016, 71(10), 1029 CAS; (e) L.-W. Zhang, Z.-G. Wen, M. Borzov and W.-L. Nie, Acta Chim. Sin., 2017, 75, 819–823 CrossRef CAS; (f) G.-F. Sun, M. Su, J. Fang, M. Borzov and W.-L. Nie, Acta Chim. Sin., 2017, 75, 824–830 CrossRef CAS; (g) Y.-Q. He, J.-W. Teng, C. Tian, M. Borzov, Q.-Sh. Hu and W.-L. Nie, Acta Chim. Sin., 2018, 76, 774–778 CrossRef CAS; (h) G.-F. Sun, Y.-Q. He, C. Tian, M. Borzov, Q.-S. Hu and W.-L. Nie, Acta Chim. Sin., 2019, 77, 166–171 CrossRef CAS.
- H.-C. Chen, L.-N. Yan and H.-Y. Wei, Organometallics, 2018, 37, 3698–3707 CrossRef CAS.
- X. Fu, W. Shen, T. Yao, W. Hou, Physical Chemistry, Higher Education Press, Beijing, 2006, Vol. 2, pp. 154–213 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS; (c) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: (1) The Gibbs free energy changes of some binary and ternary adducts relative to the corresponding single molecules. (2) The Gibbs free energy profile of path A. (3) The optimized structures of the stationary points of the reactants and products, and the stationary points of B-I′/B-I and B-II in path B. (4) Optimized Cartesian coordinates of the stationary points. See DOI: 10.1039/d1ra02399c |
| ‡ Yunqing He and Wanli Nie contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2021 |