
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConjugation of a smart polymer to doxorubicin through a pH-responsive bond for targeted drug delivery and improving drug loading on graphene oxide†
Ali Bina,
Heidar Raissi *,
Hassan Hashemzadeh and
Farzaneh Farzad*
*,
Hassan Hashemzadeh and
Farzaneh Farzad*
Department of Chemistry, University of Birjand, Birjand, Iran. E-mail: ali.bina1996@birjand.ac.ir; hraeisi@birjand.ac.ir; hashemzade_h@birjand.ac.ir; ffarzad5487@birjand.ac.ir; Tel: +98 5632502064
First published on 26th May 2021
Abstract
Polymeric nanoparticles have emerged as efficient carriers for anticancer drug delivery because they can improve the solubility of hydrophobic drugs and also can increase the bio-distribution of drugs throughout the bloodstream. In this work, a computational study is performed on a set of new pH-sensitive polymer–drug compounds based on an intelligent polymer called poly(β-malic acid) (PMLA). The molecular dynamics (MD) simulation is used to explore the adsorption and dynamic properties of PMLA–doxorubicin (PMLA–DOX) interaction with the graphene oxide (GOX) surface in acidic and neutral environments. The PMLA is bonded to DOX through an amide bond (PMLA-ami-DOX) and a hydrazone bond (PMLA-hz-DOX) and their adsorption behavior is compared with free DOX. Our results confirm that the polymer–drug prodrug shows unique properties. Analysis of the adsorption behavior reveals that this process is spontaneous and the most stable complex with a binding energy of −1210.262 kJ mol−1 is the GOX/PMLA-hz-DOX complex at normal pH. On the other hand, this system has a great sensitivity to pH so that in an acidic environment, its interaction with GOX became weaker while such behavior is not observed for the PMLA-ami-DOX complex. The results obtained from this study provide accurate information about the interaction of the polymer–drug compounds and nanocarriers at the atomic level, which can be useful in the design of smart drug delivery systems.
Introduction
Recent advances in materials science and the production of nanoparticles (NPs) with a variety of applications and non-toxic formulations are currently used as a promising approach in drug delivery systems (DDS) for the treatment of cancer.1,2 The design of new platforms, as DDSs by using different types of nanocarriers such as polymers, metal nanoparticles, nanostructures, and nanotubes, has attracted much interest in the targeted treatment of cancer.3–5 Compared to free drugs, DDSs act more efficiently and intelligently and also can diffuse more easily into cell membranes.1,2For example, graphene is one of the promising nanoparticles that has attracted a lot of attention due to its unique structure and biomedical properties.6 However, due to the poor solubility in the biological environment and low drug absorption capacity, this drug delivery system needs to be modified.7 The presence of functional groups containing oxygen (including carboxylic acid, epoxy, and hydroxy functional groups) in the surface of graphene oxide (GOX) leads to various properties such as high mechanical properties, thermal conductivity, high solubility in aqueous solutions, and a large surface area.8,9 It is noteworthy that graphene oxide nanosheets are easily synthesized by a variety of methods such as Hummers and Tour methods.10,11 Furthermore, functional groups on the GOX surface can have different interactions with drug molecules.8,12,13 Therefore, functionalized GOX can be used as an ideal system with higher solubility and more drug adsorption capacity than graphene.7 Many scientists are working to increase the efficiency of GOX in the transportation of anticancer drugs and reduce the drug's side effects.14 For example, Liu et al. synthesized graphene oxide modified with a protein to improve the therapeutic effect.15 Yang et al. were used graphene oxide as a nanocarrier to loaded doxorubicin (DOX) in a non-covalent manner.16 Furthermore, Zhang et al. experimentally showen that the DOX molecule bonded to GOX had better anti-tumor behavior.17
Many anticancer drugs can only affect intercellular organelles, such as parts of the cytoplasm, mitochondria, and nucleus. Therefore, the internal stimuli can be used to destabilize nanoparticle-based DDSs within target tissue for the targeted release of the medications.18,19 The pH level of the physiological environment is approximately 7.4 and in tumor tissues is varies from 4.5 to 6.513. Based on the significant difference in pH level between tumors and normal tissues, pH-sensitive carriers can be developed that respond to this difference.18 Yatvin et al. synthesized a pH-sensitive liposomal system, which showed the pH-dependent drug release due to this difference.20 Furthermore, most studies showen that the acidity of the biological environment is one of the effective and efficient factors in the specific secretion of anticancer drugs in tumor tissues or inside tumor cells.21,22
Polymer–drug nanocarriers are another efficient and intelligent systems that can be used in DDS design. In some of these systems, there are degradable bonds between the drug molecule and the polymer that act as a stimulus to respond to the pH level.1 These degradable bonds are sensitive to an acidic environment, which easily degraded in this media and released the drug into the cancer cell.23–26 The most common bonds easily destructible in an acidic environment are acetal, ketal, hydrazone, and imine.1,14,27 A hydrazone bond is a double bond that is formed between carbon and nitrogen atoms and easily hydrolyzed in an acidic environment.2 This bond has been linked between drugs and different polymers to study the effects of pH on the loading and distribution of anticancer drugs.28–31 Kang et al.30 developed a pH-sensitive dendritic polyurethane conjugate (PR-g-Dox), in which DOX was attached to the terminal functional groups of the polymer by hydrazone bonding. Many nanoparticles can be used as potential nanocarriers for the delivery of Doxorubicin (DOX) to minimize the side effects and destructive effects of this drug.32,33
Recently the application of smart polymers in the design of the new drug delivery systems has been promoted.34 The application of the polymer–drug complex has been used as an efficient strategy to reduce side effects, target the tumor tissue, and extend the durability of anticancer drugs.35 Tu et al.36 studied a drug–polymer delivery system based on an aliphatic dendritic polyester, which could provide a new and more effective way in cancer treatment.37 These polymers have many advantages, including low molecular weight, high solubility, targeted selection of target tissue, and enhancing drug bioavailability.2,38,39 Due to the biocompatibility of smart polymers and fast response to ambient pH, they can be conjugated to drugs or carriers and turn into smart drug delivery systems.2,40 In this way, drugs can be attached to polymers via covalent bonds that are sensitive to the pH of the environment.14 These bonds destruct rapidly in an acidic environment but are stable at the body's biological pH.26,41 Poly-β-malic acid (PMLA) is an intelligent polymer with ideal performance. This compound is an aliphatic polyester based on malic acid that can be attached to the DOX in various ways. There are suitable functional groups in the PMLA chain that can form the hydrazone bond with the DOX molecule and create a structure with high sensitivity to pH. Conjugating DOX to PMLA chain causes a significant reduction in the toxicity of the drug.22 Ljubimova et al. used the PMLA–DOX complex to target breast cancer and brain tumors, and found out that the toxicity of the drug in complex, dramaticly reduced and found out that the toxicity of the drug in the complex dramatically reduced.42
Computer simulations are used to provide a better understand from molecular structural properties of a system.43 In recent years, several computational works have been done on the development of smart drug delivery systems, which some of them are listed in Table S1.†The data obtained from these studies gain a better understanding at the atomic level. To the best of our knowledge, no computational study has been performed on pH-sensitive systems in which the drug is directly attached to the polymer and loaded onto a carrier, due to the extraordinary properties of these systems, it seems necessary to examine them. Theoretical calculations can analyze the interaction between species at the atomic level that are not empirically observable, as well as predict the interaction of different compounds in the system.44,45
In this study, a series of molecular dynamics (MD) simulations are performed to obtain quantitative and qualitative information about the chemical and physical properties of the polymer–drug complex, and its interactions with GOX. The loading and release mechanisms of free DOX and the DOX–PMLA complex at different pHs in the form of protonated and deprotonated (acidic and neutral) on the GOX surface will be investigated.
Simulation methods
Molecular models and initial structures
To study the adsorption and release of DOX and DOX–PMlA on GOX surface at two pH levels (neutral and acidic conditions), six simulation boxes are designed. In all the investigated systems, the simulation box's dimensions are 8 nm × 8 nm × 6 nm. The DOX drug structure is taken from the PubChem web-server (PubChem CID: 31703),46 and the polymer–drug compounds are created by GaussView 6.0 software.47 The PMLA chain can be attached to DOX in two ways. One way, through the formation of a hydrazone bond (sensitive to pH of ambient) between the carbonyl group of the drug and the amine group of polymer (PMLA-hz-DOX complex). Another way, the drug is attached to the polymer backbone via an amide bond (i.e., between the amine group of DOX and the hydroxyl group of the PMLA, PMLA-ami-DOX complex).22 The structure of PMLA-hz-DOX and PMLA-ami-DOX are depicted in Fig. 1.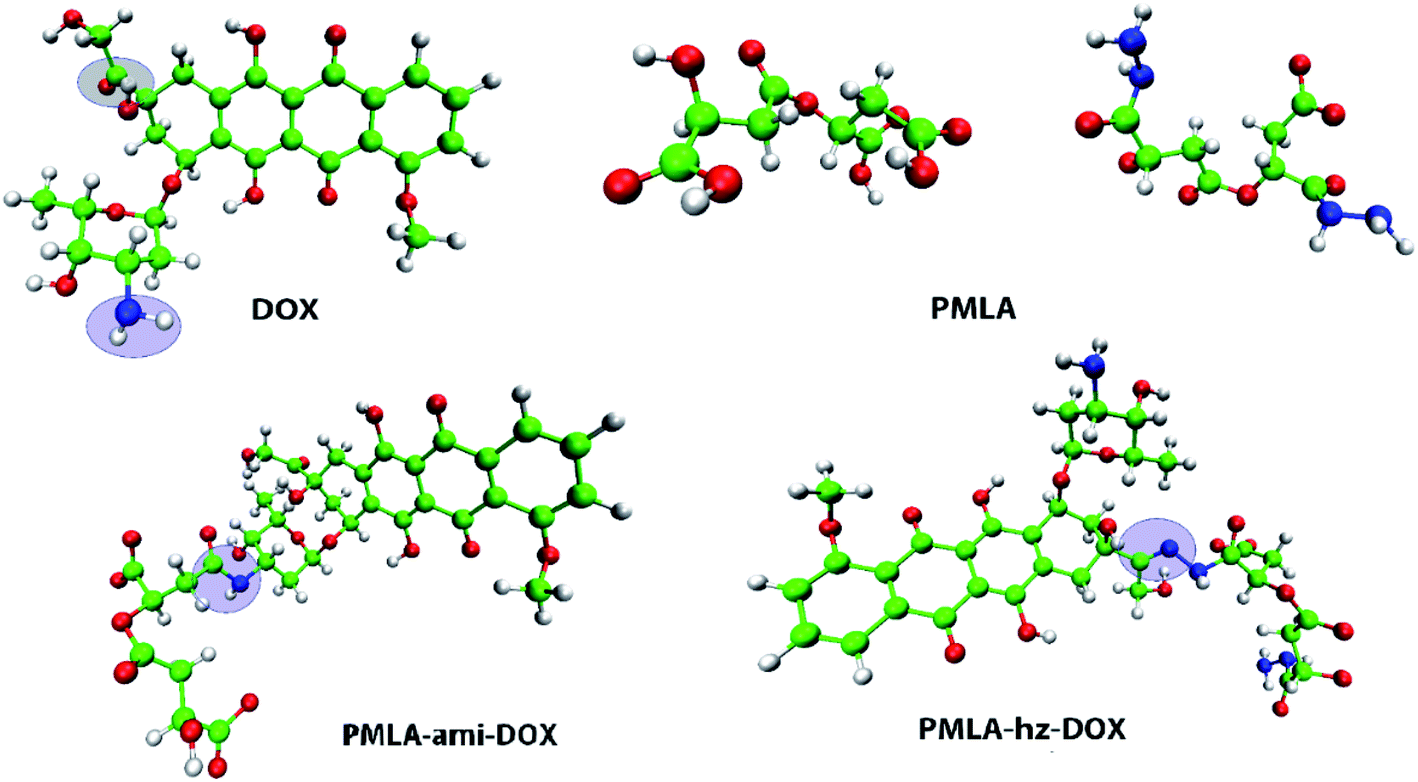 | ||
| Fig. 1 Optimized geometries of doxorubicin (DOX), poly(β-malic acid) (PMLA), and polymer-DOX complexes: PMLA-hz-DOX and PMLA-ami-DOX, the drug attached to polymer by hydrazone and amide bonds, respectively. | ||
GOX structure is constructed by decorating the graphene surface with epoxy, carboxylic, and hydroxyl functional groups. The final carbon and oxygen ratio (C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O) in the applied GOX model is about 7:2. In all of the studied systems, the GOX nanosheet is positioned at the simulation box center. On each side of the graphene oxide, the guest molecules (i.e., DOX, PMLA-hz-DOX, and PMLA-ami-DOX) are placed at an appropriate distance (this distance is long enough to prevent the initial configuration effects).
:O) in the applied GOX model is about 7:2. In all of the studied systems, the GOX nanosheet is positioned at the simulation box center. On each side of the graphene oxide, the guest molecules (i.e., DOX, PMLA-hz-DOX, and PMLA-ami-DOX) are placed at an appropriate distance (this distance is long enough to prevent the initial configuration effects).
In an acidic environment, molecules can be protonated or deprotonated, accordingly, many studies have shown that molecules can be protonated and deprotonated based on the pH they have worked on.33,48,49 For this purpose, in order to adjust the pH, we protonated and deprotonated the molecules using pKa compounds according to formulas (1) and (2).
Two simulation systems, GOX/DOX and its protonated form (pGOX/DOX) are designed for investigating the interaction of DOX with GOX at pH = 7.4 and pH = 5.0, respectively. In GOX/PMLA-ami-DOX (GOX/PMLA-hz-DOX) and pGOX/PMLA-ami-DOX (pGOX/PMLA-hz-DOX) systems, the interaction of PMLA-ami-DOX (PMLA-hz-DOX) complexes with the carrier at the neutral and acidic conditions, respectively, are investigated. The initial structures of investigated complexes are shown in Fig. 2.
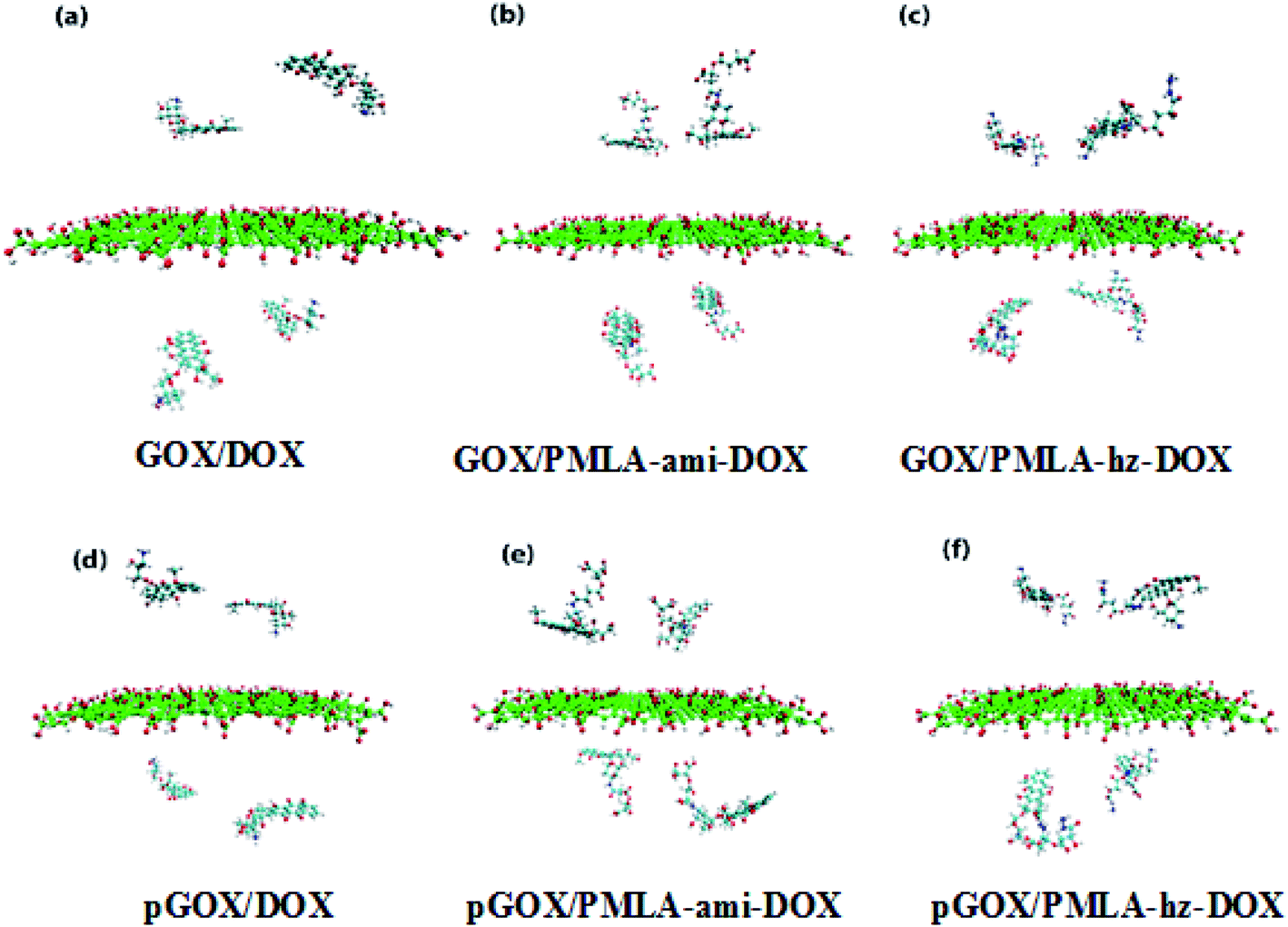 | ||
| Fig. 2 Initial snapshots of simulation box's at normal and acidic pH level. | ||
The pH effect in the simulation boxes is considered based on values pKas of the components at the pH = 7.4 and pH = 5.0. It is worth noting that the pKa of a molecule depends on its functional groups, molecular structure, neighboring groups, and resonance status.48 The pKa values for –COOH, –OH, DOX, and PMLA are 6.6, 9.0, 8.3, and 4.27, respectively.50 The number of deprotonated sites for nanoplate and PMLAs associated with DOX is calculated in pH = 7.4 according to the following equation:49
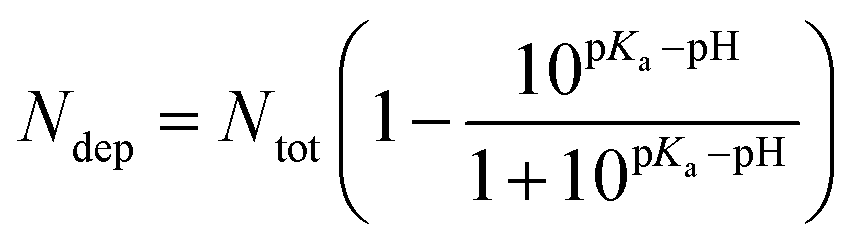 | (1) |
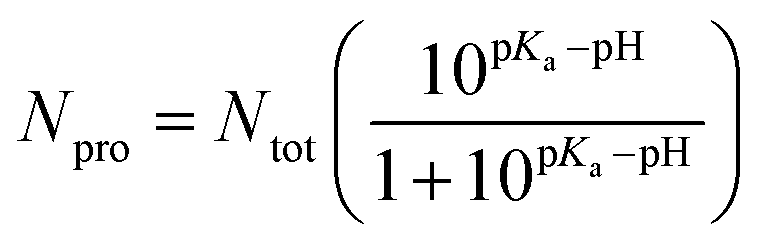 | (2) |
By varying pH from 7.4 to 5.0, the NH2 group of DOX is converted to NH3+, and in PMLA-ami, all of COO− groups are converted to COOH.
Molecular dynamics simulation
All MD simulations in this work are performed by GROMACS package,51 and force filed parameters for all of the components are extracted from charmm36-nov2018 force field.52 In all systems, the TIP3P water model is used to fill boxes.53 The number of water molecules varies in different boxes; details of all studied systems are listed in Table S1.† A certain number of ions (Na+, Cl−) were added to the simulation boxes to neutralize the system as well as reproduce a correct biological environment (i.e. 0.15 mol lit−1).54 The simulation steps (i.e., NVT, NpT, and MD), time, and time step are presented in Table S2.† Periodic boundary condition in the three directions is used. The temperature is kept constant at 310 K using the V-rescale thermostat, and to control the pressure at 1 bar during simulation, Berendsen algorithm47 is used. To visualize the absorption process, the visual molecular dynamics (VMD) software is used.55Results and discussions
To ensure that the investigated systems have reached equilibrium and stability states, the root mean square displacement (RMSD) is computed and depicted in Fig. S1.† According to the RMSD diagram, all systems after 12 ns from the beginning of the simulation have reached equilibrium.To study the adsorption and release behavior of drug molecules, in the simulation boxes, the initial and final images of the systems are shown in Fig. 2 and 3, respectively.
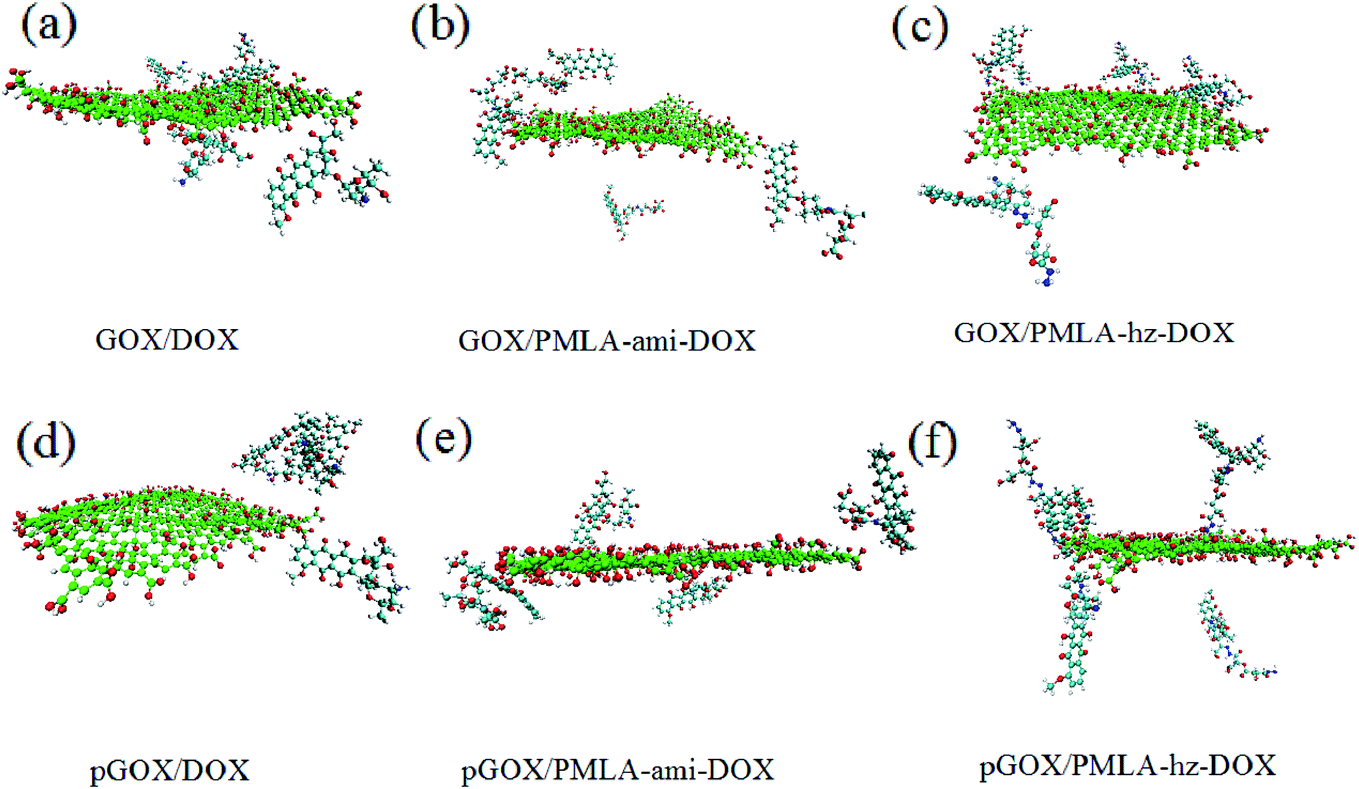 | ||
| Fig. 3 The final snapshots of simulation systems. | ||
As can be seen in the final snapshots, in all boxes, drug molecules move spontaneously towards the carrier. As absorption progresses, the distance between the drug molecules and the GOX level decreases until the monomers approach each other to reach equilibrium. The results of drug loading and its orientation concerning the nanocarrier surface show that the adsorption process is strongly dependent on pH conditions. Besides, it can be expected that the absorption process and loading capacity of anticancer drugs on GOX, in different systems, show different behavior according to the natural pH (pH = 7.4) and acidic pH (pH = 5.0). These results are in good agreement with the results of the experiments of Adnan et al., Yang et al.9,49
The final snapshots of the simulation systems are shown in Fig. 3. A comparison of Fig. 2 and 3 confirms that in all of the investigated systems, the guest molecules move spontaneously towards the carrier surface. The distance of DOX and PMLA-x-DOX (hereafter x stands for hz and ami) molecules from GOX is decreased until the host (GOX) and guest molecules form stable complexes. The process of loading drug molecules on GOX nanocarriers in different systems has shown different behaviors. Comparing the adsorption process at the neutral and acidic conditions indicated that the loading and orientation of DOX and PMLA-hz-DOX on GOX surface highly dependent on pH levels, despite the PMLA-ami-DOX case. It is found that, at the acidic condition, the interaction of DOX and PMLA-hz-DOX with GOX becomes weaker, and some of the guest molecules desorbed from the carrier surface. Another distinctive feature of GOX/PMLA-hz-DOX systems is that in the neutral environment, PMLA-hz-DOX prefers to adsorbed in the parallel orientation whereas, in acidic conditions, desorbed from the GOX surface and placed vertically on the surface.
Close-up snapshots of the formed complex between the closest guest molecule and the host are given in Fig. 4.
 | ||
| Fig. 4 Intermolecular distances between the nearest guest molecule to GOX surface. The carbon, oxygen, nitrogen, and hydrogen atoms are shown in green, red, blue, and white, respectively. | ||
As shown in Fig. 4, the drug and the polymer–drug molecules interact with the nanoplate surface through π–π stacking and hydrogen bond (HB) interactions. It should be noted that X–H⋯π (X: C, N, and O) intermolecular interactions have also been formed between drug molecules and carrier surface. Shan et al. using MD simulations, showed that π–π stacking and electrostatic interactions have the main role in the loading of DOX with GOX.56 As shown in this figure, the drug molecule can form π–π stacking and X–H⋯π intermolecular interactions with the GOX in the range of about 0.337 to 5.05 nm and 0.252 to 0.281 nm, respectively. Furthermore, several HBs between the drug and the carrier form at a distance of about 0.207–0.359 nm. A closer look at Fig. 3 and 4, indicates that the orientation of the drug and polymer–drugs in the response to pH levels is changed. In the GOX/DOX system, most of the drug molecules form strong π–π interactions with the carrier, and one of the DOX molecules is adsorbed through HB interaction. At the acidic condition, most of the DOX molecules desorbed from the GOX surface and became self-aggregated. In the GOX/PMLA-ami-DOX system, it seems that PMLA-aim-DOX does not prefer to interact with the nanosheet in such a way that, after 105 ns, almost most of them does not adsorb on the surface and remains in the aqueous phase. Unlike the DOX and PMLA-hz-DOX complexes, in acidic pH, the interaction of PMLA-ami-DOX with GOX becomes stronger, and at the end of MD simulation, three molecules adsorbed on the carrier surface. The GOX/PMLA-hz-DOX system shows similar behavior to the GOX/DOX system. All of the polymer–drug adsorbed on the GOX surface via π–π and HB interactions in the pGOX/PMLA-hz-DOX system, all of the polymer–drug molecules are slowly desorbed from the nanoplate surface. It is well known that the mechanism of drug loading and release of drug in pH-sensitive systems almost controlled through van der Walls (vdW), dipole–dipole, and hydrogen bond interactions. In the GROMACS package, interaction energy is divided into Lennard Jones (LJ) and electrostatic (elec) parts. vdW interactions such as π–π stacking can be evaluated by calculation LJ energy, and electrostatic energy can be used to assess the contribution of the dipole–dipole and HB interactions.48,57 The LJ and electrostatic energy values for the interaction of drug and polymer–drug with the carrier in studied systems are reported in Table 1.
| System | pH | LJ | Elec | Binding |
|---|---|---|---|---|
| GOX-DOX | 7.4 | −206.193 | −212.908 | −419.101 |
| pGOX-DOX | 5.0 | −4.943 | −6.922 | −11.865 |
| GOX/PMLA-ami-DOX | 7.4 | −10.725 | 5.897 | −4.828 |
| pGOX/PMLA-ami-DOX | 5.0 | −57.315 | 105.387 | 48.072 |
| GOX/PMLA-hz-DOX | 7.4 | −132.092 | −1078.17 | −1210.262 |
| pGOX/PMLA-hz-DOX | 5.0 | −37.926 | −99.723 | −137.649 |
As can be seen in this Table, the energy values for system GOX/DOX at the neutral pH are relatively high (the binding energy is about −419.101 kJ mol−1) while, in acidic pH, GOX and DOX interactions become significantly weaker. These obtained results have a good agreement with the depicted snapshots systems. In other words, in a neutral medium, by absorbing the drug molecules on the carrier (see Fig. 3a), the binding energy becomes stronger, whereas, in the acidic medium, the DOX molecules are desorbed (see Fig. 3d), which makes the binding energy more positive.
In the GOX/PMLA-ami-DOX system, binding energy values are very lower than the GOX/DOX system, which indicates the PMLA-ami-DOX does not tend to absorb on the GOX surface. This observation can be related to the presence of oxygen-containing groups in the PMLA-ami structure. Furthermore, at the pH = 5 strong repulsion between GOX and PMLA-ami leads to the electrostatic energy become more positive. In general, and according to previous research,14,48 it can be concluded that the functional groups in the structure of PMLA-ami do not show proper response to pH. A close inspection of Table 1 reveals that the highest LJ and elec energy values can be seen in the PMLA-hz-DOX system(with binding energy at about three times that of the GOX/DOX system), which confirms this polymer–drug has the most affinity to adsorb on GOX surface. The obtained energy results are in good agreement with the experimental results reported by Qiao et al.22 It can be expected that the strong interaction between the PMLA-hz-DOX and GOX causes its loading amount to be more than free DOX. In the acidic condition, the interaction energy between PMLA-hz-DOX and GOX decreases, and, therefore, this drug–polymer can release from the carrier surface. In general, by comparing different energies in two environments, it is found that the energy values in the neutral environment are higher than those in acidic media. Moreover, the comparison of the energy results and the final snapshots show that in the pGOX/DOX system, after desorption of drugs from the nanocarrier surface, they interact together and self-aggregation occurs (in spite of the pGOX/PMLA-hz-DOX system). This finding confirms that the efficiency of drug delivery and release in the PMLA-hz-DOX is more than the others.
In order to investigate the distribution and probability of the drug finding around the GOX surface, the radial distribution function has been calculated. The RDF diagrams for all of the investigated systems between guest molecules and GOX are computed and the results are shown in Fig. 5. At short distances (<0.2), the RDF is zero due to the repulsive force between the drug molecules and the graphene oxide nanoplate. According to the RDF diagrams, the DOX molecules are distributed around the surface of graphene oxide in the range of about 0.47–1.24 nm. The maximum intensity for the RDF plot of DOX appears at a distance of 0.78 nm, which indicates that the drug has more interaction with GOX at this distance. As can be seen in Fig. 5, in the neutral environment, the intensity of the RDF peak in the PMLA-hz-DOX is more than the DOX system, as well as, the width of the RDF peak in this system(0.77–1.08 nm) is narrower than DOX. This fact can be attributed to the stronger interaction of PMLA-hz-DOX with the carrier as well as its bigger size in comparison to DOX. In the PMLA-ami-DOX system, the most intense peak is observed at around (1.74–2.19 nm), which confirms the weak interaction of this drug–polymer with GOX. Investigation of the RDF diagrams in the acidic environment indicates that DOX has a lower tendency to be present and distributes around the nanoplate. Close inspection of Fig. 5 shows that no significant peaks can be observed for the pGOX/DOX system near of GOX surface. This finding has good agreement with energy results and confirms that the interactions between DOX and GOX decrease in acidic media. Similar behavior is observed in the pGOX/PMLA-hz-DOX system, where the interaction of polymer–drug and GOX is reduced, and RDF peaks appear about 1.51–2.07 nm. Furthermore, the intensity of peaks in pGOX/DOX and pGOX/PMLA-hz-DOX systems have decreased compared to its corresponding value in neutral medium. On contrary, no such behavior is observed for pGOX/PMLA-ami-DOX system. In this case, the intensity of the peak increases and locates closer to the carrier compared to GOX/PMLA-ami-DOX. Therefore, it can be claimed that polymer–drug in this form does not respond to pH levels, which is in line with the reported experimental data Qiao et al.14
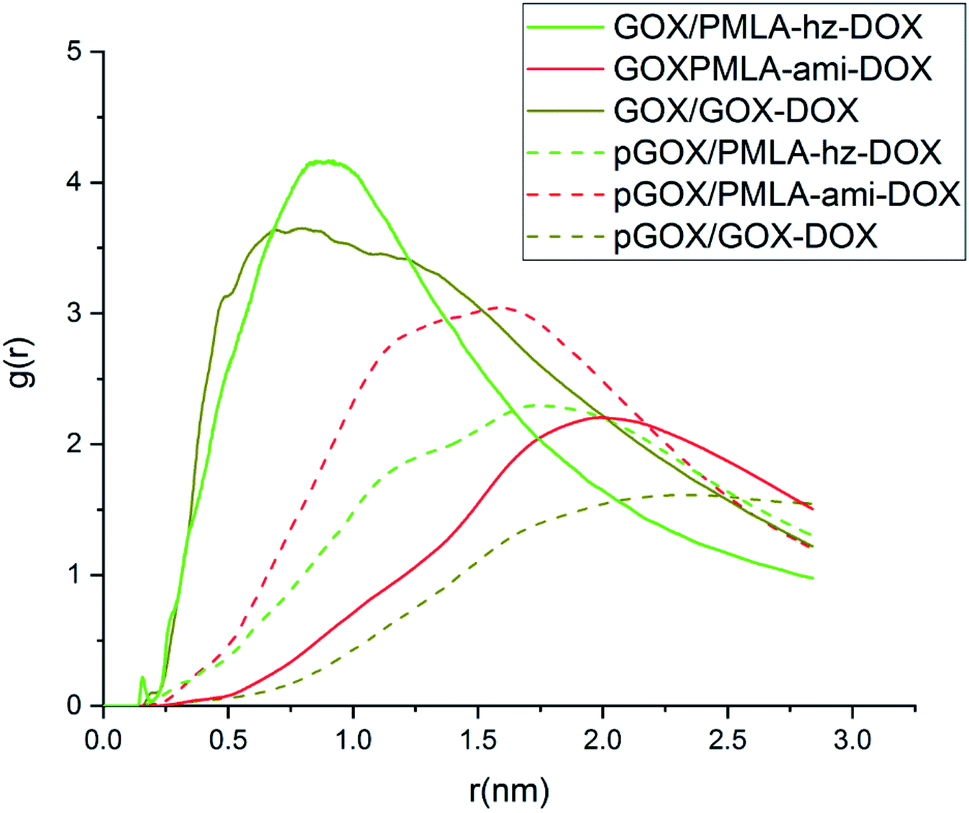 | ||
| Fig. 5 Radial distribution function (RDF) diagram between guest molecules and GOX surface. | ||
In addition to energy profiles and RDF diagrams, the number of contacts is examined to provide more insight into the effect of pH on these systems' adsorption. The number of contacts between the guest and host molecules as a function of time is shown in Fig. 6. This diagram clearly shows, when the drug and the polymer–drug molecules approach the surface of GOX, the number of contacts increases. The number of atomic contacts of DOX and PMLA-hz-DOX molecules with the carrier in the neutral condition is higher than the corresponding value for PMLA-ami-DOX. It is noteworthy that the obtained results show a good correlation between LJ energy (Fig. S2†) and the number of contacts.
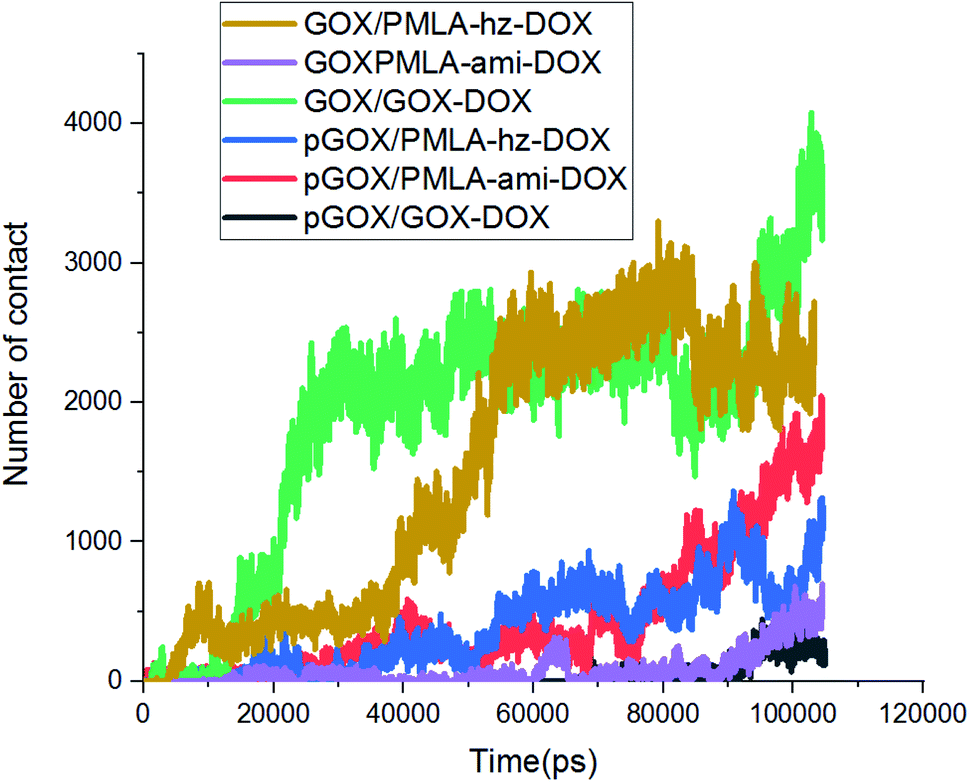 | ||
| Fig. 6 The variations of the number of contacts between the guest molecules and GOX surface. | ||
To gain deeper insight into the nature of formed complexes, the pattern of HB changes during the adsorption process is studied. The number of different HBs between (a) guest molecules and the GOX surface, (b) guest molecules and water, (c) graphene oxide and water are shown in Fig. 7. It is well known that the presence of HBs can increase the capacity of adsorption and improve the stability of complexes.58 At normal pH, the number of HBs in the GOX/DOX and GOX/PMLA-hz-DOX pairs are higher than those corresponding values in the acidic environment. Furthermore, the number of HBs during simulation time, due to adsorption of the guest molecule on GOX surface, is increased. It should be noted that the pGOX/DOX has the lowest number of HB values, but in this system, the number of HB between DOX–DOX molecules (Fig. S3†) is increased. This observation can be related to the aggregation of drugs in the pGOX/DOX system.
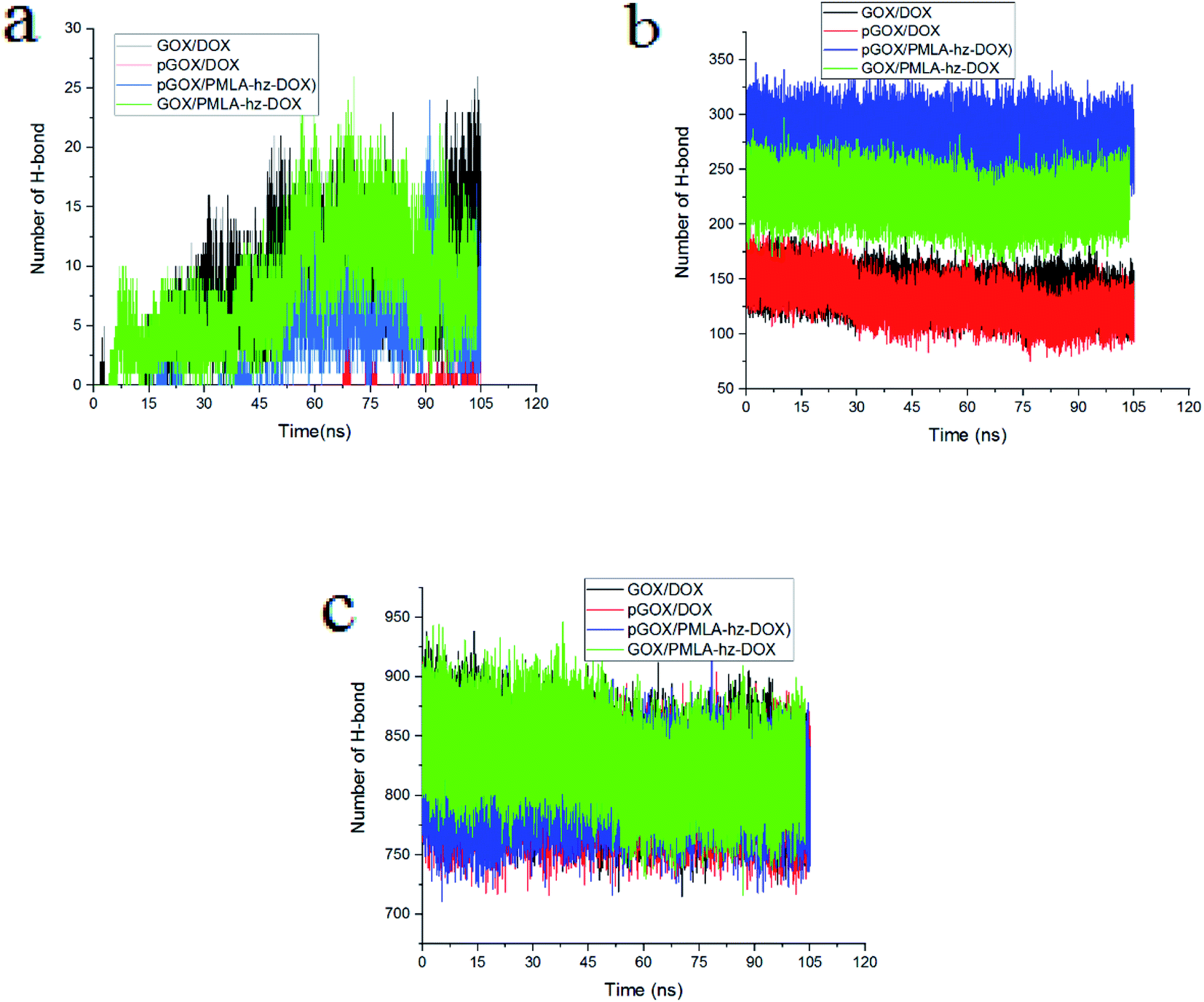 | ||
| Fig. 7 The number of hydrogen bonds between (a) guest molecules and GOX, (b) guest molecules and water, (c) GOX and water. | ||
The number of HBs formed between the drug molecules and water versus the simulation time is depicted in Fig. 7b. The number of HBs of guest molecules with the solvent molecules is almost constant. Close inspection of Fig. 7b reveals that the number of HBs for PMLA-hz-DOX in the acidic environment is more than the neutral condition. It can be concluded that the pGOX/PMLA-hz-DOX after desorption from the GOX surface tends to interact with the water molecules, and therefore self-aggregation does not occur (see Fig. 3f and 7b). Our results confirm that the number of HBs between DOX molecules and water in both pH levels are the same; however, it is not the case in DOX and GOX (compare Fig. 7 panels a and b). As observed in Fig. 7c, the number of HBs between GOX and water decreases by approaching the guest molecules to the GOX surface during the adsorption process.
Conclusions
In summary, using the MD simulation, the adsorption behavior of DOX in free form and in conjugated with the PMLA smart polymer on the GOX surface is investigated. The interaction mechanisms of these DDSs in two different pH levels (neutral and acidic conditions) are studied. The obtained results showed that in the neutral medium, the guest molecules are spontaneously adsorbed on the carrier surface, which is mainly due to the formation of π–π stacking and HBs interactions. It is found that when the PMLA is conjugated to DOX through the hydrazone bond, the interaction of the drug molecule with GOX becomes stronger and forms a more stable complex. In the acidic environment, the binding strength of the guest molecules to the carrier surface is weaker than the neutral condition, so that some of the DOX and PMLA-x-DOX molecules are desorbed from the nanosheet surface. The desorbed DOX molecules tend to self-aggregate together, while it is not the case in the PMLA-hz-DOX molecules. Moreover, it is observed that the conjugation of DOX and PMLA via an amide bond does not show the proper behavior on the adsorption and release process.Conflicts of interest
There are no conflicts to declare.References
- E. Ratemi, in Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications, Elsevier, 2018, vol. 1, pp. 121–141 Search PubMed.
- K. Ulbrich and V. Šubr, Adv. Drug Delivery Rev., 2004, 56, 1023–1050 CrossRef CAS PubMed.
- A. Haghi, H. Raissi, H. Hashemzadeh and F. Farzad, RSC Adv., 2020, 10, 44533–44544 RSC.
- A. Zaboli, H. Raissi, F. Farzad and H. Hashemzadeh, J. Mol. Liq., 2020, 301, 112435 CrossRef CAS.
- L. Razavi, H. Raissi, H. Hashemzadeh and F. Farzad, J. Biomol. Struct. Dyn., 2020, 1–10 CrossRef.
- H. Hu, J. Yu, Y. Li, J. Zhao and H. Dong, J. Biomed. Mater. Res., Part A, 2012, 100, 141–148 CrossRef.
- H. Hashemzadeh and H. Raissi, Appl. Surf. Sci., 2020, 500, 144220 CrossRef CAS.
- X. Zhang, J. Yin, C. Peng, W. Hu, Z. Zhu, W. Li, C. Fan and Q. Huang, Carbon, 2011, 49, 986–995 CrossRef CAS.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- J. Chen, Y. Li, L. Huang, C. Li and G. Shi, Carbon, 2015, 81, 826–834 CrossRef CAS.
- S.-Y. Wu, S. S. A. An and J. Hulme, Int. J. Nanomed., 2015, 10, 9 CrossRef CAS.
- Y. Yang, Y.-M. Zhang, Y. Chen, D. Zhao, J.-T. Chen and Y. Liu, Chem.–Eur. J., 2012, 18, 4208–4215 CrossRef CAS.
- Y. Zhang, T. R. Nayak, H. Hong and W. Cai, Nanoscale, 2012, 4, 3833–3842 RSC.
- H. Wang and Z. Qiu, Thermochim. Acta, 2011, 526, 229–236 CrossRef CAS.
- G. Liu, H. Shen, J. Mao, L. Zhang, Z. Jiang, T. Sun, Q. Lan and Z. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 6909–6914 CrossRef CAS.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- J.-Z. Du, X.-J. Du, C.-Q. Mao and J. Wang, J. Am. Chem. Soc., 2011, 133, 17560–17563 CrossRef CAS PubMed.
- M. B. Yatvin, W. Kreutz, B. A. Horwitz and M. Shinitzky, Science, 1980, 210, 1253–1255 CrossRef CAS.
- S. Manchun, C. R. Dass and P. Sriamornsak, Life Sci., 2012, 90, 381–387 CrossRef CAS PubMed.
- Y. Qiao, B. Liu, Y. Peng, E. Ji and H. Wu, Int. J. Mol. Med., 2019, 42, 3495–3502 Search PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- G. M. Whitesides, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826–2885 CrossRef CAS.
- P. Debbage, Curr. Pharm. Des., 2009, 15, 153–172 CrossRef CAS PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Nanomater. Neoplasms Towar. Clin. Appl., 2013, 31 Search PubMed.
- X. Guo, C. Shi, J. Wang, S. Di and S. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS.
- M. Xu, J. Qian, X. Liu, T. Liu and H. Wang, Mater. Sci. Eng., C, 2015, 50, 341–347 CrossRef CAS.
- Y. Kang, X.-M. Zhang, S. Zhang, L.-S. Ding and B.-J. Li, Polym. Chem., 2015, 6, 2098–2107 RSC.
- H. S. Yoo, E. A. Lee and T. G. Park, J. Controlled Release, 2002, 82, 17–27 CrossRef CAS.
- S. Wu, X. Zhao, Y. Li, Q. Du, J. Sun, Y. Wang, X. Wang, Y. Xia, Z. Wang and L. Xia, Materials, 2013, 6, 2026–2042 CrossRef CAS.
- M. Mahdavi, F. Rahmani and S. Nouranian, J. Mater. Chem. B, 2016, 4, 7441–7451 RSC.
- H. Zhang, T. Fan, W. Chen, Y. Li and B. Wang, Bioact. Mater., 2020, 5, 1071–1086 CrossRef PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- C. Tu, L. Zhu, F. Qiu, D. Wang, Y. Su, X. Zhu and D. Yan, Polymer, 2013, 54, 2020–2027 CrossRef CAS.
- T. Thambi, V. G. Deepagan, C. K. Yoo and J. H. Park, Polymer, 2011, 52, 4753–4759 CrossRef CAS.
- B. Chen, W. Dai, B. He, H. Zhang, X. Wang, Y. Wang and Q. Zhang, Theranostics, 2017, 7, 538 CrossRef CAS.
- J. Li, F. Yu, Y. Chen and D. Oupick\`y, J. Controlled Release, 2015, 219, 369–382 CrossRef CAS.
- Y. Chen, X.-L. Zheng, D.-L. Fang, Y. Yang, J.-K. Zhang, H.-L. Li, B. Xu, Y. Lei, K. Ren and X.-R. Song, Int. J. Mol. Sci., 2014, 15, 2761–2772 CrossRef.
- V. P. Chauhan, T. Stylianopoulos, J. D. Martin, Z. Popović, O. Chen, W. S. Kamoun, M. G. Bawendi, D. Fukumura and R. K. Jain, Nat. Nanotechnol., 2012, 7, 383–388 CrossRef CAS.
- J. Y. Ljubimova, J. Portilla-Arias, R. Patil, H. Ding, S. Inoue, J. L. Markman, A. Rekechenetskiy, B. Konda, P. R. Gangalum and A. Chesnokova, et al., J. Drug Targeting, 2013, 21, 956–967 CrossRef CAS PubMed.
- M. P. Allen, et al., Comput. soft matter from Synth. Polym. to proteins, 2004, vol. 23, pp. 1–28 Search PubMed.
- H. Hashemzadeh and H. Raissi, J. Biomed. Mater. Res., Part A, 2021, 1–10 Search PubMed.
- S. Pasban and H. Raissi, Sci. Rep., 2021, 11, 1–9 CrossRef.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen and B. Yu, et al., Nucleic Acids Res., 2019, 47, D1102–D1109 CrossRef.
- S. M. B. Castaneda, E. S. Alvarenga, A. J. Demuner and L. M. Guimaraes, Struct. Chem., 2020, 31, 599–607 CrossRef CAS.
- A. Kordzadeh, S. Amjad-Iranagh, M. Zarif and H. Modarress, J. Mol. Graphics Modell., 2019, 88, 11–22 CrossRef CAS.
- A. Adnan, R. Lam, H. Chen, J. Lee, D. J. Schaffer, A. S. Barnard, G. C. Schatz, D. Ho and W. K. Liu, Mol. Pharm., 2011, 8, 368–374 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867–872 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson and D. van der Spoel, et al., Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- S. Basith, B. Manavalan, T. H. Shin and G. Lee, Int. J. Mol. Sci., 2019, 20, 1666 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- V. Kapil, M. Rossi, O. Marsalek, R. Petraglia, Y. Litman, T. Spura, B. Cheng, A. Cuzzocrea, R. H. Meißner and D. M. Wilkins, et al., Comput. Phys. Commun., 2019, 236, 214–223 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, et al., J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- P. Shan, J.-W. Shen, D.-H. Xu, L.-Y. Shi, J. Gao, Y.-W. Lan, Q. Wang and X.-H. Wei, RSC Adv., 2014, 4, 23730–23739 RSC.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- A. Izadyar, N. Farhadian and N. Chenarani, J. Biomol. Struct. Dyn., 2016, 34, 1797–1805 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02361f |
| This journal is © The Royal Society of Chemistry 2021 |