DOI:
10.1039/D1RA02357H
(Paper)
RSC Adv., 2021,
11, 14986-14995
Fluorinated Iron(II) clathrochelate units in metalorganic based copolymers: improved porosity, iodine uptake, and dye adsorption properties†
Received
25th March 2021
, Accepted 16th April 2021
First published on 21st April 2021
Abstract
We report the synthesis of metalorganic copolymers made from the palladium catalyzed Sonogashira cross-coupling reaction between various iron(II) clathrochelate building blocks with diethynyl–triptycene and fluorene derivatives. The target copolymers CCP1–5 were isolated in excellent yield and characterized by various instrumental analysis techniques. Interestingly, investigation of the copolymers' porosity properties discloses BET surface areas up to 337 m2 g−1 for the target compounds bearing fluorinated iron(II) clathrochelate units CCP2,5. Moreover, the fluorinated copolymers display an outstanding uptake capacity of iodine with a maximum adsorption of 200 wt%. The target metalorganic copolymers CCP1–5 reveal very good adsorption of organic dyes, namely, methyl blue and methylene blue, from aqueous media.
1. Introduction
In the past two decades, there has been growing interest in designing amorphous polymers with contorted structures that induce poor three-dimensional packing in the solid state, therefore, leading to the formation of intrinsic pores which bestow the resulting materials with prominent properties allowing for their use in various applications, namely, gas storage and separation, sensing, and catalysis.1–7 Unlike highly ordered materials which require complex synthesis and isolation methodologies, such as metal–organic frameworks (MOFs)8,9 and covalent organic frameworks (COFs),10,11 microporous polymer networks (MPNs),12 in particular, polymers of intrinsic microporosity (PIMs)13,14 are renowned for their versatile synthesis and utilization in energy storage technologies, such as, (i) natural gas adsorbents for vehicles, (ii) hydrogen gas tanks for fuel cells, and (iii) vessels for CO2 separation/sequestration from flue gas.7,15–22 Amid the various synthetic methodologies to prepare microporous polymers, palladium-catalyzed cross-coupling reactions, namely, Suzuki23 and Sonogashira24 offer numerous advantages, among others, allowing the preparation of target microporous materials from hands-on synthons which are either commercially available or might require few synthetic steps. In addition, the abovementioned cross-coupling reactions permit a noticeable control over the polymerization reaction, thus, yielding highly structured copolymers. On top that, Suzuki and Sonogashira cross-coupling reactions are powerful synthetic tools for the preparation of porous materials with additional properties, such as, making conjugated polymers, introducing functional groups into the macromolecules' backbones, etc.1,25 Typically, palladium complexes in combination with copper(I) salts are employed as catalysts in Sonogashira cross-coupling reactions although alternative catalytic systems were also found to be very effective.26–28
Iron(II) clathrochelate is a robust metalorganic unit with a considerable internal free volume (IFV) and which can be easily synthesized and functionalized, especially when employed as a building block in cross-coupling reactions.29–31 Iron(II) clathrochelate complexes were tested as biosensors,32,33 catalysts for hydrogen generation,34,35 materials for electronic transport,36 organogels,37 and to make supramolecular structures of definite shapes and sizes.38–41 Recently, several iron(II) clathrochelate based polymers were reported disclosing prominent porous properties.42–44 In this prospect, we report herein the synthesis and characterization of five new iron(II) clathrochelate copolymers CCP1–5 using Sonogashira cross-coupling reaction conditions. Interestingly, the target copolymers exhibit Brunauer–Emmett–Teller (BET) surface area up to 337 m2 g−1 as well as high iodine uptake and excellent adsorption of organic dyes, namely, methyl blue and methylene blue.
2 Experimental part
2.1 General
All the reactions were carried out under inert atmosphere using dry argon. All chemical reagents were used without further purification as purchased from Aldrich, Merck, and HiMedia unless otherwise specified. 1,4-Diethynyl-9,10-dihydro-9,10-[1,2]benzenoanthracene 1a, 2,7-diethynyl-9,9-dimethyl-9H-fluorene 1b, and CC1 were prepared according to procedures reported in the literature.41,45,46 The solvents, namely, DMF, diethyl ether, methanol, hexane, DCM and THF were deoxygenated by bubbling with dry argon gas for 30 minutes. Thin-layer chromatography (TLC) was performed on aluminum sheets coated with silica gel 60 F254 and revealed using a UV lamp. NMR (1H: 600 MHz, 13C: 150 MHz) spectra were recorded on Bruker BioSpin GmbH 600 MHz spectrometer using CD2Cl2 as a solvent with the chemical shifts (δ) given in ppm and referred to tetramethylsilane (TMS). FT-IR spectra were recorded on Agilent Cary 630 FTIR instrument. UV-Vis spectra were recorded on Shimadzu UV1800 spectrophotometer. Brunauer–Emmett–Teller (BET) surface area and porosity measurements were evaluated using a Surface Area and Pore Size Analyzer (Gemini-V, Micromeritics, USA) at the boiling point of liquid nitrogen (−196 °C). Samples were degassed in VacuPrep 061 sample degassing system at a temperature of 105 °C for overnight, before the experiments. Surface areas (SBET) were calculated using the Brunauer–Emmet–Teller (BET) model of isotherms, and the adsorption of N2 at small relative pressures. Total pore volume (Vt) was determined from the specific adsorption of N2 at a p/p0 = 0.99. The t-plot method was used to estimate micropore volume (Vmic) and external surface area (Sext). Quantachrome Autosorb iQ2 analyzer was used to collect gas adsorption data. In a typical gas uptake setup, samples (120–150 mg) were loaded in a 9 mm cell and were subjected to degassing at 120 °C for 5–8 h by attaching to the degassing unit. The cells with the degassed materials were refilled with helium gas and weighed accurately. Subsequently, cells were reattached to the analysis unit of the instrument for measurements. Various temperatures of the analysis unit sample cell were maintained using KGW isotherm bath that was filled with liquid N2 (77 K), or using a temperature-controlled bath (298 K and 273 K). Polymers molecular weights were recorded on Agilent 1260 infinity II gel permeation chromatograph (GPC) against with a refractive index (RI) detector at room temperature and two columns (PL mixed-C) which are calibrated twelve monodisperse polystyrene (PS) standards with THF employed as eluent at a flow rate of 1.0 mL min−1 UV-Vis spectra were recorded on Shimadzu UV1800 spectrophotometer. X-ray Photoelectron spectroscopy (XPS) data were recorded with a Thermo ESCALAB 250 Xi using a monochromatic Al Kα-radiation source (1486.6 eV) with a spot size of 850 μm. Spectra acquisition and processing were carried out using the software Thermo Advantage Version 4.87. The base pressure in the XPS analysis chamber was in the range 10−10 to 10−9 torr. The analyzer was operated with pass energy of 20 eV, dwell time of 50 min and with a step size of 0.1 eV.
Electron impact high-resolution mass spectra (EI-HRMS) were recorded on a Thermo (DFS) with a standard PFK (perfluorokerosene) as lock mass. The analyzed data is converted to accurate mass employing X-Calibur accurate mass calculation software.
2.2 Synthesis
2.2.1 Synthesis of CC2 (procedure A). A Schlenk tube was charged with nioxime (400 mg, 2.74 mmol, 3 eq.), 4-bromo-2,6-difluorophenylboronic acid (500 mg, 2.11 mmol, 2 eq.) and anhydrous FeCl2 (115 mg, 0.91 mmol) in MeOH (20 mL) and the mixture was refluxed for 3 h under argon. The reaction mixture was allowed to cool to RT, and the resulting precipitate was isolated by filtration, washed with MeOH, diethyl ether, and dried under vacuum to yield a red solid (700 mg, 88%); 1H-NMR (600 MHz, CD2Cl2, ppm): δ 7.05–7.04 (d, J = 6 Hz, 4H, ArH), 2.92 (s, 12H, CH2), 1.83 (s, 12H, CH2); 13C-NMR (150 MHz, CD2Cl2, ppm): δ 167.98, 166.31, 152.96, 122.24,115.69, 26.75, 22.11; HRMS: m/z calculated for (M˙+) C30H28B2Br2F4FeN6O6 881.9883 found 881.9885.
2.2.2 Synthesis of CC3. CC3 was prepared following procedure A with: butyl dioxime (165 mg, 0.82 mmol, 3 eq.), 4-bromo-2,6-difluorophenylboronic acid (150 mg, 0.633 mmol, 2 eq.) and anhydrous FeCl2 (36 mg, 0.29 mmol) in MeOH (10 mL). Red solid (290 mg, 96%); 1H-NMR (600 MHz, CD2Cl2, ppm): δ 7.03–7.02 (d, J = 6 Hz, 4H, ArH), 2.82–2.79 (t, J = 6 Hz, 12H, CH2), 1.56–1.53 (m, 12H, CH2), 1.34 1.30 (m, 12H, CH2), 0.89–0.86 (t, J = 6 Hz, 18H, CH3); 13C-NMR (150 MHz, CD2Cl2, ppm): δ 168.15, 166.48, 157.94, 121.92, 115.60, 29.66, 27.69, 23.00 14.06; HRMS: m/z calculated for (M˙+) C42H58B2Br2F4FeN6O6 1056.2230 found 1056.2236.
2.2.3 Synthesis of copolymer CCP1 (procedure B). A Schlenk tube was charged with a mixture of DMF and iPr2NH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 10 mL) which was bubbled with argon for 30 min. Before the addition of CC1 (200 mg, 0.25 mmol, 1 eq.) and 1a ( 75 mg, 0.25 mmol, 1 eq.). The mixture was purged with argon for 10 min. Before adding CuI (4 mg, 0.02 mmol, 8 mol%) and Pd(PPh3)4 (23 mg, 0.02 mmol, 8 mol%) under a positive stream of argon. The reaction was stirred for 48 h at 110 °C and the precipitate was isolated by filtration while hot and washed with DMF (50 mL), water (50 mL), THF (50 mL), DCM (50 mL), acetone (50 mL), methanol (50 mL) and diethyl ether (50 mL) to yield a red solid (211 mg, 90%); FTIR (cm−1): 2955 (aliphatic –C–H stretch.), 2201 (C
:1, 10 mL) which was bubbled with argon for 30 min. Before the addition of CC1 (200 mg, 0.25 mmol, 1 eq.) and 1a ( 75 mg, 0.25 mmol, 1 eq.). The mixture was purged with argon for 10 min. Before adding CuI (4 mg, 0.02 mmol, 8 mol%) and Pd(PPh3)4 (23 mg, 0.02 mmol, 8 mol%) under a positive stream of argon. The reaction was stirred for 48 h at 110 °C and the precipitate was isolated by filtration while hot and washed with DMF (50 mL), water (50 mL), THF (50 mL), DCM (50 mL), acetone (50 mL), methanol (50 mL) and diethyl ether (50 mL) to yield a red solid (211 mg, 90%); FTIR (cm−1): 2955 (aliphatic –C–H stretch.), 2201 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretch.), 1664 (C
C stretch.), 1664 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch.), 1550 (N–O stretch.), 1431 (aliphatic –C–H bend.), and 954 (aromatic CC bend.); UV-vis: (THF, 10−8 M), λmax [nm] = 329 and 450.
N stretch.), 1550 (N–O stretch.), 1431 (aliphatic –C–H bend.), and 954 (aromatic CC bend.); UV-vis: (THF, 10−8 M), λmax [nm] = 329 and 450.
2.2.4 Synthesis of copolymer CCP2. CCP2 was prepared following procedure B with: CC2 (200 mg, 0.23 mmol, 1 eq.), 1a (69 mg, 0.23 mmol, 1 eq.), Pd(PPh3)4 (20 mg, 0.018 mmol, 8 mol%) and CuI (3 mg, 0.018 mmol, 8 mol%) in 9 mL of a 1:1 degassed mixture of DMF and iPr2NH. Red solid (200 mg, 87%). FTIR (cm−1): 2944 (aliphatic –C–H stretch.), 2201 (CC stretch.), 1618 (CN stretch.), 1545 (N–O stretch.), 1458 (aliphatic –C–H bend.), 1395 (C–F stretch.), and 964 (aromatic CC bend.); UV-vis: (THF, 10−8 M), λmax [nm] = 329 and 450.
2.2.5 Synthesis of copolymer CCP3. CCP3 was prepared following procedure B with: CC3 (250 mg, 0.24 mmol, 1 eq.), 1a (72 mg, 0.24 mmol, 1 eq.), Pd(PPh3)4 (22 mg, 0.019 mmol, 8 mol%) and CuI (4 mg, 0.019 mmol, 8 mol%) in 10 mL of a 1:1 degassed mixture of DMF and iPr2NH. The solvent was evaporated under reduced pressure and the resulting residue was extracted with ethyl acetate from an aqueous solution of 10% LiCl (100 mL). The organic layer was washed with deionized water (100 mL × 3), concentrated, and the product was precipitated by adding hexane. The precipitate was isolated by filtration under reduced pressure and washed exhaustively with hexane. Red solid (261 mg, 92%);1H-NMR (600 MHz, CD2Cl2, ppm): δ 7.60–7.53 (m, 4H, ArH), 7.31–7.09 (m, 10H, ArH), 6.03 (br, 2H, triptycene CH), 2.87 (br, 12H, butyl-CH2), 1.60 (br, 12H, butyl-CH2), 1.38 (br, 12H, butyl-CH2), 0.93 (br, 18H, butyl-CH3); 13C-NMR (150 MHz, CD2Cl2, ppm): δ 167.96, 166.32, 157.94, 148.21, 145.02, 129.68, 128.68, 126.23, 124.63, 115.58, 115.35, 114.98, 114.76, 52.62, 29.70, 27.69, 23.03, 14.11; FTIR (cm−1): 2958 (aliphatic –C–H stretch.), 2210 (CC stretch.), 1618 (CN stretch.), 1544 (N–O stretch.), 1458 (aliphatic –C–H bend.), 1400 (C–F stretch.), and 716 (aromatic CC bend.); GPC (THF): Mw (g mol−1): 14106, Mn (g mol−1): 7388, Đ: 1.9; UV-vis: (THF, 10−8 M), λmax [nm] = 329 and 450.
2.2.6 Synthesis of copolymer CCP4. CCP4 was prepared following procedure B with: CC1 (300 mg, 0.37 mmol, 1 eq.), 1b (90 mg, 0.37 mmol, 1 eq.), Pd(PPh3)4 (34 mg, 0.03 mmol, 8 mol%) and CuI (5 mg, 0.03 mmol, 8 mol%) in 15 mL of a 1:1 degassed mixture of DMF and iPr2NH. Red solid (301 mg, 90%); FTIR (cm−1): 2955 (aliphatic –C–H stretch.), 2201 (CC stretch.), 1602 (CN stretch.), 1492 (N–O stretch.), 1434 (aliphatic –C–H bend.), and 960 (aromatic CC bend.); UV-vis: (THF, 10−8 M), λmax [nm] = 354, 371, 400 and 450.
2.2.7 Synthesis of copolymer CCP5. CCP5 was prepared following procedure B with: CC2 (180 mg, 0.21 mmol, 1 eq.), 1b (50 mg, 0.21 mmol, 1 eq.), Pd(PPh3)4 (19 mg, 0.017 mmol, 8 mol%) and CuI (3 mg, 0.017 mmol, 8 mol%) in 8 mL of a 1:1 degassed mixture of DMF and iPr2NH. Red solid (188 mg, 95%); FTIR (cm−1): 2942 (aliphatic –C–H stretch.), 2207 (CC stretch.), 1618 (CN stretch.), 1542 (N–O stretch.), 1436 (aliphatic –C–H bend.), 1396 (C–F stretch.), and 959 (aromatic CC bend.); UV-vis: (THF, 10−8 M), λmax [nm] = 354, 371, 400 and 450.
3. Results and discussion
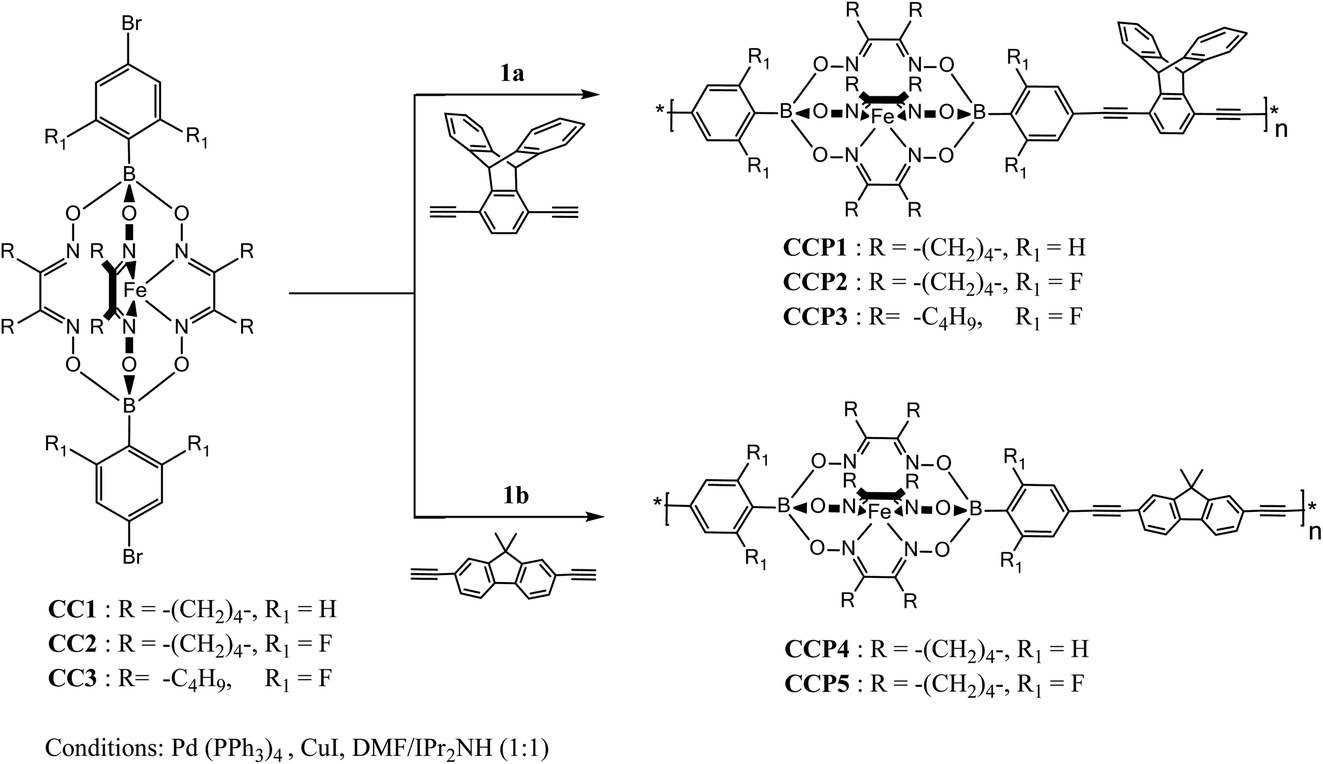
Scheme 1 depicts the synthetic strategy which was devised to prepare the target clathrochelate copolymers CCP1–5 via a typical Sonogashira cross-coupling reaction condition using the dibrominated iron(II) clathrochelate synthons CC1–3 with each of 1,4-diethynyl triptycene 1a and 2,7-diethynyl-9,9-dimethyl fluorene 1b. The desired copolymers CCP1–5 were isolated after 2 days of reaction at 110 °C by simple filtration while hot followed by exhaustive sequential washings of the precipitates with hot solvents, thus, affording the desired target materials in excellent yields (87–95%). The copolymers whose clathrochelate units bear cyclohexyl side groups (i.e. CCP1,2,4,5) were found to be highly insoluble in common organic solvents. Consequently, the hitherto mentioned four copolymers were characterized using FTIR and XPS spectroscopy (Fig. S8–S15 in the ESI† file). Nevertheless, copolymer CCP3, which contains clathrochelate units with the more flexible butyl side groups, was found to be soluble in most common organic solvents (e.g. DCM, THF, chloroform, acetone, and DMF), therefore, allowing for its thorough structural analysis by 1H- and 13C- NMR, FTIR, and XPS spectroscopy (Fig. 1, 2, 4 and S5 in the ESI† file).
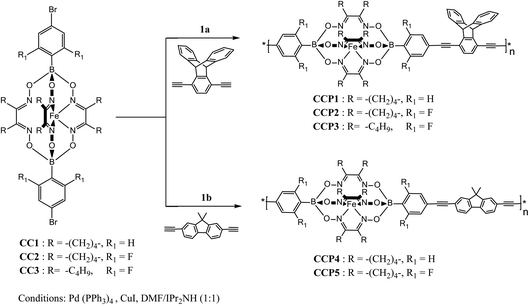 |
| | Scheme 1 Synthesis of copolymers CCP1–5. | |
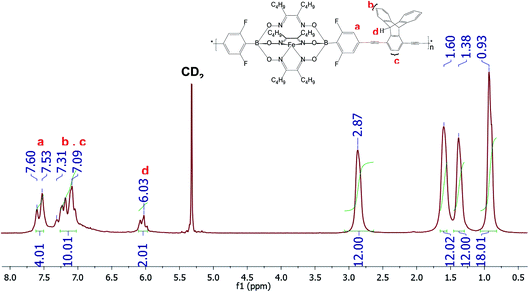 |
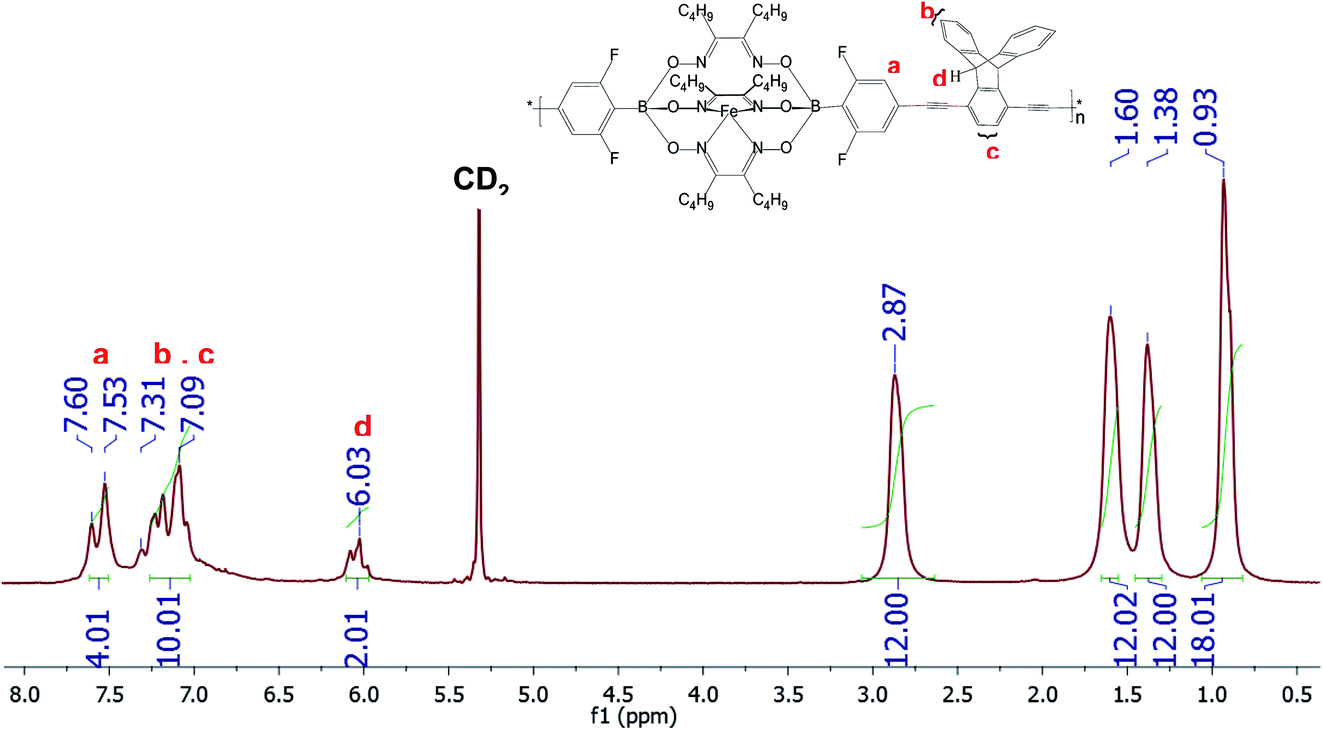
| | Fig. 1 1H-NMR spectrum of CCP3 recorded in CD2Cl2. | |
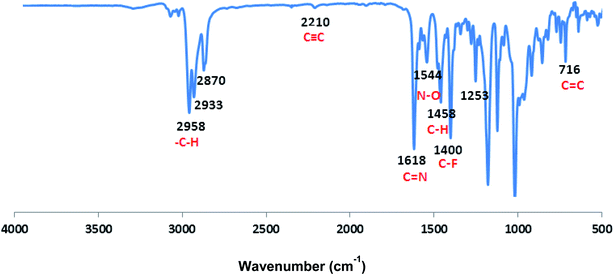 |
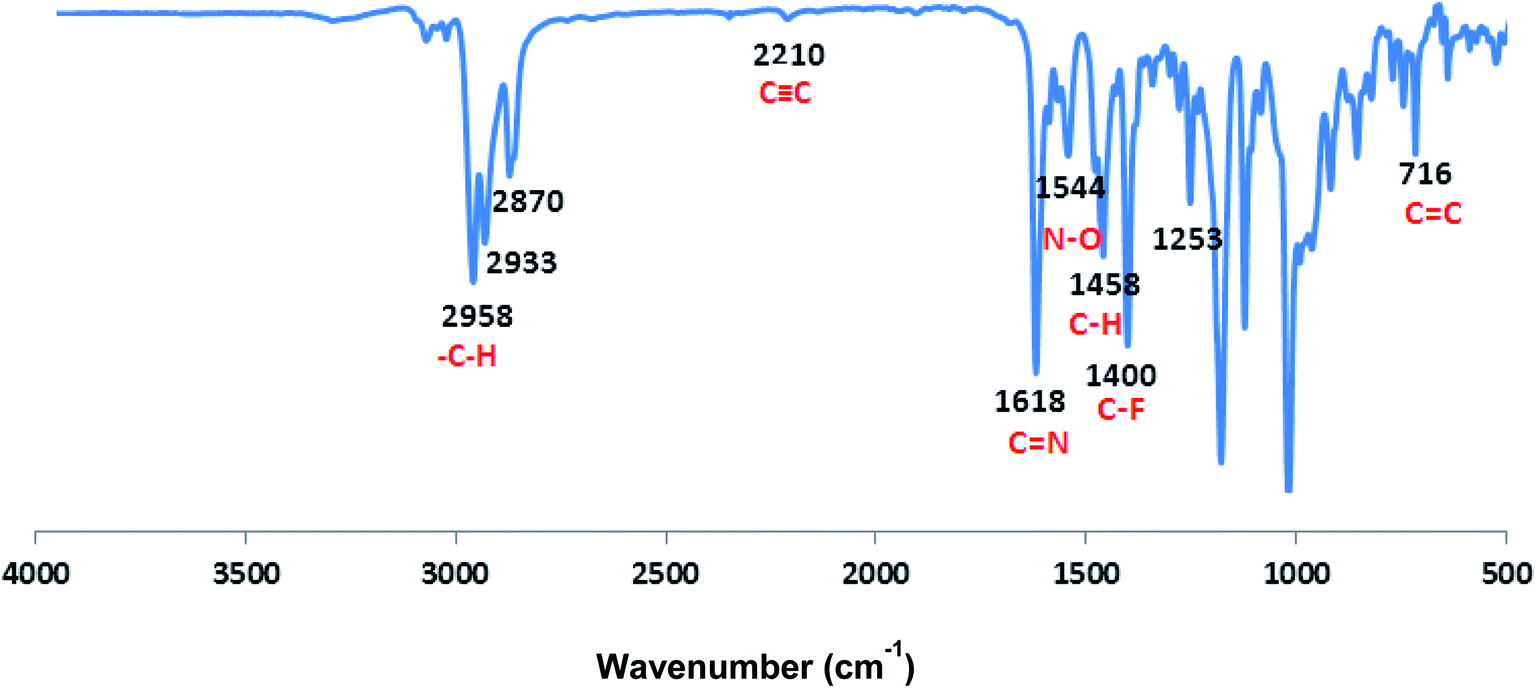
| | Fig. 2 FTIR spectrum of CCP3. | |
Fig. 1 below portrays the 1H-NMR spectrum of CCP3 in CD2Cl2 where the chemical shifts ranging from 7.60 ppm to 7.53 ppm are attributed to the four aromatic protons of the difluorobenzene boronate groups of the iron(II) clathrochelate unit whereas the four peaks observed at 2.87 ppm, 1.6 ppm, 1.38 ppm, and 0.93 ppm are assigned to the butyl side chains. Furthermore, the characteristic chemical shift of the sp3 protons in the triptycene unit is detected at 6.03 ppm whereas the aromatic protons of this latter are identified in the range of 7.31–7.09 ppm. In addition, 13C-NMR spectrum of CCP3 displays all the characteristic chemical shifts, which further confirms its successful formation in high purity (Fig. S5 in the ESI† file).
The FT-IR absorption spectrum shown in Fig. 2 divulges all the desired peaks of target copolymer CCP3 which clearly confirms its formation. Hence, the peaks observed at 1618 cm−1 and 1400 cm−1 are attributed to the characteristic stretching vibrations of CN and C–F, respectively. Similarly, the peaks detected at 2958 cm−1 and 1544 cm−1 are assigned to the stretching vibrations of the aliphatic C–H and N–O groups, respectively. Moreover, the absorption peaks identified at 1458 cm−1 and 716 cm−1 correspond to the bending vibrations of the aliphatic C–H and aromatic CC, respectively. It is noteworthy that the CC stretching vibrations is also spotted at 2210 cm−1. Similarly, all the other target copolymers disclose the characteristic peaks, thus, confirming their successful synthesis (Fig. S8–S11 in the ESI† file).
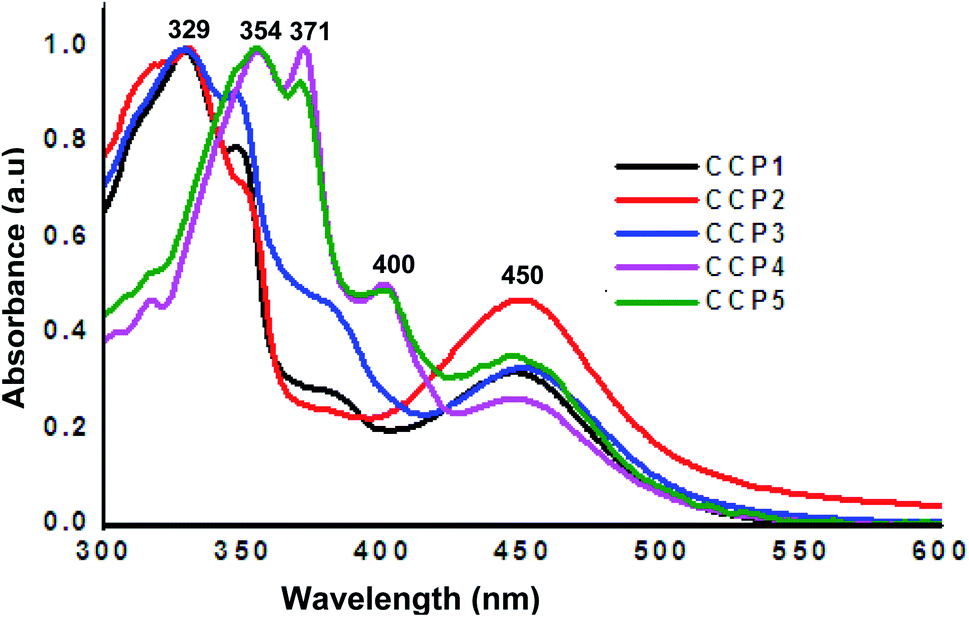
The photophysical properties of CCP1–5 were investigated by means of UV-Vis absorption spectroscopy using THF as a solvent (Fig. 3). Iron(II) clathrochelate copolymers with triptycene units CCP1–3 display analogous absorption bands with two peaks maxima at ∼329 nm and 450 nm, where the former can be attributed to the absorption of triptycene47 while the latter corresponds to the clathrochelate moiety.47 On the other hand, clathrochelate copolymers with fluorene units CCP4,5 display three main absorption bands maxima, two of which are attributed to fluorene at ∼354 nm and 371 nm,48 whereas the one detected at 450 nm corresponds to the clathrochelate unit.
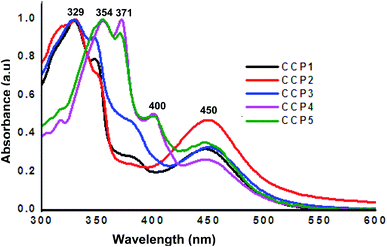 |
| | Fig. 3 Normalized UV-Vis absorption (CM = 10−8 M in THF) spectra of CCP1–5. | |
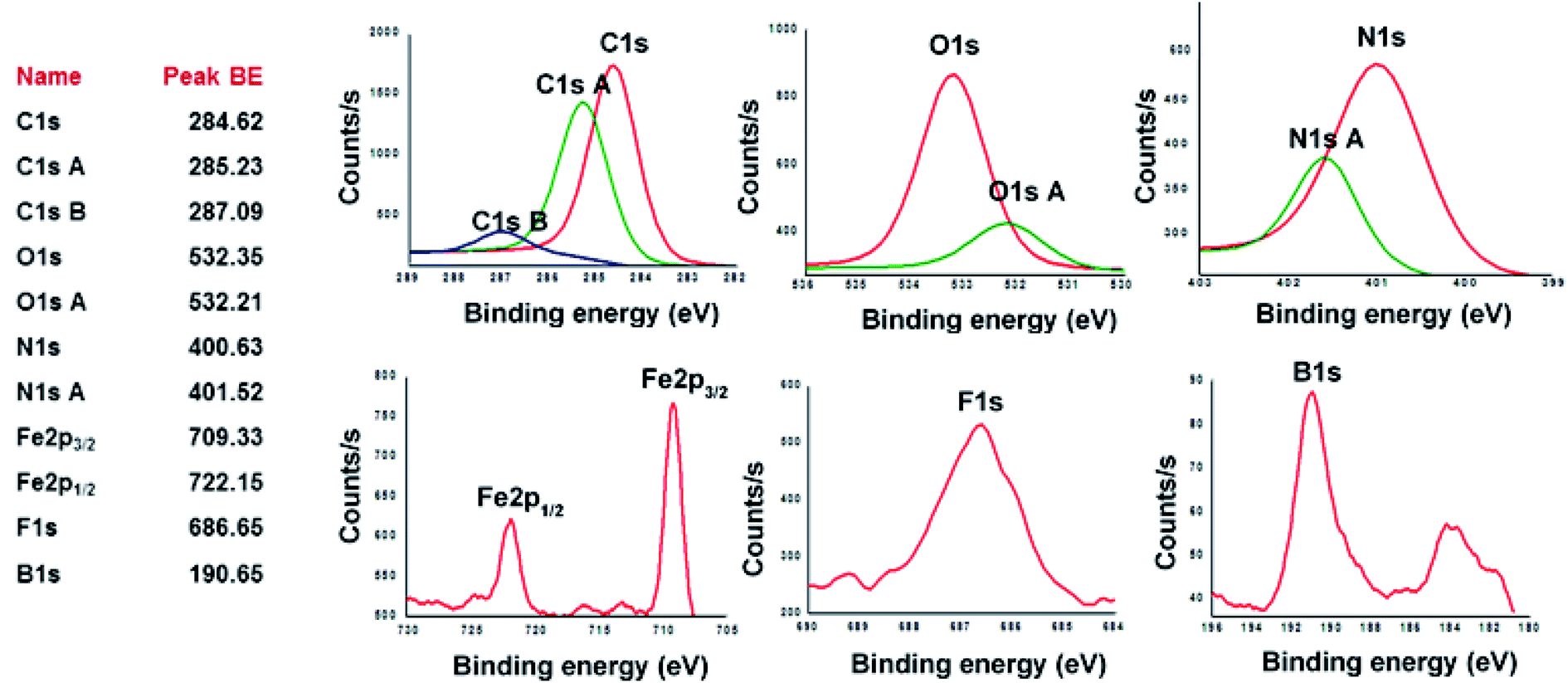
X-ray photoelectron spectroscopy (XPS) was utilized to analyze the elemental composition of copolymers CCP1–5. XPS survey-scan spectrum of CCP3, shown in Fig. 4 below, confirms the presence of all the constituting elements i.e. carbon, oxygen, nitrogen, iron, fluorine, and boron.49 C1s peak of CCP3 can be fitted into two main binding energy values at ∼284.62 eV and 285.23 eV where the former is assigned to the aromatic carbon groups (CC) whereas the latter corresponds to that of imine carbons (CN). The binding energy for oxygen detected at ∼532.3 eV relates to a hydrocarbon bonded to boron and nitrogen. On the other hand, N1s spectrum exhibits two peaks at 400.63 eV and 401.52 eV, which correspond to carbon–nitrogen (C–N) and nitrogen–oxygen (N–O), respectively. F1s core-level spectrum was detected at 685.65 eV, thus, indicating the presence of C–F.50 B1s core-level spectrum was detected at 190.65 eV, which clearly divulges the presence of boron oxide (B–O) usually observed above 190 eV.51 Fig. 4 also reveals the XPS peak for Fe2p with binding energy values of 709.33 eV and 722.15 eV, which are attributed to Fe(II)–N compounds.52 It is noteworthy that the other target copolymers CCP2–5 portray similar XPS binding energy values, which undoubtedly confirm their structures (Fig. S12–S15 in the ESI† file).
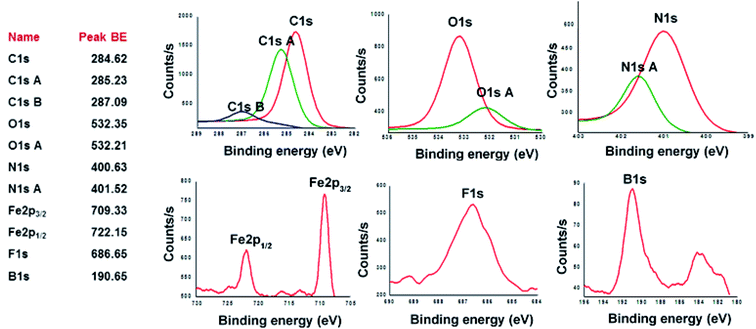 |
| | Fig. 4 High-resolution XPS spectra of C1s, O1s, N1s, Fe2p, F1s and B1s of CCP3. | |
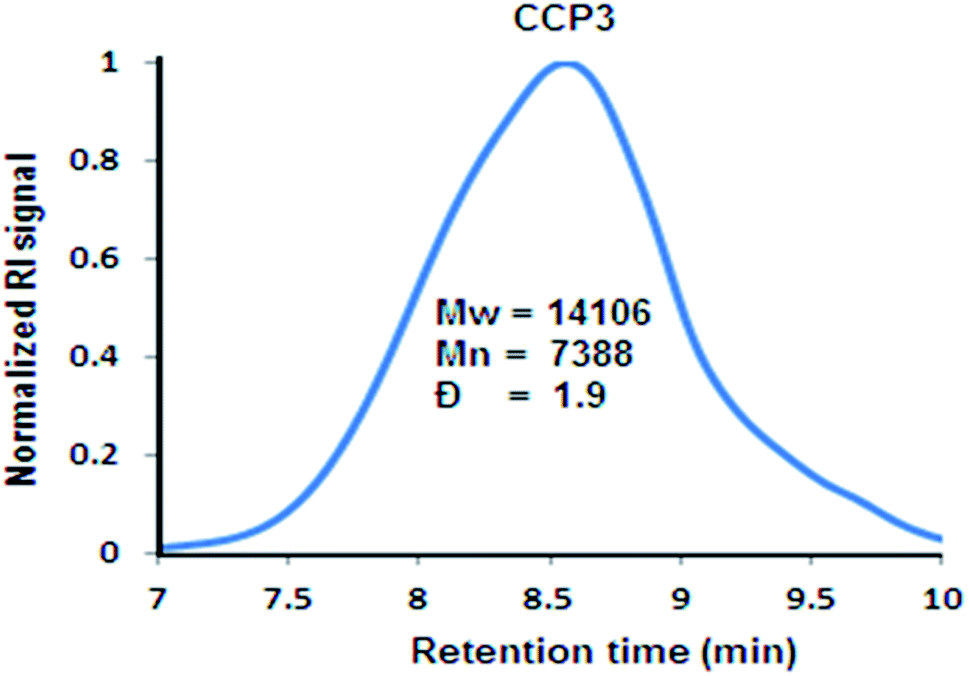
As it could be noticed from Fig. 5, the relatively good solubility of clathrochelate copolymer CCP3 in common organic solvents has allowed for the determination of its molar weight by gel permeation chromatography (GPC), revealing a weight average molar mass Mw of ∼14 kDa and number average molar mass Mn of ∼7 kDa, thus, showing a polydispersity index (PDI = Mw/Mn) of ∼1.9. Nevertheless, the scarce solubility of the triptycene-containing copolymers CCP1,2 and those bearing dimethyl fluorene units CCP4,5 in common organic solvents prevented recording their GPC chromatograms.
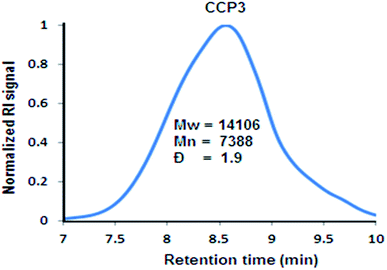 |
| | Fig. 5 Normalized GPC chromatograph of CCP3. | |
4. Surface area and porosity analysis
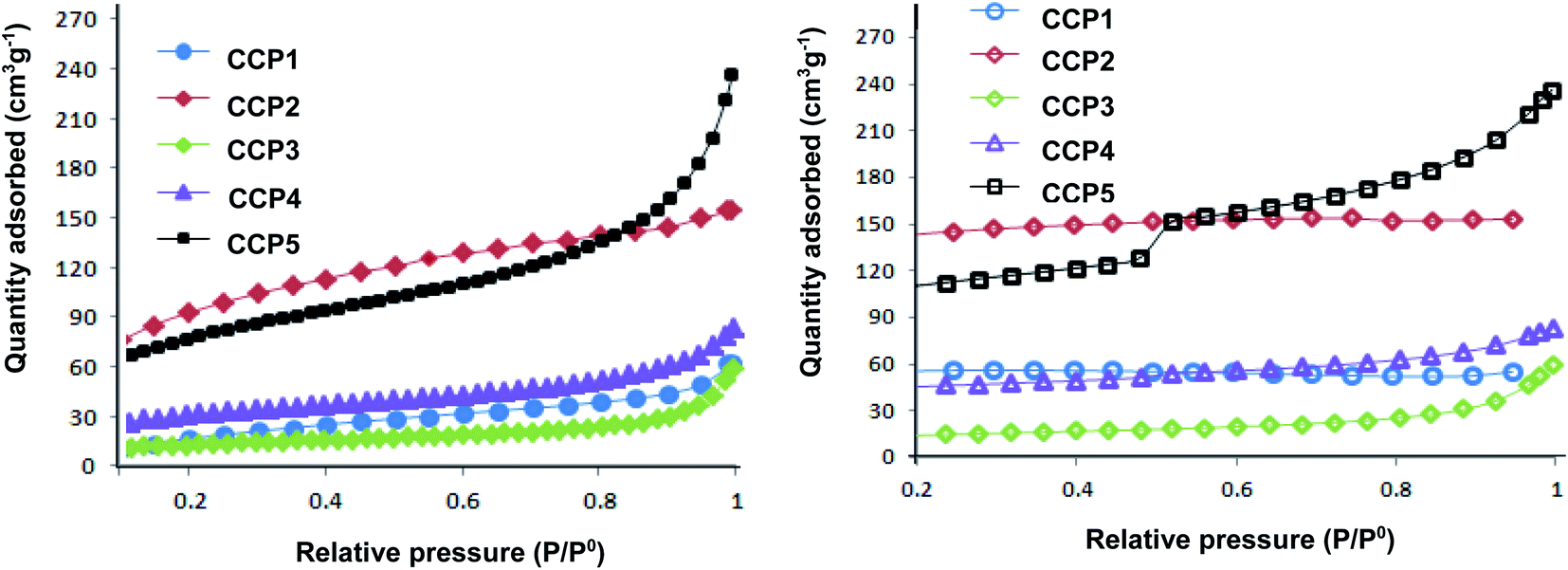
The surface areas and porous properties of copolymers CCP1–5 were investigated by carrying out N2 adsorption experiments at 77 K and low relative pressure (Fig. 6). Table 1 below summarizes the Brunauer–Emmett–Teller (BET) surface areas and pore volumes derived from the nitrogen sorption isotherms. Unsurprisingly, CCP3 divulges the lowest BET surface area (∼45 m2 g−1) which can be explained by the presence of the flexible butyl side chains which block the intrinsic pores.44 The clathrochelate copolymers bearing triptycene CCP1 and dimethyl fluorene CCP4 units portray surface areas of ∼79 m2 g−1 and 111 m2 g−1 CCP1,3, respectively. Interestingly, the replacement of the two hydrogen atoms located at the ortho positions with respect to boron with the bulkier and more electronegative fluorine atoms in the iron(II) clathrochelate moiety of both copolymers i.e. the one with dimethyl fluorene units CCP5 and that with triptycene derivatives CCP2 resulted in a noticeable increase of the surface areas of 279 m2 g−1, for the former and 337 m2 g−1, for the latter. All five target copolymers were found to have pore volumes ranging between 0.039 cm3 g−1 and 0.222 cm3 g−1 (Table 1).
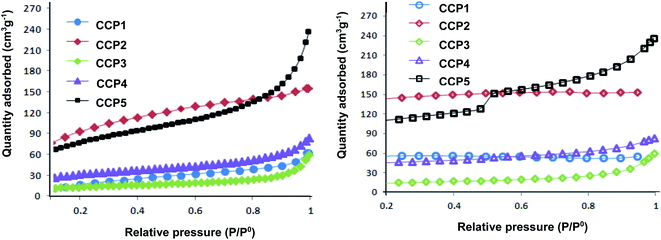 |
| | Fig. 6 Nitrogen adsorption (left) and desorption isotherms (right) of CCP1–5 measured at 77 K. | |
Table 1 Summary of the microporosity properties of polymers CCP1–5
| Entry |
Polymer |
BET surface area (m2 g−1) |
Pore volume (cm3 g−1) |
| 1 |
CCP1 |
79 |
0.086 |
| 2 |
CCP2 |
337 |
0.214 |
| 3 |
CCP3 |
45 |
0.039 |
| 4 |
CCP4 |
111 |
0.085 |
| 5 |
CCP5 |
279 |
0.222 |
5. Iodine uptake study
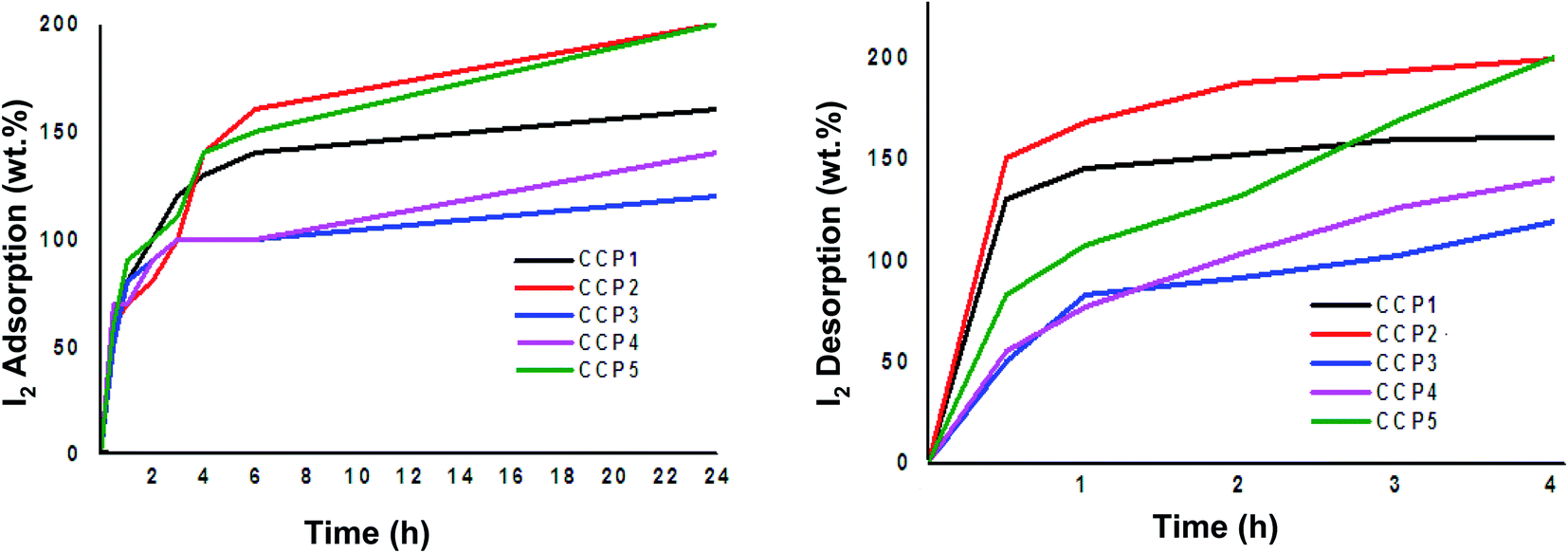
Copolymers CCP1–5 were explored as potential iodine adsorbents using a standard gravimetric analysis protocol reported in the literature.53,54 The iodine uptake tests were conducted by placing a 10 mg sample for each of copolymers CCP1–5 in an open glass vial, which was in turn put inside a sealed glass vessel that contained excess solid iodine at 80 °C under atmospheric pressure and the gravimetric analysis was subsequently recorded at different time intervals (Fig. 7). Evidently, the fluorinated iron (II) clathrochelate copolymers which revealed superior BET surface areas, i.e. CCP2 and CCP5, display the highest iodine uptake reaching a maximum of 200 wt%. It is noteworthy that iodine adsorption of CCP2 reached ∼160 wt% after 6 hours of exposure while that of CCP5 attained 150 wt% for the same period time. On the other hand, the maximum uptake capacities of copolymers CCP1 and CCP4 were found to reach 160 wt% and 140 wt%, respectively, after 24 hours of exposure to I2 (Table 2). The 200 wt% uptake for CCP2 and CCP5 to iodine is promising because of the several advantages these copolymers present, namely, their versatile synthesis from commercially available synthons and the ease of their isolation, especially when compared to the polymer networks reported in the literature which require intricate synthetic and/or purification steps and whose iodine adsorption values are either lower or approximately equal to the ones presented herein.53–65
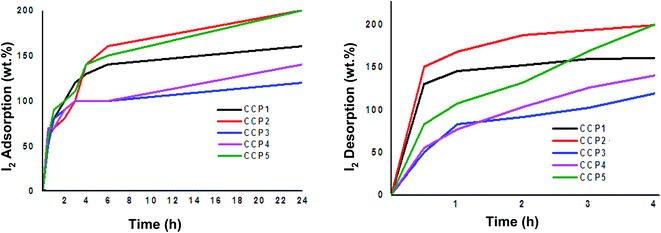 |
| | Fig. 7 Wt% iodine adsorption (left) and desorption (right) graphs of CCP1–5. | |
Table 2 Summary of iodine adsorption and desorption of copolymers CCP1–5
| Entry |
Time (h) |
Wt% I2 adsorption after 24 h |
Wt% I2 desorption after 4 h |
| 1 |
CCP1 |
160 |
160 |
| 2 |
CCP2 |
200 |
199 |
| 3 |
CCP3 |
120 |
119 |
| 4 |
CCP4 |
140 |
140 |
| 5 |
CCP5 |
200 |
200 |
Iodine-containing copolymers CCP1–5 (I2@CCP1–5) underwent desorption by simple heating in air at 120 °C. The complete wt% of iodine released from I2@CCP1–5 samples was recorded at different time intervals (Fig. 7 and Table 2). In addition, reusability tests of the copolymers were carried out using CCP2 as a model sample because it disclosed the maximum uptake capacity for iodine. Hence, a sample of CCP2 fully loaded with iodine i.e. I2@CCP2 was heated at 120 °C for 24 hours, to ensure the complete release of the adsorbate. The reactivated sample CCP2 was subsequently exposed to iodine vapors and its uptake values were recorded gravimetrically using the procedure described above, which revealed similar results to a freshly prepared copolymer after three iodine adsorption–desorption cycles.
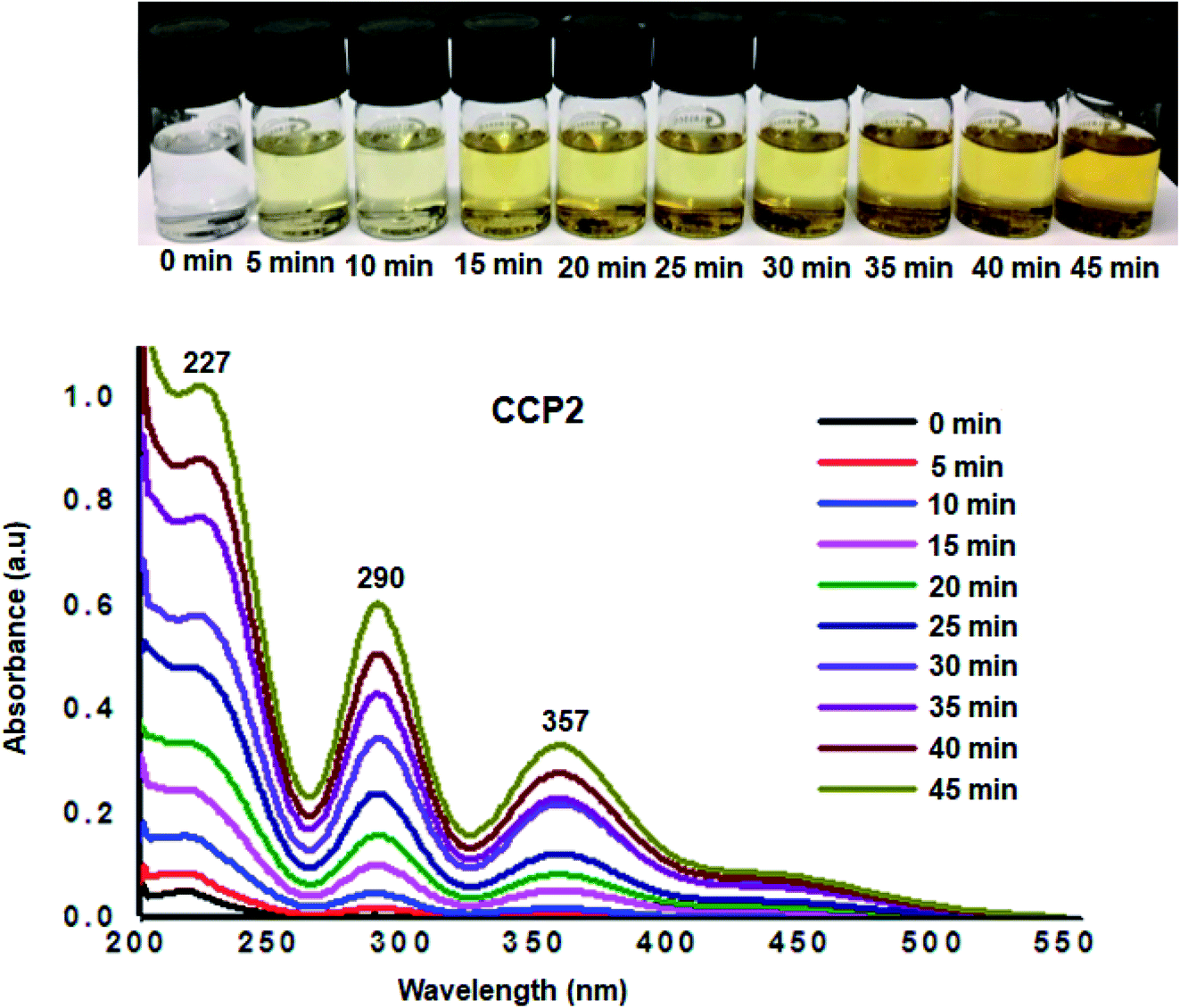
Additional desorption tests were investigated by immersing fully loaded samples of I2@CCP1–5 in ethanol, where the latter is an excellent solvent for iodine. The release of iodine from I2@CCP1–5 samples suspended in ethanol was analyzed by recording the UV-visible absorbance spectra of ethanol at different time intervals (Fig. 8 and S16–S19 in the ESI† file). A conspicuous increase was observed in the absorbance intensity maxima which correspond to iodine, namely, at ∼227 nm (due to I2), ∼290 nm and ∼357 nm (due to polyiodide ions), which confirms the release of the adsorbate from CCP1–5 samples under ambient conditions. It is noteworthy that the amount of iodine released from CCP1–5 reached equilibrium after 45 minutes with a clear change in the color intensity of the solution from colorless to yellow (Fig. 8). These experimental observations strongly suggest that copolymers CCP1–5 can be employed as efficient iodine sorbents and can be easily regenerated either by simple heating or soaking in ethanol, therefore, rendering the recycling process very practical.
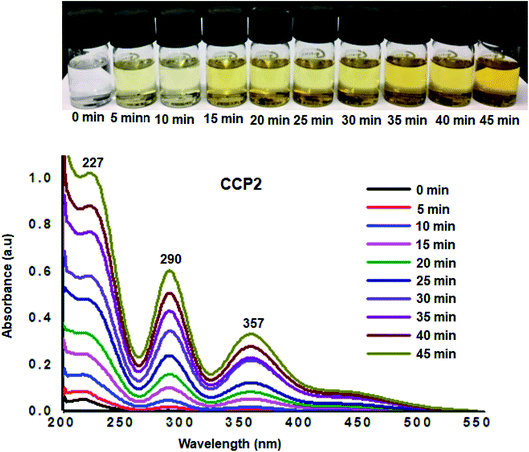 |
| | Fig. 8 UV-Vis absorption spectra upon immersion of I2@CCP2 in ethanol. Inset: photos of the solutions showing the color change upon immersion in ethanol. | |
6. Dye adsorption
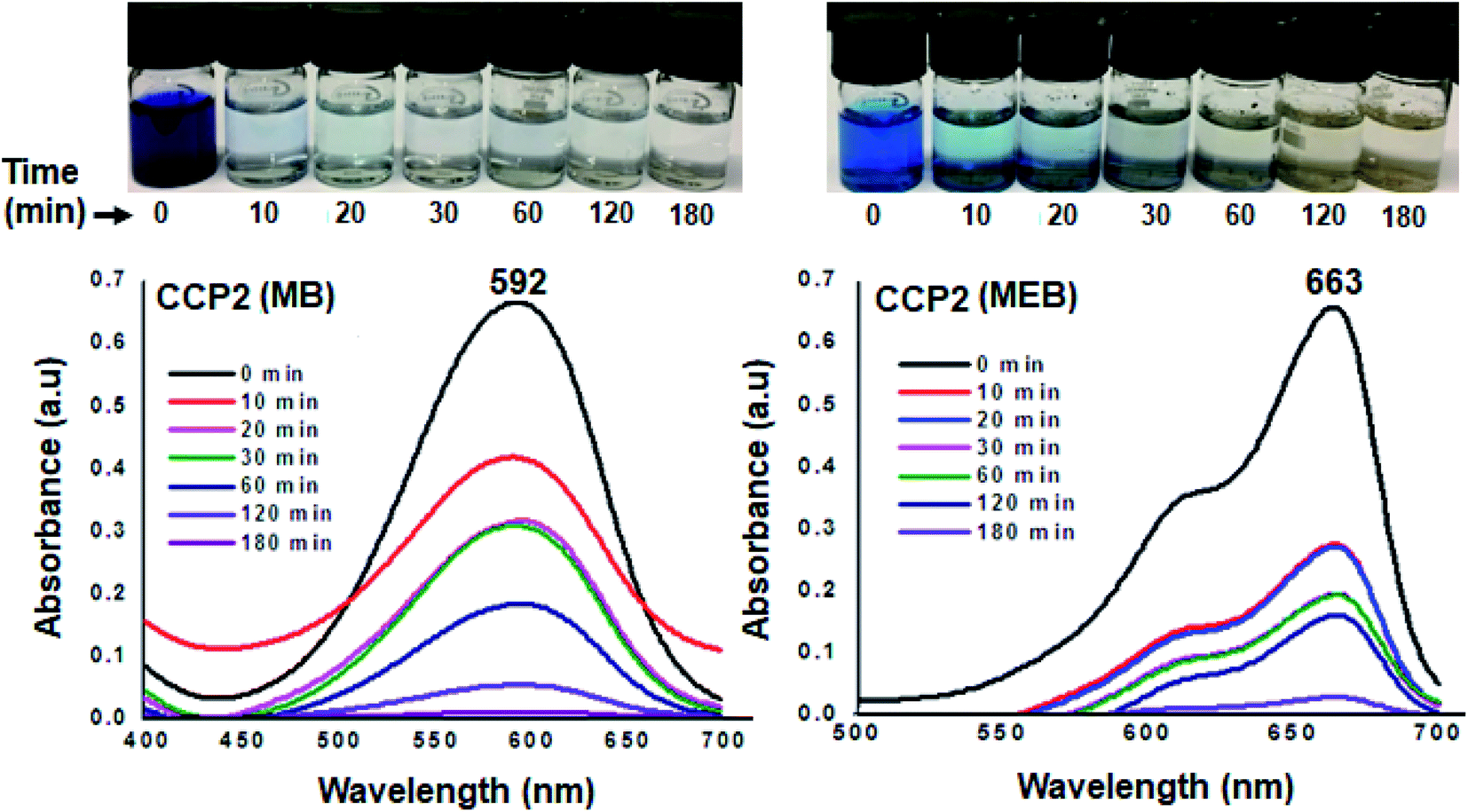
The efficiency of the clathrochelate-based copolymers CCP1–5 to adsorb dyes from aqueous solution was carried out using methyl blue (MB, anionic dye) and methylene blue (MEB, cationic dye). The former is an important reagent for various biological and industrial applications whereas the latter is considered to be a primary source of pollution in water resources.8,66–69 The capacity of CCP1–5 to adsorb MB and MEB was investigated by stirring at ambient temperature a 4 mg sample of a target copolymer in a 5 mL of a 5 mg L−1 aqueous solution of either MB or MEB dyes. The adsorption tests were performed by recording the UV-visible absorbance spectra at different time intervals (Fig. 9 and S20–S27 in the ESI† file). Interestingly, all the clathrochelate-based copolymers CCP1–5 demonstrated an excellent adsorption capacity towards MB where more than 95% of the latter was removed within 2 h of reaction and a 100% adsorption was detected after 3 h of reaction. On the other hand, target copolymers CCP1–5 revealed a less efficient adsorption capacity towards MEB except for CCP2 which disclosed the adsorption of ∼70% of the dye in 10 minutes and ∼100% after 3 h (Fig. 9). Target copolymers CCP1,3–5 adsorbed only ∼70–80% of MEB even after long reaction times (Fig. S20–S27 in the ESI† file).
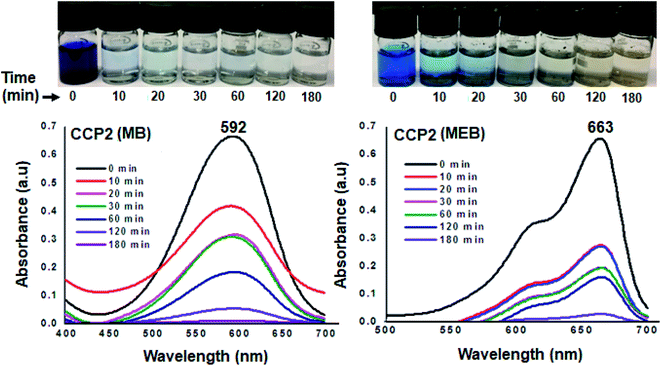 |
| | Fig. 9 UV-Vis absorption spectra of aqueous solution MB (left) and MEB (right) in the presence of CCP2 at various time intervals (inset: photographs showing the color change upon dye adsorption). | |
7. Conclusion
Five metalorganic copolymers CCP1–5 were prepared in very good yields via a one-step Sonogashira cross-coupling reaction between various dibrominated iron(II) clathrochelate units with 1,4-diethynyl triptycene and 2,7-diethynyl-9,9-dimethyl fluorene synthons. Gel permeation chromatography of the soluble target copolymer CCP3 disclosed weight average molar mass Mw of ∼14 kDa and number average molar mass, Mn of ∼7 kDa. Porosity investigation divulged BET surface areas ranging from 45 to 337 m2 g−1 with greater porosity values for the fluorinated copolymers CCP5 bearing fluorene derivatives and CCP2 containing triptycene units, which disclosed surface areas of 279 m2 g−1 for the former copolymer and 337 m2 g−1 for the latter. Likewise, iodine uptake studies depicted adsorption capacities up to 200 wt% for the fluorinated copolymers CCP2,5. Furthermore, CCP1–5 demonstrate efficient adsorption of both methyl blue and methylene blue from aqueous solution with a higher preference towards the former. It is worth mentioning that the metalorganic copolymers CCP1–5 present several advantages, among others, their ease of synthesis and isolation, superior stability, in addition to the possibility for their utilization in the treatment of emission of toxic iodine vapors and organic dyes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was partially supported by Kuwait Foundation for the Advancement of Sciences (KFAS) under project codes P314-34SC-01 and PN18-14SC-03. N. D. thanks the Indian Institute of Technology Patna (IIT Patna) for providing instrumental facilities required for the research. A. H. thanks IIT Patna for providing a research fellowship. We would like to thank the general facilities projects GS01/03, GS01/05, GS03/01, and GS03/08 at Kuwait University.
References
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef CAS.
- X. Yu, Z. Yang, B. Qiu, S. Guo, P. Yang, B. Yu, H. Zhang, Y. Zhao, X. Yang, B. Han and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 632–636 CrossRef CAS PubMed.
- J. Guo, L. Yu and H. Yue, React. Funct. Polym., 2019, 135, 58–64 CrossRef CAS.
- Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J.-X. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- M. Kalaj, K. C. Bentz, S. Ayala, J. M. Palomba, K. S. Barcus, Y. Katayama and S. M. Cohen, Chem. Rev., 2020, 120, 8267–8302 CrossRef CAS PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- R. K. Sharma, P. Yadav, M. Yadav, R. Gupta, P. Rana, A. Srivastava, R. Zbořil, R. S. Varma, M. Antonietti and M. B. Gawande, Mater. Horiz., 2020, 7, 411–454 RSC.
- G. Kupgan, L. J. Abbott, K. E. Hart and C. M. Colina, Chem. Rev., 2018, 118, 5488–5538 CrossRef CAS PubMed.
- A. B. Foster, M. Tamaddondar, J. M. Luque-Alled, W. J. Harrison, Z. Li, P. Gorgojo and P. M. Budd, Macromolecules, 2020, 5, 569–583 CrossRef.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- S. Shetty, N. Baig, A. Hassan, S. Al-Mousawi, N. Das and B. Alameddine, Microporous Mesoporous Mater., 2020, 303, 110256 CrossRef CAS.
- H. Hong, Z. Guo, D. Yan and H. Zhan, Microporous Mesoporous Mater., 2020, 294, 109870 CrossRef CAS.
- Z. Zhu, J. Zhu, J. Li and X. Ma, Macromolecules, 2020, 53, 1573–1584 CrossRef CAS.
- M. A. Abdulhamid, H. W. H. Lai, Y. Wang, Z. Jin, Y. C. Teo, X. Ma, I. Pinnau and Y. Xia, Chem. Mater., 2019, 31, 1767–1774 CrossRef CAS.
- S. Neyertz and D. Brown, Macromolecules, 2018, 51, 7077–7092 CrossRef CAS.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesana-Gandara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–937 CrossRef CAS PubMed.
- M. Galizia, W. S. Chi, Z. P. Smith, T. C. Merkel, R. W. Baker and B. D. Freeman, Macromolecules, 2017, 50, 7809–7843 CrossRef CAS.
- M. D. Waseem Hussain, S. Bandyopadhyay and A. Patra, Chem. Commun., 2017, 53, 10576–10579 RSC.
- N. Miyaura, T. Yanagai and A. Suzuki, Synth. Commun., 1981, 11, 513–519 CrossRef CAS.
- S. Takahashi, Y. Kuroyama, K. Sonogashira and N. Hagihara, Synthesis, 1980, 627–629 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, React. Funct. Polym., 2019, 139, 153–161 CrossRef CAS.
- M. Schilz and H. Plenio, J. Org. Chem., 2012, 77, 2798–2807 CrossRef CAS PubMed.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed.
- H. Plenio, Angew. Chem., Int. Ed., 2008, 47, 6954–6956 CrossRef CAS PubMed.
- I. N. Denisenko, O. A. Varzatskii, R. A. Selin, A. S. Belov, E. G. Lebed, A. V. Vologzhanina, Y. V. Zubavichus and Y. Z. Voloshin, RSC Adv., 2018, 8, 13578–13587 RSC.
- O. A. Varzatskii, S. V. Shul’ga, A. S. Belov, V. V. Novikov, A. V. Dolganov, A. V. Vologzhanin and Y. Z. Voloshin, Dalton Trans., 2014, 43, 17934–17948 RSC.
- M. D. Wise, A. Ruggi, M. Pascu, R. Scopelliti and K. Severin, Chem. Sci., 2013, 4, 1658–1662 RSC.
- R. O. Selin, I. Klemt, V. Y. Chernii, M. Y. Losytskyy, S. Chernii, A. Mular, E. Gumienna-Kontecka, V. B. Kovalska, Y. Z. Voloshin, A. V. Vologzhanina, P. V. Dorovatovskii and A. Mokhir, RSC Adv., 2021, 11, 8163–8177 RSC.
- V. Kovalska, S. Vakarov, M. Losytskyy, M. Kuperman, N. Chornenka, Y. Toporivska, E. Gumienna-Kontecka, Y. Voloshin, O. Varzatskii and A. Mokhir, RSC Adv., 2019, 9, 24218–24230 RSC.
- G. E. Zelinskii, A. A. Pavlov, A. S. Belov, I. G. Belaya, A. V. Vologzhanina, Y. V. Nelyubina, N. N. Efimov, Y. V. Zubavichus, Y. N. Bubnov, V. V. Novikov and Y. Z. Voloshin, ACS Omega, 2017, 2, 6852–6862 CrossRef CAS PubMed.
- S. El Ghachtouli, M. Fournier, S. Cherdo, R. Guillot, M.-F. Charlot, E. Anxolabéhère-Mallart, M. Robert and A. Aukauloo, J. Phys. Chem. C, 2013, 117, 17073–17077 CrossRef CAS.
- S. M. Jansze and K. Severin, Acc. Chem. Res., 2018, 51, 2139–2147 CrossRef CAS PubMed.
- B. Alameddine, S. Shetty, R. S. Anju, F. Al-Sagheer and S. Al-Mousawi, Eur. Polym. J., 2017, 95, 566–574 CrossRef CAS.
- O. M. Planes, P. A. Schouwink, J. L. Bila, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Cryst. Growth Des., 2020, 20, 1394–1399 CrossRef CAS.
- G. E. Zelinskii, S. V. Dudkin, A. S. Chuprin, A. A. Pavlov, A. V. Vologzhanina, E. G. Lebed, Y. V. Zubavichus and Y. Z. Voloshin, Inorg. Chim. Acta, 2017, 463, 29–35 CrossRef CAS.
- G. Cecot, M. Marmier, S. Geremia, R. D. Zorzi, A. V. Vologzhanina, P. Pattison, E. Solari, F. F. Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2017, 39, 8371–8381 CrossRef PubMed.
- G. Cecot, B. Alameddine, S. Prior, R. D. Zorzi, S. Geremia, R. Scopelliti, F. T. Fadaei, E. Solari and K. Severin, Chem. Commun., 2016, 52, 11243–11246 RSC.
- J. L. Bila, J. Pijeat, A. Ramorini, F. Fadaei-Tirani, R. Scopelliti, E. Baudat and K. Severin, Dalton Trans., 2019, 48, 4582–4588 RSC.
- N. Baig, S. Shetty, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, Polymer, 2018, 151, 164–170 CrossRef CAS.
- B. Alameddine, S. Shetty, N. Baig, S. Al-Mousawi and F. Al-Sagheer, Polymer, 2017, 122, 200–207 CrossRef CAS.
- Z. Zhu and T. M. Swager, Org. Lett., 2001, 3, 3471–3474 CrossRef CAS PubMed.
- M. Morel, C. Aguilera, V. Jiménez, L. Basáez, P. Gebhardt and S. Heggie, Liq. Cryst., 2012, 39, 847–856 CrossRef CAS.
- B. Alameddine, N. Baig, S. Shetty, S. Al-Mousawi and F. Al-Sagheer, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 931–937 CrossRef CAS.
- J. C. Sanchez and W. C. Trogler, J. Mater. Chem., 2008, 18, 3143–3156 RSC.
- S. Shetty, N. Baig, S. Al-Mousawi, F. Al-Sagheer and B. Alameddine, Polymer, 2019, 178, 121606 CrossRef CAS.
- S. Peng, S. Yan, N. Wang, W. Nan, J. Wang, X. Chen, C. Wang, X. Qi and S. Dai, RSC Adv., 2018, 8, 12701–12707 RSC.
- A. Velamakanni, K. J. Ganesh, Y. Zhu, P. J. Ferreira and R. S. Ruoff, Adv. Funct. Mater., 2009, 19, 3926–3933 CrossRef CAS.
- Y.-C. Lin, J. Hong, C.-N. Yen, S.-Y. Tong, M.-J. Tung, H.-W. Shiu, C.-H. Chen and M.-T. Lin, Jpn. J. Appl. Phys., 2015, 54, 033002 CrossRef CAS.
- N. Baig, S. Shetty, S. Al-Mousawi and B. Alameddine, Polym. Chem., 2020, 11, 3066–3074 RSC.
- Y. H. Abdelmoaty, T.-D. Tessema, F. A. Choudhury, O. M. El-Kadri and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2018, 10, 16049–16058 CrossRef CAS PubMed.
- M. Ansari, A. Alam, R. Bera, A. Hassan, S. Goswami and N. Das, J. Environ. Chem. Eng., 2020, 8, 103558 CrossRef CAS.
- H. Guan, D. Zou, H. Yu, M. Liu, Z. Liu, W. Sun, F. Xu and Y. Li, Frontiers in Materials, 2019, 6, 12 CrossRef.
- D. Shetty, T. Skorjanc, J. Raya, S. K. Sharma, I. Jahovic, K. Polychronopoulou, Z. Asfari, D. S. Han, S. Dewage, J.-C. Olsen, R. Jagannathan, S. Kirmizialtin and A. Trabolsi, ACS Appl. Mater. Interfaces, 2018, 10, 17359–17365 CrossRef CAS PubMed.
- D. Chen, Y. Fu, W. Yu, G. Yu and C. Pan, Chem. Eng. J., 2018, 334, 900–906 CrossRef CAS.
- M. Liu, C. Yao, C. Liu and Y. Xu, Sci. Rep., 2018, 8, 14071 CrossRef PubMed.
- T. Geng, W. Zhang, Z. Zhu, G. Chen, L. Ma, S. Ye and Q. Niu, Polym. Chem., 2018, 9, 777–784 RSC.
- G. Li, C. Yao, J. Wang and Y. Xu, Sci. Rep., 2017, 7, 13972 CrossRef PubMed.
- X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- K. C. Park, J. Cho and C. Y. Lee, RSC Adv., 2016, 6, 75478–75481 RSC.
- R. M. Weiss, A. L. Short and T. Y. Meyer, ACS Macro Lett., 2015, 4, 1039–1043 CrossRef CAS.
- L. Wang, P. Chen, X. Dong, W. Zhang, S. Zhao, S. Xiao and Y. Ouyang, RSC Adv., 2020, 10, 44679–44687 RSC.
- M. Shahnawaz Khan, M. Khalid and M. Shahid, Adv. Mater., 2020, 1, 1575–1601 RSC.
- M. Saghian, S. Dehghanpour and M. Sharbatdaran, RSC Adv., 2020, 10, 9369–9377 RSC.
- A. Ahmad, S. Mohd-Setapar, S. C. Chuo, A. Khatoon, W. Wani, R. Kumar and M. Rafatullah, RSC Adv., 2015, 5, 30801–30818 RSC.
- N. Baig, S. Shetty, S. Al-Mousawi and B. Alameddine, Polym. Chem., 2021 10.1039/D1PY00193K.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02357h |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Saleh Al-Mousawid,
Neeladri Das
c,
Saleh Al-Mousawid,
Neeladri Das