
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir activation of charcoal monoliths for capacitive energy storage†
Yu Ma ab,
Hanqin Lianga,
Jinwei Yina,
Dongxu Yaoa,
Yongfeng Xiaa,
Kaihui Zuoa and
Yu-Ping Zeng*a
ab,
Hanqin Lianga,
Jinwei Yina,
Dongxu Yaoa,
Yongfeng Xiaa,
Kaihui Zuoa and
Yu-Ping Zeng*a
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China. E-mail: yuping-zeng@mail.sic.ac.cn; my@student.sic.ac.cn; hqliang@mail.sic.ac.cn; yinjinwei@mail.sic.ac.cn; yaodongxu@mail.sic.ac.cn; yfxia@mail.sic.ac.cn; zuokh@mail.sic.ac.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
First published on 22nd April 2021
Abstract
Charcoal monoliths derived from waste wood were activated with air for the application of electrochemical capacitor electrodes and an insight was given into the activation mechanism. The mild air activation is effective and pollution-free compared to the common chemical activation using KOH etc. for the preparation of crack-free carbon monoliths. The activation process was controlled by altering the activation temperature and time, and their effects on the nanostructure of charcoal monoliths were studied. As the activation temperature or time increased, air eroded the defective surface of charcoal layer-by-layer, with the oxygen atoms being introduced by chemisorption and oxidation reactions and removed by dehydration and decomposition reactions. Meanwhile, micro-pores were produced. The electrode activated at 300 °C for 1 h, with a specific surface area of 567 m2 g−1 and a high micro-porosity of 86%, exhibited a specific capacitance of 203 F g−1 and 35.5 F cm−3. Moreover, it presented a higher total capacitance of 3.6 F cm−2 than most reported pellet electrodes. These findings give a reasonable picture of the air activation process and are instructive to prepare activated carbon monoliths under an oxidizing environment.
1. Introduction
The concept of sustainable development for dealing with the problem of declining fossil fuels across the world spawns ongoing research on renewable energy resources, environmentally friendly materials and green applied technologies.1–3 Electrochemical energy as one form of renewable energy resource has gained more and more attention in order to relieve the increasingly intense energy and environmental crisis.4 There are mainly three electrochemical energy conversion/storage technologies, including electrochemical capacitors (i.e., supercapacitors or ultracapacitors), batteries and fuel cells.4 Compared to the other two models, electric double layer capacitors store the energy through electrostatic adsorption charge and thus feature a high-power output and superlong cycle life, which are especially applicable to rail transportation and renewable energy production (electric power grid).5 In pseudocapacitors, which are made of metal oxide or conducting polymer electrodes, a high amount of pseudocapacitance can be introduced by faradaic electron charge-transfer with redox reactions, intercalation or electrosorption.6,7 Electrode materials, the key component in the construction of capacitors, are also highly required from sustainable renewable precursors and green fabrication technology.8Many carbon-based materials have been investigated as electrode materials, such as modified carbon materials, surface modification of carbonaceous materials, mesoporous carbon nanofibers, hierarchical nanostructured carbons with meso-macroporosity and biomass-derived carbon.9–12 Biomass-derived carbon is the most used electrode materials in capacitors, Li ion batteries etc., owing to the high surface area, rich surface functional groups and renewability.2,4,13 The commercial carbon electrodes derived from coconut shell etc. are common in the form of pellets, which suffer from a limited thickness with a mass loading of <10 mg cm−2 because of some introduced binders.14,15 In fact, thicker electrodes possess higher total capacitance.16 Ultra-thick wood carbon monoliths with hierarchically porous structure arouse increasing researchers' interest in recent years, due to their directional macro-porous channels derived from wood which provide an unobstructed pathway for charge transport.3
The carbon directly pyrolyzed from biowaste (e.g., rice husk, nut shell) and wood is called biochar and charcoal, respectively. They are known as activated carbon when an activation process is applied.17 Generally, two methods, including chemical activation and physical activation, are applied to prepare activated carbon materials.18 Chemicals (such as potassium hydroxide, phosphoric acid, zinc chloride) introduced in the chemical activation method are harmful to the environment. In contrast, physical activation by carbon dioxide, air or steam although taking a long time is recognized in the view of little environmental impacts. The air, an available anywhere activation agent, is inevitable or introduced on purpose in the preparation of activated carbon, but with a problem of high “burn off” or a risk of combustion.19,20 However, it is proposed that the pore structure and surface chemical properties of carbon materials can be improved as long as the activation temperature and time are well controlled to avoid combustion.21 For example, Xuan et al.22 enhanced electrochemical performance of the air-activated carbon spheres via selecting the activation temperature as 900 °C and time as 6 h in a muffle furnace. Despite that lots of works have been done to confirm the flexibility of air activation to prepare activated carbon powders,23–25 few reports about production of activated carbon monoliths with air activation have been done. Even though the ultrathick activated wood-carbon is obtained by air activation at 450 °C for supercapacitor electrodes,26 the detailed information about activation effects on the properties of charcoal monoliths is far from clear.
In this paper, cotton rose wood, with a unique hierarchical macro-meso-microporous structure,27 was selected as the raw materials to prepare charcoal monoliths. Subsequently, a simple and mild air activation method was adopted to prepare activated carbon monoliths for the application of electrochemical capacitor electrodes. The effects of activation temperature and time on the physicochemical properties and pore structure were elaborated, in order to clarify the air activation mechanism and improve the electrochemical properties of the electrodes. Finally, the electrochemical performance is evaluated using the obtained activated carbon electrodes.
2. Materials and methods
2.1. Materials
Cotton rose wood was selected in the field, Jiangxi Province, China. Hydrochloric acid (HCl, AR) and potassium hydroxide (KOH, AR) were purchased from Sinopharm Chemical Regent Co., Ltd., China.2.2. Preparation of activated carbon monoliths
After natural drying, the wood was cut into desired thickness (2–3 mm) along radial direction and then dried overnight at 80 °C. The dried wood was pyrolyzed at 800 °C with a 5 °C min−1 heating rate for 3 h under vacuum conditions with the pressure at 5–20 Pa. After cooling down, the obtained charcoal monoliths were polished to a certain size (1 cm × 1 cm × 1 mm) with a piece of metallographic abrasive paper and polished charcoal monoliths were ultrasonic cleaned for 30 min to remove attached powders. Finally, the samples were purified by 2 mol L−1 HCl solution at 60 °C for 3 h and washed by distilled water until pH 7. The activation treatment for the cleaned charcoal monoliths were carried out at 300 °C, 350 °C and 400 °C for 1 h in a muffle furnace with an air environment. Above 400 °C, cracks generated in the activated carbon monoliths resulting of severe shrinkage. To study the effect of activation time, the charcoal monoliths were activated at 300 °C for 0.5 h to 10 h in the muffle furnace.2.3. Characterizations
The degree of disorder of these samples was evaluated by Raman spectrum (inVia) using a 532 nm laser. The surface functional groups were detected by Fourier transformed infrared spectroscopy (FTIR, Spotlight400). The microstructure was observed using a field emission scanning electron microscopy (FE-SEM, SU8220). The pore structure was characterized by N2 adsorption/desorption method at 77 K (DEMO TriStar II 3020 V1.0). Before the test, the activated carbon monolith was grounded and passed through a 60-mesh screen. The specific surface area (SBET) indicated the Brunauer Emmett Teller (BET) surface area. Total pore volume (V) was the N2 adsorption capacity at relative pressure (P/P0) of 0.98 and micropore volume (Vmicro) was calculated using t-plot method. The microporosity (P) was the ratio of (Vmicro/V) × 100%. Pore size (d) was the adsorption average pore diameter (4 V/SBET by BET). To analyze the elemental compositions and distinguish the chemical state of oxygen, X-ray photoelectron spectroscopy (XPS) analysis was carried out on an ESCAlab250 spectrometer for both the outer and inner (etching 10 s with Ar+) surface of the activated carbon monoliths. In addition, bulk density (ρ) was the average ratio of weight to volume of at least five electrodes. The carbon loss indicated the weight loss caused by activation of the charcoal monolith.The electrochemical performance was evaluated on a CHI 660E electrochemical workstation (Shanghai Chenhua, China) in a three-electrode setup. The activated carbon monoliths were directly used as the working electrodes, with a mass loading (m) of about 18 mg cm−2. A platinum plate and Hg/HgO electrode were applied as the counter and reference electrode, respectively. KOH solution (2 mol L−1) was selected as the electrolyte. The specific capacitance Cg (F g−1) was calculated according to the formula  , where I (A) denotes the galvanostatic discharge current in the GCD measurement, Δt (s) denotes the discharge time, M (g) is the mass of the working electrode, and ΔV (V) is the potential window excluding the IR drop. The areal specific capacitance (Cs, μF cm−2) was evaluated from
, where I (A) denotes the galvanostatic discharge current in the GCD measurement, Δt (s) denotes the discharge time, M (g) is the mass of the working electrode, and ΔV (V) is the potential window excluding the IR drop. The areal specific capacitance (Cs, μF cm−2) was evaluated from  The total capacitance (C, F cm−2) was obtained by the formula: C = m × Cg. The volumetric specific capacitance (Cv, F cm−3) was the result of multiplying Cg by ρ.
The total capacitance (C, F cm−2) was obtained by the formula: C = m × Cg. The volumetric specific capacitance (Cv, F cm−3) was the result of multiplying Cg by ρ.
3. Results and discussion
3.1. The effects of activation temperature
The Raman spectra of the samples activated at 300 °C, 350 °C and 400 °C in the air, as presented in Fig. 1a (left), are typical patterns of partial graphitized carbon with D band (sp3 carbon) at ∼1350 cm−1 and G band (sp2 carbon) at ∼1590 cm−1. The degree of disorder is estimated by the ratio of the intensity of D band to G band (ID/IG).28 As observed in Fig. 1a (right), the plot of ID/IG ratio vs. increasing activation temperature exhibits a downward trend overall. The sample at 0 °C indicates the non-activation charcoal monolith and the value was obtained from our previous work.29 We proposed that improved graphitization is mainly caused by the removal of tarry gradients. In detail, the ratios drop sharply after activation at 300 °C, increase subsequently when the temperature rises up to 350 °C, and again decrease at 400 °C. It is believed to be related to the migration process of the introduced oxygen atoms during activation in air, which is further verified by FTIR analysis. | ||
| Fig. 1 (a) Raman spectra (left) and the variation of ID/IG ratio vs. activation temperature (right), (b) FTIR spectra, (c) survey XPS spectra and (d) deconvolved O 1s spectra for the charcoal monoliths activated at different temperatures. | ||
The form of introduced oxygen is mainly reflected on the surface functional groups, as revealed in Fig. 1b. For all samples, the appearance of broad band at 3200–3700 cm−1 is contributed to the O–H bond vibration in hydroxylic groups and chemisorbed water,30 the intensity of which declines with the increase of activation temperature. Moreover, alcoholic C–O bond (1050 cm−1 and 1090 cm−1) and C–H bond (881 cm−1) from out of plane deformation vibrations in benzene derivatives disappear above 300 °C.31,32 By contrast, the intensity of the bonds at 1627 cm−1 (quinone or conjugated ketone groups), 1750 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration in anhydride, lactone and ketene), and the accompanied broad peak at 1500–1250 cm−1 (C–O stretching and O–H bending modes in ether, lactone, carboxyl, phenolic structure, etc.) enhance.30,33,34 The following reactions caused by the activation are deduced according to the above results: (1) dehydration reactions between hydroxyl groups, carboxyl groups or hydroxyl and carboxyl groups; (2) oxidation reactions that aromatic compounds (e.g., benzene derivatives, alcohol organics) turn to be quinone or ketone species and further to be CO2 or CO gases.29 In addition, the peaks at 2880 (or 2850) cm−1 and 2975 (or 2925) cm−1 that correspond to C–H stretching vibrations in alkane and alkene groups of aliphatic species weaken at 350 °C and disappear at 400 °C, which indicate that some small organic molecules were gradually removed.14,35 In summary, the migration of oxygen atoms referring to the way of introduction or elimination is involved in these reactions.
O vibration in anhydride, lactone and ketene), and the accompanied broad peak at 1500–1250 cm−1 (C–O stretching and O–H bending modes in ether, lactone, carboxyl, phenolic structure, etc.) enhance.30,33,34 The following reactions caused by the activation are deduced according to the above results: (1) dehydration reactions between hydroxyl groups, carboxyl groups or hydroxyl and carboxyl groups; (2) oxidation reactions that aromatic compounds (e.g., benzene derivatives, alcohol organics) turn to be quinone or ketone species and further to be CO2 or CO gases.29 In addition, the peaks at 2880 (or 2850) cm−1 and 2975 (or 2925) cm−1 that correspond to C–H stretching vibrations in alkane and alkene groups of aliphatic species weaken at 350 °C and disappear at 400 °C, which indicate that some small organic molecules were gradually removed.14,35 In summary, the migration of oxygen atoms referring to the way of introduction or elimination is involved in these reactions.
Furthermore, chemical state of the introduced oxygen in the activated carbon was investigated by XPS analysis. The peaks of carbon and oxygen dominate the XPS survey spectra (Fig. 1c). Their contents of the outer and inner surface of the activated carbon monoliths are listed in Table 1. The trend of oxygen contents vs. activation temperature is similar to that of ID/IG ratios as shown in Fig. 1a (right). It is demonstrated that the degree of disorder of the activated carbon is directly related to the oxygen content. Subsequently, the O 1s spectra are deconvoluted to identify the chemical status of oxygen atoms, as shown in Fig. 1d. The peak at ∼531.3 eV indicates the existence of CO bond in carbonyl or quinone groups and the peak at ∼533.3 eV refers to C–O bond in esters, ether, phenol or hydroxyls.30,36–38 The peaks at above 534 eV are assigned to chemisorbed oxygen (–COOH) or water molecular.30 The contribution of cover area of each peak is listed in Table 1. On one hand, the content of chemisorbed O increases with the increase of activation temperature, which indicates the generation of more and more new surface. On the other hand, the contents of CO groups decease rapidly because high temperature promotes their decomposition to form CO2 gases.39 In comparison, the C–O groups change slightly because they are quite stable. In summary, chemisorbed oxygen (e.g., carboxyl groups) that is the most sensitive to activation temperature is the initial form of introduced oxygen whose content changes quickly with the activation temperature. Subsequently, oxidation and dehydration reactions occur accompanying with the generation of ether and ester species as these groups are more stable than hydroxyl groups and carboxylic acid at 300–450 °C.39 At last, these formed groups decompose to CO2 and/or CO gases and new pores generate simultaneously, which is confirmed by the variation of carbon loss from about 3 wt% both at 300 °C and 350 °C to 13 wt% at 400 °C. In addition, the enhanced O2 diffusion along the defect sites initially also promotes the formation of pores in the oxidation process.
| Conditions (°C–h) | Etched surface | Outer surface | Outer surface O 1s | ||||
|---|---|---|---|---|---|---|---|
| C (at%) | O (at%) | C (at%) | O (at%) | CO (%) |
C–O (%) | Chemisorbed oxygen (%) | |
| 300–0.5 | 92.35 | 6.44 | 89.03 | 9.81 | 50.2 | 38.9 | 10.8 |
| 300–1 | 91.42 | 7.17 | 86.59 | 12 | 53.7 | 41.4 | 5.0 |
| 300–2 | 92.56 | 6.48 | 87.46 | 11.47 | 44.8 | 43.5 | 11.7 |
| 300–3 | 92.11 | 6.68 | 86.87 | 11.85 | 49.2 | 45.1 | 5.8 |
| 350–1 | 89.28 | 9.81 | 85.13 | 14.03 | 49.5 | 40.2 | 10.3 |
| 400–1 | 89.46 | 9.62 | 85.81 | 13.32 | 39.2 | 43.5 | 17.3 |
Fig. 2 and S3† display the SEM micrographs of the channel surface at various activation temperatures and the activated carbon monolith (activated at 300 °C for 1 h), respectively. The activated carbon monoliths exhibit a directional hierarchical porous structure with a honey-combed configuration in the transverse section (Fig. S3a†) and abreast channels with many interconnected pores in the vertical section (Fig. S3b†). Fig. 2a shows the cell surface of charcoal monolith obtained from vacuum pyrolysis without activation. The bulges generate as a result of the rapid removal of volatiles from defective sites. By contrast, bulges evolve to be pits in Fig. 2b–d and these pits become larger and deeper with gradually rising activation temperature. Furthermore, much more secondary pits are observed on the surface of those initial pits, as shown in Fig. 2c and d. The O2 diffuses along the defect locations of the charcoal surface accompanying with the activation process proceeding layer by layer along the defect edge, which is consistent with the reported “reaming” effect by Xiao et al.40 This explains why the oxygen content contained in the activated carbon increases and decreases repeatedly. The surface tends to absorb oxygen when new surface exposes and excludes oxygen through dehydration and decomposition reactions when the chemisorbed oxygen is saturable.
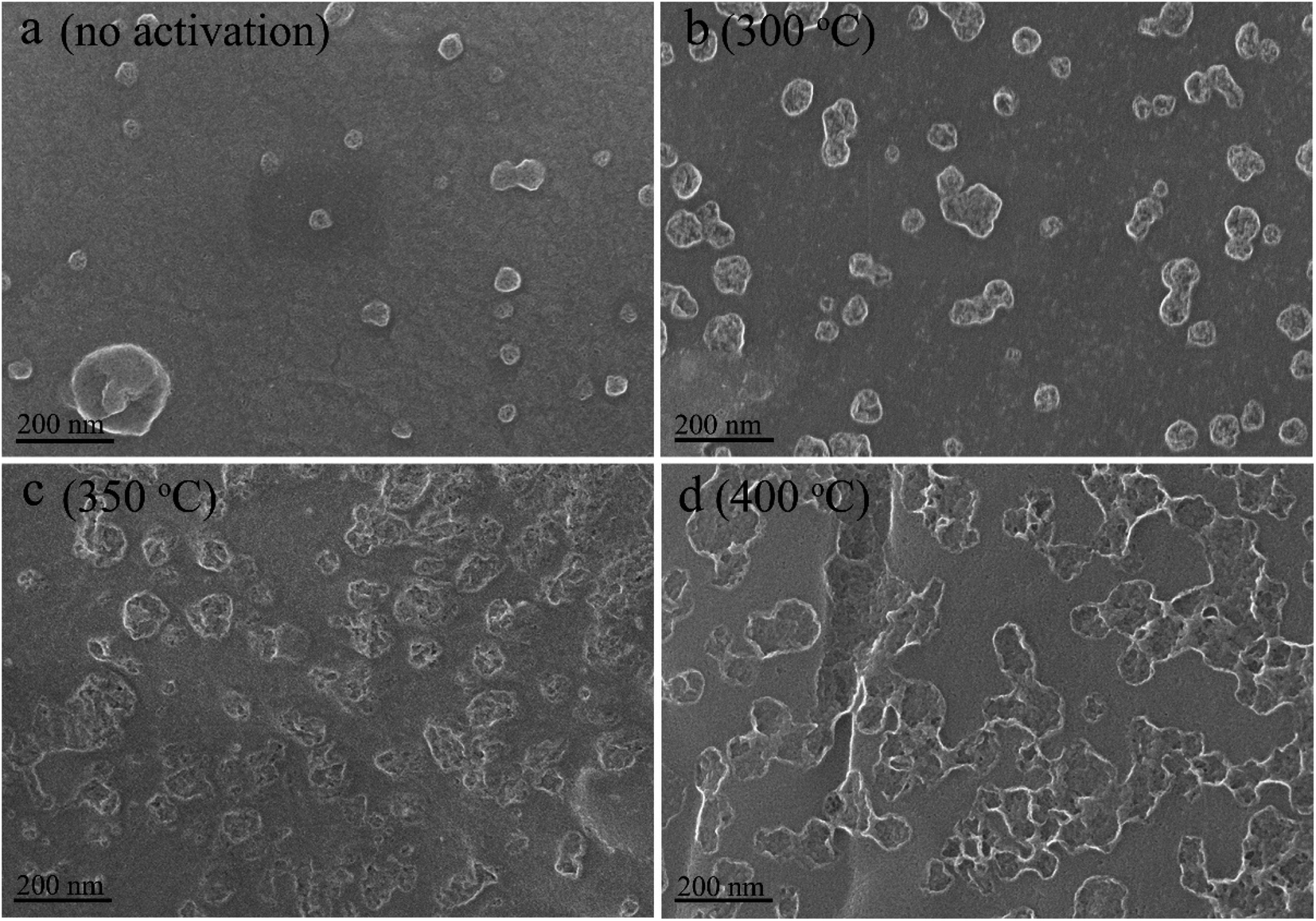 | ||
| Fig. 2 SEM micrographs of cell surface of the charcoal monoliths activated at (a) none, (b) 300 °C, (c) 350 °C, and (d) 400 °C. | ||
The N2 adsorption/desorption isotherms and pore size distributions are shown in Fig. 3. The samples activated at 300 °C and 350 °C exhibit a type I N2 adsorption/desorption isotherms with a steep adsorption of N2 at the very low relative pressure and a weak capillary condensation at very high relative pressure (Fig. 3a), which correspond to microporous dominated structure.41 The sample activated at 350 °C shows a higher adsorption capacity of N2, possessing a specific surface area of 601 m2 g−1 and a pore volume of 0.25 cm3 g−1, which is higher than that of the sample activated at 300 °C (Table 2). Additionally, the micro-porosity for both of them are above 80%. When the activation temperature rises up to 400 °C, the N2 adsorption/desorption isotherms turn to be type IV with a hysteresis loop at the relative pressure of above 0.4, indicating a mesoporous structure.42 The specific surface area and the micro-porosity are 469 m2 g−1 and 64%, respectively. As shown in Fig. 3b, the obtained activated carbon all present a wide pore size distribution. The pore size distribution for the samples activated at 300 °C and 350 °C is alike in the mesopore range, except for a slight increase in the range of 2–3 nm for the latter. However, the quantity of macropores increase evidently for the sample activated at 350 °C. The sample activated at 400 °C features richer mesopores, especially the pores with a diameter of smaller than 10 nm, but the macropores are not salient. This demonstrates that micropores are etched to be meso-macropores and then micropores form again on the surface of the previous meso-macropores with increasing temperature, in agreement with above mentioned description about the air activation processes.
 | ||
| Fig. 3 (a) N2 adsorption/desorption isotherms and (b) pore size distribution for the charcoal monoliths activated at different temperatures. | ||
| Conditions (°C–h) | ρ (g cm−3) | SBET (m2 g−1) | V (cm3 g−1) | P (%) | d (nm) |
|---|---|---|---|---|---|
| 300–0 | 0.182 ± 0.005 | 226 | 0.12 | 68 | 2.15 |
| 300–0.5 | 0.180 ± 0.001 | 493 | 0.20 | 84 | 1.63 |
| 300–1 | 0.175 ± 0.003 | 567 | 0.23 | 86 | 1.61 |
| 300–2 | 0.180 ± 0.004 | 565 | 0.23 | 85 | 1.61 |
| 300–3 | 0.183 ± 0.004 | 617 | 0.25 | 84 | 1.63 |
| 350–1 | 0.171 ± 0.001 | 601 | 0.25 | 84 | 1.67 |
| 400–1 | 0.169 ± 0.002 | 469 | 0.22 | 64 | 1.84 |
3.2. The effects of activation time
Raman spectra of the samples activated at 300 °C for different time are presented in Fig. 4a (left) and S1,† which display that these samples are all partial graphitized. The ratios for ID/IG experience a process of repeated rise and fall from 0.5 h to 10 h, but a downward trend on the whole (Fig. 4a, right). As demonstrated above, the ratio has a direct relationship with the oxygen content contained in the activated carbon. Therefore, the activation time on the activated carbon have similar effect with activation temperature that induces a dynamic oxygen migration process. | ||
| Fig. 4 (a) Raman spectra (left) and the variation of ID/IG ratio vs. activation time (right), (b) FTIR spectra, (c) deconvolved O 1s spectra for the charcoal monoliths activated for different time. | ||
Fig. 4b presents the FTIR spectra of the samples activated for various time. As observed, the bond intensity of hydrogen groups at 3200–3700 cm−1 decreases obviously with the gradually extended activation time. In addition, those peaks at 2880 cm−1, 2975 cm−1, 1050 cm−1, 1090 cm−1 and 881 cm−1 disappear until for 3 h, indicating that the organic molecules, adhering to carbon skeleton, are removed or oxidized completely. The peak at 1750 cm−1 (CO vibration in lactone etc.) generates after activation for 1 h and continuously enhances as the activation time is prolonged to 3 h. By contrast, the bond at 1627 cm−1 derived from quinone species always exists, denoting that the dehydration reaction between carboxyl groups is, to some extent, more difficult to occur than the oxidation reactions.
The XPS analyzing results of the carbon and oxygen contents contained in the activated carbon monoliths are listed in Fig. S2† and Table 1. As expected, changes of oxygen content are consistent with the changes of ID/IG ratio. It is confirmed that oxygen content directly influences the degree of disorder of the activated carbon. Further, high-resolution O 1s XPS spectra are shown in Fig. 4c and the contents of various forms of oxygen are listed in Table 1. The results indicate that the introduced oxygen in the charcoal exists in the forms of C–O, CO and chemisorbed oxygen. The content of chemisorbed oxygen proceeds a periodical process of falling and rising, accompanying with an opposite trend for the contents of CO. Therefore, it is proposed that the chemisorbed oxygen is expected to turn to be carbonyl containing groups (CO) via dehydration reaction. That is, the charcoal surface mainly adsorbed oxygen from the air in a short activation time of 0.5 h, subsequently dehydration reaction between surface functional groups dominated with the activation time extending to 1.0 h (Fig. 4c). Once these unstable CO groups decompose to CO2, new active surface is exposed and chemisorbed reaction to oxygen starts again.
Fig. 5 exhibits the cell surface morphology of activated carbon monoliths with different activation time. Increasing activation time from 0.5 h to 3 h, the number of pits increases with the average size scaling up from 20 μm to 50 μm. Compared with the surface morphologies in Fig. 2, the edges of pits become smoother, indicative of milder activation effect than that of activation temperature. For instance, the pits on the cell surface of the samples activated for a long time (4–10 h, Fig. S4†) is being deeper and larger, similar to the surface morphology of the sample activated at 350 °C (Fig. 2c). Moreover, TEM micrographs confirm that much more micro-mesopores are created on the carbon sheet of activated carbon (Fig. 5e) than the non-activation one (Fig. 5f), as shown in the marking areas by yellow lines.
 | ||
| Fig. 5 SEM micrographs of cell surface for the charcoal monoliths activated for (a) 0.5 h, (b) 1 h, (c) 2 h and (d) 3 h; TEM micrographs of carbon sheets activated for (e) none, (f) 1 h. | ||
Type I N2 adsorption/desorption loop are observed for the samples activated for different time, as shown in Fig. 6. Air activation promotes the specific surface area of the original charcoal from 226 m2 g−1 to 617 m2 g−1 after activation for 0.5 h to 3 h, and upgrades the pore volume from 0.12 cm3 g−1 to 0.25 cm3 g−1 (Table 2). In comparison, the micro-porosity reaches the maximum value of 86% for 1 h, which is related to the variation of pore size. With the increasing of activation time, more and more micropores generate on the surface of pits and then these micropores are enlarged to be meso-macropores, as the description for SEM images (Fig. 5a–d). Notably, there is merely change for pore structure parameters between the samples activated for 1 h and for 2 h. This indicates that the reaction of chemisorbing oxygen from the air dominate this period, with few pores being generated. Nevertheless, this phenomenon does not occur with the increase of activation temperature whose effects are so drastic that the pore structure varies fast even in the process of absorbing oxygen. Additionally, altering the activation time, the bulk density of activated carbon monolith does not change much, but decreases obviously with the increasing of activation temperature. It is also suggestive of the prominent role of activation temperature.
 | ||
| Fig. 6 (a) N2 adsorption/desorption isotherms and (b) pore size distribution for the charcoal monoliths activated for 0.5 to 3 h. | ||
3.3. Mechanism of activation reactions and pore formation
Based on the results mentioned above, there are four types of reactions taking place during the air activation process, including absorbing reactions, oxidation reactions, dehydration reactions and decomposition reactions. The representative reactions are shown in formulas (1)–(4), respectively.| C + O2 + H2O → COOH | (1) |
|
C–OH + O2 → CO + H2O
| (2) |
| COOH + C–OH → COOC + H2O | (3) |
| COOC → CO2 | (4) |
In general, the oxygen in air is introduced by formulas (1) and (2) and removed by formulas (3) and (4). Meanwhile, pores generate on the surface of charcoal. Here, a layer-by-layer model is proposed for the pore forming process, as illustrated in Fig. 7. The defect locations on the surface of charcoal tend to adsorb oxygen at the elevated temperatures. Subsequently, enhanced O2 diffusion and reactions between oxygen related surface functional groups erode the surface carbon to generate pores. When fresh surface is exposed, the process of chemisorbing oxygen dominates again. Furthermore, this layer-by-layer pore forming mechanism is expected to explain the pore forming process by any other oxidizing gases (e.g., CO2, steam).
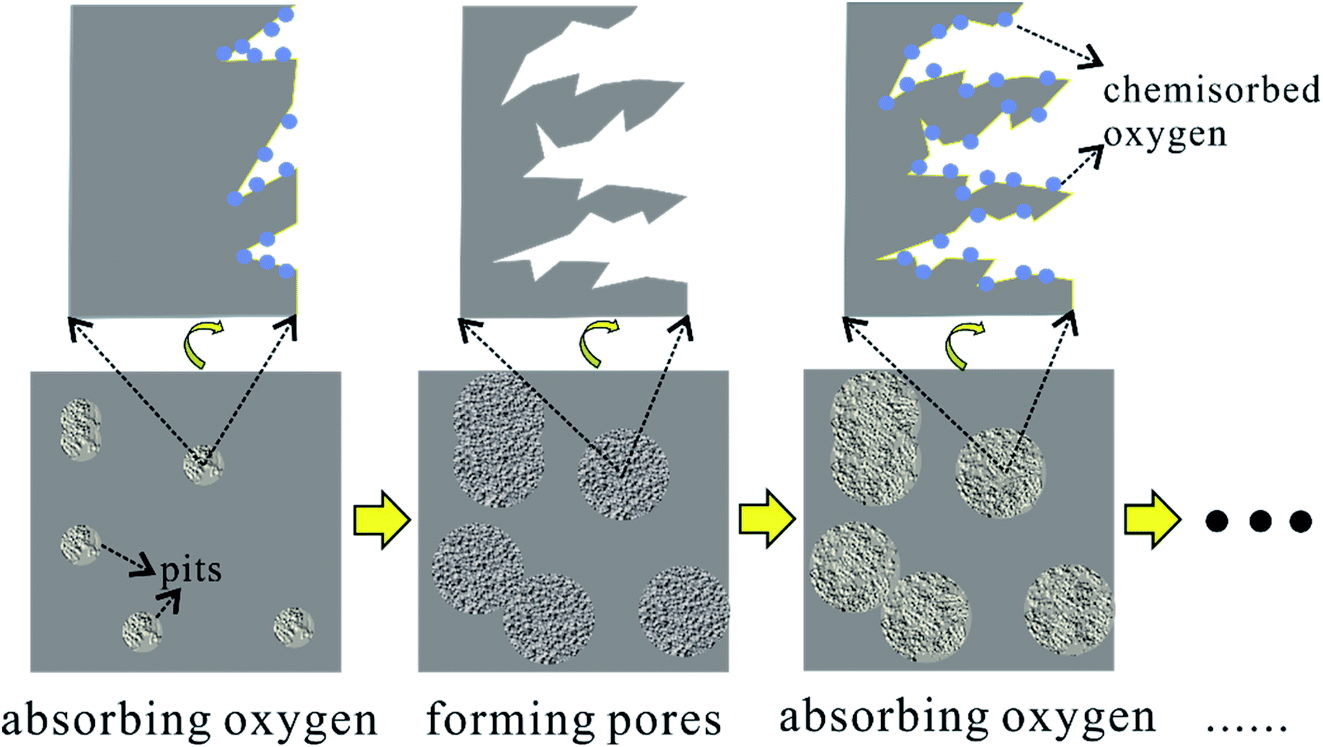 | ||
| Fig. 7 Schematic diagram of the layer-by-layer pore forming mechanism of air activation. | ||
3.4. Evaluation of electrochemical performance
As shown in Fig. 8, the effects of activation temperature on the electrochemical performance were studied by CV, GCD and EIS measurements. The area covered by CV loop denotes the capacitive ability of an electrode. Judging from Fig. 8a, the capacitive performance of electrode gets worse with the increasing of activation temperature. Accordingly, the discharge time of GCD curve (Fig. 8b) becomes shorter at the same current density. Moreover, the IR drop increases, indicating an increasing ion diffusion resistance within the hierarchically porous structure of the electrode.43 The calculated specific capacitances as a function of current density are plotted in Fig. 8c. The specific capacitances are 203 F g−1, 209 F g−1 and 70 F g−1 at 5 mA cm−2 and retain 157 F g−1, 10 F g−1 and 0 F g−1 at 50 mA cm−2 for the samples activated at 300 °C, 350 °C and 400 °C, respectively. Generally, the electrochemical properties deteriorate with the increasing of activation temperature whatever from the point of view of energy density or the rate performance. Only at 5 mA cm−2, the specific capacitance of the sample activated at 350 °C is highest owing to its highest microporosity. | ||
| Fig. 8 The effects of activation temperature on the electrochemical performance. (a) CV curves at 10 mV s−1, (b) GCD plots at 10 mA cm−2, (c) EIS spectra, (d) comparison of specific capacitances. | ||
Furthermore, EIS measurement is performed to study the charge conduction and ion diffusion behaviors through the electrode. The intercept of EIS spectra on Z′ axis denotes the equivalent series resistance (ESR), including the resistance of electrode and electrolyte as well as the contact resistance between them. The diameter of semicircle in the high frequency region is controlled by the charge transfer resistance and the length of Warburg curve with a slope of 45° in the middle frequency region reflects the ion diffusion rate.44 In low frequency region, a steeper line means a better capacitive behavior. As shown in Fig. 8d, the ESR of these electrodes are approximate, but the charge transfer resistance and ion diffusion resistance both increased with the increase of activation temperature, which damage the rate capability. As mentioned above (Fig. 1b), the lack of surface acidic group (carboxyl and hydroxyl) leads to a weak interaction between the activated carbon surface and alkaline electrolyte, since surface acidic groups provide sites that can form exceptionally strong hydrogen bonds with weak acids and bases.21 It appears surface active carboxyl/hydroxyl groups play a much more important role for determining the electrochemical performance, considering that the sample activated at 350 °C has the highest microporosity.
The effects of activation time are not as intense as the activation temperature, as seen from Fig. 9. The covered area by CV loop (Fig. 9a) and the IR drop shown in GCD curves (Fig. 9b) change insignificantly until the activation time increases to 3 h. The EIS spectra in Fig. 9c shows that all of the ESR resistance, the charge transfer resistance and the ion diffusion resistance increase with the prolonged activation time, especially for the sample activated for 3 h. The specific capacitances of all samples at various current density are shown in Fig. 9d. The capacitances at low current densities of 5 mA cm−2 and 10 mA cm−2 present an upward trend with the increase of activation time, but a downward trend at high current density range. Therefore, the activation time has a positive effect on the energy density, but a negative effect on the rate performance. On one hand, the enhanced specific surface area generated by air activation promote the energy density. On the other hand, the loss of surface acidic groups, caused by gradually intensive activation effect, results in the damage of the electrochemical performance of the electrode. Overall, the optimum condition is 300 °C of the activation temperature and 1 h of the activation time. Fig. 9e shows the GCD curves for the sample activated for 1 h at 300 °C from 5 mA cm−2 to 50 mA cm−2. The capacitance increases 18% compared with the non-activation sample.29 The cycle stability is tested at 50 mA cm−2 for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, as plotted in Fig. 9f. The average specific capacitance is 154 F g−1 in the first 2000 cycles and rises to about 180 F g−1 later. After 10000 cycles, the electrode loses 4% (172 F g−1) of the capacitance, exhibiting an excellent cycling stability.
000 cycles, as plotted in Fig. 9f. The average specific capacitance is 154 F g−1 in the first 2000 cycles and rises to about 180 F g−1 later. After 10000 cycles, the electrode loses 4% (172 F g−1) of the capacitance, exhibiting an excellent cycling stability.
 | ||
| Fig. 9 The effects of activation time on the electrochemical performance. (a) CV curves at 10 mV s−1, (b) GCD plots at 10 mA cm−2, (c) EIS spectra, (d) comparison of specific capacitances. (e) GCD plots at various current densities and (f) cycling test at 50 mA cm−2 for 10000 cycles for the electrode activated for 1 h. | ||
To obtain a comprehensive evaluation of the electrode, areal specific capacitance and total capacitance should be considered apart from the gravimetric specific capacitance.45,46 Table 3 lists various capacitances of biomass-derived carbon electrode in pellet or monolith form as well as some polymer-derived monolithic carbon electrodes. The electrodes in pellet form possess a higher gravimetric specific capacitance (200–400 F g−1) than the monolithic electrodes (100–300 F g−1), due to their high specific surface area. However, the areal specific capacitance is only one-third of the capacitance of monolithic electrodes because of the limited accessible surface area by electrolyte in pellet electrodes. The introduced additives in the preparation of pellet electrodes hinder the infiltration of the electrolyte, which further lead to a limited mass loading of the active electrode materials. The total capacitance is thus at a level of <1 F cm−2. Owing to the hierarchically porous structure of monolithic electrode, the total capacitance reaches to a value of >3 F cm−2. For the monolithic electrode, volumetric specific capacitance is another important indicator as the space for power device is usually limited.2 The volumetric specific capacitance of the prepared electrodes in this work is comparable to other reported monolithic electrodes. Given that the air activation method excluding corrosive chemicals and complicated preparation process is mild and simple, the cotton rose-derived carbon electrodes outperform others.
| Raw materials | Activation agents | Cg (F g−1) | Cs (μF cm−2) | Cv (F cm−3) | C (F cm−2) | Test conditions | m (mg cm−2) | Form | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Bamboo | K2FeO4 | 222 | 12.8 | — | 0.7 | 6 M KOH | 3 | Pellet | 47 |
| 0.5 A g−1 | |||||||||
| Cornstalk | K2C2O4 | 337 | 17.6 | — | 0.1 | 1 M H2SO4 | 0.28 | Pellet | 48 |
| 1 A g−1 | |||||||||
| Corn husk | KOH | 356 | 41.1 | — | 0.7 | 6 M KOH | 2 | Pellet | 36 |
| 1 A g−1 | |||||||||
| Celtuce leaves | KOH | 421 | 12.4 | — | 3.4 | 2 M KOH | 8 | Pellet | 49 |
| 0.5 A g−1 | |||||||||
| Wood tar | KOH | 338.5 | 17.8 | — | 1.0 | 6 M KOH | 3 | Pellet | 50 |
| 1 A g−1 | |||||||||
| Mushroom | H3PO4/KOH | 306 | 10.2 | — | 0.6 | 6 M KOH | 2 | Pellet | 43 |
| 1 A g−1 | |||||||||
| Pomelo peel | KOH | 230 | 13.3 | — | — | 2 M KOH | — | Pellet | 51 |
| 1 A g−1 | |||||||||
| Cornstalk | K4[Fe(CN)6] | 213 | 27.0 | — | — | 6 M KOH | 3.75 | Pellet | 52 |
| 1 A g−1 | |||||||||
| Tealeaves | KOH | 330 | 11.6 | — | 2.6 | 2 M KOH | 8 | Pellet | 53 |
| 1 A g−1 | |||||||||
| Coconut shell | ZnCl2/FeCl3 | 268 | 14.3 | — | 0.1 | 6 M KOH | — | Pellet | 54 |
| 1 A g−1 | |||||||||
| Poplar wood | HNO3 | 235 | 56.5 | 35.3 | 3.5 | 2 M KOH | 15 | Monolith | 55 |
| 5 mA cm−2 | |||||||||
| Cedar wood | HNO3 | 115 | 36.3 | — | 1.4 | 5 M H2SO4 | Monolith | 56 | |
| 0.5 A g−1 | |||||||||
| Chinese fir | CO2/KOH/HNO3 | 285.6 | 40.6 | — | — | 1 M Na2SO4 | — | Monolith | 57 |
| 10 mA cm−2 | |||||||||
| Lignin | — | 208.4 | 26.0 | 97.1 | 3 | 6 M KOH | 14.4 | Monolith | 45 |
| 0.1A g−1 | |||||||||
| Phenolic resol | N-doped | 147.9 | — | 21.5 | — | 6 M KOH | — | Monolith | 58 |
| 1 A g−1 | |||||||||
| Resorcinol-formaldehyde | — | 159.6 | — | 28.7 | — | 6 M KOH | — | Monolith | 59 |
| 0.2 A g−1 | |||||||||
| Cotton rose-1 h | Air | 203 | 35.8 | 35.5 | 3.6 | 2 M KOH | 17.5 | Monolith | This work |
| 5 mA cm−2 |
4. Conclusion
Ultra-thick activated carbon monoliths, with a unique hierarchically porous structure and a high micro-porosity, have been prepared by air activation of charcoal, using cotton rose wood as raw materials. The properties of activated carbon monoliths are controlled by the activation temperature and time. Particular attention is paid to give an insight for the activation mechanism. It is demonstrated that the oxygen atoms are introduced by chemisorption and oxidation reactions and removed by dehydration and decomposition reactions through which the active carbon atoms on the surface are consumed layer by layer. Pores are thus produced on the surface of the charcoal channel. The results show that appropriate activation promotes the generating of micro-pores, but excessive activation leads the micropores to be meso-macropores and fast loss of acidic functional groups that are beneficial for absorbing electrolyte. The activated carbon monolith, activated at 300 °C for 1 h, shows the highest micro-porosity of 86% and a specific surface area of 567 m2 g−1. The specific capacitance is 203 F g−1 at 5 mA cm−2 and still remains 157 F g−1 at 50 mA cm−2, with 96% retention of its highest capacitance after 10000 cycles at 50 mA cm−2. Furthermore, a comprehensive evaluation for the properties of electrodes shows that the as obtained activated carbon monolith outperforms other biomass-derived carbon materials considering the accessible available micro-porosity, total capacitance and green preparation method. These findings give a reference for preparing activated carbon monoliths using oxidizing gas for surface activation which is a green and sustainable technology.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2017YFB0406200, 2018YFF01013605), Natural Science Foundation of China (No. 51902327), the Youth Innovation Promotion Association CAS (No. 2019254), Science Foundation for Youth Scholar of State Key Laboratory of High Performance Ceramics and Superfine Microstructures (SKL201701).References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- K. Fic, A. Platek, J. Piwek and E. Frackowiak, Mater. Today, 2018, 21, 437–454 CrossRef CAS.
- J. L. Huang, B. T. Zhao, T. Liu, J. R. Mou, Z. J. Jiang, J. Liu, H. X. Li and M. L. Liu, Adv. Funct. Mater., 2019, 29, 1902255 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- J. R. Miller, J. Power Sources, 2016, 326, 726–735 CrossRef CAS.
- D. Z. Wang, Y. Y. Xiao, X. N. Luo, Z. Z. Wu, Y. J. Wang and B. Fang, ACS Sustainable Chem. Eng., 2017, 5, 2509–2515 CrossRef CAS.
- B. Muthulakshmi, D. Kalpana, S. Pitchumani and N. G. Renganathan, J. Power Sources, 2006, 158, 1533–1537 CrossRef CAS.
- J. Deng, M. M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- B. Fang, Y. Z. Wei and M. Kumagai, J. Power Sources, 2006, 155, 487–491 CrossRef CAS.
- B. Fang, Y. Z. Wei, K. Suzuki and M. Kumagai, Electrochim. Acta, 2005, 50, 3616–3621 CrossRef CAS.
- Y. L. Xing, B. Z. Fang, A. Bonakdarpour, S. C. Zhang and D. P. Wilkinson, Int. J. Hydrogen Energy, 2014, 39, 7859–7867 CrossRef CAS.
- B. Fang, J. H. Kim, M. S. Kim and J. S. Yu, Acc. Chem. Res., 2013, 46, 1397–1406 CrossRef CAS PubMed.
- J. L. Yong, F. Chen, Q. Yang, J. L. Huo and X. Hou, Chem. Soc. Rev., 2017, 46, 4176–4190 RSC.
- D. C. Martinez-Casillas, I. Mascorro-Gutierrez, C. E. Arreola-Ramos, H. I. Villafan-Vidales, C. A. Arancibia-Bulnes, V. H. Ramos-Sanchez and A. K. Cuentas-Gallegos, Carbon, 2019, 148, 403–412 CrossRef CAS.
- H. Q. Gao, D. Zhang, H. T. Zhou, J. C. Wu, G. J. Xu, Z. L. Huang, M. H. Liu, J. H. Yang and D. Chen, Appl. Surf. Sci., 2020, 534, 147613 CrossRef CAS.
- C. J. Chen, Y. Zhang, Y. J. Li, J. Q. Dai, J. W. Song, Y. G. Yao, Y. H. Gong, I. Kierzewski, J. Xie and L. B. Hu, Energy Environ. Sci., 2017, 10, 538–545 RSC.
- J. Marousek, O. Strunecky and V. Stehel, Clean Technol. Environ., 2019, 21, 1389–1395 CrossRef CAS.
- M. Danish and T. Ahmad, Renewable Sustainable Energy Rev., 2018, 87, 1–21 CrossRef CAS.
- K. Tomkow, A. Jankowska, F. Czechowski and T. Siemieniewska, Fuel, 1977, 56, 101–106 CrossRef CAS.
- J. Yin, W. L. Zhang, N. A. Alhebshi, N. Salah and H. N. Alshareef, Small Methods, 2020, 4, 1900853 CrossRef CAS.
- F. Xiao and J. J. Pignatello, Environ. Sci. Technol., 2016, 50, 6276–6283 CrossRef CAS PubMed.
- H. Q. Xuan, Y. L. Wang, G. X. Lin, F. Wang, L. Zhou, X. P. Dong and Z. Chen, RSC Adv., 2016, 6, 15313–15319 RSC.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- R. Bardestani and S. Kaliaguine, Biomass Bioenergy, 2018, 108, 101–112 CrossRef CAS.
- Z. Li, L. Zhang, B. S. Amirkhiz, X. H. Tan, Z. W. Xu, H. L. Wang, B. C. Olsen, C. M. B. Holt and D. Mitlin, Adv. Energy Mater., 2012, 2, 431–437 CrossRef CAS.
- W. J. Zhang, T. Liu, J. R. Mou, J. L. Huang and M. L. Liu, Phys. Chem. Chem. Phys., 2020, 22, 2073–2080 RSC.
- Y. Ma, D. X. Yao, H. Q. Liang, J. W. Yin, Y. F. Xia, K. H. Zuo and Y. P. Zeng, Electrochim. Acta, 2020, 352, 136452 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50–58 CrossRef CAS.
- Y. Ma, Y. S. Li and Y. P. Zeng, J. Mater. Sci., 2021, 56, 8588–8599 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- A. C. Lua and T. Yang, J. Colloid Interface Sci., 2004, 276, 364–372 CrossRef CAS PubMed.
- C. Moreno-Castilla, M. V. Lopez-Ramon and F. Carrasco-Marin, Carbon, 2000, 38, 1995–2001 CrossRef CAS.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Orfao, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- H. Deng, G. X. Li, H. B. Yang, J. P. Tang and J. Y. Tang, Chem. Eng. J., 2010, 163, 373–381 CrossRef CAS.
- K. Wang, N. Zhao, S. W. Lei, R. Yan, X. D. Tian, J. Z. Wang, Y. Song, D. F. Xu, Q. G. Guo and L. Liu, Electrochim. Acta, 2015, 166, 1–11 CrossRef CAS.
- S. J. Song, F. W. Ma, G. Wu, D. Ma, W. D. Geng and J. F. Wan, J. Mater. Chem. A, 2015, 3, 18154–18162 RSC.
- A. F. Perez-Cadenas, F. J. Maldonado-Hodar and C. Moreno-Castilla, Carbon, 2003, 41, 473–478 CrossRef CAS.
- S. Liu, J. Q. Tian, L. Wang, Y. W. Zhang, X. Y. Qin, Y. L. Luo, A. M. Asiri, A. O. Al-Youbi and X. P. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- G. delaPuente, J. J. Pis, J. A. Menendez and P. Grange, J. Anal. Appl. Pyrolysis, 1997, 43, 125–138 CrossRef CAS.
- F. Xiao, A. H. Bedane, J. L. X. J. Zhao, M. D. Mann and J. J. Pignatello, Sci. Total Environ., 2018, 618, 276–283 CrossRef CAS PubMed.
- Z. F. Yu, X. Z. Wang, X. D. Song, Y. Liu and J. S. Qiu, Carbon, 2015, 95, 852–860 CrossRef CAS.
- X. Deng, B. T. Zhao, L. Zhu and Z. P. Shao, Carbon, 2015, 93, 48–58 CrossRef CAS.
- P. Cheng, S. Y. Gao, P. Y. Zang, X. F. Yang, Y. L. Bai, H. Xu, Z. H. Liu and Z. B. Lei, Carbon, 2015, 93, 315–324 CrossRef CAS.
- Y. X. Xu, Z. Y. Lin, X. Zhong, X. Q. Huang, N. O. Weiss, Y. Huang and X. F. Duan, Nat. Commun., 2014, 5, 4554 CrossRef CAS PubMed.
- H. Li, D. Yuan, C. H. Tang, S. X. Wang, J. T. Sun, Z. B. Li, T. Tang, F. K. Wang, H. Gong and C. B. He, Carbon, 2016, 100, 151–157 CrossRef CAS.
- J. Pang, W. F. Zhang, J. L. Zhang, G. P. Cao, M. F. Han and Y. S. Yang, Green Chem., 2017, 19, 3916–3926 RSC.
- Y. N. Gong, D. L. Li, C. Z. Luo, Q. Fu and C. X. Pan, Green Chem., 2017, 19, 4132–4140 RSC.
- J. M. Li, Q. M. Jiang, L. S. Wei, L. X. Zhong and X. Y. Wang, J. Mater. Chem. A, 2020, 8, 1469–1479 RSC.
- R. T. Wang, P. Y. Wang, X. B. Yan, J. W. Lang, C. Peng and Q. J. Xue, ACS Appl. Mater. Interfaces, 2012, 4, 5800–5806 CrossRef CAS PubMed.
- J. Wu, M. W. Xia, X. Zhang, Y. Q. Chen, F. Sun, X. H. Wang, H. P. Yang and H. P. Chen, J. Power Sources, 2020, 455, 227982 CrossRef CAS.
- G. Qu, S. F. Jia, H. Wang, F. Cao, L. Li, C. Qing, D. M. Sun, B. X. Wang, Y. W. Tang and J. B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 20822–20830 CrossRef CAS PubMed.
- L. Wang, G. Mu, C. G. Tian, L. Sun, W. Zhou, P. Yu, J. Yin and H. G. Fu, ChemSusChem, 2013, 6, 880–889 CrossRef CAS PubMed.
- C. Peng, X. B. Yan, R. T. Wang, J. W. Lang, Y. J. Ou and Q. J. Xue, Electrochim. Acta, 2013, 87, 401–408 CrossRef CAS.
- L. Sun, C. G. Tian, M. T. Li, X. Y. Meng, L. Wang, R. H. Wang, J. Yin and H. G. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 RSC.
- M. C. Liu, L. B. Kong, P. Zhang, Y. C. Luo and L. Kang, Electrochim. Acta, 2012, 60, 443–448 CrossRef CAS.
- J. H. Jiang, L. Zhang, X. Y. Wang, N. Holm, K. Rajagopalan, F. L. Chen and S. G. Ma, Electrochim. Acta, 2013, 113, 481–489 CrossRef CAS.
- S. Zhang, C. L. Wu, W. Wu, C. Zhou, Z. W. Xi, Y. Y. Deng, X. Wang, P. Quan, X. J. Li and Y. F. Luo, J. Power Sources, 2019, 424, 1–7 CrossRef CAS.
- Y. R. Liu, J. Porous Mater., 2014, 21, 1009–1014 CrossRef.
- Y. F. Yu, J. Du, L. Liu, G. X. Wang, H. L. Zhang and A. B. Chen, J. Nanopart. Res., 2017, 19, 119 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02192c |
| This journal is © The Royal Society of Chemistry 2021 |