
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChlorophylls in thin-film photovoltaic cells, a critical review
O. I. Koifmana,
P. A. Stuzhin a,
V. V. Travkinab and
G. L. Pakhomov*ab
a,
V. V. Travkinab and
G. L. Pakhomov*ab
aIvanovo State University of Chemistry and Technology (ISUCT), 7 Sheremetievskiy Ave., Ivanovo 153000, Russian Federation
bInstitute for Physics of Microstructures of the Russian Academy of Sciences (IPM RAS), GSP-105, Nizhny Novgorod 603950, Russian Federation. E-mail: pakhomov@ipmras.ru
First published on 22nd April 2021
Abstract
Chlorophyll dyes are widely spread in nature, and some of their semisynthetic analogs are available on the market. It is, therefore, very tempting to use these molecules in artificial photoconverting systems such as solar cells, thus avoiding problems with a labor-intensive synthesis of new photoactive components, their toxicity, biocompatibility and photostability. However, similar to a situation when mimicking a bird's flight does not necessarily lead to creation of a reliable aircraft, the attempts to make photovoltaically effective mimics of, for example, chlorophyll-based photosynthetic antenna have not so far ended up in commercial breakthroughs. This particularly concerns multilayer thin-film cells with planar or bulk molecular heterojunction(s), the type of devices that have been demonstrating a significant growth in power conversion efficiency over the past few decades. In this review we focus on those aspects of optical, self-assembling and bulk electrical properties of chlorophyll-type dyes that are of importance in solid-state photovoltaics. Starting from the very basics and tracing the developments in this field retrospectively, we come to a conclusion that the morphology of thin solution-processed films is a critical problem that arises immediately at the stage of manufacturing of a laboratory prototype. The measures that have been taken to date to improve the mobility of charge carriers both in the chlorophyll aggregates and throughout the photoconductive layer seem insufficient by comparison with their more successful counterparts like phthalocyanines.
1. Introduction
The number of publications in the field of bio-inspired device engineering is growing tremendously. In particular, introduction of biologically important or biocompatible molecules into optoelectronic devices has become a very popular area of science over the past years, attracting the attention of researchers from materials physics, chemistry, engineering and biology. The reader interested in a broader understanding of the state-of-the-art in this field should address the recent reviews.1–3 In the narrow context of this paper, we will focus on the works related to the thin-film photovoltaic cells (PVC), where chlorophyll-like molecules are utilized as active materials – Fig. 1. Activity here means ability of the molecules in the bulk phase to not only absorb photons from sunlight, but also participate in the generation of free charge carriers and then funnel them to the electrodes. This rules out the need to photosensitize, functionalize or mix organic or inorganic matrices with dye molecules and allows us to turn to the purely “excitonic” organic solar cells incorporating planar or bulk heterojunctions (types (a) and (b) in Fig. 2), or their combinations. | ||
| Fig. 1 Examples of the chlorophyll-like molecules utilized in PVC. | ||
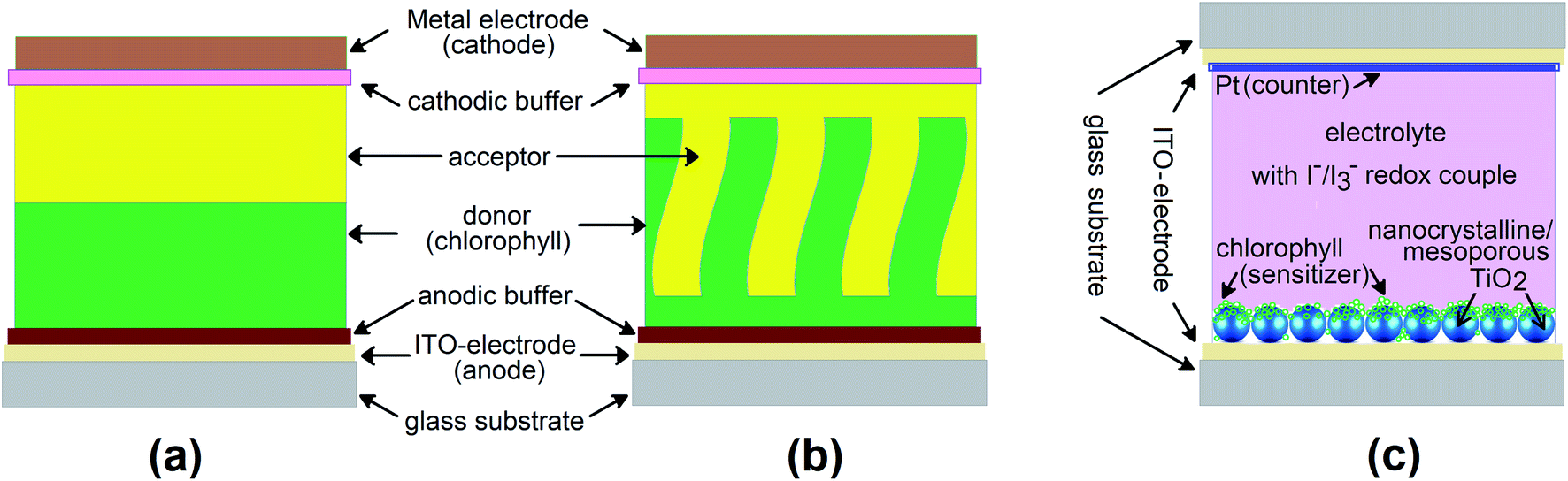 | ||
| Fig. 2 Schematic of photovoltaic devices: (a) thin-film cell with planar heterojunction, PHJ; (b) thin-film cell with bulk (or structured) heterojunction, BHJ; (c) dye-sensitized solar cell, DSSC. | ||
Chlorophylls are the only naturally occurring organic semiconductors. With chlorophylls, the path from a precursor with an almost infinite resource like spinach or spirulina to a solid-state device exhibiting typical photovoltaic characteristics is probably the shortest and lightest in modern optoelectronics.4–6 The types of chlorophyll dye solar cells commonly assembled in laboratories are shown in Fig. 2. In the thin-film devices of types (a) and (b), where photogenerated hole–electron pair separates at the heterojunction formed by the donor and acceptor molecules (here, equivalent of p-type and n-type semiconductors), chlorophylls act normally as electron donors, although the exceptions are known. The second component of this junction (acceptor) is another organic semiconductor, the choice of which is often a mixture of theoretical judgment and experimental luck. In the more bulky DSSC (type (c)), the photoexcited chlorophyll molecules serve to inject the electrons into wide bandgap inorganic semiconductor, like TiO2 or ZnO, while the transport of charges is mediated by a liquid (sometimes solid) electrolyte – Fig. 2.
In this review, we give a brief description of the molecular structure and optical properties of chlorophyll-type metal complexes, and then proceed to consider their organization consecutively at the supramolecular and at thin-film level. After the readers become more familiar with the topic, a historical overview of work on thin-film electronic devices employing these molecules to convert the light energy will be made. The parameters characterizing the DC conductivity in thin films, as well as the device efficiency, will be summarized. Since the inherent chemical fragility of chlorophylls has long shifted the attention of researchers and engineers to their analogs phthalocyanines, we will constantly compare these natural and synthetic dyes.
2. Molecules and visible absorption spectra
Chlorophylls lie at the core of the natural photosynthetic systems found in cyanobacteria and chloroplasts of plants. The transfer of the photoexcitation energy to the reaction centers proceeds through antenna structures formed by porphyrins and carotenoid arrays that can effectively absorb solar irradiation.7 This implies that the aggregation motifs must play a key role also in the artificial photoabsorbing systems consisting of these molecules, like thin-film PVC. In addition to the fully natural compounds that can be isolated from the bio-raw materials, a large number of “semisynthetic” chlorophyll dyes are available.4,8–12 They can be produced by a mere chemical modification of natural precursors, for instance by replacement of the peripheral groups, de- or remetallation.8–11 Such protocols serve to adapt the target product to the practical applications in various fields of medicine, biochemistry or optoelectronics.13Molecular structures of some chlorophyll dyes that are used as functional components of PVC are shown in Fig. 1. The nomenclature and possible modifications of chlorophylls are described in ref. 8 and 14. Their common feature is a nearly planar tetrapyrrole macrocycle with a 18 π–electron aromatic chromophore that determines the optical properties of a molecule. The chelate window can accommodate divalent metal atoms without loosing the overall planarity. The presence of a metal in the centre enhances the coordination activity towards small molecules, particularly O2.15 In the naturally occurring derivatives the metals are magnesium (chlorophyll), iron (heme) and cobalt (cobalamin). Complexes with other heavy metals can be obtained synthetically, particularly with zinc, copper, nickel, iron, cadmium, tin (rare earths are questionable).1,7,8,15–20 The best studied macroheterocycle is variously substituted pheophorbide a (Pheo), probably due to close similarity of its core with natural chlorophylls; full numbering of carbon atoms is given in Fig. 3.
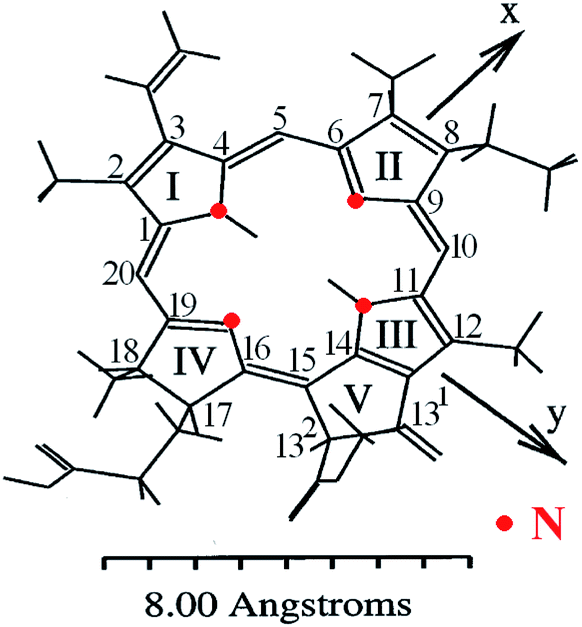 | ||
| Fig. 3 Conformation of Pheo molecule and orientation of the dipole moments calculated with the molecular mechanics force field, with usual atom numbering.4,21 | ||
However, in certain cases the intramolecular distortion of the porphyrinic skeleton due to metal coordination, introduction of heteroatoms or moieties can be quite perceptible. Fig. 4 shows the actual shapes of the nickel(II) pyro-pheophorbide a molecule resolved from single crystal X-ray measurements compared to non-metallated molecule. This derivative exhibits a ruffled core structure, clearly the result of the Ni–N bond contraction.17–19 Intermolecular interactions can also be pronounced and directional. So, zinc(II) methyl-pyro-pheophorbide a derivative with an oxime moiety at 31-position (Fig. 3) forms in chloroform a mutually coordinated dimeric structure with a pyrrole-over-pyrrole overlap, which transforms to a polymeric staircase structure in the solid state.17–19 Intermolecular distances [(C1)⋯C(pyrrole II) and (C-1)⋯N(pyrrole II)] range from 0.368 to 0.381 nm, indicating at least weak π-contact. Adjacent chains appear to be linked further by an oxime-to-ester hydrogen bond [(O⋯O) = 0.285 nm]. The differences in the structure of porphyrin and chlorin ligand are shown in Fig. 5.
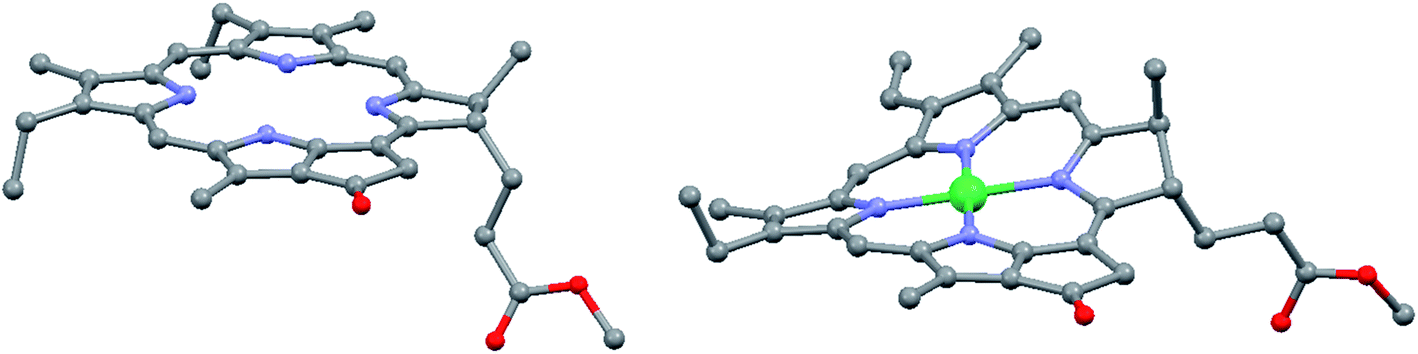 | ||
| Fig. 4 View of the molecular structures of metal-free and nickel pyro-pheophorbide in the crystal, hydrogen atoms have been omitted for clarity. Redrawn from the data published in ref. 17–19. | ||
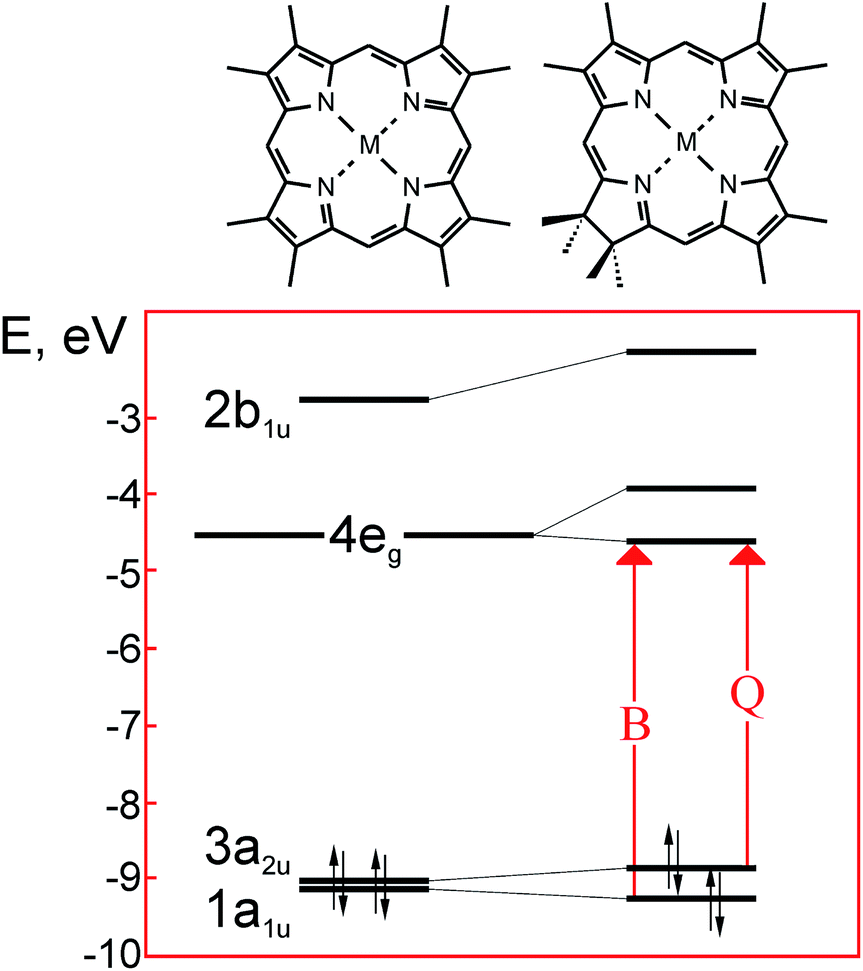 | ||
| Fig. 5 Scheme of the frontier molecular orbitals in the metal complexes of porphyrin (left) and its reduced form – chlorin (right). Redrawn from the data published in ref. 23. | ||
A significant deviation from planarity occurs in the axially substituted complexes, such as (chloro)iron(III)complex of phytochlorin methyl ester,20 but the overall type of its conformation is similar to the conventional Mg or Zn complexes.22 This derivative crystallizes with two crystallographically independent, but chemically equivalent, molecules in the asymmetric unit, mainly distinguished by the orientation of the axial ligand. The axial Fe–Cl bond lengths are about 0.223 nm in the two independent molecules, respectively, whereas the Fe–Cl vector is not orthogonal to the molecular plane: the Cl–Fe–N angles range from 97.8 to 107.2(3)°. Authors did not find any evidence of intermolecular contacts <0.4 nm, except for short intermolecular non-bonding contacts H⋯Cl (≈0.29 nm) and H⋯O (≈0.25 nm).20 Similarly, the crystal structure revealed no evidence for π–π interactions between the adjacent molecules.
Very recently, the synthesis of a tin complex bearing two axial halogen ligands was reported.24 As known, metallation causes a slight shift of the redmost absorption band in absorption spectra of chlorophylls (Qy in Fig. 6(a)) towards the shorter wavelengths.15,24,27 Upon the axial halogenation, this shift is enhanced and accompanied by a bathochromic displacement of the Soret-band,24 which makes the “green gap” in the chlorophyll spectrum narrower – Fig. 6(b).
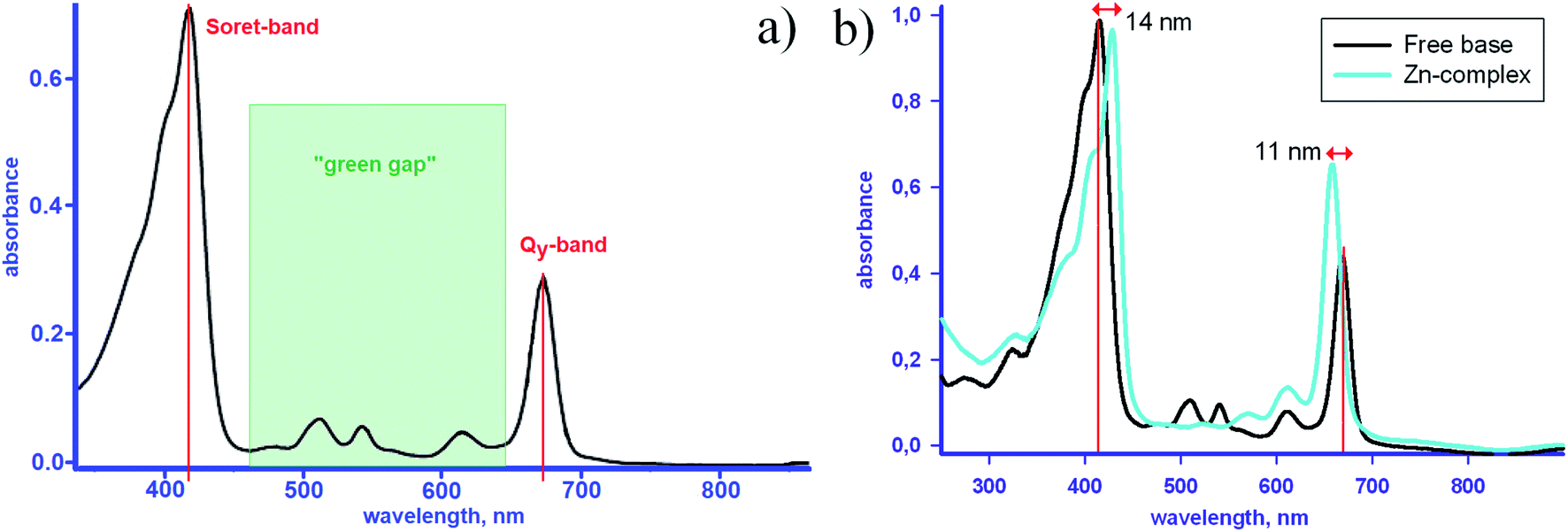 | ||
| Fig. 6 (a) Absorption spectra of metal-free methyl pyro-pheophorbide a in pyridine13 and (b) in dichloromethane, in the latter case the spectrum of the Zn-complex is plotted for comparison – see ref. 25, 26, and references therein. | ||
In the free bases, two of four inner nitrogens are bound with protons in place of a metal atom – Fig. 1 and 3. As seen from Fig. 6(b), this does not alter the electronic structure of the main porphyrinic chromophore principally,7,27 but could be of some significance for the output characteristics of PVC based on fully molecular bulk heterojunctions.28 Transformations of molecular orbitals in the chlorin as compared to the parent symmetrical porphyrin macroheterocycle are sketched in – Fig. 5.
As shown in the early works of Gouterman,23 reduction of one of the pyrrole rings in the metal porphyrins leading to a chlorin-type macrocycle not only removes the degeneration of LUMO 4eg, but also causes considerable differentiation of energy in the quasi-degenerated pairs of HOMO 3a2u and 1a1u. As a result, the intensity of the Q-band in the chlorin derivatives strongly increases. It should be noted that the values of HOMO–LUMO gap cannot be correctly reproduced by self-consistent molecular orbital calculations used in ref. 23. Unfortunately, we only found a couple of works, in which the modern calculation methods, such as DFT, were applied to describe the electronic structure of the Pheo derivatives.29,30 In contrast, the structure of the frontier orbitals in the conventional tetrapyrrolic macroheterocycles has been rigorously studied both theoretically and experimentally, due to the significance of this issue in dyeing, catalysis and medicine.16,23
The photoexcitation of chlorophylls in the solar activity range leads to formation of the two lowest singlet exited states. Among them, the π–π* electronic transition from the highest occupied molecular orbital HOMO with a1u symmetry to the lowest unoccupied molecular orbital LUMO with symmetry e1g yields a weaker band in the longer visible wavelengths (Q-band), while the transition from HOMO−1 with symmetry a2u to LUMO yields a stronger band close to the UV-range (Soret or B-band) – Fig. 5 and 6. The electronic density of these orbitals is localized essentially on the same carbon and nitrogen atoms for the metal-free, peripherally substituted or metal complexes.16 Such persistence originates from a facile formation of a π–cation radical that plays an important role in the biochemical function of porphyrins.7 The understanding of the electronic configuration of various porphyrins rests on the Gouterman's four orbital model, with many subsequent extensions and refinements (for more details see, e.g. ref. 16 and 23).
An example of the absorption spectra of methyl pyro-pheophorbide a, one of the members of the pheophorbide family,8,13,21,25,26,31–35 is shown in Fig. 6(a and b). The spectrum consists of a diffuse Soret band peaked at about 418 and a series of Q-bands, the most intensive of which is located at 673 nm. The C2v symmetry is responsible for the splitting of two Q-bands into four ones.21,34 The bands at 673 and 615 nm correspond to the vibrational levels of the first excited electronic state and are polarized in the y-direction of the molecule (defined by the clockwise order of pyrrole rings – Fig. 3). The Soret band consists of more than 10 electronic transitions with different orientations and sizes of the dipole moments.21,34 The intensity of Q-band is less than that of Soret, their maximums weakly depend on the solvent type or complexation (Fig. 6).32 The low-intensity bands located at 508 and 541 nm are polarized in the x-direction and correspond to the vibrational levels of the second excited electronic state.
The Yablonsky diagram summarizing the excitation/emission parameters of the parent monomeric Pheo a is drawn in ref. 31. For this compound, the absorption maximum of the Qy band corresponding to the S1–S0 transition was observed at λmax = 667 nm in ethanol, the extinction coefficient ε = 44![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 L mol−1 cm−1.34 The steady-state fluorescence measurements have revealed typical emission peaks at 675 and 720 nm, with the fluorescence quantum yield of 0.28 and decay time of ∼6 ns.
500 L mol−1 cm−1.34 The steady-state fluorescence measurements have revealed typical emission peaks at 675 and 720 nm, with the fluorescence quantum yield of 0.28 and decay time of ∼6 ns.
Two intensive absorption bands of chlorophylls lie within the spectral domain of high solar activity, but a broad transparency window, referred to as “the green gap”, exists in the middle10 – Fig. 6. In the natural photoconverting systems this gap is partly filled with the absorption edge of the carotenoid companion, which stretches to ∼500 nm.33,36,37 When designing an efficient PVC, one therefore needs to find a complementary photoabsorber in order to harvest maximum photons from the solar spectrum and thus achieve the high photocurrent.28 For any chlorophyll derivative the position and intensity of absorption bands may depend on the environment, e.g. ambient humidity or matrix type,5,14,22,31,38,39 but most importantly – on the aggregation of molecules.4,9–12,21,27,32,34,38,40–49 The optical spectroscopy, due to convenience of use and availability in laboratories, serves as the most frequently applied tool for assessing the aggregation state of chlorophylls in solutions and films.
3. Molecular aggregation
Assembly or aggregate is a supramolecular non-covalently bound system whose properties can “often drastically differ from those of their monomeric constituents. Thus, ensemble characteristics are dominant and novel functions emerge”.14 This classical definition originating from the supramolecular chemistry, “a chemistry beyond the molecule”, may cause misunderstanding as to how to construct an efficient thin-film device. It intuitively guides the researchers to a concept of 1D stacks or, rarely, 2D sheets – Fig. 7. However, most electronic devices deal with the ordered films, which are essentially 3D (bulk) structures. Ultimately, a single van der Waals crystal is a perfect 3D molecular aggregate, where individual properties of a molecule give way to the collective ones, such as electrical conductivity. In this sense, a thin film is a system “beyond the aggregate”, and there should be no gap between the morphology/properties of molecular aggregate and the morphology/properties of film. Yet, it does exist.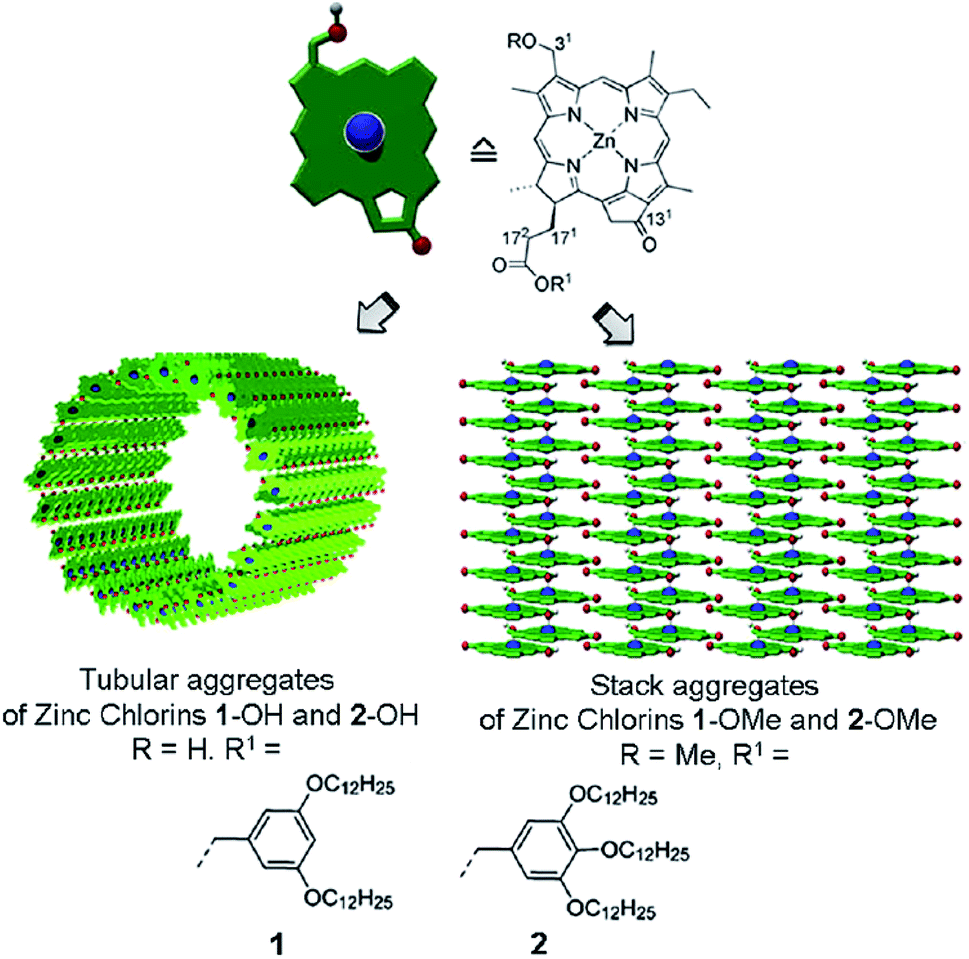 | ||
| Fig. 7 Chemical structures and self-assembled architectures of semisynthetic 31-hydroxy (1-OH, 2-OH) and 31-methoxy (1-OMe, 2-OMe) zinc chlorins. Reprinted with permission from ref. 9 copyright 2012 American Chemical Society. | ||
Formation of even a simple dimer of Pheo in aqueous solutions causes material changes in the optical characteristics.21,34,45,49 Particularly, λmax(Qy) shifts bathochromically to 685 nm, being accompanied by a decrease in ε to 30000 mol−1 cm−1, whereas the Soret band experiences the hypsochromic shift and splitting (in other works, however, a rise in the Q-band intensity for aggregates was detected – see, below). Roughly speaking, any long wavelength forms of chlorophylls result from a cooperative interaction, either by a direct interaction of the chlorophyll molecules, or interaction mediated by a bifunctional ligand such as water.22 Investigation of fluorescence from the films containing different numbers of monolayers shows that the interaction of excited molecules in the neighboring layers is even more intense than in the ground state.21,34
Slipped stacking of molecules leads to the formation of the so-called J-aggregates, 1D supramolecular systems that are similar, but not equal, to natural photosynthetic organisms.4,5,7,9–12,14,27,32,45,49,50 The distinct spectral features of aggregation are shown in Fig. 8(a) for two zinc complexes synthesized from a methyl pyro-pheophorbide d precursor. Both Qy around 650 nm and Soret band at ∼424 nm shift bathochromically upon aggregation. All of the resulting peaks become at least twice as wide as that of monomers, but their fine structure changes insignificantly. These features indicate the formation of self-assembled aggregates with the J-type excitonic coupling.4,9–12,27,46–48,51 The J-type of aggregation dominates in the supramolecular arrays of most chlorophylls, but in some cases the shifts of absorption bands in the optical spectra might suggest formation of H-aggregates, too.5,28
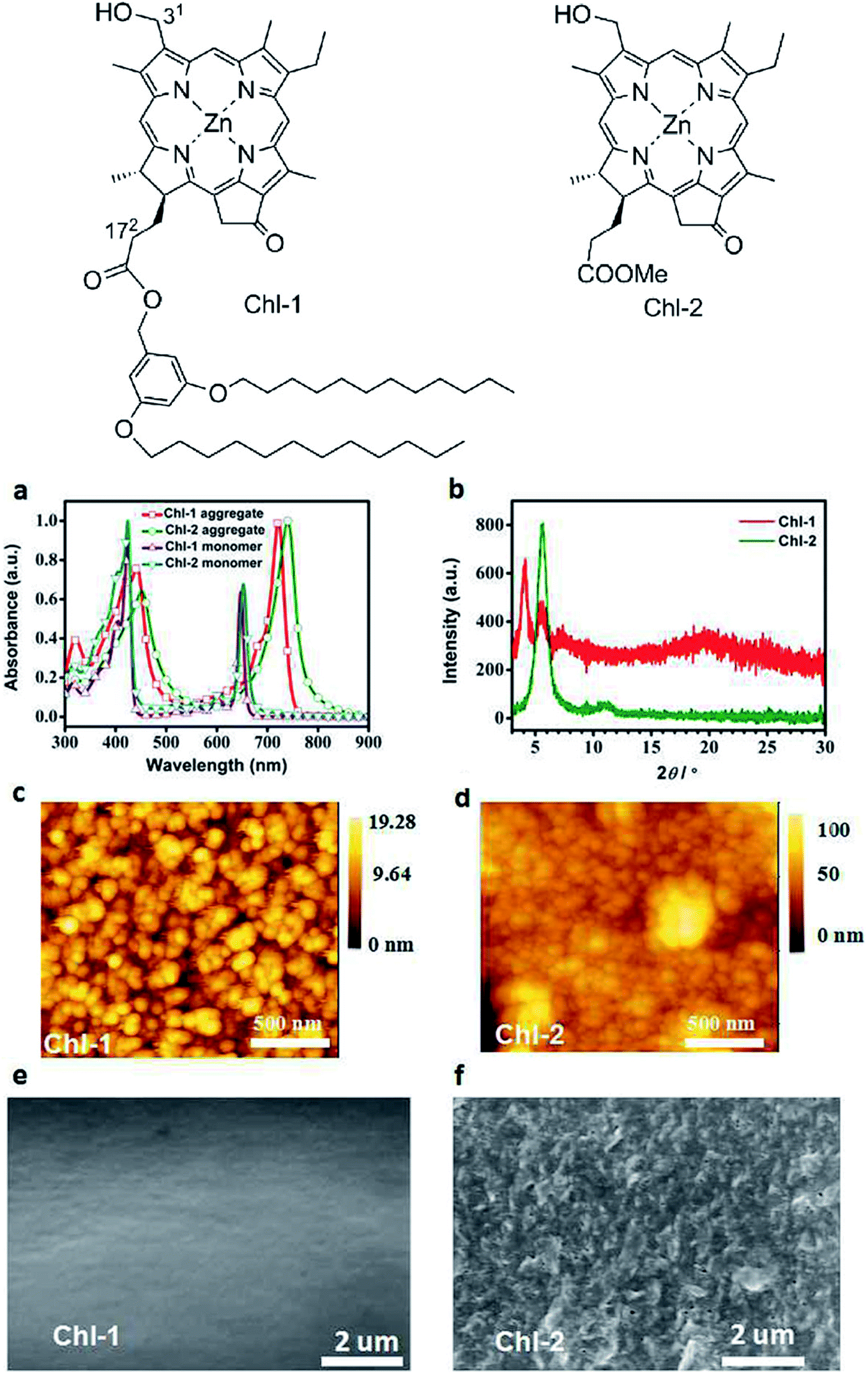 | ||
| Fig. 8 (a) UV/Vis absorption spectra of Chl-1,2 monomers in THF and aggregates spin coated film on glass; (b) XRD patterns of the spin-coated Chl-1 and Chl-2 films on glass; (c) and (d) tapping-mode AFM images of aggregates of Chl-1 and Chl-2; (e) and (f) SEM images of Chl-1 Chl-2 film surfaces, respectively.† Top – chemical structures of Chl-1 and Chl-2. Reprinted with permission from ref. 4 copyright 2016 Wiley. | ||
In the stacks of metallated chlorophyll derivatives the molecules align not only through vertical π–π overlap but also through a more accentuated interaction between a metal atom in one and a specific atom (or group) in another molecule. The overwhelming majority of works on the semisynthetic chlorin or Pheo derivatives deal with the zinc complexes.3–5,9–12,27,38,40,42,43,45–47,49,50,52 Only Sasaki et al. frequently use metal-free derivatives,5,6,27,32,33,40,43,50,52,53 which could potentially be helpful for elucidating the role of metal ligation in the charge transport throughout the aggregate films.
Review14 summarizes the hierarchy of interactions through which the molecules build up their aggregates, in order of weakening:
(1) Metal ligation. The strongest non-covalent bonding due to which the metallo-chlorins, as well as other metallated tetrapyrroles, possess a very rich coordination chemistry. Fig. 9 shows schematically the superposition of molecular interactions for the bacteriochlorophyll c.
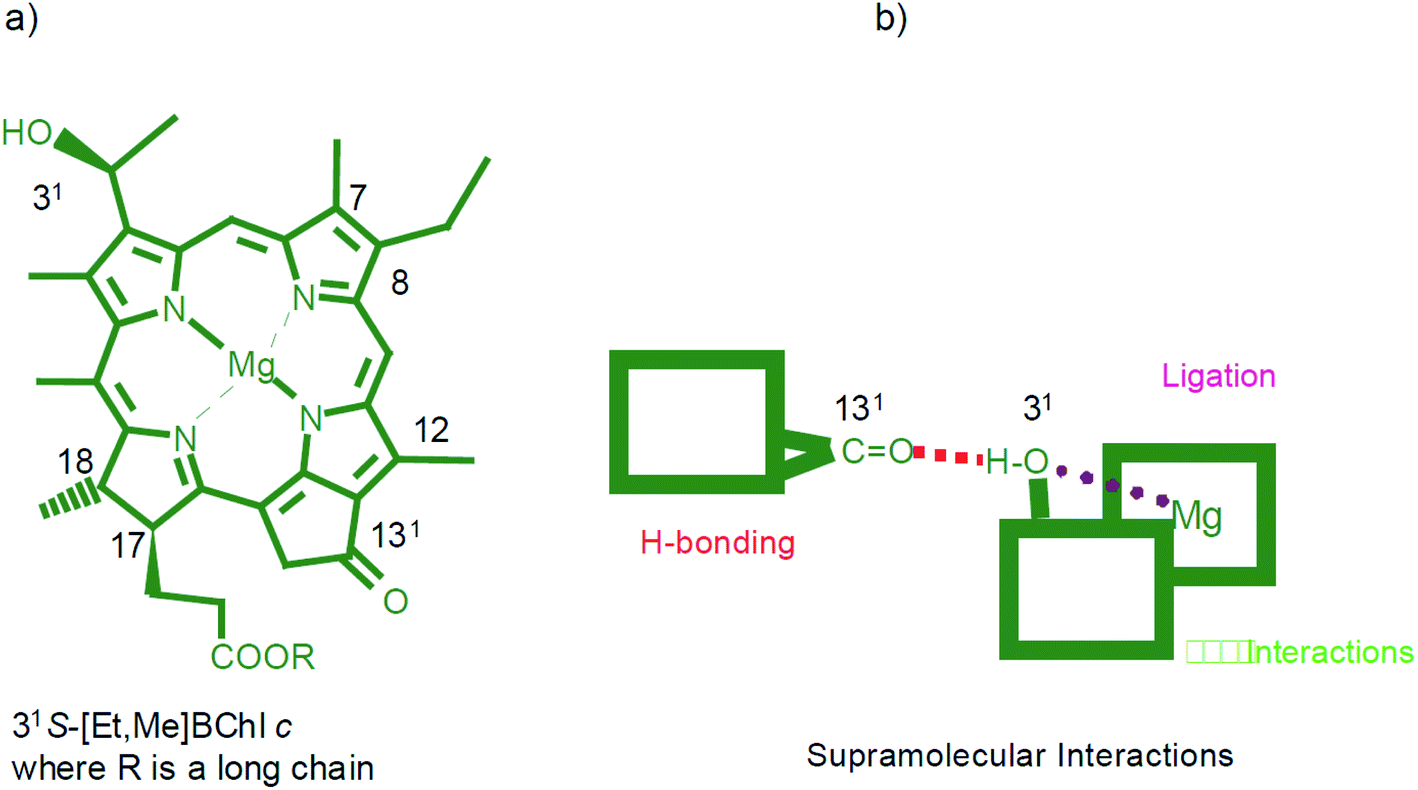 | ||
| Fig. 9 (a) Molecular formula and atom numbering for a typical natural bacteriochlorophyll c – the 31-carbon atom may have either R or S configuration while the 17- and 18-carbon atoms are both S. Different substituents in the 8- and 12-positions (depicted are ethyl and methyl) as well as various long chain alcohols (such as farnesyl, stearyl, cetyl, oleyl, etc.) may esterify the 17-propionic acid side chain. The different configurations (R or S) of the hydroxethyl groups induce the different chirality in the self-assembled species. (b) The three important supramolecular interactions are presented schematically. Reprinted with permission from ref. 49 copyright 2003 Wiley. | ||
(2) Hydrogen bonding. Next strongest supramolecular interaction in this hierarchy, provides tight and directional binding. All (bacterio)chlorophylls carry the 13-carbonyl group in the fifth ring which can act as an acceptor group for hydrogen bonding and most compounds, except for BChl c, d, and e, they also possess a methoxycarbonyl group in the 132 position whose carbonyl group is often involved in hydrogen bonding. For bacteriochlorophyll c (Fig. 9(a)) the ligation of the central magnesium atom by the 31-hydroxy group of another molecule overlaps with hydrogen bonding of the same OH group to the 131-carbonyl group of a third molecule. The methoxycarbonyl group at the 132 position may contribute to an intermolecular hydrogen bond, however, in the chlorosomal J-aggregate of bacteriochlorophylls c, d, e the substituent on the ring V (Fig. 3) would disturb the stacking of the tetrapyrrolic pigment.54 In the ethyl chlorophyllide a dihydrate crystals one water molecule coordinates the central magnesium atom while a second structural water molecule is doubly hydrogen bonded between the first water and the 132-methoxycarbonyl group.
(3) π–π interaction. Typical of conjugated macrocycles, such as planar porphyrins, coupling of closely located (stacked) chromophores, a kind of strong van der Waals bonding in molecular solids. The π–π stacked distances generally found in the crystalline porphyrins and other related π-stacked compounds correspond to ∼0.35 nm.55 This explains the frequent appearance of a peak at 25° in the XRD patterns obtained from the ordered thin films of chlorophylls – Fig. 8(b). In ref. 49, the free bases were metallated by using zinc acetate in a chloroform/methanol (4:1) mixture at room temperature with almost quantitative yields. Compounds with Zn are the fully synthetic mimics of magnesium bacteriochlorophyll c that can self-assemble. The three structural elements responsible for the self-assembly, namely a hydroxy group, a central metal atom with affinity for a fifth ligand, and a carbonyl group are allowed to interact so that the algorithm leading to self-assembly is able to become operative – Fig. 9(b).
(4) Hydrophobic effects. Long chain fatty alcohols esterify the 17-propionic acid residue of all (bacterio)chlorophylls. These large residues are highly flexible and can thus adapt to occupying voids within hydrophobic pockets of protein matrices, or may help to solvate (B)Chls in non-polar solvents.14
(5) Positive cooperativity of these interactions leads to thermodynamic stabilization of the supramolecular assemblies beyond that yielded by the mere sum of the individual non-covalent bonding contributions – see ref. 45, for example.
Depending on the molecular properties of the units building up the 1D supramolecular array, the absorbed excitation can be delocalized over at least 10–15 aggregated pigments even at room temperature, RT.56 These aggregates can further be organized into the multi-level 3D structures, the size and other properties of which can be accounted for by stereochemistry of the chlorophyll molecules,9–11 examples are shown in Fig. 7. The optical and energy-transfer characteristics of some of these structures are remarkably similar to those of the light harvesting chlorophyll assemblies in vivo, particularly of photosynthetic bacteria.
The liquid media enable a larger degree of freedom for the slow self-assembly process. Both natural bacteriochlorin (BChl) and its synthetic analogs with 3,5-di-tert-butylphenyl groups form macroscopic aggregates in non-polar solvents visible to the eye as “fluffs”.49 In the absence of atmospheric oxygen these fluffs are thermodynamically stable even when exposed to the light and can reversibly de-assemble to nanoaggregates of about 100 nm upon gentle shaking and then re-assemble in a few hours. In thin solid films grown on the substrates the Chl aggregates also form fractal structures that can easily be observed using the optical microscopy.9,10,44,55
Such freedom, or lability of solution-mediated intermolecular interactions, manifested itself in the competition of kinetically and thermodynamically controlled pathways of self-assembling in the water solutions of inherently chiral Zn chlorin derivative with a bulky side substituent.45 A thermodynamically-driven pathway yields stable, extended 1D helical fibers consisting of extended J-aggregates. However, an increasing content of water in the MeOH:H2O mixture or concentration of dissolved dye increases stability of the off-pathway nanoparticles consisting of excitonically coupled J-dimers formed upon a competing, kinetically-driven process. The trapped off-pathway nanoparticle products do not transform to the more ordered thermodynamic product within the time scale of several months. This, or course, adds complexity to the mechanisms of self-assembly, but at the same time opens up new possibilities in varying the aggregation length/shape, which could be useful.
This line of research was recently continued in ref. 57 with a semi-synthetic zinc 31-methoxy-chlorophyll derivative having amide and urea groups in the 17-substituent (Fig. 3). The gradual transformation from kinetically formed dimeric species into the more thermodynamically stable J-aggregates in the solid state was observed by combination of optical and microscopic methods. The kinetically and thermodynamically formed self-assemblies have particle-like and sheet-like supramolecular nanostructures, respectively.57 These two-dimensional nanomaterial photophysically and nanostructurally mimics chlorosomal bacteriochlorophyll c–f J-aggregates.
From the viewpoint of application of organic dyes as photoabsorbers in solid-state PVC, high molar extinction coefficients in the visible range become necessary to reduce the thickness of active layers. By modifying the chromophore from a porphyrin to a chlorin an increase in the extinction coefficients of both the Soret and Q bands should be observed (Fig. 5), in different proportions.14,16,23 However, after self-assembly, broad bands of about the same intensity in the visible region were obtained from both the chlorin and the porphyrins.
Pheophytin a is the metal-free chlorophyll dye also well-spread in plants. Films of pheophytin a show a remarkably large red shift; the red maximum that appears at 668 nm for pheophytin in hydrocarbon solutions was found at 695 nm in the films. This large red shift in anhydrous films of pheophytin may result from a strong plane–plane interaction, as observed for other porphyrins. The chlorophylls, however, because of the presence of the central magnesium atom, have much greater difficulty in assuming parallel configurations and thus exhibit much more variable behavior.22
Dimensionality of a single chlorophyll aggregate determines the experimental setup required for measuring its photoconductive properties, particularly the size and nature of the electrical contacts to be used. One-dimensional self-assemblies on a length scale of 10–100 nm can easily be obtained on functionalized surfaces by deposition from solutions. We failed to find in the literature the number of units in such assemblies, whereas the averaged numbers of the bacteriochlorophyll c molecules contained in a single chlorosome, which is a much more complex system, has been estimated. Length of tubular aggregates (rods, wires) up to several micrometers9–11,58 makes possible direct two-terminal measurements. However, we did not find any information on planar nanogap devices with two- or three terminals bridged by a chlorophyll aggregate.59 The electrical measurements using lithographically pre-patterned electrodes with the lateral dimensions on the order of a few tens nanometers, which exactly coincides with the dimensions of stacked 1D aggregates, are well known for the biologically important macromolecules and metal nanoparticles,59 but not for chlorophylls. Such measurements could shed more light on a steady-state or field-modulated conductivity owing to a free choice of electrode geometry and material.
The molecules of a zinc-metallated chlorin derivative with a long side chains assemble in the 1D stacks (nanowires) that can be deposited from a cyclohexane solution onto a highly ordered pyrolytic graphite and visualized using the tapping-mode atomic force microscopy (AFM) technique, as demonstrated by Würthner and coworkers.9–11,46–48,55 The width of the nanowire coincides with the size of two closely packed chlorin molecules with the tiles out. Such nanowires separated by their alkyl chains create 2D domains (lamella) with different packing motifs on the graphite surface, which depend on the initial concentration of molecules in the solution. Judging from the AFM images shown in the article, the maximum domain size seems to coincide with the length of a single 1D molecular stack and exceeds 200 nm for the more densely packed domains. Notably, more densely packed and extended domains were obtained from a less concentrated solution, which indicates that the intermolecular interactions inside and between the aggregates dominate the possible quazi-epitaxial growth of molecules on a highly regular surface, although the authors adhere to a different opinion.46–48 In ref. 60 the size of molecular aggregates in either crystalline or micelle chlorophyll a was estimated to be within the range of tens to hundred of nm in the electrodeposited films with thickness of about 100 nm.
In ref. 58, zinc complexes of chlorins with esterified long alkyl chains at the 17-propionate residue were found to form chlorosomal self-aggregates in a nonpolar organic solvent. Their chlorosomal rod/wire nanostructures with a 5 nm diameter in the solid state were observed on the substrates with different hydrophobicity, indicating domination of intermolecular interaction over other factors. The fine supramolecular structure and stability of these rods on the examined substrates depended on the length of an oligomethylene chain at the 17-propionate residue, while their microscopic-level organization (density, full length, shape) depended also on the surface properties.
More sophisticated tubular aggregates (nanorods) can also be obtained on the pyrographite using another zinc chlorin derivative equipped with hydrophobic dendron wedges – Fig. 10. When composed of zinc chlorin-naphthalene bisimide dyads, such aggregates have dimensions of ∼300 ± 97 nm in length and ∼5.8 ± 0.4 nm in height, or ∼93 ± 75 nm in length and 7.3 ± 0.2 nm in height.46,47 Anyway, the nanorods are randomly distributed over the deposition surface, largely vary in length and usually deviate from the shape of a straight line, in contrast with the 2D assemblies discussed in the previous paragraph. This corroborates the unimportance of the surface forces and strong ability of the chlorin molecules to organize themselves in accordance with their geometry and mutual interactions.
 | ||
| Fig. 10 HR-TEM micrograph of aggregates drop-cast from water/methanol (100:1; v/v) (c ∼ 0.5 × 10−5 M) stained with 0.5% aqueous uranyl acetate at an accelerating voltage of 160 kV. The hollow channels in the nanotubular aggregates are clearly visible in the inset (top right) of this figure. Reprinted with permission from ref. 11 copyright 2012 Wiley. | ||
Then, by using the conductive AFM tip as one electrode, pre-deposited conductive polymer strip as the other electrode and an SiO2 insulator as substrate,9 the conductivity of the single nanorod was measured (∼4.8 × 10−3 S cm−1 in average, under ambient conditions). Charge carrier mobility was found to reversibly increase from 0.017 to 0.043 cm2 V−1 s−1 with temperature increasing from −25 °C to +100 °C – cf., Table 1. This indicates that the scattering of carriers induced by the thermally activated molecular motions negligibly disturbs the charge transport along the stack.
| μ, cm2 V−1 s−1 | Carrier type | Compound type | Sample type | Method | Ref. |
|---|---|---|---|---|---|
| ∼10−3–10−2 | Σ(h + e) | Zinc chlorin derivatives functionalized with dendron wedges | Solution-processed bulk mesophases with liquid crystals columns | Pulsed radiolysis time-resolved microwave conductivity (PR-TRMC) | 10 |
| 0.07–0.28 | h ? | Zinc chlorin derivatives with C31-hydroxyl- and methoxy-groups | Solution-processed bulk mesophases with 2D stacked assemblies | PR-TRMC | 9 |
| 0.03 | h | Zinc chlorin derivative with hydrophilic dendron wedges | Self-assembled nanorods (nanotubes) | PR-TRMC and dc measurements with conductive tip (C-AFM) | 9 |
| 6.49 × 10−3 | h | Zinc chlorophyll derivative with two dodecyl groups (Chl-1) | Spin-coated films | Space charge-limited current (SCLC) | 4 |
| 1.55 × 10−3 | h | Zinc chlorophyll derivative with a single methyl group (Chl-2) | Spin-coated films | SCLC | 4 |
| 6.2 × 10−4 | h | Zinc methyl 3-devinyl-3-hydroxymethyl-pyropheophorbide a | Spin-coated films (J-aggregates) | Field-effect transistor (FET) | 27 |
| 4.6 × 10−6 | e | Methyl 32,32-dicyano-pyropheophorbide a | Spin-coated films | SCLC | 33 |
| 3.0 × 10−5 | e | Methyl 131-deoxo-131-(dicyanomethylene)pyropheophorbide a | Spin-coated films | SCLC | 33 |
| 4.11 × 10−3 | h | Zinc chlorin with C31-hydroxy group | Spin-coated films with well-ordered structure of J-type aggregates | SCLC | 5 |
| 6.08 × 10−7 | h | trans-32-Carboxy-purpurin-18 methyl ester (H2P) | Spin-coated films | SCLC | 40 |
| 1.21 × 10−5 | h | Zinc-trans-32-carboxy-purpurin-18 methyl ester (ZnP) | Spin-coated films | SCLC | 40 |
| 9.5 × 10−7 | h | 32,32-Dicyano-pyropheophorbide-a | Spin-coated films | SCLC | 41 |
| 4.6 × 10−6 | e | ||||
| 2.0 × 10−6 | h | Methyl-131-deoxo-131-(dicyanomethylene)-pyropheophorbide a | Spin-coated films | SCLC | 41 |
| 3.0 × 10−5 | e | ||||
| 2.49–7.93 × 10−6 | h | Bacterio-chlorophyll-fullerene dyads (DAs) | Spin-coated films (mobility depends on the DAs structure) | SCLC | 53 |
| 2.15–7.95 × 10−6 | e | ||||
| 2.7 × 10−5 | h | Trimethyl ester of chlorin e6 | Spin-coated films | SCLC | 6 |
| 2.2 × 10−4 | h | Cu-metallated trimethyl ester of chlorin e6 | Spin-coated films | SCLC | 6 |
| 1.71 × 10−4 | h | Zinc dodecyl 131-deoxo-3-devinyl-131-dicyanomethylene-3-hydroxymethyl-pyropheophorbide a blended with [6,6]-phenyl-C71-butyric acid methyl ester | Spin-coated films | SCLC | 42 |
| 1.03 × 10−4 | e | ||||
| 5.13 × 10−5 | h | Zinc dodecyl 131-deoxo-3-devinyl-131-dicyanomethylene-3-hydroxymethyl-pyropheophorbide a | Pure aggregate films, untreated (spin-coated) | SCLC | 42 |
| 5.53 × 10−5 | e | ||||
| 5.84 × 10−5 | h | Zinc dodecyl 131-deoxo-3-devinyl-131-dicyanomethylene-3-hydroxymethyl-pyropheophorbide a | Pure aggregate films, solvent treated | SCLC | 42 |
| 7.30 × 10−5 | e | ||||
| 5.22–26.7 × 10−4 | h | Zinc 3′,5′-bis(dodecyloxy)benzyl-3-devinyl-3-hydroxymethyl-pyropheophorbide a | Spin-coated films (mobility depends on the solvent type) | SCLC | 43 |
| 5.80 × 10−2 | ? | Zinc chlorin derivatives with alkyl chains (ZnChl-C6,C18) | Self-assembled nanowire on SiO2 surface | C-AFM | 61 |
| 10−6 | h | Chlorophyll a | Electrodeposited films (300 nm) | Time-of-flight technique (TOF) | 62 |
| ∼6 × 10−5 up to 2.14 × 10−3 in absence of disorder (=μ0) | h | Chlorophyll a | Electrodeposited films (5–10 μm) | TOF | 63 |
| 5.8 × 10−3 | h | Chlorophyll a | Spin-coated films | FET | 64 |
| 2.2 × 10−3 | e | Chlorophyll a | Spin-coated films | FET | 64 |
|
|||||
| Zinc phthalocyanines (ZnPc) | |||||
| 6.85 × 10−4 | h | Hexamethyl-mono-n-butyl-substituted-ZnPc | Spin-coated films | SCLC | 65 |
| 8.63 × 10−5 | h | Mono-n-butyl-substituted-ZnPc | Spin-coated films | SCLC | 65 |
| 1.8 × 10−4 | h | Tetra-substituted ZnPc with dendron wedges (ZnPcR4) | Spin-coated films with liquid crystalline structure | FET | 66 |
| 2–4 × 10−4 | TOF | ||||
| 1.7 × 10−4 | h | ZnPc (unsubstituted) | Vacuum-deposited films | FET | 67 |
| 1.0 × 10−4 | e | Zinc octa-fluorinated derivative (F8ZnPc) | Vacuum-deposited films | FET | 67 |
| 1.3 × 10−2 | h | ZnPc (unsubstituted) | Structured vacuum-deposited films on tri-layer dielectric | FET | 68 |
In addition to the AFM technique, the authors obtained fine images of the objects in question by means of the high resolution transmission electron microscopy, HR-TEM. Fig. 10 shows the ‘life’ of tubular aggregates on a carbon-coated copper grid used as a substrate for HR-TEM measurements and calls forth a couple of comments.
First, it is clear that every nanorod contains perfectly linear sectors, typically <100 nm long. But on a large scale, all nanorods in Fig. 10 bend or consist of several straight sectors. This is indicative of their defective growth resulting in the inhomogeneity of molecular packing. The curvature of elongated nanorods has been repeatedly mentioned, although at a scale of <100 nm (and from computed 3D drawings) they look perfectly straight.46,47,61 The length of this perfect section, which indicates the absence of defects, depends on the nature of aggregating molecules (the energy of intermolecular interaction), but for a particular compound can be, to some extent, determined by the growth conditions, including solvent and substrate type. In thin-film PVC, the length of the aggregates is not as important as their mutual arrangement in a film even provided that the energy/charge transport within each aggregate is perfect. Meanwhile, the measurements of electrical conductivity value should best be taken within a straight sector of an aggregate, which most likely was not the case. Otherwise the data would suffer from the defect states that could serve either as a source of charge carriers (holes) or, on the contrary, as capture centers lowering the actual mobility along the nanorod.
Secondly, the conductivity parameters were calculated on the approximation of a 3D rod-like conductor with two flat face/end contacts. In fact, however, the resistor was a ∼6 nm thick tube with a ∼2 nm hollow inside, i.e. a molecular wall measured with two arbitrary point contacts, and so the resistance should be have been treated as 2D (sheet).
The keen interest in mechanisms of self-assembly is inspired by the aspiration to better understand, reproduce and, eventually, exploit the photoelectrical processes running in the natural chlorophylls. This relates to a ‘biomimetic’ approach that intends to accommodate the semi- or fully synthetic dyes whose self-assemblies can mimic the light-harvesting system in photosynthetic bacteria for use in the PVC.2,3,9–11,14,50 However, it is worth knowing that the “analogs of chlorophyll” and natural chlorophylls are structurally not identical. Even little changes in the nature of peripheral substituents can cause variations in conformation, stereochemistry, additional pathways of growth etc.45 and the solid-state arrangement will be essentially not the same. For instance, the structure of the chlorophyll a antenna is not similar to the stacks found in the chlorophyllide crystals.22 Thus there is a high risk of a situation when the molecular ordering in the aggregates consisting of “analogs” imitates the ordering of the natural dyes, but the resulting (anticipated) bulk properties, like photoconduction, do not.
The question of whether or not a lengthy peripheral chain (phytyl, dodecyl) attached to the porphyrinic macrocycle facilitates the long-distance ordering of molecules remains unclear. On the one hand, the largest reported aggregates in the form of rods or tubes were composed by the molecules with a tail, and the most outstanding results on the conductivity9–11 and photovoltaic efficiency42 were obtained on them. From this it follows that hydrophobicity of the side chains seems to play a role during the assembling of molecules from a solution and/or during separation of these assemblies for subsequent analytical studies.
On the other hand, the absence of such a tail cannot surely be regarded as detrimental for the efficient self-assembling. First, there are numerous examples of highly ordered phases of unsubstituted porphyrins and phthalocyanines, the length of aggregation in which is greater than in natural chlorophylls – see, Sections 6 and 7. Second, if a couple of appropriate (small) solubility-inducing groups are appended to the chlorin core, the thus obtained semisynthetic molecules will assemble to form macroaggregates in n-heptane or n-hexane, just like the natural BChls c, d and e do.49 Remarkably, the spin-cast films (∼60 nm) obtained in ref. 49 from self-assembling Zn-complex show very broad, accreted absorption peaks covering entirely the range of ∼400–650 nm and non-quenched fluorescence, which is untypical of aggregated chromophores. Earlier studies also suggest that a phytyl chain does not influence the photo- and semiconducting properties of chlorophyll-type dyes,69–71 which are primarily mediated by π–π (or π–metal) contacts of the stacked porphyrinic chromophores.
For structurally identical dye molecules, the following factors may distort the structure of the aggregates (a) solution chemistry; (b) substrate material; (c) deposition method; (d) deposition conditions. For instance, morphology of the thin films of Chl aggregates depends on the application of DC voltage during the casting from the water solution.44 The sensitivity of a supramolecular organization to the deposition conditions reduces the reproducibility of the results and has thus to be minimized by choosing molecular structures having an invariant route of assembly.
4. Thin solid films
First of all, it is important to differentiate between the morphology of thin films of chlorophylls subjected to standard photoelectrical measurements or standard procedures necessary involved in fabrication of PVC, and the morphology of individual aggregates discussed in Section 3.Due to the presence of alkyl, hydroxyl, carboxyl or acetic moieties (Fig. 1) attached to the macrocycle, the chlorophyll molecules are soluble in many common organic solvents or mixtures thereof. A long phytyl side chain in natural chlorophylls enhances solubility in the alcohol/water mixtures and nonpolar aliphatic solvents. As such, thin films can be prepared by electrodeposition, layer-by-layer self-assembly, Langmuir–Blodgett (LB), spin-coating and casting techniques, all methods are exemplified in the literature. For the same reason, these compounds tend to decompose at elevated temperatures,26 whereas thin films of simple unsubstituted porphyrins are easy to grow by thermal evaporation on practically any kind of substrate.
Each deposition method has its own restrictions. Spin-coating requires an accurate balance between adhesion to the substrate, solution viscosity and a desired film quality. The latter means that the film should be homogeneous enough to prevent pinholes (electrical shorts), dense enough to withstand the deposition of following layers and thin enough to possess a reasonably low resistivity. The substrates on which the films are to be deposited should be the opaque or transparent electrodes, in most cases chemically functionalized to match the work function of the adjacent molecular semiconductor, i.e., roughly the redox potentials of a concrete chlorophyll derivative. As a result, the films in the real PVC do not necessarily contain those extended 1D–3D assemblies or tubular aggregates that are shown in Fig. 7 and 10. Instead, the AFM images6,32,40–42 of the film surface obtained by spin-coating display a surface roughness on the order of a few nanometers at the nominal film thickness being on the order of several tens nanometers. Absence of large-scaled objects indicates that the film represents, most likely, a mixture of monomeric and oligomeric forms of chlorophyll molecules,38,46,48,72 which can be confirmed by the optical spectroscopy – Fig. 8(a).
Interesting data are reported in ref. 14. Assuming that all procedures and methods were strictly identical for two chlorophyll derivatives shown in Fig. 8, it turns out that the presence of two long dodecyl chains severely affects the morphology of films obtained by spin-coating. Firstly, the Chl-1 molecules form clearly defined grains with size ∼50 nm according to Fig. 8(c), whereas the grains of Chl-2 are smaller and less defined although tending to coalesce in the big irregular objects (Fig. 8(d)) so that on the large-area SEM images the surface looks as a crumpled paper – Fig. 8(f). Secondly, a larger grain size of Chl-1 can be presumed from the narrow absorption peaks in Fig. 8(a), X-ray analysis and, with a great care, from the mobility measurements – Section 6. The appearance of a low-angle peak in the XRD spectra of Chl-1 is consistent with the increased intermolecular distances in the crystals of chlorins having long side alkyl chains.9,10 Thereby, the chlorophyll-type molecules with a tail of two dodecyl groups have demonstrated a better ability to assemble during spin-coating, and their assemblies form thin films with a 4 times greater hole mobility.
In Fig. 8(c–f) one can see molecular films formed by the intergrown 3D objects with ordered molecules inside – grains, nanocrystals, domains. The lateral dimensions of these objects seem to be comparable with the typical film thicknesses in PVC (confusing experimental conditions in ref. 4 and 12 do not allow us to come to a more certain conclusion). By analogy with the above described nanorods/sheets, one could expect a good electronic/excitation transport within the regular domain. If this is the case, it is reasonable to maintain the thickness of films to be used in the devices at a level corresponding to the size of a single domain and simultaneously to make these domains as large as possible for the maximal light collection.
For the electrodeposited Chl films, Leblanc et al.73 estimated the grain size to vary from 30 to 100 nm. Such films featured a polycrystalline order with many defects, which correlates with the electric properties.62 The authors have deduced that, in order to improve the organization of microcrystalline Chl films, it is crucial to better understand the mechanism of microcrystal formation.73
The surface of thin films of various chlorophyll derivatives was examined using AFM/STM and/or SEM techniques in ref. 4, 5, 12, 27, 32, 40–44, 52, 64 and 73. However, the published figures cannot provide sufficient insight into the film morphology and usually serve as supplementary experimental facts in the description of samples. For example, the cross-sectional SEM images in ref. 5, 27, 42, 43, 52 and 64 do not yield quantitative information and are corrupted by the underlying materials, such as TiO2, without aerial views of the reference (pure) films on atomically flat substrates. We have not found any phenomenological model describing how the aggregates actually behave in a thin film: size or number of molecular units, orientation with respect to the substrate, density etc. For individual aggregates of several Zn-metallated Pheo derivatives, such a model has been successfully developed by Würthner, Sengupta et al.9–11,55 but it has never been extended to thin films. The XRD studies on the chlorophyll films are rare and fail to bridge the gap between the lattice parameters derived from the single-crystal analysis and the diffraction pattern of a film.4,12,42
5. Historical background and key works in the field
Parent natural chlorophyll a (Chl), a green magnesium porphyrin, is the oldest photovoltaic material among the organic dyes. The systematic experimental investigation of its photo-induced conductivity in solid samples was initiated by acad. A. N. Terenin, A. T. Vartanyan and co-workers (St. Petersburg) in a huge series of works published regularly from the late 1930s through 1960s,69–71,74–76 and continued by V. A. Benderskii and co-workers (Moscow) in 1960–1970s.77,78 It was experimentally proved that Chl behaves as a good photoconductor in the solid state (films or pressed pellets). The key electrical properties of a family of natural and synthetic tetrapyrrolic dyes were described for the first time: conductivity (σ) in both DC and AC mode in the dark and under continuous or monochromatic illumination, photovoltaic effect and luminescence, thermally activated and time-resolved processes, effects of annealing, doping and mixing, solvent treatment and many other. As early as 1953 Terenin et al. succeeded in attempts to photosensibilize the inorganic semiconductors like ZnO, CdS with chlorophylls and porphyrins.75Using the Arrhenius plot, the dark and photoconduction activation energy Eact of ∼1.7 and 0.15 eV respectively was derived in vacuum for thin Chl films deposited from chloroform.75 These issues closely intersect with the comprehensive study of Rosenberg and Camiscoli (1961) on the compressed crystals of chlorophyll a and b in the different environments (air, vacuum, oxygen, nitrogen, argon, hydrogen), in which such an important question as formation of a Chl:O2 complex and its role in the “thermal spike” on the σ = f(t0) dependence was raised.79
In 1975, C. W. Tang and A. C. Albrecht (New York) published their widely cited results on the photovoltaic effect in single-layer two-terminal cells where an electrodeposited microcrystalline Chl film was sandwiched between two metallic electrodes.80–82 They tested a number of evaporated (Al, Au, Cr, Cu, Ni, Ag) and liquid (Hg, Ga, Hg/In) metallic electrodes and found out that the sign and magnitude of the photovoltage are due to appearance of a Schottky barrier at the interface between the p-type Chl semiconductor and the low work function metal. The photovoltaic effect was discussed in terms of charge separation in the electrostatic field at the non-ohmic contact and supported by the relevant band diagram. The maximum achieved power conversion efficiency (PCE) lay in the range of 10−3% for samples with the evaporated electrodes and 5 × 10−2% for Cr/Chl/Hg cells; open circuit voltage (Voc) was about 0.3 V. In these works, a detailed device characterization was carried out, including dark and light J–V curves (plus numerical modeling), time-dependent and flux-dependent measurements, photocurrent action spectra and quantum efficiency of the charge photogeneration. Thus, the standards in the experimental organic photovoltaics were established for many years to come.83
However, in addition to the low efficiency, the behavior of the devices was extremely materials/procedure dependant, and the basic photovoltaic parameters were estimated at the incident light intensity of less than 0.1 mW cm−2, since higher intensities caused deterioration of the cells.80,82 Later, C. W. Tang has overcome these problems by using a pair of synthetic dyes, Cu-phthalocyanine/perylene tetracarboxylic derivative, instead of the natural one and a molecular PHJ instead of a Schottky junction.84 Such a concept assisted in achieving a PCE of about 1% and stimulated a new wave of interest towards the small-molecule based solar cells, which has not subsided until now.28,83
Next, we should mention a series of articles published by R. M. Leblanc and coworkers (Quebec) who studied the photovoltaic effects in the Chl films obtained by either electrodeposition62,63,73,85–91 or LB36,90 method. In their earlier work,36 the parameters of the sandwich-type devices with two dissimilar metallic electrodes were analyzed at the varying illumination by halogen–tungsten lamp. It was noticed that Chl in LB films behaves as a p-type molecular semiconductor, just like many carotenoids.3 Very interestingly, the authors have noticed that a PVC having the composition Al/Cantha/Cantha(0.2):Chl(0.8)/Chl/Ag, where Cantha = carotenoid canthaxanthin, perform better than the cells with bi- or single layer photoabsorbers, all devices being treated as the Schottky-type. The contribution of both natural dyes to the photocurrent was detected, while the cells with only the Cantha photoabsorber were inactive. This led the authors to conclude that carotenoid molecules in the bottom layer are unable to generate photoinduced charge carriers acting rather as sensitizers for the energy or electron transfer to the Chl molecules in the overlying layer.36 However, given the thickness of the mixed Cantha:Chl layer and the schematic of those cells, it was probably the earliest known demonstration of a mixed (or BHJ) junction composed by small molecules – cf.28 Maximum efficiencies of thus made PVC corresponded to the range of 5–6 × 10−2%, with Voc up to ∼0.7 V.
In 1991, these authors fabricated a Chl-based PVC with a hybrid PHJ, where inorganic n-type semiconductor was cadmium sulfide.86 The full photovoltaic characterization of the CdS/Chl/Ag heterostructure on glass/ITO substrates was carried out (ITO is a transparent indium–tin oxide), including calculation of fill factor FF, series Rs and shunt Rp resistance, and monochromatic efficiency at 740, 680 and 560 nm, i.e. in the regions of minimum and maximum optical activity of Chl (Fig. 6). Modest advantage over the single-layer Al/Chl/Ag devices was accomplished, such as FF = 0.45 and Voc = 0.4 V (see, also ref. 62). Degradation of the Chl material under prolonged illumination was observed, too.
Later, the authors carried out the measurements of capacitance and DC current in the Chl films at different temperatures below RT using the sandwich cells with one barrier contact.87 This is the second attempt known to us after the earlier studies of the 1950–1960s, when the temperature sweeps of conductivity were systematically studied to extract Eact. The vast majority of experiments available in literature refers to the heating cycles in a relatively narrow interval of temperatures, ranging from RT to 60–90 °C. Elevated temperatures provoke several undesired, and often instrumentally uncontrolled, processes such as sintering, decomposition or loss of structural water in the aggregates, thin films and bulky crystals of chlorophylls.9–11,23,39,44,69,71,72,75,79 This, in turn, leads to uncertainty and possible misinterpretation of the electrical conduction mechanisms by different authors since the parameters can be calculated or compared adequately only when the samples are chemically and structurally equivalent. Looking to the future, we fear that the (photo-)thermal stability issues will dominate the other when developing the PVC devices based on chlorophylls, as it happened with the hybrid perovskites in 2010s.4,12,50,65 The intramolecular structures however can be relatively stable: the thermal destruction of methyl Pheo was observed only at t0 > 351 °C.26 Any kind of derivatization reduces stability, for example destruction of chlorin e6 begins at t0 > 307 °C.
To avoid the risks associated with the increase in temperature, Leblanc et al.87 used a lower the temperature range, down to −150 °C. Of course, such low temperatures have nothing to do with the practical application in PVC, but can shed light on the mechanisms of charge transport in solid phases with a mixed type of conductivity. The capacitance of the cells at various frequencies was measured and discussed with the focus on the electro-chemical processes in the depletion layer at the Chl/Al interface and with less attention to the bulky processes, despite the film thickness of 300 nm.87 A transition from a voltage-dependent depletion layer capacitance at RT to a voltage-invariant geometric capacitance at −150 °C and vice versa was detected. From this, it was assumed that the temperature facilitates mobilization/immobilization of the charge carriers (holes) from the non-uniformly distributed traps due to the structural imperfections and/or impurities. Further,89 from the J–V curves recorded on 30 nm thick films in the range of −20 to 50 °C, authors revealed “a change in the conduction mechanism in the Chl film around 15 °C”, which was attributed to presence of traps. The structural changes in this range were ruled out judging solely from the UV/Vis spectra.89
Several notable articles describing photophysical properties of porphyrinoids, including Chl and Pheo, in the condensed state (LB films), in solutions and in the form of dyads were published by B. Roeder et al. (Berlin) in 1997–2006.21,31,34 Later, they have extended application of the optical methods and quantum chemical calculations to the zinc phthalocyanines and resolved three N⋯H tautomers in the annulated dinuclear free bases.91 Understanding the electronic transitions in the porphyrins, where symmetry is reduced to D2h due to a couple of central hydrogens, is of practical importance for the identification of the absorption bands in the visible and near UV range.16
A nice and brief introduction to the history of organic photovoltaics including chlorophyll-based devices, as of 2004, was published by Spanggaard and Krebs.83 The fundamentals of the device operation and the basics of heterojunction design can also be found there and in ref. 2, 3, 28, 40 and 92.
An essential progress in the study of aggregation of natural chlorophylls in thin solid films was achieved in the laboratory of A. A. Krasnovsky, whose activity reached its peak in the mid-70th.38 Many aspects of self-assembling, chirality and aggregation of various chlorophyll-like molecules with the accent on the light-harvesting and charge-transporting properties of their micro- and mesoscopic phases are addressed in the reviews.10,14,50,93 We would like to highlight the latest essay of Schmidt and Würthner,93 in which the authors introduce the concept of supramolecular building blocks (or elements) in the description and prediction of the electronic properties.
It goes without doubt saying that the most impressive contribution to the development of variously designed PVC utilizing semi-synthetic chlorophyll derivatives has been made over the past decade by the groups of H. Tamiaki (Kusatsu) and S. Sasaki (Nagahama), often in collaboration with the group of X.-F. Wang (Jilin).4,5,12,27,32,33,35,40–43,52–54,57,58 Very recently, an overview of their results was published in Solar RRL.50 Numerous works containing an extensive collection of experimental data cover almost all possible functions of these molecules in the processes of generation and transport of charge carriers in the modern PVC devices and are repeatedly cited throughout this text.
6. Conductivity in thin films
We dare to assert that, after many decades of research, the electrical properties of chlorophylls in the solid state are still little known. Surprisingly, the fundamental conductivity measurements on the single-crystal samples are not described in the modern literature, in spite of the fact that the preparation of high purity, ribbon-like crystals of Chl with a length of ∼0.1 mm was described as early as in the 1950th (ref. 22, 94, 95 and references therein). The conductivity parameters reported by the different authors vary depending on the sample type, history (purity), measurement method etc. Often, they appear as the supplementary part to the main research theme and suffer from the authors' intention to support the trend they put forward.Usually, thin solid films composed of chlorophyll molecules are poorly conductive (σ < 10−8 S cm−1). This is due to the low charge carrier mobility (μ); the literature values are summarized in Table 1. The majority charge carriers are the holes, but there is no clear indication of whether natural chlorophylls are intrinsic or extrinsic semiconductors.96 In the earlier works,39,69–71,74–83 it was mostly believed that conventional organic dyes with an extended π-conjugated system behave as extrinsic p-type semiconductors due to the oxygen being never fully desorbed from the samples during measurements. Loss of this accepting admixture usually leads to a decrease in the dark conductivity and, less evident, photoconductivity that was thought to be purely electronic primary photophysical process. Continual evacuation or purging with a compensating gas (e.g., hydrogen) would reverse the conductivity type by eliminating oxygen or make the material an insulator. Nonetheless, this concept was vulnerable even at that time. From various evidences, the sign of majority charge carriers may also depend on the aggregation form, presence of a metal atom in the molecular core, another admixture (e.g., water) or the implicit photo-electrochemical processes at the electrodes.
Inclusion of the water molecules in form of a monohydrate or dihydrate affects the structure of the crystalline chlorophyll a films,22,38,39,50,60,63,69,70,76,80,86–88,94,95 so that they electrically resemble molecularly doped polymers rather than vapor deposited glasses.63 Especially for the chlorophylls isolated from the natural raw precursors, there is a question of ‘bound water’ vs. ‘free water’ which have different origin, desorption mechanisms and locations in the bulk sample. The role of water in the supramolecular chemistry of chlorophylls has been intensively debated since the first results of the X-ray analysis of their crystals appeared,22,95 and is thoroughly described in the topical surveys.14,38 We would only like to mention here the unequal coordination activity of metallated/metal-free derivatives with respect to H2O molecules. Thus, hydration of dry Chl films leads to reorganization of molecules relative to each other and is accompanied by a large red shift of Q-band by ∼62 nm. The Soret-band shifts by only 9 nm in the same direction. At the same time, the spectrum of pheophytin a stays unchanged when the film is exposed to water vapors.22
Solvents commonly used to deposit a chlorophyll film by spin-coating are chloroform, alcohols, toluene, chlorobenzene, acetone, dichloromethane and mixtures thereof. Replacement of the organic solvents a film was cast from, or variations in their mixing ratios, can stimulate morphological changes and, consequently, affect the light-harvesting and charge-transporting properties.43,52,97
Though the traces of polar solvents nearly inevitably exist in the films, and the molecules themselves can easily be reduced or oxidized, the absence of ionic motion (e.g., protonic conductivity3) under an applied electric field is disputable. When measuring J–V characteristics of PVC in air, Leblanc et al.62,86,89,90 observed the polarity-dependant dark voltages up to 0.5–0.6 V, which were attributed to the electrochemical reactions at the Chl/metal electrode interface. Significant changes in these dark voltages were observed with a varying sample temperature, residual pressure and over the batch of samples. Keeping in mind the sensitivity of both the dark- and photoconduction to the measurement time (duration, sample age) and conditions in general, repeatedly noted in the literature, it is not unrealistic to assume that some ionic transport may occur in the solution-processed chlorophyll films and contribute to the inertial drift and/or irreversibility of their electrical characteristics.
The more advanced AC measurements on chlorophyll films, termed as electrochemical impedance spectroscopy (EIS), did not reveal the low-frequency signs typical of the ion diffusion.12,42,52 However, the frequency sweep in those studies was limited to 1 MHz to 1 Hz, whereas in such multilevel phases featuring different conduction pathways and grain boundaries, it is always useful to look at the fastest and slowest possible processes. The Warburg-type processes were not taken into account in the equivalent circuit model in ref. 12, 42 and 52. As known, the movement of ionic charge carriers can be detected in the low frequency domain by EIS and quantified by introducing the Warburg diffusion impedance as constant phase element.98,99 A study of the thermally-activated charge transport would also be useful here; unfortunately the measurements of DC or AC conductivity in thin films or individual aggregates of chlorophylls at low temperatures are very scarce – Section 5.
On the other hand, according to Corker and Lundström72 the standard Chl film obtained by the same technique and similarly characterized in Cr(or Au)/Chl/Hg sandwich cells as in Tang's or Leblanc's works should be regarded as an insulator with a large number of traps, rather than a semiconductor. Unlike ref. 87–89, these authors differentiated between the bulk and interface regions for the approximately 0.5 μm thick Chl film. In the bulk region, holes are in excess of their thermal equilibrium concentration due to the presence of trapped electrons. The interfacial region is depleted of holes. In the freshly prepared films the conductivity in the bulk of a Chl film is high, and the resistance of the cell is primarily due to the interfaces. As the film discharges, the depletion region extends across the cell and the resistance increases by orders of magnitude. In the cells having the rectifying Cr contact (Au was ohmic), the interfacial barrier width was estimated at about 45 nm.
Authors argued that their molecular films are not at the equilibrium when as-prepared or after the illumination cycle.72 In the dark, changes within the films cause the DC and AC impedances of the sandwich cells to increase with age. Apart from (or together with) electronic processes, soft annealing slightly above 30 °C or simply ageing initiates structural transformation. In this work, as well as in many others, the coexistence of monomeric, oligomeric and bulky (here, microcrystalline) forms of chlorophyll molecules in the film was hypothesized.
Noteworthy results were obtained by Li et al.,4,12 who compared three structurally similar zinc-complexes of bacteriochlorin, chlorin, and porphyrin that feature different degree of conjugation of the central chromophore, while all peripheral groups remain the same. Among them, thin films of a highly conjugated porphyrin derivative showed the highest degree of crystallinity and highest broadening of the Soret band concomitant with the strongest increase in the intensity of Q-band relative to the monomeric state. This suggests that the extension of the π-conjugation contour is another and quite promising tool for managing the properties of aggregates. Unfortunately, the authors aimed at inserting these materials as a hole-transporting component in the perovskite-based PVC, and a hasty conclusion about “a weak p-type characteristic” was made for the porphyrin derivative, based on the estimation of the relative HOMO–LUMO positions.
It is practically important to look into the mobility of charge carriers in the closest analogues and competitors, zinc phthalocyanines. Their ordered thin-film phases obtained from a solution exhibit slightly greater μ values than those measured for the films of chlorophyll aggregates – Table 1. With the use of the vacuum evaporation technique, epy mobility of both holes and electrons in the film can be tuned by optimization of the growth surface and conditions during the deposition in a wide range up to μh > 10−2 cm2 V−1 s−1 (Table 1). For certain metal-phthalocyanine complexes, quite impressive values of 1–3 cm2 V−1 s−1 were obtained in the FET configuration – see, ref. 51 for comparison of epy data over several classes of organic dyes.
The trump card of the chlorophylls is their ability to organize themselves in such a way as to ensure an efficient transfer of energy (but not exactly charge) from one molecule to another, which is not easily achievable with the vacuum-deposited phthalocyanines. For instance, the conductivity along the hollow nanotubes formed by the self-organized semisynthetic bacteriochlorophyll aggregates amounts to 0.009 S cm−1.11 Note, that the latter are very specific objects randomly distributed on the SiO2 wafer, like in Fig. 10, which are accessible for characterization solely by the conductive AFM tip.9–11,61 There are no indications that it is possible to obtain such organized structures as compact thin films for use in the conventional solid-state PVC.
Metal-free Pheo molecules bearing two cyano-groups at the periphery can function as acceptors when paired with carotenoids (lycopene) or phthalocyanines,33,41 i.e. their thin films should behave as n-type semiconductors. The devices containing lycopene:131-deoxo-131-(dicyanomethylene)pyropheophorbide a BHJ with mixing ratio 1:4 generated a fairly decent photovoltage of 0.85 V – Table 2. The absorption spectrum of this tetrapyrrolic dye showed an expanded Soret-band, and an increased photon-to-electron conversion efficiency was measured in the corresponding devices. However, the authors33 used calcium directly deposited on an organic film as the top electrode without subtracting/appending the contribution of this highly rectifying interface from the output characteristics, which makes their photovoltaic records ambiguous. The same concerns the inverted PVC with a scheme ITO/Ca (1 nm)/Pheo derivative (8 nm)/CuPc (20 nm)/MoO3 (5 nm)/Al (100 nm), which showed illuminated J–V plots typical of a Schottky-type cell.108 It is no wonder that the FF values in such devices were less than 0.28, with the PCE of only a few tens of a percent.
| Photovoltage, V | Junction typea | Compounds in junction | Ref. |
|---|---|---|---|
| a As claimed by the authors.b [6,6]-Phenyl C71 butyric acid methyl ester. | |||
| 0.50 | PHJ | Methyl-bacteriopheophorbide a/C70 | 32 |
| 0.36 | PHJ | Methyl-pyrobacteriopheophorbide a/C70 | 32 |
| 0.66 | PHJ | Methyl-pheophorbide a/C70 | 32 |
| 0.64 | PHJ | Methyl-pyropheophorbide a/C70 | 32 |
| 0.54–0.55 | BHJ | Lycopene:32,32-dicyano-pyropheophorbide a | 33 |
| 0.81–0.85 | BHJ | Lycopene:131-deoxo-131-(dicyanomethylene)-pyropheophorbide a | 33 |
| 0.64 | PHJ | H2P (see, Table 1)/C70 | 40 |
| 0.60 | PHJ | ZnP/C70 | 40 |
| 0.83–0.90 | BHJ | H2P:PC71BMb | 40 |
| 0.83–0.76 | BHJ | ZnP:PC71BM | 40 |
| 0.53(0.63) | PHJ | CuPc/32,32-dicyano-pyropheophorbide a (methyl-131-deoxo-131-dicyanomethylene-pyropheophorbide a) | 41 |
| 0.87(0.71) | PHJ | 32,32-dicyano-pyropheophorbide a (methyl-131-deoxo-131-dicyanomethylene-pyropheophorbide a)/C70 | 41 |
| 0.66–0.81 | Single layer | Synthetic dyad with fullerene derivatives, e.g. metal-free (bacterio)chlorin-C3-methyl, phenyl-C61-butylate | 53 |
| 0.49–1.07 | BHJ | Dyad:PCBM | 53 |
| 0.33(0.49) | PHJ | Cu-metallated trimethyl ester of chlorin e6 (metal-free trimethyl ester of chlorin e6)/C70 | 6 |
| 0.73–0.80 | BHJ | Cu-metallated trimethyl ester of chlorin e6 (metal-free trimethyl ester of chlorin e6):PC71BM | 6 |
| 0.70–0.74 | PHJ | Zinc dodecyl 131-deoxo-3-devinyl-131-dicyanomethylene-3-hydroxymethyl-pyropheophorbide a/PC71BM | 42 |
| 0.72–0.74 | BHJ | Zinc dodecyl 131-deoxo-3-devinyl-131-dicyanomethylene-3-hydroxymethyl-pyropheophorbide a:PC71BM | 42 |
| 0.55–0.70 | Combined BHJ/PHJ | TiO2:methyl-trans-32-carboxy-pyropheophorbide a/zinc 3,5-bis-(dodecyloxy)-benzyl, 3-devinyl-3-hydroxymethyl-pyropheophorbide a | 43,52 |
| ∼0.2–0.5 | Schottky | Chl/metals | 80–82 |
| 0.25–0.40 | Schottky | CdS/Chl//Ag | 86 |
| 0.4–1.1 | Schottky | Chl/Al | 90 |
| 0.48 | Schottky | Sodium copper chlorophyllin/Al | 100 |
| 0.7 | PHJ | P3HT:Chl | 101 |
| ∼0.17 | BHJ | Ferrocene:Chl | 102 |
| 0.6 | BHJ | P3HT:PCBM:Chl | 103 |
| 0.55–0.64 | PHJ or BHJ | Various ZnPc/PCBM or ZnPc:PCBM mixtures (vacuum deposition and solvent treatment) | 97 |
| 0.507–0.517 | Graded HJ | Various ZnPc:C60 ratios in the mixed/graded heterojunctions containing ZnPc aggregates |
104 |
| 0.75–0.84 | BHJ | 1,4,8,11,15,18,22,25-Octahexylphthalocyanine:PCBM | 105 |
| 1.11–1.3 | PHJ or BHJ | Boron chloride subphthalocyanine:C60 (or C70) vacuum-deposited bilayer or blended junctions | 106 |
| 0.53 | PHJ | Glass/ITO/MoO3/ZnPc:C60/Mg:Ag | 107 |
Another hint at a possible n-type conductivity in chlorophylls can be found in ref. 101. Poly-3-hexyl-thiophene (P3HT) served as an electron donor for the spin-coated PHJ or BHJ, while Chl was claimed to be an acceptor – Table 2. The PHJ-based devices with the top Al electrodes had poor characteristics except probably for Voc, and those with BHJ were photovoltaically inactive. Authors did not consider the intrinsic absorption of the photons in a tetrapyrrolic dye and discussed the photoconversion process only in terms of transfer of the photoinduced electrons from P3HT to Chl. The Schottky-type contact at the Chl/Al interface, possibly responsible for rectification and photovoltaic activity of thus made devices, remains ignored.101 Even worse results were obtained for the devices with the ferrocene:Chl blend.102
Introduction of the electron-withdrawing groups or atoms on the periphery of the macroheterocycle is a common approach used to obtain n-type conductivity in molecular solids, which has been studied in great detail for the halogen-substituted phthalocyanines (ref. 51, 67, 92 and references therein). There are no such credible and in-depth studies on the chlorophyll dyes. For a deeper insight into their (semi-)conducting properties, the effects of electronegativity, position and number of peripheral substituents on the electronic structure of the solid should be addressed in the future research.
The concentration (or density) of the mobile charge carriers N, another parameter contributing to the σ value,3 is by far less frequently reported for the semiconductors of chlorophyll-type than μ. In various sources, this value is reported to be ∼1021 m−3,9 2.5 × 1023 m−3,37 ∼1024 m−3,61 2 × 1023 m−3.88 The density of molecules calculated from XRD is equal to 1021 cm−3 for the crystals of ethyl chlorophyllide dehydrate; the Chl crystals are less densely packed (1.08 g cm−3 vs. 1.30 g cm−3).22 Therefore, the amount of mobile charges is, very roughly, at least four orders of magnitude less than the number of molecules able to generate them, and the impurity mechanism of conduction could be expected.72 Comparing the conductivity in the crystals of chlorophyll a and b, authors in ref. 79 postulated that the mobility of the charge carriers is the same, and the density of dark current through the samples is governed by the number of charge carriers, which is larger in chlorophyll a due to the lower activation energy. The precisely controlled introduction of an electrically active impurity (dopant) has not been implemented in any work, which greatly complicates the discussion.
To sum up, a widely publicized penchant of chlorophylls for self-assembly does not clarify the picture of electrical conduction in their 3D phases, such as films. Spontaneously formed assemblies of various shapes and dimensionality are likely to build up the conductive pathways within themselves, but their mosaic mutual orientation and physically indefinite interfaces with the electrodes reduce this effect to zero. That is probably why many authors do not go beyond speculations on “less ordered” or “better ordered aggregates” based on conventional AFM, SEM or XRD studies of films in an attempt to link their conductivity with morphology. As a first step towards more reliable results, it would be better to start with amorphous (fully disordered, monomeric) films and then gradually induce ordering in them with epy help of some external trigger as it is done, e.g. for phthalocyanines by functionalizing the surface of a channel in FETs or by annealing.51
7. Photovoltaic devices
In contrast to the ample data on the Graetzel-type cells (or DSSC) incorporating chlorophylls, there are few works dealing with the thin-film PVC that use classical PHJ or BHJ based on these molecules (Table 2). The possible reasons for this are as follows:• Low mobility of charge carriers in the bulk phase, as follows from Section 6 – Table 1. It is a general drawback of molecular solids formed by the weak van der Waals forces, which lessens the density of currents leaking both in the dark and under illumination and, therefore, deteriorates the output parameters of PVC. The most common measure that will help to overcome this problem is to create ordered molecular structures containing channels with high mobility of charge carriers, such as closely packed 1D molecular stacks.
• A meager selection of materials/methods for the device fabrication. Although the problem of electrical contacts to the supramolecular aggregates was evident right from the pioneering works of Tang and Leblanc on the Schottky-type cells, not enough attention is paid to the inorganic (or organic) materials that are interfaced with the chlorophylls in a particular device scheme. Many authors borrow the functional envelope, i.e., charge transporting layers or electrode materials, from the parallel works on organic PVC employing conventional small-molecule donors like phthalocyanines or thiophenes. However, those PVC layouts have been developed with account for structural, redox and conducting properties of the specific donor, and are not necessarily suitable. Development of new interfacial materials adapted for chlorophylls might increase the efficiency of devices.
• Few tools were applied to manage the device parameters at the molecular level. Semi-synthetic methods yield metal-free, zinc or magnesium complexes. We did not find any works in the literature on the PVC with, for example, non-planar transition metal chlorophylls – Tables 1 and 2. By analogy with phthalocyanines, conically-shaped molecules with heavy metal atoms should exhibit nontrivial crystalline ordering16,51 and, hence, a prominent (photo-)conductivity. A systematic study of such complexes as photoabsorbers will complement the picture of the synthetic approach to engineering of solid-state PVC.
• Nothing is said about the stability of the photovoltaic performance as a function of storage or operation time, which was assessed just as low in the above works as in the studies dating back to the 1960–70s.
Maximum photovoltage is the fundamental, albeit not decisive, parameter of organic PVC, as it reflects the offset of the energy levels necessary for separation of the charge carriers at the junction and, thus, the proper selection of a D/A-pair.38,39,63,64,92,106,109,110 In Table 2, the data found in the literature for the devices with Chl, Pheo and their derivatives are collected, including the pairing material, either acceptor or donor. The relevant references should be carefully examined to see the full device architecture, because the schematic of cells chosen by the authors may not only downgrade the measured Voc, but also interfere with the understanding of the observed photovoltaic effect.92,106,108–110
The photovoltage in the majority of PVC fluctuates within the range of 0.6–0.8 V, with no apparent correlation with the junction type – Table 2. The presence of a metal atom in the tetrapyrrolic ring has an ambivalent effect. Metal-free chlorin e6 trimethyl ester paired with fullerene in either PHJ or BHJ gives greater Voc than its complex with copper, while the mobility of charge carriers in its pure phase is lower; the same is valid for the purpurin complex with/without zinc6,40,42 – Section 6. This could be explained by the overlap between the chlorophyll molecule ability to form regular aggregates with good conduction and the interaction in the D/A pair at the heterojunction.
The most critical parameter of a solar cell, short circuit current Jsc, depends on the device scheme to such an extent that correlation with the intrinsic material properties is hardly possible. The champion PCE of 5.2% showed the devices with a solution-processed PHJ where the Zn-metallated Pheo derivative is paired with the fullerene derivative PC71BM as acceptor42 (Tables 1 and 2). The chlorin macrocycle here was modified by an electron-accepting dicyanomethylene group at 131 position and by a long dodecyl chain at the C17-propionate residue (Fig. 3). The boost of the overall efficiency was attributed to an enlarged photocurrent contribution of PC71BM and to a better aggregation retention in the PHJ structure compared to the analogous BHJ-based devices.
Several somewhat odd and contradictory facts are reported in this work.42 First, the devices with a bilayer D/A structure (PHJ) outperformed the devices with a photoactive blend (BHJ) (normal scheme: ITO/MoO3 (10 nm)/PHJ or BHJ/BCP (10 nm)/Ag (100 nm), PHJ = Pheo (22 nm)/PC71BM (50 nm); BHJ = 69 nm). Notwithstanding this fact the viability of a PHJ layout for the all-solution processed devices raised doubts and rightfully so.
It has long been clear that sequential deposition of molecular films by spin-coating inevitably leads to at least partial dissolution of the uppermost layers in the film that was deposited first. This was nicely illustrated in the earlier research of Brett et al.,97 in which the same solvent used to spin-coat the overlying fullerene layer was also used to treat the underlying ZnPc layer in order to trace its possible transformation during the PHJ fabrication (see below). That is why the truly PHJs are usually made from vacuum-deposited organics.
And indeed, if the bottom layer of Pheo in ref. 42 was deposited from tetrahydrofuran:chloroform 3:1 solution and the top layer of PC71BM was deposited from chloroform:chlorobenzene 2:1 solution, then no sharp D/A interface in the resulting bilayer film was found. Instead, there was a compression of the bilayer film relative to the nominal sum of the thicknesses of individual films. From this it follows that the “bilayer film”42 contains very likely an unwittingly formed “graded” heterojunction, the concept of in-between PHJ and BHJ.104 For the small molecule organics, the graded HJ seems to have been the most effective approach in the thin-film photovoltaic devices to date. It requires appropriate D/A intermixing along the vertical concentration gradient at the level of individual molecules, but not excluding their aggregation.104
Therefore, the aggregation state of any component of the junction should be evaluated.111,112 The authors in ref. 42 found very insignificant spectral changes that might suggest an enhancement of the Pheo aggregation in the PHJ relative to BHJ and an untreated chlorin film but left the second component, PC71BM unattended. Meanwhile, it is well-known that a properly selected solution chemistry increases the dispersion degree of the fullerene in the blend, which promotes a better D/A phase separation throughout a BHJ.111,112 The BHJs in ref. 42 were prepared by mixing the chlorin dissolved in tetrahydrofuran and PC71BM dissolved in chlorobenzene. The devices based on these BHJ showed PCE of 3.5–4.0%, which is less than that for devices with PHJs. It is hence very likely that, Pheo itself does not contribute to the improvement of performance of the devices with PHJ. The real reason could be an inhibited aggregation of PC71BM in the pure solvents that were used for making bilayer films. In fact, the authors observed an increased signal from the fullerene photoabsorber in the photocurrent action spectra but assigned it to the “shallow pores” on the surface of the solvent-treated Pheo film seen with AFM (surface roughness does not change).28 Note, that the surface of the bottom film before depositing the acceptor layer is irrelevant for the graded HJ since it is dissolved; the emphasis should be on the distribution/intermixing of both components throughout the photoactive layer (depth profiles).97,113
Second, replacement of the interfacial materials (both cathode and anode buffers) did not practically change the output parameters of PVC including, very surprisingly, Voc42 (inverted scheme: ITO/ZnO (25 nm)/PHJ or BHJ/MoO3 (10 nm)/Ag (100 nm), see above). This was unexpected even for the authors themselves, who explained such insensitivity by “ambipolar characteristics of the chlorophyll aggregate”. However, when they replace Pheo derivative by its analog bearing 131-oxo group and lacking 131-dicyanomethylene group (that is the feature responsible for ambipolarity!) the insensitivity of the Pheo/PC71BM junction to the buffers persists.
This situation deserves attention because it illustrates how little is known about interfaces of chlorophyll aggregates with other functional materials. While the standard planar interface between fullerene and MoO3 should give a Schottky junction,6,106,110 the n-ZnO/p-Pheo interface is of a rather complex nature that has been ignored in ref. 42. As known, ZnO helps to realize an ohmic contact between the electrode and n-type semiconductor-fullerene107 but it is supposed to act in the opposite way when interfaced with porphyrins, which are mainly p-type. We may very tentatively assume that the observed anomaly is due either to serious experimental inconsistencies or a much higher degree of intermixing within the entire organic/inorganic stack between the charge-collecting electrodes. In the latter case, this would result in a p–i–n-like structure described by Hiramoto et al.110 assuming that both molecular semiconductors in ref. 42 are unintentionally doped by the respective buffer materials.
A typical misconception here and in some other cited articles is that the authors rely on the SCLC technique to extract the value of μ and to conclude on the conductivity type, but deal with the samples where none of the contacts is ohmic.96 For example, MoO3 (in fact, non-stoichiometric MoOx) is a degenerate n-type semiconductor with deeply lying energy levels and work function of about 6.8 eV.106,108,113 Its conductivity and energy structure drastically change with the thickness and deposition/measurement conditions, so the ohmicity must be proven in every single case. The same concerns ZnO, BCP, reactive metals etc. In ref. 6 and 41 the authors are aware of this problem admitting the presence of Schottky barrier at the MoO3/chlorophyll interface, but disregarding it elsewhere. Next, when measuring the sandwiches with a two-component D/A film, the homogeneity of the blend must be documented. Otherwise, a truly, phase-separated BHJ will contain different conductive paths for the charge carriers92 and, hence, will be different mobilities within the respective phases. Last, the mobility measured in a bilayer film sandwiched between two flat electrodes has no physical meaning.
A few months earlier the same authors investigated the ITO/MoO3 (5 nm)/PHJ(BHJ)/BCP (8 nm)/Al (100 nm) devices, where PHJ = chlorin derivative (5 nm)/C70 (40 nm), and BHJ = chlorin derivative:PC71BM blend (40 nm).6 It was found out that for the PHJ-based devices PCE does not exceed 1%, while for the BHJ-based devices PCE increases to >2%. The progress was explained by the enlargement of the D/A interface at strongly non-uniform blending ratio of 1:8. Perhaps, a distinct feature here is that the compact, disk-like chlorin molecules without side chain were used. As can be presumed from the broadening and shift of the bands in the electron absorption spectra,6 such molecules aggregate worse (unfortunately, the reference spectra for the tile-free chlorin derivatives have been recorded in chloroform, while for Zn-pyropheophorbide a derivative with a dodecyl side chain – in tetrahydrofuran, in the former the solvent-induced aggregation may distort the reference spectrum). Again, the structure of chlorophyll dyes seems to affect the output characteristics of the solution-processes PVC through different aggregation states. More efforts should be spent to isolate the effect of the molecular architecture on the length of an aggregate from the morphological aspects related to the entire (bilayer or blended) heterojunction.
In ref. 16 all components of PVC except for methyl(bacterio)Pheo were deposited by thermal evaporation in vacuum. In thus made ITO/MoO3 (5 nm)/(B)Pheo (10 nm)/C70 (40 nm)/BCP (8 nm)/Al (100 nm) device a moderate efficiency in the range of 0.96–1.63% was obtained. An amazing point is the thickness of donor. The authors insist that the exciton and the charge carrier diffusion length in their chlorophyll aggregates is short,32,50 despite the lack of convincing experimental evidences (except for the flatness of molecular film approaching 0.31 nm, by AFM). Beware of this, they maintain the thickness of the first photoabsorber layer at the level of 10 nm, thereby losing a fraction of the incoming photons. For comparison, the maximum lateral size of a zinc chlorin is 3.5 nm with dodecylalkoxy chains and 1.9 nm without them.48 From geometrical considerations, a 10 nm thick film would consist of about 5 and 9 lateral stacks of vertically aligned molecules, respectively. For the standing-on stacks, thickness of 10 nm corresponds to ∼30 horizontally lying molecules with typical distance of 0.34 nm between flat neighboring porphyrin units.10 Given the random distribution of molecular stacks, a certain fraction of the stacked assemblies consisting of more than 30 molecules should cross the nominal layer and bridge the electrodes. Yet, even for such a short aggregation length authors speculate about good or bad ordering in the films refusing to believe that excitation transfer occurs on a film thickness scale. Curiously enough, in their earlier work4 the same authors referred to the films of similar chlorophylls (Fig. 8) as “highly crystallized”, and the films with a thickness of 20 nm served as “efficient hole transporters” in solar cells.
In the case of DSSC, an interesting attempt to tie the output parameters to the chemical structure of the compounds from the pheophorbide family is described.35 When comparing a set of molecules having slightly different structures and physicochemical properties, a clear dependence of the photocurrent Jsc on the Qy absorption as well as on the one-electron oxidation potential was observed. Unluckily, the obtained results and suggestions they make concern the electron transfer to/from TiO2,35 which is the basic component of DSSC – Fig. 2. The stability tests revealed an approximately two-fold drop in the Qy-band intensity during 1 h illumination with simulated sunlight for the Pheo sensitizer deposited on an 8 μm TiO2 film and a decrease in PCE by a quarter after 24 h.
In general, the photovoltaic efficiency of the chlorophyll-based PVC does not differ much from the analogous devices that incorporate their nearest congeners – semiconducting zinc phthalocyanine films.97,104,107,109 Below we offer a couple of the interesting examples.
The mesogenic peripherally modified phthalocyanine without metal was paired with the phenyl-C61-butyric acid methyl ester (PCBM) acceptor to obtain a solution-processed BHJ.105 In the pure crystalline phase composed of discotic phthalocyanine molecules, the drift mobility of holes and electrons amounts to 1.4 and 0.5 cm2 V−1 s−1, respectively (by TOF, at −15 °C), with the strong temperature dependence for μh. These values cannot directly be compared with the values shown in Table 1, since they were obtained for an array of crystals in the bulk sandwich cells with two ITO electrodes spaced 12.5 μm apart, i.e., not for thin films. In the thin-film devices, the best performance was reached through the reinforced crystallinity and phase separation in the blended films caused by the treatment with toluene and chloroform as main solvents and 1,8-diiodo(chloro, bromo)octanes as processing additives.105 Maximum PCE of 4.2% was reached in the normal ITO/MoOx/BHJ/LiF/Al cells at Voc = 0.78 V – Table 2. Materials and methods used in this work closely resemble the above mentioned works on the aggregates of Pheo derivatives, and the performance metrics are quite similar.
Solvent treatment was also engaged in fabrication of both PHJ and BHJ with a ZnPc/PCBM pair.97 By using the glancing angle deposition in vacuum (GLAD), authors succeeded in growing films composed of slanted nanorod arrays of ZnPc, with a nanorod diameter of nominally 40 nm and length up to 450 nm – cf.10,11,46,47,55. Since the conditions of vacuum deposition are strongly nonequilibrium, the classical (slow) “self-assembling” scenario cannot be applied. However, the high symmetry and compact lattice structure of the dye allow a long-range ordering of molecules to occur, as shown in Fig. 11(a). The nanorod arrays entirely cover the substrate surface in a “grass lawn” manner,97,114 which makes future fabrication of a thin-film PVC much easier than in the case of randomly distributed chlorophyll nanorods (Fig. 10). Moreover, the shape of the nanorods in Fig. 11(a) begins to mimic an ideal comb-like structure of a heterojunction predicted from the viewpoint of balance between the exciton diffusion length and maximum light harvesting.
 | ||
| Fig. 11 Scanning electron microscopy images: (a) aerial and cross-sectional view of the slanted post (SP) ZnPc thin film preceded by a 10 nm planar layer grown at normal incidence immediately prior to GLAD deposition, α = the angle of incidence. Substrate is a glass/ITO slide coated with a 40 nm poly(3,4-ethylenedioxythiophene) polystyrene sulfonate (PEDOT:PSS) layer. (b–d) Aerial views of the solvent-treated ZnPc thin films: (b) 65 nm ZnPc film growth in a planar configuration and treated by placing four drops (∼200 μL) of dichlorobenzene on the film surface from a glass pipet and spinning the solvent off at 1000 rpm; (c) 65 nm ZnPc film treated by bathing the sample in 5 mL of chlorobenzene in a glass Petri dish for 10 min and then allowing the solvent to volatilize off the film under ambient conditions after removal from the bath; (d) the same treatment of 10 nm planar + 90 nm SP films. Reprinted with permission from ref. 97 copyright 2011 Elsevier. | ||
The mobility of charge carriers in such vacuum-deposited ZnPc nanorods was not evaluated.97 We may refer to ref. 114, where visually similar nanorods on the ITO surface were growth from the methanol/tetrahydrofuran solution of the amide armed ZnPc–COOH molecules. The σ value in the straight-standing nanorods assessed by C-AFM corresponds to ∼10−3 S cm−1 and μh to ∼10−3 cm2 V−1 s−1 (by SCLC) – cf. Table 1.
Next, the vacuum-deposited ZnPc films were subjected to treatment by various solvents, which corresponds to a rather steady-state regime of crystallization.97 ZnPc recrystallizes regardless of the initial morphology, yielding a variety of possible nanoscale objects whose shape depends on the type of treatment, solvent used and exposure time – Fig. 11(b–d). The absorption spectra of solvent-treated films showed even greater changes in the Q-band profile than those seen for the chlorophylls in various states of aggregation, but the XRD pattern changes slightly. Similar pictures were observed elsewhere for other metallo-phthalocyanines that initially were deposited by thermal evaporation in vacuum under controlled equilibrium conditions.51 It should be kept in mind that, unlike intrinsically soluble chlorophylls, conventional phthalocyanines have limited solubility in the organic solvents used for treatment. This, again, indicates a higher degree of freedom in the short-range molecular motion due to, say, partial dissolution facilitates growth of sub-micron assemblies of metallo-porphyrin dyes, in the form of regular columns, flakes or more complex fractal objects.
Finally, variously prepared ZnPc/PCBM heterojunctions were tested in PVC with the ‘ITO/PEDOT:PSS/PHJ(BHJ)/BCP/Al’ scheme,97 where PEDOT:PSS is a transparent conducting polymer, poly(3,4-ethylenedioxythiophene) polystyrene sulfonate, BCP is bathocuproine. A larger thickness of the structured/treated donor layer was assumed to elevate the series resistance in the devices, which limits FF and the resulting efficiency. The best results were obtained for the thinnest, structured, but solvent-untreated ZnPc films. PCE of 3.0% at Voc = 0.63 V (Table 2) was measured for devices based on the ZnPc layer with a composition of 10 nm planar + 20 nm SP at α = 75 and PCBM deposited from dichlorobenzene, in accordance with the morphological model of a structured heterojunction.97 This PCE value appears lower than might be expected, due to the non-optimal combination of the functional materials in the device.
The most effective porphyrinoids for photovoltaics are non-planar phthalocyanines, basically near-IR absorbing transition-metal complexes with axial ligation or boron complexes with contracted macrocycle – subpthalocyanines (SubPcs). Several laboratories successfully work on the development of vacuum-deposited and solution-processed devices, both can easily be realized with SubPcs.92,106,109 The photovoltages in the single-junction PVC with SubPc as either donor or acceptor usually exceed 1 V, provided that other functional materials have correctly been adapted – Table 2. Clear understanding of the conduction mechanisms in SubPcs, including doping, intensified by a good knowledge of molecular properties and phase structure, including thin-film morphology, allows fabrication of PVC prototypes with efficiencies exceeding 6% for the normal single-junction cells and 8% for the cells with a cascade junction. For chlorophylls, the control over the morphology of the photoactive layer still seems rather accidental, the possibilities of solvent treatment, annealing, co- or re-crystallization and templating should be investigated in more detail.
8. Concluding remarks
Our findings regarding the current accomplishments in the field of photovoltaic cells based on chlorophylls in the form of thin films are of a rather negative nature. In spite of apparent compliance of natural tetrapyrrolic dyes with the idea of photovoltaic conversion of solar energy, the manufacturing of the ready-to-use solid-state PVC remains a challenging task.Isolation of single molecules from raw materials and their further introduction as an active semiconductor layer does not seem to have brought competitive results so far. Most likely, the reason is a very peculiar microscopic organization of chlorophylls in vivo through self-assembling of molecules from the (semi)liquid phase. Thus formed assemblies make the energy transfer and charge separation, which are the steps following the excitation of a molecule by light, more efficient and targeted. Without such a smart assembling, electronic conduction and excitonic transport in the bulk phases of chlorophyll molecules become inferior to those of the well-ordered artificial (fully synthetic) dyes belonging to the same porphyrin family, e.g. phthalocyanines.
On the other hand, the attempts to construct more or less “biomimetic” chlorophyll assemblies and then integrate them ‘in whole’ into the classical PVC scheme with a planar or bulk heterojunction encounter serious experimental difficulties. (Properly speaking, natural photosynthetic organisms do not contain pure dye systems, the cell dyes are bound with proteins and lipids in a network for serving energy transport and distribution). The fragile structure of supramolecular aggregates tends to break down when undergoing standard thin-film deposition and/or processing procedures; and the morphology of the semiconductor layer becomes rather vague. An effect of residual solvents, primarily water, is not understood well enough. The temperature and current-induced stresses should be studied adequately, since they always happen during the PVC operation under load.
The next open issue is a narrow-band absorption in the blue and red. These molecules are ideally synthesized by the Nature to harvest irradiation of the Sun due to the expanded but stable cyclic chromophore that provides intensive absorption of the incident photons. However, there is a green gap, the existence of which is important for all leaving organisms. Even with broadening of the main bands due to aggregation, natural chlorophylls and their synthetic analogs cannot provide themselves with a panchromatic photoconversion that is desired for use in PVC. If so, their combination with other photoabsorbers becomes necessary. Here, the problem of compatibility between pairing materials in the heterojunction arises, since the complementary photoabsorber has its own structural features and redox activity. Friendly carotenoids cannot help because of the narrow absorption bands and poor solid-state properties. An ideal case would be a PVC with a single photoabsorber: a dyad or hybrid system reconciling a broad-band optical absorption and dual structure that enables the effective exciton dissociation/charge transport (what was once the advantage of the hybrid perovskites). In this respect, development of completely new, robust and affordable synthetic routes would be rewarding.
The demonstration of a working and stable PVC with the chlorophyll-like molecules as an active material, in any junction geometry and with any conductivity type, would be the most elegant solution in the field of the green energy at the current stage of technological evolution. This challenge stimulates further studies of not only “soft” methods for processing of organic or hybrid photovoltaic systems, but also of the fine supramolecular and mesoscopic structure of these fascinating macroheterocycles.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was supported by RSF grant #20-13-00285. Authors express their sincere gratitude to Prof. D. V. Belykh (Komi Science Center UB RAS) for providing the UV/Vis spectra and to A. I. Koptyaev for many helpful discussions.References
- M. Irimia-Vladu, E. D. Glowacki, N. S. Sariciftci and S. Bauer, Green materials for electronics, Wiley, Weinheim, 2018 Search PubMed.
- V. Vohra, Materials, 2018, 11, 2579 CrossRef CAS PubMed.
- N. Amdursky, E. D. Głowacki and P. Meredith, Adv. Mater., 2019, 31, 1802221 CrossRef PubMed.
- M. Li, Y. Li, S. I. Sasaki, J. Song, W. Chen, H. Tamiaki, W. Tian, G. Chen, T. Miyasaka and X.-F. Wang, ChemSusChem, 2016, 9, 1–9 CrossRef.
- Y. Li, W. Zhao, M. Li, G. Chen, X. F. Wang, X. Fu, O. Kitao, H. Tamiaki, K. Sakai, T. Ikeuchi and S. I. Sasaki, Chem.–Eur. J., 2017, 23, 10886–10892 CrossRef CAS PubMed.
- S. Wang, S. Duan, Y. Wang, C. Sun, X. F. Wang and S. I. Sasaki, J. Energy Chem., 2019, 38, 88–93 CrossRef.
- G. Calogero, G. Di Marco, S. Caramori, S. Cazzanti, R. Argazzi and C. A. Bignozzi, Energy Environ. Sci., 2009, 2, 1162–1172 RSC.
- V. Y. Pavlov and G. V. Ponomarev, Chem. Heterocycl. Compd., 2004, 40, 393–425 CrossRef CAS.
- S. Patwardhan, S. Sengupta, L. D. A. Siebbeles, F. Würthner and F. C. Grozema, J. Am. Chem. Soc., 2012, 134, 16147–16150 CrossRef CAS PubMed.
- S. Sengupta and F. Würthner, Acc. Chem. Res., 2013, 46, 2498–2512 CrossRef CAS PubMed.
- S. Sengupta, D. Ebeling, S. Patwardhan, X. Zhang, H. Berlepsch, C. Böttcher, V. Stepanenko, S. Uemura, C. Hentschel, H. Fuchs, F. C. Grozema, L. D. A. Siebbeles, A. R. Holzwarth, L. Chi and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6378–6382 CrossRef CAS PubMed.
- M. Li, N. Li, W. Hu, G. Chen, S. I. Sasaki, K. Sakai, T. Ikeuchi, T. Miyasaka, H. Tamiaki and X.-F. Wang, ACS Appl. Energy Mater., 2018, 1, 9–16 CrossRef CAS.
- A. Y. Rybkin, A. Y. Belik, P. A. Tarakanov, K. R. Taziev, A. V. Kozlov, N. S. Goryachev, I. V. Sulimenkov, V. I. Kozlovskiy, Y. V. Romanenko, O. I. Koifman and A. I. Kotelnikov, Macroheterocycles, 2019, 12, 181–186 CrossRef CAS.
- T. S. Balaban, H. Tamiaki and A. R. Holzwarth, Top. Curr. Chem., 2005, 258, 1–38 CrossRef CAS.
- B. Grimm, R. J. Porra, W. Ruediger and H. Scheer, Chlorophylls and bacteriochlorophylls: biochemistry, biophysics, functions and applications, Springer, Dordrecht, 2006, pp. 67–77 Search PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, Handbook of porphyrin science, World Scientific Press, Singapore, 2010, vol. 9, pp. 1–650 Search PubMed.
- K. M. Kadish, K. M. Smith and R. Guilard, Handbook of porphyrin science, World Scientific Press, Singapore, 2010, vol. 13, pp. 253–97 Search PubMed.
- K. M. Smith and M. O. Senge, Z. Kristallogr. - Cryst. Mater., 1992, 199, 239–248 CrossRef.
- M. O. Senge and K. M. Smith, Photochem. Photobiol., 1991, 54, 841–846 CrossRef CAS.
- M. O. Senge, K. R. Senge K and K. M. Smith, Z. Naturforsch., B: J. Chem. Sci., 1995, 50, 139–146 CrossRef CAS.
- I. Eichwurzel, H. Stiel and B. Roeder, J. Photochem. Photobiol., B, 2000, 54, 194–200 CrossRef CAS.
- K. Ballschmiter and J. J. Katz, Biochim. Biophys. Acta, 1972, 256, 307–327 CrossRef CAS.
- C. Weiss, H. Kobayashi and M. Gouterman, J. Mol. Spectrosc., 1965, 16, 415–450 CrossRef CAS.
- Y. Kawamoto, Y. Kinoshita and H. Tamiaki, Tetrahedron, 2020, 76, 130948 CrossRef CAS.
- L. I. Mazaletskaya, N. I. Sheludchenko, I. S. Tarabukina and D. V. Belykh, Pet. Chem., 2014, 54, 309–315 CrossRef CAS.
- D. B. Berezin, V. V. Makarov, S. S. Guseinov, Y. V. Romanenko, I. S. Khudyaeva, O. M. Startseva, D. V. Belykh and A. V. Kustov, Russ. J. Gen. Chem., 2017, 87, 1557–1561 CrossRef CAS.
- Y. Li, S. I. Sasaki, H. Tamiaki, C. L. Liu, J. Song, W. Tian, E. Zheng, Y. Wei, G. Chen, X. Fu and X.-F. Wang, J. Power Sources, 2015, 297, 519–524 CrossRef CAS.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 CrossRef.
- N. R. Kim, S. J. Kim, J. D. Kim, D. S. Huh and Y. K. Shim, Bull. Korean Chem. Soc., 2009, 30, 205–213 CrossRef CAS.
- J. Seminario, Challenges and advances in computational chemistry and physics, Springer, Dordrecht, 2010, vol. 16, pp. 217–247 Search PubMed.
- B. Roeder, T. Hanke, S. Oelckers, S. Hackbarth and C. Symietz, J. Porphyrins Phthalocyanines, 2000, 4, 37–44 CrossRef.
- D. Shengnan, C. Guo, L. Mengzhen, C. Gang, W. Xiao-Feng, H. Tamiaki and S. I. Sasaki, J. Photochem. Photobiol., A, 2017, 347, 49–54 CrossRef.
- T. Zhuang, S. I. Sasaki, T. Ikeuchi, J. Kido and W. Xiao-Feng, RSC Adv., 2015, 5, 45755 RSC.
- O. Korth, T. Hanke and B. Roeder, Thin Solid Films, 1998, 320, 305–315 CrossRef CAS.
- X. F. Wang, Y. Koyama, H. Nagae, Y. Wada, S. I. Sasaki and H. Tamiaki, J. Phys. Chem. C, 2008, 112, 4418–4426 CrossRef CAS.
- A. Diarra, S. Hotchandani, J. J. Max and R. M. Leblanc, J. Chem. Soc., Faraday Trans. 2, 1986, 82, 2217–2231 RSC.
- A. Diarra, S. Hotchandani, H. Kassi and R. M. Leblanc, Appl. Surf. Sci., 1991, 49, 567–571 CrossRef.
- A. A. Krasnovsky and M. I. Bystrova, BioSystems, 1980, 12, 181–194 CrossRef CAS PubMed.
- S. Ichimura, Biophys. J., 1960, 1, 99–109 CrossRef CAS PubMed.
- X. Wang and Z. M. Wang, High-efficiency solar cells: physics, materials, and devices, Springer, Switzerland, 2014, pp. 319–355 Search PubMed.
- Y. W. Wang, S. I. Sasaki, T. Zhuang, H. Tamiaki, J. P. Zhang, T. Ikeuchi, Z. Hong, J. Kido and X.-F. Wang, Org. Electron., 2013, 14, 1972–1979 CrossRef CAS.
- S. Duan, Q. Zhou, C. Dall'Agnese, G. Chen, X. F. Wang, H. Tamiaki, K. Sakai, T. Ikeuchi and S. I. Sasaki, Sol. RRL, 2019, 3, 1900203 CrossRef CAS.
- W. Zhao, S. I. Sasaki, H. Tamiaki, Y. Sanehira, Y. Wei, G. Chen and X. F. Wang, Org. Electron., 2018, 59, 419–426 CrossRef CAS.
- G. C. Pedro, F. D. S. Gorza, R. J. Silva, N. C. Souza and J. R. Silva, Thin Solid Films, 2019, 692, 137608 CrossRef CAS.
- S. Ogi, C. Grzeszkiewicz and F. Würthner, Chem. Sci., 2018, 9, 2768–2773 RSC.
- V. Huber, M. Katterle, M. Lysetska and F. Würthner, Angew. Chem., Int. Ed., 2005, 44, 3147–3151 CrossRef CAS.
- C. Roeger, M. G. Mueller, M. Lysetska, Y. Miloslavina, A. R. Holzwarth and F. Würthner, J. Am. Chem. Soc., 2006, 128, 6542–6543 CrossRef CAS.
- V. Huber, M. Lysetska and F. Würthner, Small, 2007, 3, 1007–1014 CrossRef CAS PubMed.
- T. S. Balaban, A. D. Bhise, M. Fischer, M. Linke-Schaetzel, C. Roussel and N. Vanthuyne, Angew. Chem., Int. Ed., 2003, 42, 2140–2144 CrossRef CAS PubMed.
- S. Duan, Q. Zhou, A. Li, X. F. Wang, S. I. Sasaki and H. Tamiaki, Sol. RRL, 2020, 4, 2000162 CrossRef CAS.
- M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- W. Zhao, X. F. Wang, C. Dall'Agnese, Y. Wei, G. Chen, H. Tamiaki, Y. Sanehira and S. I. Sasaki, J. Photochem. Photobiol., A, 2019, 371, 349–354 CrossRef CAS.
- S. I. Sasaki, S. Duan, W. D. Hu, X. F. Wang and H. Tamiaki, J. Porphyrins Phthalocyanines, 2019, 23, 1–11 CrossRef.
- T. Oba and H. Tamiaki, Photosynth. Res., 1999, 61, 23–31 CrossRef CAS.
- S. Sengupta, S. Uemura, S. Patwardhan, V. Huber, F. C. Grozema, L. D. A. Siebbeles, U. Baumeister and F. Würthner, Chem.–Eur. J., 2011, 17, 5300–5310 CrossRef CAS.
- V. I. Prokhorenko, A. R. Holzwarth, M. G. Müller, K. Schaffner, T. Miyatake and H. Tamiaki, J. Phys. Chem. B, 2002, 106, 5761–5768 CrossRef CAS.
- S. Shoji, T. Ogawa, S. Matsubara and H. Tamiaki, Sci. Rep., 2019, 9, 14006 CrossRef.
- S. Shoji, T. Hashishin and H. Tamiaki, Chem.–Eur. J., 2012, 18, 13331–13341 CrossRef CAS PubMed.
- X. Chen, Z. Guo, G. M. Yang, J. Li, M. Q. Li, J. H. Liu and X. J. Huang, Mater. Today, 2010, 13, 28–41 CrossRef CAS.
- L. Tugulea, Nanostructured Films and Coatings, Springer, Dordrecht, 2000, vol. 78, pp. 347–354 Search PubMed.
- E. Harputlu, K. Ocakoglu, F. Yakuphanoglu, A. Tarnowska and D. T. Gryko, Nano-Struct. Nano-Objects, 2017, 10, 9–14 CrossRef CAS.
- A. Oueriagli, H. Kassi, S. Hotchandani and R. M. Leblanc, J. Appl. Phys., 1992, 71, 5523–5530 CrossRef CAS.
- H. Kassi, S. Barazzouk, M. Brullemans, R. M. Leblanc and S. Hotchandani, Thin Solid Films, 2010, 518, 5345–5348 CrossRef CAS.
- Y. Yu, Y. Zhang, L. Jin, Z. Chen, Y. Li, Q. Li, M. Cao, Y. Che, H. Dai, J. Yang and J. Yao, Org. Electron., 2019, 65, 381–385 CrossRef CAS.
- Q. Hu, E. Rezaee, L. Dong, Q. Dong, H. Shan, Q. Chen, M. Li, S. Cai, L. Wang and Z.-X. Xu, Sol. RRL, 2019, 3, 1900182 CrossRef CAS.
- N. B. Chaure, T. Basova, M. Zahedi, A. K. Ray, A. K. Sharma, M. Durmuş and V. Ahsen, J. Appl. Phys., 2010, 107, 114503 CrossRef.
- P. R. Warren, J. F. M. Hardigree, A. E. Lauritzen, J. Nelson and M. Riede, AIP Adv., 2019, 9, 035202 CrossRef.
- A. Dey, A. Singh, D. Das and P. K. Iyer, ACS Omega, 2017, 2, 1241–1248 CrossRef CAS PubMed.
- A. Terenin, E. Putzeiko and I. Akimov, Discuss. Faraday Soc., 1959, 27, 83–93 RSC.
- Y. K. Putseyko, Dokl. Akad. Nauk SSSR, 1961, 138, 1381–1384 Search PubMed.
- A. T. Vartanyan, Dokl. Akad. Nauk SSSR, 1963, 149, 812–815 CAS.
- G. A. Corker and I. Lundström, J. Appl. Phys., 1978, 49, 686–700 CrossRef CAS.
- S. Boussaad, J. A. DeRose and R. M. Leblanc, Chem. Phys. Lett., 1995, 246, 107–113 CrossRef CAS.
- A. T. Vartanyan, Zhur. Fiz. Khim. USSR, 1948, 22, 769–782 CAS.
- Y. K. Putseiko and A. N. Terenin, Dokl. Akad. Nauk SSSR, 1953, 90, 1005–1008 Search PubMed.
- A. N. Terenin, Y. K. Putseyko, I. A. Akimov and A. M. Meshkov, Dokl. Akad. Nauk SSSR, 1964, 155, 900–903 CAS.
- I. S. Meilanov, V. A. Benderskii and L. A. Bliumenfeld, Biofizika, 1970, 15, 822–827 CAS.
- I. S. Meilanov, V. A. Benderskii and L. A. Bliumenfeld, Biofizika, 1970, 15, 958–962 CAS.
- B. Rosenberg and J. F. Camiscoli, J. Chem. Phys., 1961, 35, 982–991 CrossRef CAS.
- C. W. Tang and A. C. Albrecht, J. Chem. Phys., 1975, 62, 2139–2149 CrossRef CAS.
- C. W. Tang and A. C. Albrecht, J. Chem. Phys., 1975, 63, 953–961 CrossRef CAS.
- C. W. Tang and A. C. Albrecht, Nature, 1975, 254, 507–509 CrossRef CAS.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125–146 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- H. Kassi, R. M. Leblanc and S. Hotchandani, Phys. Status Solidi B, 2000, 220, 931–939 CrossRef CAS.
- J. Segui, S. Hotchandani, D. Baddou and R. M. Leblanc, J. Phys. Chem., 1991, 95, 8807–8812 CrossRef CAS.
- T. Taleb, C. Nasr, S. Hotchandani and R. M. Leblanc, J. Appl. Phys., 1996, 79, 1701–1706 CrossRef CAS.
- C. Nasr, S. Hotchandani, H. Kassi, S. Nsengiyumva and R. M. Leblanc, Sol. Energy Mater. Sol. Cells, 1995, 36, 261–270 CrossRef CAS.
- M. Mabrouki, A. Oueriagli, A. Outzourhit, E. Ameziane, S. Hotchandani and R. M. Leblanc, Phys. Status Solidi A, 2002, 191, 345–354 CrossRef CAS.
- A. Desarmeaux, J. J. Max and R. M. Leblanc, J. Phys. Chem., 1993, 97, 6670–6678 CrossRef.
- C. Litwinski, I. Corral, E. A. Ermilov, S. Tannert, D. Fix, S. Makarov, O. Suvorova, L. Gonzalez, D. Woehrle and B. Roeder, J. Phys. Chem. B, 2008, 112, 8466–8476 CrossRef CAS PubMed.
- G. Pakhomov, V. Travkin and P. Stuzhin, Recent Advances in Boron-containing Materials. InTechOpen, London, 2020 Search PubMed.
- H. W. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 2–12 CrossRef.
- F. Bertinelli, C. Zauli and A. Riva, Mol. Cryst. Liq. Cryst., 1974, 28, 9–19 CrossRef CAS.
- E. E. Jacobs, A. E. Vatter and A. S. Holt, Arch. Biochem. Biophys., 1954, 53, 228–238 CrossRef CAS PubMed.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- J. G. Van Dijken, M. D. Fleischauer and M. J. Brett, Org. Electron., 2011, 12, 2111–2119 CrossRef CAS.
- T. Q. Nguyen and C. Breitkopf, J. Electrochem. Soc., 2018, 165, E826–E831 CrossRef CAS.
- G. L. Pakhomov, V. I. Shashkin, D. E. Pozdnyaev, C. Muller and J. M. Ribo, Org. Electron., 2002, 3, 93–103 CrossRef CAS.
- M. E. Aydin, A. A. M. Farag, M. Abdel-Rafeac, A. H. Ammar and F. Yakuphanoglu, Synth. Met., 2012, 161, 2700–2707 CrossRef.
- J. J. Yun, H. S. Jung, S. H. Kim, E. M. Han, V. Vaithianathan and S. A. Jenekhe, Appl. Phys. Lett., 2005, 87, 123102 CrossRef.
- S. Ahmad, M. I. Misrun, M. Isa and N. A. M. Affendi, J. Phys.: Conf. Ser., 2020, 1432, 012039 CrossRef CAS.
- B. Bita, S. Iftimie, A. Radu, V. A. Antohe, D. Coman, C. Miron, D. Staicu, L. Dan, L. Ion and S. Anthone, Proc. Rom. Acad. Math. Phys. Tech. Sci. Inf. Sci., 2019, 20, 51–57 Search PubMed.
- B. Beyer, R. Pfeifer, J. K. Zettler, O. R. Hild and K. Leo, J. Phys. Chem. C, 2013, 117, 9537–9542 CrossRef CAS.
- Q. D. Dao, T. Hori, K. Fukumura, T. Masuda, T. Kamikado, A. Fujii, Y. Shimizu and M. Ozaki, Org. Electron., 2013, 14, 2628–2634 CrossRef CAS.
- G. Williams, S. Sutty, R. Klenkler and H. Aziz, Sol. Energy Mater. Sol. Cells, 2014, 124, 217–226 CrossRef CAS.
- W. Riedel, S. Wiesner, D. Greiner, V. Hinrichs, M. Rusu and M. C. Lux-Steiner, Appl. Phys. Lett., 2014, 104, 173503 CrossRef.
- M. Zhang, Irfan, H. Ding, Y. Gao and C. W. Tang, Appl. Phys. Lett., 2010, 96, 183301 CrossRef.
- S. Sutty, G. Williams and H. Aziz, J. Photonics Energy, 2014, 4, 040999 CrossRef CAS.
- M. Hiramoto, M. Kubo, Y. Shinmura, N. Ishiyama, T. Kaji, K. Sakai, T. Ohno and M. Izaki, Electronics, 2014, 3, 351–380 CrossRef CAS.
- Y. Zheng, G. Wang, D. Huang, J. Kong, T. H. Goh, W. Huang, J. Yu and A. D. Taylor, Sol. RRL, 2018, 2, 1700144 CrossRef.
- T. F. Guo, T. C. Wen, G. L. Pakhomov, X. G. Chin, S. H. Liou, P. H. Yeh and C. H. Yang, Thin Solid Films, 2008, 516, 3138–3142 CrossRef CAS.
- V. V. Travkin, A. Y. Luk’yanov, M. N. Drozdov, E. A. Vopilkin, P. A. Yunin and G. L. Pakhomov, Appl. Surf. Sci., 2016, 390, 703–709 CrossRef CAS.
- Z. Lu, C. Zhan, X. Yu, W. He, H. Jia, L. Chen, A. Tang, J. Huang and J. Yao, J. Mater. Chem., 2012, 22, 23492–23496 RSC.
Footnote |
| † The substrate material, film thickness, and grain size cannot be specified due to a slapdash description of the experimental conditions and errors in supplementary info. |
| This journal is © The Royal Society of Chemistry 2021 |