
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen versus carbon in planar pentacoordinate environments supported by Be5Hn rings†
Jian-Hong Bian a,
Bo Jina,
Xue-Feng Zhaob,
Rui Suna,
Caixia Yuana,
Cheng-Yong Zhoub and
Yan-Bo Wu*ab
a,
Bo Jina,
Xue-Feng Zhaob,
Rui Suna,
Caixia Yuana,
Cheng-Yong Zhoub and
Yan-Bo Wu*ab
aKey Laboratory of Materials for Energy Conversion and Storage of Shanxi Province, Institute of Molecular Science, Shanxi University, Taiyuan 030006, People's Republic of China. E-mail: wyb@sxu.edu.cn
bDepartment of Chemistry, Changzhi University, Changzhi, 046011, People's Republic of China
First published on 28th April 2021
Abstract
NBe5Hnn−3 (n = 0–5) (0A–5A) species with a novel planar pentacoordinate nitrogen (ppN) were designed by the isoelectronic substitution of the C atom in planar pentacoordinate carbon (ppC) species CBe5Hnn−4 (n = 0–5) with an N atom. The highly flexible H atoms found in ppC species CBe5H22− and CBe5H3− were fixed upon the nitrogen substitution, as mirrored by the non-flexible H atoms in their ppN analogues NBe5H2− (2A) and NBe5H3 (3A). Moreover, the N atom was found to fit the H-surrounded Be5 rings better than the C atom because the ppC species CBe5H4 and CBe5H5+ adopted non-planar structures due to size-mismatch between the C atom and the H-surrounded Be5 ring, but their ppN analogues NBe5H4+ (4A) and NBe5H52+ (5A) adopted perfect planar structures. The electronic structure analyses revealed that the N atoms in 0A–5A were involved in four doubly occupied orbitals, including three six-center two-electron (6c-2e) σ bonds and one 6c-2e π bond. Therefore, these ppN species not only obey the octet rule, but also possess the interesting σ and π double aromaticity, which contributes to the stabilization. Consequently, 2A, 4A, and 5A are charged kinetically viable global energy minima, and are suitable for the gas phase generation and spectroscopic characterization.
Introduction
In general, a typical main group nonmetal can have the maximum number of four closely connected atoms and they often adopt a tetrahedral arrangement as suggested by the van't Hoff/Le Bel rule. Therefore, planar pentacoordination is especially interesting in that it violates both the highest number and the suggested arrangement of closely connected atoms. Such a non-classical bonding has mainly been observed, heretofore, in the planar pentacoordinate carbon1 (ppC) species. Note that though ppC stemmed from the planar tetra-2 and hexa-coordinate carbon3 (ptC and p6C), it played a more important role in planar carbon chemistry recently because a dozen ppC species were found to be the global energy minima (GEM),1b,4 suitable for experimental realization. To our knowledge, the ppC GEMs represent more than half of known GEM planar carbon species. Their GEM nature may be the result of geometric and electronic good fitting between C atom and five-membered rings. However, exceptions can be found in the CBe5Hnn−4 (n = 2–5) system (see Fig. S1 in ESI†), where CBe5H4 and CBe5H5+ species are non-planar, suggesting that carbon is too big to stay at the center of Be5H4 and Be5H5 rings preferentially.5As the heavier neighbor of carbon, nitrogen has rich chemistry, too. In particular, many non-classical bonding patterns of carbon had been extended to nitrogen.6 For instance, the planar tetracoordinate nitrogen (ptN) had been observed in NSiAl3, NAl4/NAl4−, NM4H4+ (M = Cu, Ni), NXAl3+ (X = N, P, As), etc.7 Noticeably, the higher electronegativity of nitrogen than that of carbon leads to the higher preference for the localized bonding, which is unbeneficial to the design of planar hypercoordinate nitrogen species. Nevertheless, Ding et al. proposed and demonstrated that this bonding preference could be utilized to design the ptC and ptN species.7d,8
Nonetheless, the planar pentacoordinate nitrogen (ppN) was less familiar to chemists. To the best of our knowledge, two ppN species (NCu5H5+ and NBe5Au52+)9,10 had been reported, but none of them were convincingly demonstrated to be the GEM, which lowers their possibility to be observed in the experiments. Can a ppN species be a GEM and what is the difference between ppN and ppC species possessing the similar geometrical skeletons? Through design and characterization of the ppN species NBe5Hnn−3 (n = 0–5) by substituting the C atom in CBe5Hnn−4 (n = 0–5)5 with N atom, we reported in the present work the first examples of the GEM ppN species. Simultaneously, we found that nitrogen fits more nicely with the Be5Hn rings than carbon and the nitrogen substitution for the carbon atom in ppC species CBe5H22− and CBe5H3− can fix the highly flexible H atoms.
Computational methods
For ppN species NBe5Hnn−3 (n = 0–5) (0A–5A), the geometry optimization and harmonic vibrational frequency analysis were performed at the B3LYP/aug-cc-pVTZ level. The B3LYP functional was calibrated using ab initio calculations at the MP2/aug-cc-pVTZ level, which gave the optimized structures of 1A–5A not different in essential from those optimized using B3LYP functional, suggesting that it is safe to use B3LYP to study the current system. To better understand the electronic structures, the natural bond orbital (NBO)11 and the adaptive natural density partitioning (AdNDP)12 were analyzed at the B3LYP/aug-cc-pVTZ and B3LYP/6-31G levels, respectively. As an extension of NBO analysis, AdNDP shows the electronic structure of a molecular system in terms of n-center two-electron (nc-2e) bonds (n ranges from one to total number of atoms in the molecule). Thus, AdNDP recovers not only the Lewis elements (lone pairs and 2c-2e bonds), but also the delocalized nc-2e bonds. To assess the aromaticity, the electron localization function (ELF)13 and the nucleus-independent chemical shifts (NICS)14 analyses were carried out at the B3LYP/aug-cc-pVTZ level. To assess the thermodynamic stability of the design ppN species, the potential energy surfaces (PESs) of NBe5Hnn−3 (n = 1–5) components were explored by the stochastic search algorithm.15 Note that both singlet and triplet surfaces were considered in the PES explorations. The generated random structures were initially optimized at the B3LYP/6-31G(d) level. Then, ten identified lowest isomers were re-optimized at the B3LYP/aug-cc-pVTZ level. The energies were further improved at the CCSD(T)/aug-cc-pVTZ level based on the B3LYP/aug-cc-pVTZ optimized geometries. The relative energies of the isomers were compared at the CCSD(T)//B3LYP level, where CCSD(T)//B3LYP denoted the sum of CCSD(T)/aug-cc-pVTZ single point energy and B3LYP/aug-cc-pVTZ zero-point energy correction. To evaluate the kinetic stability, the Born-Oppenheimer molecular dynamic (BOMD)16 simulations were run at the B3LYP/6-31G(d) level and concerned temperatures, including 4, 298, and 500 K. The simulations were performed adiabatically, the initial velocity was not fixed but related to the temperatures considered, and the Hessian was recalculated every 5 gradient points. The structural evolution during the simulations was evaluated by the root-mean-square deviation (RMSD) relative to the B3LYP/6-31G(d)-optimized geometries. The vertical detachment energy (VDE) and the vertical electron affinity (VEA) were calculated using the outer valence Green's function (OVGF)17 at the OVGF/aug-cc-pVTZ level. The stochastic search algorithm was performed using GXYZ 2.0 program,18 the AdNDP was analyzed using AdNDP program,19 the CCSD(T) calculations were carried out using MolPro 2012.1,20 and all other calculations were performed using the Gaussian 09 package.21Results and discussion
Design of NBe5Hnn−3 (n = 0–5)
The ppN species NBe5Hnn−3 (n = 0–5) (0A–5A, see Fig. 1) could be designed simply by isoelectronic substitution of C atom in ppC species CBe5Hnn−4 (n = 0–5) with N+ cation. Note that 2A and 3A both have an isomer, i.e. 2A′ and 3A′ shown in Fig. 1, whose structural difference lies in the distribution of H atoms. According to our calculations at the CCSD(T)//B3LYP level, 2A′ and 3A′ are 10.9 and 9.5 kcal mol−1 higher in energy than 2A and 3A, respectively, thus they are discarded in the following. As shown in Fig. 1, the energy minima 0A–5A adopt the perfect planar structures at the B3LYP/aug-cc-pVTZ level. In particular, the planar ppN species 4A and 5A are in sharp contrast to their quasi-planar analogues CBe5H4 and CBe5H5+, suggesting the better match of N atom than C atom with the Be5H4 and Be5H5 rings. The N–Be interatomic distances range from 1.557 to 1.781 Å in 0A–5A, which is short enough for each Be atom to be considered as a coordination to N atom, so 0A–5A are eligible ppN species.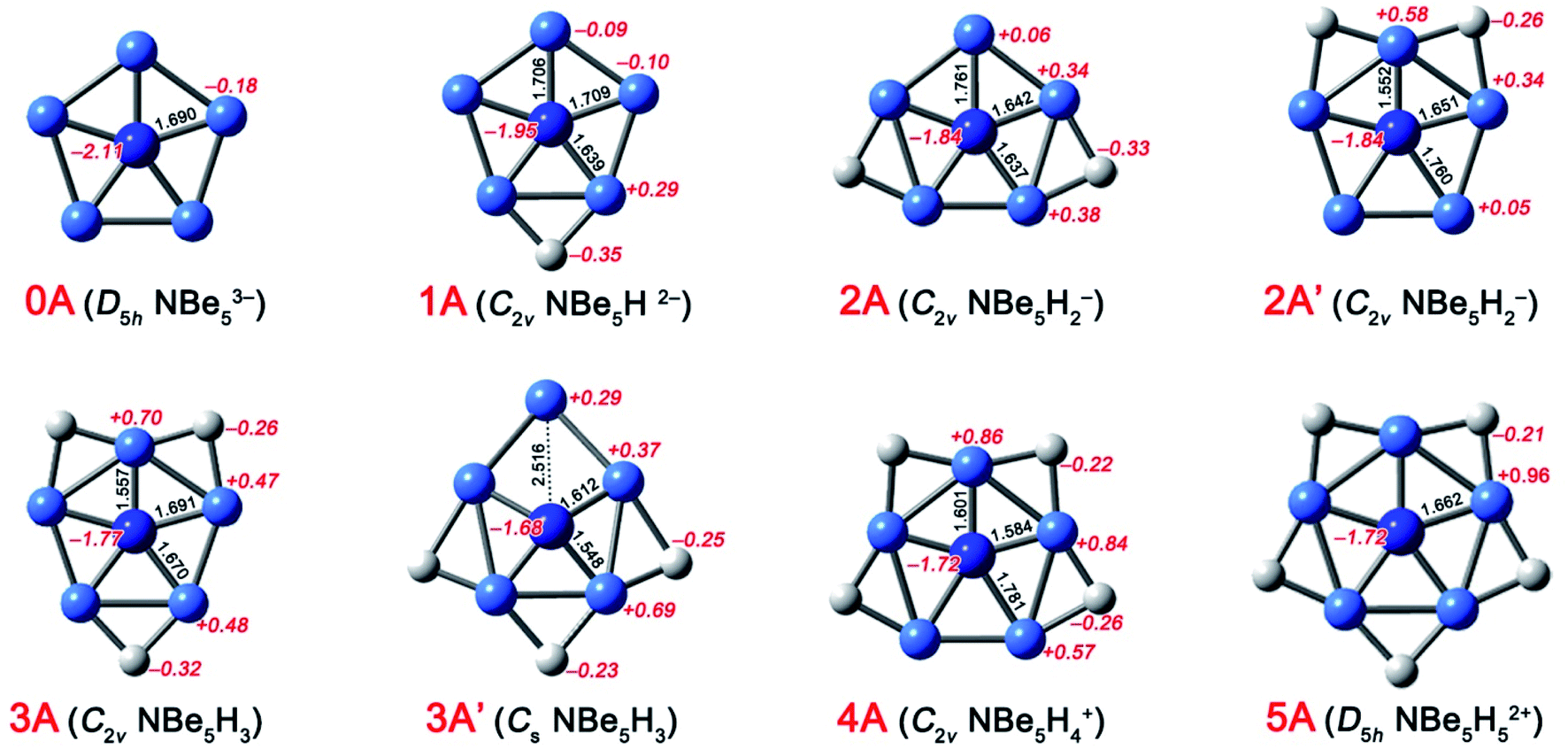 | ||
| Fig. 1 Optimized structures of ppN species at the B3LYP/aug-cc-pVTZ level. Bond distances (in Å) and NBO charges (in |e|) are given in black and italic red fonts, respectively. | ||
Electronic structure analyses
The chemical bonding in ppN species 0A–5A are similar to their analogue ppC species CBe5Hnn−4 (n = 0–5). Fig. 2 shows the AdNDP view of chemical bonding in 0A–5A. As shown in Fig. 2, for Be–Be edge without bridging H atom, there is a 2c-2e Be–Be σ bond with occupation numbers (ONs) ranging from 1.96 to 1.98 |e| (see the first column). Exposing such diffused metal–metal σ bonds may be negative to the stabilization of ppN species. In contrast, for Be–Be edge with a bridging H atom, there is a Be–H–Be three-center two-electron (3c-2e) σ bond (ON = 1.91–1.99 |e|, see the second column). Though such bonds involve three centers, they are less diffused than the Be–Be 2c-2e bonds, which may be positive to the stabilization from the electronic structure point of view. | ||
| Fig. 2 AdNDP bonding patterns of 0A–5A with occupation numbers (ONs). | ||
As shown in Fig. 3, the advantage to possess the Be–H–Be 3c-2e bonds can be demonstrated by the increasing HOMO–LUMO gaps from 0.44 eV for NBe5 without bridging H to 7.93 eV with all Be–Be edges bridged by the H atoms. Note that the most obvious variation occurs at n = 4 to n = 5, which corresponds to the complete elimination of diffused Be–Be 2c-2e bond, verifying the beneficial effect of Be–H–Be 3c-2e bonds to the stabilization of the ppN structures. We also note that the HOMO–LUMO gaps of ppN species NBe5Hnn−3 (n = 0–5) are all higher than their analogue ppC species CBe5Hnn−4 (n = 0–5), which indicates the electronically better match of N atom than that of C atom with Be5Hn rings.
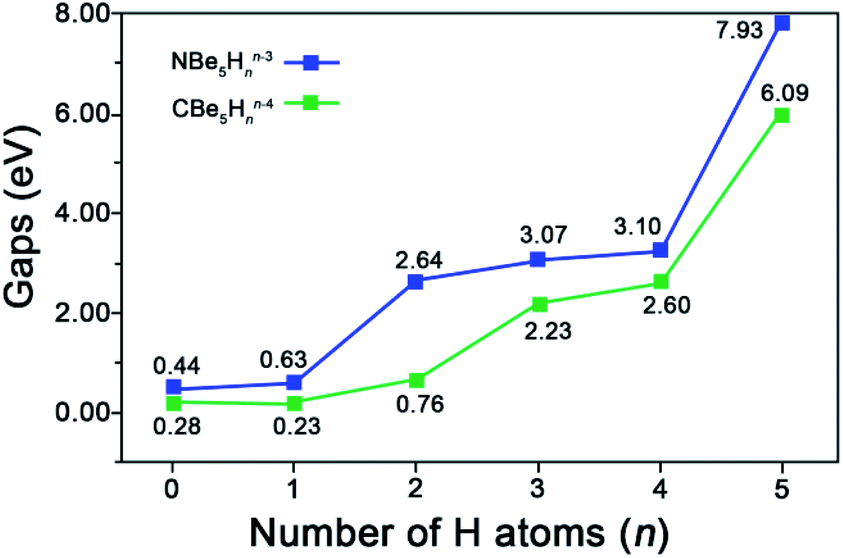 | ||
| Fig. 3 Variation of HOMO–LUMO gaps (gaps, in eV) with the increasing number of H atoms (n) in NBe5Hnn−3 and CBe5Hnn−4 (n = 0–5). | ||
Bonding patterns shown in the third column of Fig. 2 depict the interesting delocalized six-center two-electron (6c-2e) σ bonds, while those in the last column illustrate the delocalized 6c-2e π bonds. These four 6c-2e bonds are very similar in all NBe5Hnn−3 species despite of n values and they are also similar to those of their ppC analogues CBe5Hnn−4 (n = 0–5). The existence of these four bonds builds the basis for the stabilization of ppN arrangement from the electronic structure point-of-view. First, these four bonds fill eight electrons to the valence shell of central nitrogen atoms, i.e. the ppN species obey the octet rule. Second, three delocalized σ bonds and one delocalized π bond meet the Hückel's 4n+2 rule for n = 1 and 0, respectively, suggesting the novel 6σ + 2π double aromaticity.
The aromaticity within NBe5 moieties of 2A–5A was evaluated using ELF analysis. As shown in Fig. S2,† the bifurcation values for σ bonding (ELFσ) range from 0.936 to 0.944, while that for π bonding (ELFπ) are all 0.999, thus giving the average bifurcation values (ELFav) from 0.968 to 0.972. Since the ELFσ, ELFπ and ELFav values are all much larger than 0.7, the threshold value for ELF criterion of aromaticity judgement,13,22 the ppN species 2A–5A should be the σ, π and overall aromatic molecules.
Taking 5A as an example, we studied aromaticity by performing the NICS analysis. As shown in Fig. 4, the NICS values for almost all the examined points near the molecule are obviously negative, suggesting aromatic nature for 5A. An exception with the positive NICS value of +3.7 ppm can be found at the point located 0.5 Å above the beryllium atom, but it does not influence the aromatic nature of 5A as a whole. Note that the points with largest negative NICS values (−52.9 and −21.4 ppm) locate 0.5 and 1.0 Å above the nitrogen atom, suggesting the π electron delocalization, while the points with the second largest negative NICS values (−18.8 and −16.1 ppm) locate in the molecular plane within Be5 ring, indicating the σ electron delocalization. The NICS values become close to zero when the distance between the examined point and the molecule becomes long. The NICS results verify the existence of aromaticity, consistent with the conclusion from the AdNDP and ELF analyses.
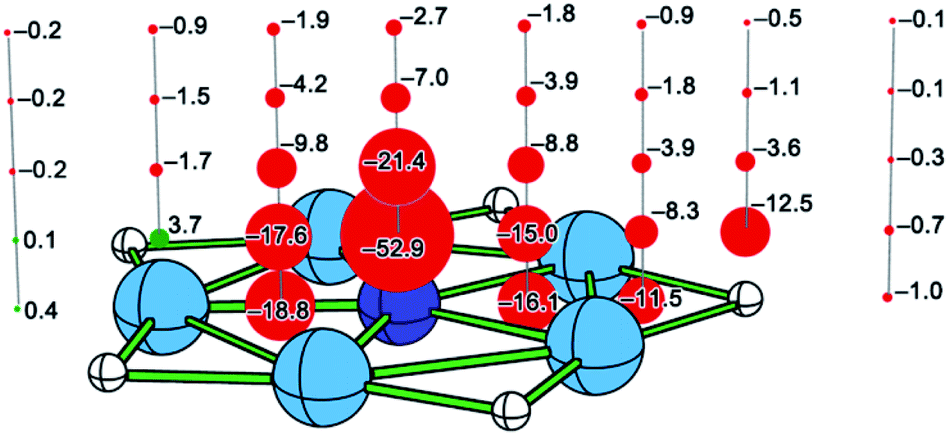 | ||
| Fig. 4 NICS results for 5A. The points with negative and positive NICS values are shown in red and green, respectively. | ||
The conventional NBO analyses were also performed to qualitatively describe the chemical bonding in 0A–5A. The electrostatic interactions can be described by the natural charge distribution. As shown in Fig. 1, the N and Be atoms in 0A both possess the negative charges (−2.11 and −0.18 |e|, respectively), indicating the obvious Coulomb explosion effect, which would be the most important factor to destabilize 0A. We note that the bridging H atoms in 1A–5A all possess the negative charges (−0.21 to −0.35 |e|). Simultaneously, the natural charges on Be atoms are highly affected by the bridging H atoms. For example, the Be atoms linking one bridging H atom bear the positive charges ranging from +0.29 to +0.57 |e|, while those linking two bridging H atoms bear the charges ranging from +0.70 to + 0.96 |e|. Correspondingly, for the molecular region where the Be–Be edge was bridged by an H atom, the N, Be, and H atoms possess the negative, positive, and negative charges, respectively, representing a kind of favorable Coulomb attractive interactions, which is similar to the pattern found in CBe5Hnn−4 (n = 0–5).
The covalent bonding was evaluated by the Wiberg bond indices (WBI, see Table 1) of concerned atom–atom bonds. The WBIBe–H values range from 0.36 to 0.55, signifying the formation of Be–H–Be 3c-2e σ bonds. The WBIN–Be values range from 0.28 to 0.58, suggesting the non-local bonding between N and Be atoms, which is in line with the AdNDP results (revealing the existence of delocalized σ and π bonds). The Be–Be bonding can be classified into two types. For Be–Be edges without H atom, the WBIBe–Be values range from 0.93 to 0.96, pronouncing the standard single bonds. In contrast, for Be–Be edges with a H atom, the WBIBe–Be values range from 0.15 to 0.19, suggesting the absence of direct Be–Be covalent bonding, which is consistent with the formation of H-bridged Be–H–Be 3c-2e bonds. The covalent bonding is also akin to the bonding pattern found in CBe5Hnn−4 (n = 0–5).
| νmin | WBI | |||
|---|---|---|---|---|
| N–Be | Be–Be | Be–H | ||
| NBe53− (0A) | 218 | 0.35 | 0.17/0.94 | N/A |
| NBe5H2− (1A) | 196 | 0.32–0.44 | 0.94 | 0.43 |
| NBe5H2− (2A) | 192 | 0.29–0.44 | 0.16/0.93/0.96 | 0.42/0.46 |
| NBe5H3 (3A) | 167 | 0.37–0.58 | 0.15/0.17/0.96 | 0.36–0.55 |
| NBe5H4+ (4A) | 117 | 0.28–0.52 | 0.15/0.19/0.96 | 0.36–0.55 |
| NBe5H52+ (5A) | 77 | 0.42 | 0.18 | 0.46 |
Stability consideration
The experimental viability of a theoretically designed species is closely related to its stability. The small clusters designed in this work are expected to be realized in the gas-phase generation and characterized in the followed spectroscopy.Since the net charges on 0A is too high to deter it from being realized experimentally, 0A was excluded during the stability studies. Herein, the thermodynamic stability of 1A–5A was examined by the extensive exploration of their PESs. At the final CCSD(T)//B3LYP level, 2A, 4A, and 5A are proved to be the GEMs, being 0.2, 8.4, and 28.6 kcal mol−1, respectively, lower in energy than their second lowest isomers (see Fig. S3 in ESI†). As a comparison, 1A and 3A are 7.7 and 11.5 kcal mol−1 higher in energy at the same level than the located lowest-lying isomers, respectively.
Next, the kinetic stability of 2A–5A was studied by BOMD simulations. Fig. 5 shows the RMSD plots for a set of 100 pico-second (ps) BOMD simulations running at 4, 298, and 500 K, respectively. As the figure shows, the variation of RMSD values suggested that the structures of 2A, 3A, and 5A can be well-maintained for the simulations at all three examined temperatures, as reflected by the small RMSD values with upward jump. The RMSD plots of 4A also indicate the well-maintained structure at 4 and 298 K. However, the structure of 4A is not kinetically stable at 500 K, as mirrored by a reversible upward jump of RMSD plot at about 10 ps. The structural sampling revealed the reversible isomerization to 4C. Nevertheless, the molecules exist in the form of 4A for about 90% of simulation time, suggesting that 4A will determine the spectroscopic signals at 500 K.
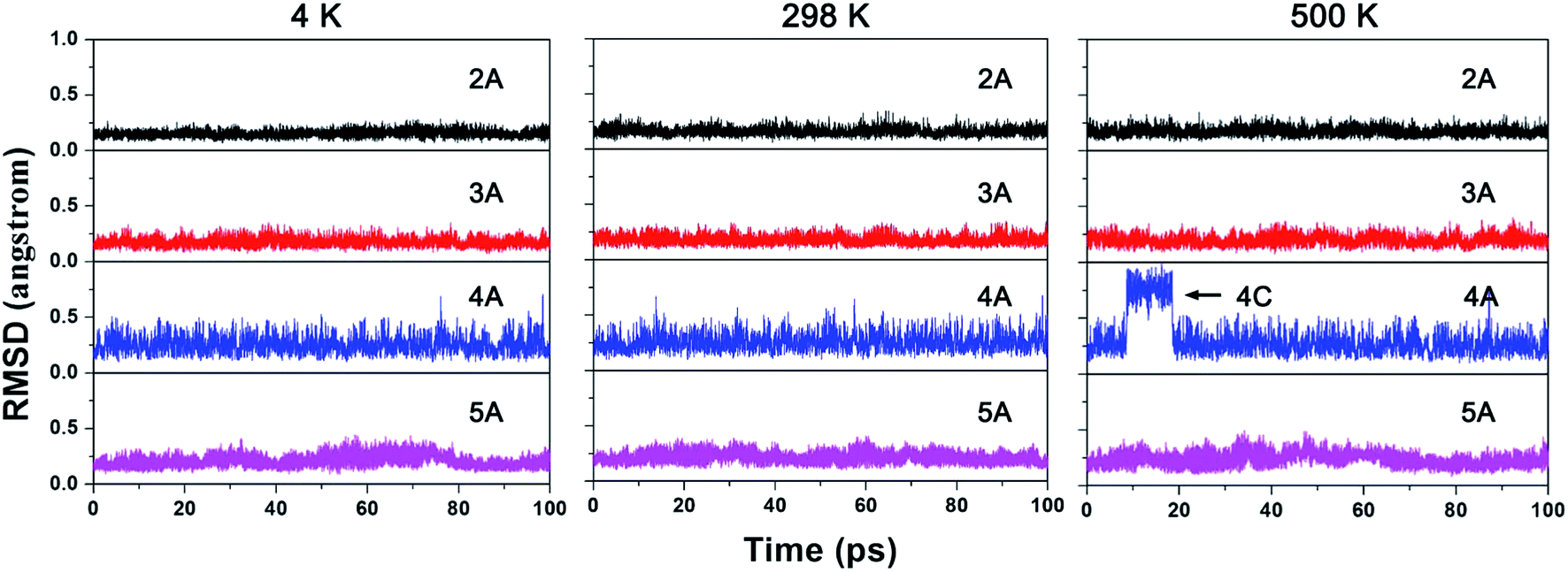 | ||
| Fig. 5 The RMSD plots for the BOMD simulations of 2A–5A at 4 K, 298 K and 500 K from left to right, respectively. | ||
To avoid the influence from stochastic behaviors of BOMD simulations to the conclusion of kinetic stability, we selected the 500 K as the test temperature to run additional five independent simulations for 20 ps and corresponding RMSD plots are given in ESI as Fig. S4.† As the figure shows, the structures of 2A, 3A, and 5A can also be well-maintained, while that of 4A shows the reversible instantaneous upward jump. The detailed structural sampling revealed the transient existence of 4B and 4C. Nevertheless, in majority of simulation time, 4A is the main existence form, which is consistent with the above 100 ps simulation. So 4A is also kinetically viable.
Herein, we would like to compare the dynamic behaviors of ppN species NBe5Hnn−3 (n = 2−5) with that of ppC analogue species CBe5Hnn−4 (n = 2–5). Our previous studies revealed that the H atoms in CBe5H22− and CBe5H3– were both highly flexible, rotating around the CBe5 core moiety by moving from one Be–Be edge to another.5 However, when the ppC was replaced by ppN, the flexibility of H atoms disappeared, as reflected by the RMSD plots of 2A and 3A without upward jump. Simultaneously, 4A and 5A inherit the good kinetic stability of CBe5H4 and CBe5H5+. Therefore, the dynamic simulations not only demonstrated the good kinetic viability of 2A–5A, but also revealed that the N-substitution can fix the highly flexible H atoms found in CBe5H22− and CBe5H3−.
Stability about gaining and losing electron
To evaluate the electron-gaining and losing properties, the VDEs and VEAs of 2A–5A were calculated. The VDE for mono-anion 2A is 2.52 eV, which is reasonably high for photoelectron spectroscopy, while the VEA of 2A is positive, suggesting the endothermic electron-gaining process for 2A. For neutral molecule 3A, the VDE and VEA values are 7.10 and 0.22 eV, respectively, revealing its low tendency to lose or gain an electron. For mono-cation 4A, it would be very difficult to lose an electron due to the rather high VDE of 11.93 eV and it would be not easy to gain an electron, as reflected by the VEA value (−4.41 eV) close to K+ cation (−4.34 eV). For dication 5A, it is not curious that the VDE is as high as 22.35 eV. However, its VEA value of −9.23 eV is actually very low, being lower than the VEA of Ba2+ (−10.00 eV, the lowest VEA value for any atomic dication in the Periodic Table), suggesting its relatively low tendency to gain an electron in dications. Therefore, 5A is eligible to be regarded as the so-called “superalkaline earth metal dication”, which might be stable enough to exist as the positive portion of an ionic salt.Conclusion
In summary, we have demonstrated based on the extensive computational explorations that nitrogen is more suitable than carbon to fit the H-surrounded Be5 rings both geometrically and electronically. The nitrogen substitution for the planar pentacoordinate carbon in CBe5Hnn−4 (n = 0–5) leads to the perfect planar structure for n = 4 and 5 and fixes the highly free H atoms for n = 2 and 3. Remarkably, when the diffused Be–Be 2c-2e σ bonds were gradually replaced by much less diffused Be–H–Be 3c-2e bonds, the structures were gradually stabilized from the electronic structure point of view, as reflected by the increasing HOMO–LUMO gaps. In six planar pentacoordinate nitrogen species designed in this work, NBe5H52+ (5A) possesses the best stability, which would be promising to obtain it as the positive portion of an ionic salt.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by NSFC (Grant Nos. 21720102006 and 22073058), the Natural Science Foundation of Shanxi Province (Grant Nos. 201901D111018 and 201901D111014), the OIT Program, the Shanxi “1331 Project” Engineering Research Center (PT201807), the Shanxi 1331KIRT, and the HPC of Shanxi University.References
- (a) L.-M. Yang, E. Ganz, Z. Chen, Z.-X. Wang and P. v. R. Schleyer, Angew. Chem., Int. Ed., 2015, 54, 9468–9501 CrossRef CAS PubMed; (b) V. Vassilev-Galindo, S. Pan, K. J. Donald and G. Merino, Nat. Rev. Chem., 2018, 2, 0114 CrossRef CAS.
- (a) K. Sorger and P. v. R. Schleyer, J. Mol. Struct.: THEOCHEM, 1995, 338, 317–346 CrossRef; (b) D. Röttger and G. Erker, Angew. Chem., Int. Ed. Engl., 1997, 36, 812–827 CrossRef; (c) L. Radom and D. R. Rasmussen, Pure Appl. Chem., 1998, 70, 1977–1984 CAS; (d) W. Siebert and A. Gunale, Chem. Soc. Rev., 1999, 28, 367–371 RSC; (e) R. Keese, Chem. Rev., 2006, 106, 4787–4808 CrossRef CAS PubMed; (f) G. Merino, M. A. Méndez-Rojas, A. Vela and T. Heine, J. Comput. Chem., 2007, 28, 362–372 CrossRef CAS PubMed.
- K. Exner and P. v. R. Schleyer, Science, 2000, 290, 1937–1940 CrossRef CAS PubMed.
- (a) P. Liu, J.-H. Bian, Q. Wang, F. Huang, D. Li and Y.-B. Wu, Phys. Chem. Chem. Phys., 2018, 20, 12642–12649 RSC; (b) X.-F. Zhao, J.-H. Bian, F. Huang, C. Yuan, Q. Wang, P. Liu, D. Li, X. Wang and Y.-B. Wu, RSC Adv., 2018, 8, 36521–36526 RSC; (c) S. Pan, J. L. Cabellos, M. Orozco-Ic, P. K. Chattaraj, L. Zhao and G. Merino, Phys. Chem. Chem. Phys., 2018, 20, 12350–12355 RSC; (d) J.-C. Guo, L.-Y. Feng, C. Dong and H.-J. Zhai, J. Phys. Chem. A, 2018, 122, 8370–8376 CrossRef CAS PubMed; (e) J.-C. Guo, Y.-X. Cheng and X.-F. Wu, Comput. Theor. Chem., 2020, 1180, 112824 CrossRef CAS; (f) J.-C. Guo, L.-Y. Feng, J. Barroso, G. Merino and H.-J. Zhai, Chem. Commun., 2020, 56, 8305–8308 RSC; (g) M.-h. Wang, X. Dong, Y.-h. Ding and Z.-h. Cui, Chem. Commun., 2020, 56, 7285–7288 RSC; (h) R. Sun, X.-F. Zhao, B. Jin, B. Huo, J.-H. Bian, X.-L. Guan, C. Yuan and Y.-B. Wu, Phys. Chem. Chem. Phys., 2020, 22, 17062–17067 RSC.
- J.-C. Guo, G.-M. Ren, C.-Q. Miao, W.-J. Tian, Y.-B. Wu and X. Wang, J. Phys. Chem. A, 2015, 119, 13101–13106 CrossRef CAS PubMed.
- (a) X. Li and L. S. Wang, Eur. Phys. J. D, 2005, 34, 9–14 CrossRef CAS; (b) B. B. Averkiev, A. I. Boldyrev, X. Li and L.-S. Wang, J. Chem. Phys., 2006, 125, 124305 CrossRef PubMed; (c) B. Song, C.-H. Yao and P.-l. Cao, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 035306 CrossRef; (d) B. B. Averkiev, A. I. Boldyrev, X. Li and L.-S. Wang, J. Phys. Chem. A, 2007, 111, 34–41 CrossRef CAS PubMed; (e) B. B. Averkiev, S. Call, A. I. Boldyrev, L.-M. Wang, W. Huang and L.-S. Wang, J. Phys. Chem. A, 2008, 112, 1873–1879 CrossRef CAS PubMed; (f) W.-Q. Zhang, J.-M. Sun, G.-F. Zhao and L.-L. Zhi, J. Chem. Phys., 2008, 129, 064310 CrossRef PubMed; (g) L.-M. Wang, W. Huang, L.-S. Wang, B. B. Averkiev and A. I. Boldyrev, J. Chem. Phys., 2009, 130, 134303 CrossRef PubMed; (h) H. Wang, Y. J. Ko, K. H. Bowen, A. P. Sergeeva, B. B. Averkiev and A. I. Boldyrev, J. Phys. Chem. A, 2010, 114, 11070–11077 CrossRef CAS PubMed.
- (a) P. v. R. Schleyer and A. I. Boldyrev, J. Chem. Soc., Chem. Commun., 1991, 1536–1538 RSC; (b) S. K. Nayak, B. K. Rao, P. Jena, X. Li and L. S. Wang, Chem. Phys. Lett., 1999, 301, 379–384 CrossRef CAS; (c) S. D. Li, G. M. Ren, C. Q. Miao and Z. H. Jin, Angew. Chem., Int. Ed., 2004, 43, 1371–1373 CrossRef CAS PubMed; (d) Z.-h. Cui and Y.-h. Ding, Phys. Chem. Chem. Phys., 2011, 13, 5960–5966 RSC.
- Z.-h. Cui, C.-b. Shao, S.-m. Gao and Y.-h. Ding, Phys. Chem. Chem. Phys., 2010, 12, 13637–13645 RSC.
- S. D. Li, C. Q. Miao and G. M. Ren, Eur. J. Inorg. Chem., 2004, 2232–2234 CrossRef.
- J.-C. Guo, L.-Y. Feng, X.-Y. Zhang and H.-J. Zhai, J. Phys. Chem. A, 2018, 122, 1138–1145 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- J. C. Santos, J. Andres, A. Aizman and P. Fuentealba, J. Chem. Theory Comput., 2005, 1, 83–86 CrossRef PubMed.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed; (b) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- (a) M. Saunders, J. Comput. Chem., 2004, 25, 621–626 CrossRef CAS PubMed; (b) P. P. Bera, K. W. Sattelmeyer, M. Saunders, H. F. Schaefer and P. v. R. Schleyer, J. Phys. Chem. A, 2006, 110, 4287–4290 CrossRef CAS PubMed.
- (a) J. M. Millam, V. r. Bakken, W. Chen, W. L. Hase and H. B. Schlegel, J. Chem. Phys., 1999, 111, 3800–3805 CrossRef CAS; (b) X. S. Li, J. M. Millam and H. B. Schlegel, J. Chem. Phys., 2000, 113, 10062–10067 CrossRef CAS.
- J. V. Ortiz, V. G. Zakrzewski and O. Dolgounircheva, Conceptual Perspectives in Quantum Chemistry, Kluwer Academic, 1997 Search PubMed.
- (a) Y.-B. Wu, H.-G. Lu, S.-D. Li and Z.-X. Wang, J. Phys. Chem. A, 2009, 113, 3395–3402 CrossRef CAS PubMed; (b) H. G. Lu and Y. B. Wu, in GXYZ 2.0, A Random Search Program, Shanxi University, Taiyuan, 2015 Search PubMed.
- The AdNDP program was downloaded freely at, http://ion.chem.usu.edu/%7Eboldyrev/adndp.php Search PubMed.
- H.-J. Werner, et al., in MolPro 2012.1, University College Cardiff Consultants Limited, Cardiff U.K., 2012 Search PubMed.
- M. J. Frisch, et al., in Gaussian 09 Revision D.01, Gaussian Inc.,Wallingford CT, 2013 Search PubMed.
- (a) J. C. Santos, W. Tiznado, R. Contreras and P. Fuentealba, J. Chem. Phys., 2004, 120, 1670–1673 CrossRef CAS PubMed; (b) R. Islas, E. Chamorro, J. Robles, T. Heine, J. C. Santos and G. Merino, Struct. Chem., 2007, 18, 833–839 CrossRef CAS; (c) J. C. Santos and P. Fuentealba, Chem. Phys. Lett., 2007, 443, 439–442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures of CBe5Hnn−4 (n = 2–5), the results of ELF analysis for 2A–5A, optimized structures and the relative energies of 1A–5A and their four lowest isomers, the five independent 20 ps BOMD simulations of 2A–5A at 500 K, the energy profile for the generation of 4C from 4A, and Cartesian coordinates for the species reported in this work. See DOI: 10.1039/d1ra02178h |
| This journal is © The Royal Society of Chemistry 2021 |