
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomo–hetero/core–shell structure design strategy of NaYF4 nanocrystals for superior upconversion luminescence
Raheel Ahmed Janjua *a,
Obaid Iqbalb,
Muhammad Aqeel Ahmedc,
Abdullah A. Al-Kahtani*d,
Sara Saeede,
Muhammad Imranf and
Abdul Ghafar Wattoo*g
*a,
Obaid Iqbalb,
Muhammad Aqeel Ahmedc,
Abdullah A. Al-Kahtani*d,
Sara Saeede,
Muhammad Imranf and
Abdul Ghafar Wattoo*g
aNational Engineering Research Center for Optical Instruments, College of Optical Science and Engineering, Zhejiang University, Hangzhou 310058, China. E-mail: raheel@mail.ustc.edu.cn
bNational Synchrotron Radiation Laboratory, Anhui Provincial Engineering Laboratory of Advanced Functional Polymer Film, CAS Key Laboratory of Soft Matter Chemistry, University of Science and Technology of China, Hefei 230026, China
cDepartment of Electronics, Quaid-i-Azam University, Islamabad, Pakistan
dChemistry Department, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
eDepartment of Physics, University of Science and Technology of China, Hefei, Anhui, China
fDepartment of Chemistry, Faculty of Science, King Khalid University, P.O. Box 9004, Abha 61413, Saudi Arabia
gDepartment of Physics, Khwaja Fareed University of Engineering and Information Technology, Rahim Yar Khan 64200, Pakistan
First published on 10th June 2021
Abstract
A comprehensive strategy has been developed to construct nano-sized homogeneous and heterogeneous core/shell structures of NaYF4 host. Synthesis conditions of cubic phase/α-NaYF4 and hexagonal phase/β-NaYF4 are discussed. Pure cubic NaYF4:Yb,Er nanocrystals were synthesized with different average sizes extending from 7 nm to 15 nm by varying the reaction time. Temperature and time thresholds of hexagonal nucleation were determined and utilized for controlled core/shell structures of different phases. α-NaYF4:Yb,Er@α-NaYF4, α-NaYF4:Yb,Er@β-NaYF4, β-NaYF4:Yb,Er@α-NaYF4, and β-NaYF4:Yb,Er@β-NaYF4 core/shell structures were prepared by adopting the required conditions to achieve the desired phase. Excess sodium was used to grow hexagonal shell over metastable cubic core under controlled conditions of reaction time and temperature to prevent the structural transition of the core. Upconversion emission spectra have also been obtained. UCL integrated intensities demonstrated about 5-fold enhancement for α-shell over α-core as compared to the core alone and 22-fold enhancement with β-shell. On the other hand, α-shell over β-core exhibited 5-fold enhancement and β-shell over β-core exhibited 6-fold enhancement.
Introduction
Up-converting nano-materials has attracted considerable attention due to their broad utility in bio-applications,1–7 3D display devices,8–11 printing,12–14 lasers15,16 and storage devices.17 Up-converters are normally excited by NIR, which allows deep penetration through living cells without damaging them, prevents auto-fluorescence, has a high signal to noise ratio, and is a fast thermal imaging technique.18–22 UCL studies mainly revolve around Ln-doped halides23–25 and oxides.26–28 Among these, Ln-NaYF4 has been the focus of most studies due to its efficient UCL properties.29,30 However, high efficiency has always been an urgent demand, for which several strategies have been adopted to improve the efficiency, such as co-doping,31–35 plasmonic effect and surface modification.36–39 One of the principle of engineering with nanocrystals is tailoring their surfaces.40–44 Nanocrystalline cores have suffered some issues due to high surface to volume ratio, boundary defects, and ligands.45–47 These issues tend to quench the luminescence of the core. Surface passivation by inert shell structures has been a proven tool to enhance luminescence.40,42–44 Therefore, the formation of a shell solves this problem. Core–shell structures are also used for multiple functions such as drug delivery, photodynamic therapy,18,20,22 and multicolor displays.24Several studies have shown that the growth of a shell around the NaYF4 core enhances the UCL efficiency by improving the transfer of excitation and emission energy to the inner core from the boundaries. A thin shell can passivate the surface of the core by reducing the defects, and control UCL quenching caused by ligands and other impurities.37,42,43,48–50 NaYF4 exists in cubic and hexagonal phases at ambient conditions. So, different groups proposed different strategies to synthesize core–shell structures with different combinations of phases. Yan et al.51 adopted a two-step growth method to synthesize core–shell NaYF4 with cubic core/cubic shell and hexagonal core/cubic shell. They observed a significant enhancement in the intensity of green emission and further attributed this enhancement to the repair of surface defects and ligands by shell formation. Zhang et al.52 reported hexagonal phase core and hexagonal phase shell and concluded that the red to green ratio of emission bands was significantly affected by excitation power in the core but remained unchanged in core/shell. van Veggel et al.53 floated a novel strategy to synthesize cubic core and hexagonal shell. Zhao et al.54 also grew hexagonal phase shells induced by the heterogeneous cubic core as a strategy of controlling the small size in the hexagonal phase. Different combinations of core/shell on the basis of structure exhibited their own advantages. Few contributions have been made, but a detailed study is still required that provides better control to synthesize UCL core/shell structures with all possible combinations of phases. In this work, we proposed a comprehensive strategy to design homogeneous and heterogeneous core/shell structures. Every possible combination of core/shell with respect to structural symmetry requires its own synthesis conditions. Homo-structures of NaYF4 is recommended under similar conditions of temperature to avoid secondary phase impurities. NaYF4 with hetero-structure fabrications has more difficulties due to the thermally motivated transition of the cubic phase to the hexagonal phase.55 If the hexagonal shell is grown over the cubic core at a certain temperature suitable for hexagonal nucleation, then the metastable core also undergoes a phase transition to the hexagonal phase. Our previous approach56 of sub-10 nm synthesis of hexagonal phase by the use of excess sodium has been used as a key to synthesizing hexagonal shell over cubic core under controlled condition of temperature, which prevents the transformation of the cubic phase core to hexagonal phase. Also, cubic phase core nanocrystals have been synthesized with different sizes to check the stability of the cubic phase for different durations of heating. Therefore, in this effort, we have addressed different synthesis conditions to achieve better control over the fabrication of core–shell structures. NaYF4 in this study may act as a prototype for the synthesis of other similar structures.
Results and discussion
Effect of heating on the size of the cubic phase
Fig. 1a shows TEM images of cubic phase NaYF4:Yb,Er. Nanocrystals are mono-dispersed and spherical in shape with an average size of 7 nm to 15 nm. The size distributions are also shown in the inset of Fig. 1a. The particles were prepared by adopting the reported procedure with slight modification. All the samples were prepared with a fixed composition of precursors in 1-octadecene and oleic acid at a temperature of 280 °C. | ||
| Fig. 1 (a) TEM images with inset size distributions of monodispersed nanocrystals of cubic phase NaYF4:Yb,Er synthesized at T = 280 °C for different reaction time (i) 30 min, (ii) 1 h, (iii) 1.5 h, (iv) 2 h, (v) 2.5 h and (vi) 3 h. (b) XRD patterns of NaYF4:Yb,Er synthesized at T = 300 °C for different reaction time (i) 30 min, (ii) 60 min, (iii) 90 min, (iv) 120 min, (v) 150 min and (vi) 180 min. (c) Shows the growth of particle size with increasing time of reaction. | ||
Only the heating time has been varied from 30 min to 180 min. When the solution was heated at 280 °C for 30 min, pure cubic phase NaYF4:Yb,Er was obtained, which is confirmed by X-ray diffraction measurement, as shown in Fig. 1b. The nanoparticles exhibited an average size of 7 nm.
When the heating time was increased to 1 hour, the average size of the particles was 9 nm. Similarly, as the heating time was further increased to 1.5 hours, 2 hours, 2.5 hours, and 3 hours, the average size of the obtained particles was 10 nm, 11 nm, 13 nm and 15 nm, respectively. All the reactions yielded a pure cubic phase without the formation of secondary phases. The crystallinity and phase of all the samples were determined by the X-ray diffraction patterns shown in Fig. 1b. Normalized intensities of six samples were plotted against 2θ from 10 to 70. The planes labeled as (111), (200), (220), (311), (222) and (400) corresponded to the cubic symmetry of the JCPDS file no. 77-2042.57 Crystallite sizes measured using the Debye–Scherrer formula were also consistent with the particle sizes calculated from the TEM images shown in Table 1.
| Reaction time (min) | Particle size calculated from TEM | Crystallite size by calculated Scherrer's equation |
|---|---|---|
| 30 | 7 | 7.060 |
| 60 | 9 | 8.509 |
| 90 | 10 | 9.808 |
| 120 | 11 | 10.790 |
| 150 | 13 | 12.826 |
| 180 | 15 | 14.563 |
Effect of temperature on the α- to β-phase transition
A series of experiments were performed with increasing temperature to investigate the hexagonal nucleation temperature threshold. When the solution was heated at 300 °C for 30 min, a pure cubic phase was obtained. When the reaction time was prolonged to 1 hour, 1.5 hours, 2 hours and 2.5 hours with the reaction temperature of 300 °C, the little hexagonal phase peaks appeared at 90 min and became sharper after 120 min of heating. The XRD peaks corresponding to the hexagonal phase according to JCPDS file no. 16-0334 (ref. 57) are marked with an asterisk, as shown in Fig. 2a. After the reaction time of 150 min, pure hexagonal phase was obtained. These observations of reaction time and temperatures were very useful to synthesize homo–hetero core–shell structures.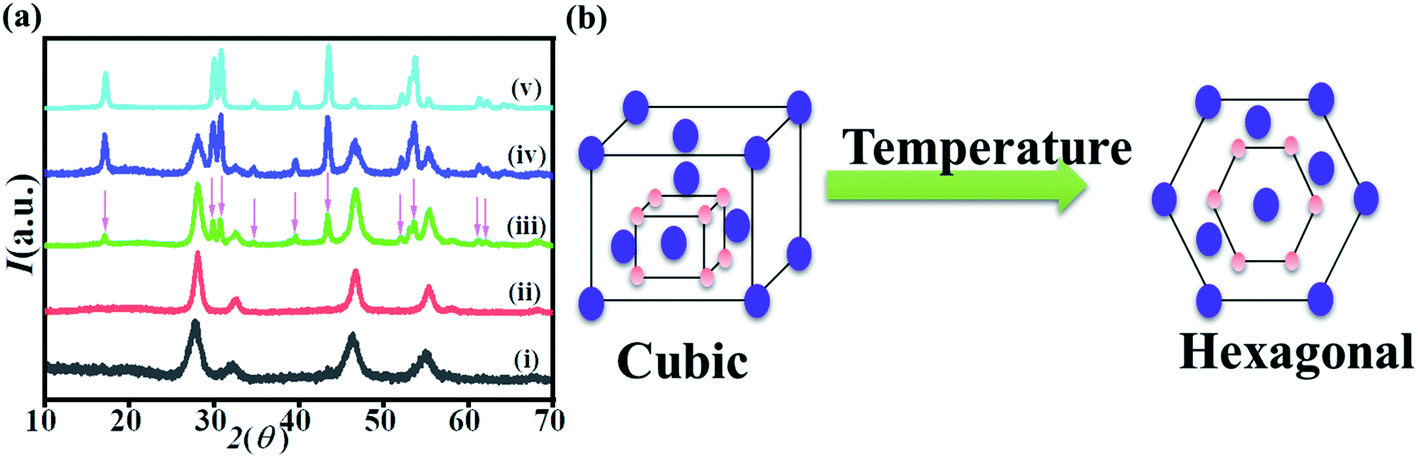 | ||
| Fig. 2 (a) XRD patterns of NaYF4:Yb,Er synthesized at T = 300 °C for different reaction time (i) 30 min, (ii) 60 min, (iii) 90 min, (iv) 120 min and (v) 150 min. (b) Schematic figure shows the phase transition from cubic to hexagonal after prolonged heating. | ||
Synthesis of α-NaYF4:Yb,Er@α-NaYF4
Cubic phase NaYF4:Yb,Er prepared at 280 °C for 60 min was used as the core. The dispersed α-NaYF4:Yb,Er nanocrystals were introduced to the solution at 70 °C until hexane was evaporated completely. Then, the temperature of the reaction was raised to 280 °C and kept for 30 min. α-NaYF4:Yb,Er served as seed, and the shell was formed around α-NaYF4:Yb,Er. TEM images in Fig. 3c show the core/shell NaYF4:Yb,Er@NaYF4. The thickness of the shell was approximately 2 nm. X-ray diffraction patterns in Fig. 3a confirmed that the shell was pure cubic phase. | ||
| Fig. 3 (a) XRD patterns of core/shell NaYF4 with Yb and Er doped in core. The phase combination consist of (i) α-NaYF4:Yb,Er, (ii) α-NaYF4:Yb,Er@α-NaYF4, (iii) α-NaYF4:Yb,Er@β-NaYF4, (iv) β-NaYF4:Yb,Er, (v) β-NaYF4:Yb,Er@α-NaYF4, and (vi) β-NaYF4:Yb,Er@β-NaYF4. (b) Shows the combinations of the core/shell structure. (c) TEM images of core/shell and the size distributions inset with (i) α-NaYF4:Yb,Er, (ii) α-NaYF4:Yb,Er@α-NaYF4, (iii) α-NaYF4:Yb,Er@β-NaYF4, and FESEM images of (iv) β-NaYF4:Yb,Er, (v) β-NaYF4:Yb,Er@α-NaYF4, and (vi) β-NaYF4:Yb,Er@β-NaYF. | ||
Synthesis of α-NaYF4:Yb, Er@β-NaYF4
α-NaYF4 nanocrystals prepared at 300 °C for 30 min were used as the core for the β-NaYF4 shell. The β-shell is very critical to synthesize because the nucleation of β-NaYF4 occurs at high temperature for a longer time, and if the reaction proceeds at high temperature for a longer time, then the α-phase core particles that are thermally metastable will naturally transform to β-phase particles by the Ostwald ripening process. Therefore, a different strategy is required here to synthesize the β-shell around the α-core at the minimum possible temperature within a short time period so as to prevent the phase transition of α-seeds. Keeping all situation under consideration, we employed our recent reported strategy of Na+ driven nucleation of β-NaYF4:Yb,Er. Excess sodium precursor has been used to lower the nucleation threshold for the NaYF4 shell, keeping the α-core stable. Therefore, the solution for the shell was prepared with excess sodium instead of a stoichiometric amount. The solution was heated for 30 min at 300 °C to grow a shell around the α-core. Since the temperature was low enough for a shorter time, it will not affect the phase of the α-core. X-ray diffraction of the α-core/β-shell is shown in Fig. 3a. The patterns show the peaks of the α-phase and β-phase. TEM images in Fig. 3c show uniform morphology of the core–shell structure with the size distributions in the inset.Synthesis of β-NaYF4:Yb,Er@α-NaYF4
α-Shell over β-core was grown by using the general precursors. The reaction temperature was 300 °C for 30 min. FESEM image in Fig. 3b(v) shows the β-core–α-shell morphology with an average size of 35 nm. Uniform clear core/shell structures are formed. X-ray diffraction analysis confirms the formation of α-shell over pure β-core.Synthesis of β-NaYF4:Yb,Er@β-NaYF4
β-Shell was grown over β-shell by heating the standard concentration of the precursor solution at 300 °C for 1.5 hours in the presence of the β-core. The core–shell morphology is shown in Fig. 3c(vi). The estimated core/shell thickness was 35 nm. X-ray diffraction patterns confirm that pure β-shell has been grown.Upconversion luminescence
Fig. 4a shows the up-conversion emission spectra of the cubic phase NaYF4:Yb,Er nanocrystals of varying sizes. All nanocrystals exhibited up-conversion emission in green and red when excited with 980 nm NIR radiation. Green emissions were observed at about 540 nm, which can be ascribed to the 2H11/2, 4S3/2 → 4I15/2 transitions. Red emission band centered at 660 nm has been obtained due to the 4F7/2 → 4I15/2 transitions. It is apparent that the luminescence of cubic nanocrystals increases with increasing particle size, which is consistent with the previous study. | ||
| Fig. 4 (a) UC luminescence spectra of α-NaYF4:Yb,Er with sizes from 7 nm to 15 nm. (b) UC luminescence spectra of α-NaYF4:Yb,Er, α-NaYF4:Yb,Er@α-NaYF4, and α-NaYF4:Yb,Er@β-NaYF4. (c) UC luminescence spectra of β-NaYF4:Yb,Er, β-NaYF4:Yb,Er@α-NaYF4, and β-NaYF4:Yb,Er@β-NaYF4. | ||
Fig. 4b and c demonstrate the up-conversion emission enhancement in the core/shell structure of NaYF4:Yb,Er as compared to the core alone. The β-shell exhibits more intense luminescence as compared to the α-shell. The β-shell serves as a better shield of defects as compared to the α-shell. The plausible reason for better enhancement in the case of the β-shell is a high density of β-phase NaYF4. The high-density layer provided a better cure to surface defects. Core/shell structures also have shown the enhancement of up-conversion luminescence with the β-core.
α-Shell over β-core proves to be a good protector of defects and other impurities by enhancing the luminescence. The β-shell comes out superior to the α-shell as it enhances the luminescence more than the α-coating. The enhancement factor for core–shell structures has been calculated from the integrated intensities of emission. α-Core/α-shell yielded about 5-fold enhancement as compared to the α-core, whereas α-core/β-shell exhibited 22-fold enhancement. In the case of the β-core, α-shell with β-core produced 5-fold enhancement and β-shell over β-core produced 6-fold enhancement. All the UCL for core/shell structures as compared to the core alone are summarized in Table 2.
| Structure of particle | UCL enhancement factor |
|---|---|
| α-NaYF4:Yb,Er | 1Φ |
| α-NaYF4:Yb,Er@α-NaYF4 | 5Φ |
| α-NaYF4:Yb,Er@β-NaYF4 | 22Φ |
| β-NaYF4:Yb,Er | 1Ψ |
| β-NaYF4:Yb,Er@α-NaYF4 | 5Ψ |
| β-NaYF4:Yb,Er@β-NaYF4 | 6Ψ |
Conclusion
NaYF4 has been used as a prototype to propose a detailed strategy for synthesizing core/shell structures at nanoscale. The synthesis conditions of two structures of NaYF4, which are cubic and hexagonal, were discussed in detail. Each crystal symmetry had its own synthesis conditions, which were determined in this work. Size-dependent cubic phase nanocrystals were synthesized by varying the reaction time. A longer heating time yielded larger particles. The UCL intensities were also stronger for larger particles. Core/shell structures with the following combinations were prepared: α-NaYF4:Yb,Er@α-NaYF4, α-NaYF4:Yb,Er@β-NaYF4, β-NaYF4:Yb,Er@α-NaYF4, and β-NaYF4:Yb,Er@β-NaYF4. The cubic shell was grown over the cubic core and hexagonal core by using suitable conditions. Hexagonal shell over cubic core was grown using excess sodium to lower the nucleation threshold of hexagonal symmetry. This helped to prevent the conversion of the metastable cubic core to the hexagonal phase. Upconversion luminescence spectra of all prepared samples were determined. Enhancement was observed after shell formation.Methods
Synthesis section
Synthesis of hexagonal phase NaYF4:Yb,Er
For the synthesis of sub-10 nm of hexagonal phase NaYF4:Er,Yb, 1 mmol of RE-oleates (0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.18:0.02/Y:Yb:Er ratio) and 7 mmol of sodium acetate were dissolved in 10 mL ethanol in a three-necked glass flask. 5 mL of 1-octadecene mixed with 10 mL of oleic acid was then added to the above solution, which was heated at 60 °C until all the ethanol was evaporated. The solution was further heated to an elevated temperature of 160 °C for 30 min under the steady flow of Ar gas. The solution was then cooled down to room temperature when the residual gases were removed. Next, 4 mmol of NH4F and 2.5 mmol of NaOH were mixed in 10 mL of methanol and then transferred to the three-necked flask. The flask containing the mixture was heated to 300 °C with a revolution rate of 20 °C per min for 30 min. It was then left to cool naturally to ambient temperature and the product precipitated in acetone. The precipitate was centrifuged to get the sample and then washed several times with ethanol. The samples were collected after centrifugation and dispersed in hexane.
:0.18:0.02/Y:Yb:Er ratio) and 7 mmol of sodium acetate were dissolved in 10 mL ethanol in a three-necked glass flask. 5 mL of 1-octadecene mixed with 10 mL of oleic acid was then added to the above solution, which was heated at 60 °C until all the ethanol was evaporated. The solution was further heated to an elevated temperature of 160 °C for 30 min under the steady flow of Ar gas. The solution was then cooled down to room temperature when the residual gases were removed. Next, 4 mmol of NH4F and 2.5 mmol of NaOH were mixed in 10 mL of methanol and then transferred to the three-necked flask. The flask containing the mixture was heated to 300 °C with a revolution rate of 20 °C per min for 30 min. It was then left to cool naturally to ambient temperature and the product precipitated in acetone. The precipitate was centrifuged to get the sample and then washed several times with ethanol. The samples were collected after centrifugation and dispersed in hexane.
Synthesis of cubic phase NaYF4:Yb,Er
A similar synthesis procedure was adopted for the sub-10 nm cubic phase NaYF4:Er,Yb by varying concentrations of the reactants and reaction time. 1 mmol of rare-earth oleates (0.8:0.18:0.02/Y:Yb:Er ratio) were dissolved in 10 mL of ethane. 5 mL of 1-octadecene and 10 mL of oleic acid were used. 4 mmol of NH4F and 2.5 mmol of NaOH were mixed in 10 mL of methanol. The mixture was heated to 280 °C at the rate of 20 °C per min for 30 min. The samples were collected after centrifugation and dispersed in hexane. When the reaction time was varied from 30 min to 60 min, the particle sizes changed from 7 nm to 9 nm. Similarly, as the heating time was raised to 1.5 hours, 2 hours, 2.5 hours, and 3 hours, the average size of the obtained particles became 10 nm, 11 nm, 13 nm and 15 nm, respectively.
Sample characterization
Morphological measurements were performed with High-Resolution Transmission Electron Microscopy (JEOL-2010) to measure the particle size and identify the shape. The phase and structure identification were achieved by X-ray diffraction studies (Rigaku SmartLab diffractometer) by employing Cu-Kα radiation (λ = 0.154056 nm). Emission spectra were obtained on a fluorescence spectrometer (Princeton Instruments Acton SP 2750) equipped with an excitation source of 980 nm laser. Pellets of each nanocrystal were prepared to measure the spectra.Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for funding this work through Research Group No. RG-1441-5070. M. Imran expresses appreciation to the Deanship of Scientific Research at King Khalid University Saudi Arabia for funding through research group program under grant number R.G.P. 2/28/42.Conflicts of interest
There are no conflicts to declare.References
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3432–3448 CrossRef PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- J. Liang, F. Li and H. M. Cheng, Energy Storage Materials, 2017, 7, A1–A3 CrossRef.
- B. Scrosati, J. Solid State Electrochem., 2011, 15, 1623–1630 CrossRef CAS.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 18013 CrossRef.
- L. Wang, Q. Wang, W. Jia, S. Chen, P. Gao and J. Li, J. Power Sources, 2017, 342, 175–182 CrossRef CAS.
- Y. Cheng, J. Huang, J. Li, Z. Xu, L. Cao, H. Ouyang, J. Yan and H. Qi, J. Alloys Compd., 2016, 658, 234–240 CrossRef CAS.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854–3859 CrossRef CAS PubMed.
- X. Xie, Z. Ao, D. Su, J. Zhang and G. Wang, Adv. Funct. Mater., 2015, 25, 1393–1403 CrossRef CAS.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- M. Wilkening and P. Heitjans, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–13 CrossRef.
- M. Wilkening, W. Küchler and P. Heitjans, Phys. Rev. Lett., 2006, 97, 1–4 CrossRef PubMed.
- A. van Der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216–1225 CrossRef CAS PubMed.
- G. H. Newman and L. P. Klemann, J. Electrochem. Soc., 1980, 2097 CrossRef CAS.
- D. A. Winn, J. M. Shemilt and B. C. H. Steele, Mater. Res. Bull., 1976, 11, 559–566 CrossRef CAS.
- Y. Liu, H. Wang, L. Cheng and N. Han, Nano Energy, 2016, 20, 168–175 CrossRef CAS.
- Z. Hou, C. Li, P. Ma, Z. Cheng, X. Li, X. Zhang, Y. Dai, D. Yang, H. Lian and J. Lin, Adv. Funct. Mater., 2012, 22, 2713–2722 CrossRef CAS.
- L. Liang, Y. Liu and X. Z. Zhao, Chem. Commun., 2013, 49, 3958 RSC.
- X. Liu, M. Zheng, X. Kong, Y. Zhang, Q. Zeng, Z. Sun, W. J. Buma and H. Zhang, Chem. Commun., 2013, 49, 3224–3226 RSC.
- A. Podhorodecki, M. Banski, A. Noculak, B. Sojka, G. Pawlik and J. Misiewicz, Nanoscale, 2013, 5, 429–436 RSC.
- Q. Xiao, Y. Ji, Z. Xiao, Y. Zhang, H. Lin and Q. Wang, Chem. Commun., 2013, 49, 1527–1529 RSC.
- J. C. Boyer, F. Vetrone, L. A. Cuccia and J. A. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS PubMed.
- S. Heer, K. Kömpe, H. U. Güdel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- H. U. Gu, M. Pollnau, D. R. Gamelin and S. R. Lu, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, Chem. Mater., 2003, 15, 3650–3655 CrossRef CAS.
- A. Patra, C. S. Friend, R. Kapoor and P. N. Prasad, J. Phys. Chem. B, 2002, 106, 1909–1912 CrossRef CAS.
- Q. Lu, Y. Hou, A. Tang, H. Wu and F. Teng, Appl. Phys. Lett., 2013, 102(23), 233103 CrossRef.
- D. Kumar, K. Verma, S. Verma, B. Chaudhary, S. Som, V. Sharma, V. Kumar and H. C. Swart, Phys. B, 2018, 535, 278–286 CrossRef CAS.
- R. A. Janjua, U. Farooq, R. Dai, Z. Wang and Z. Zhang, Opt. Lett., 2019, 44, 4678 CrossRef CAS PubMed.
- Y. Wang, R. Cai and Z. Liu, CrystEngComm, 2011, 13, 1772–1774 RSC.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- P. Qiu, N. Zhou, H. Chen, C. Zhang, G. Gao and D. Cui, Nanoscale, 2013, 5, 11512–11525 RSC.
- H. Na, K. Woo, K. Lim and H. S. Jang, Nanoscale, 2013, 5, 4242–4251 RSC.
- Q. Dou and Y. Zhang, Langmuir, 2011, 27, 13236–13241 CrossRef CAS PubMed.
- Y. Ma, H. Liu, Z. Han, L. Yang, B. Sun and J. Liu, Analyst, 2014, 139, 5983–5988 RSC.
- S. Schietinger, T. Aichele, H. Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134–138 CrossRef CAS PubMed.
- D. M. Wu, A. García-Etxarri, A. Salleo and J. A. Dionne, J. Phys. Chem. Lett., 2014, 5, 4020–4031 CrossRef CAS PubMed.
- H. Zhang, Y. Li, I. A. Ivanov, Y. Qu, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2010, 49, 2865–2868 CrossRef CAS PubMed.
- H. Guo, Z. Li, H. Qian, Y. Hu and I. N. Muhammad, Nanotechnology, 2010, 21(12), 125602 CrossRef PubMed.
- J. Pichaandi, F. C. J. M. van Veggel and M. Raudsepp, ACS Appl. Mater. Interfaces, 2010, 2, 157–164 CrossRef CAS.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- G. S. Yi and G. M. Chow, Chem. Mater., 2007, 19, 341–343 CrossRef CAS.
- J. Zhao, Y. Sun, X. Kong, L. Tian, Y. Wang, L. Tu, J. Zhao and H. Zhang, J. Phys. Chem. B, 2008, 112, 15666–15672 CrossRef CAS PubMed.
- K. W. Krämer, D. Biner, G. Frei, H. U. Güdel, M. P. Hehlen and S. R. Lüthi, Chem. Mater., 2004, 16, 1244–1251 CrossRef.
- J. Shan, M. Uddi, R. Wei, N. Yao and Y. Ju, J. Phys. Chem. C, 2010, 114, 2452–2461 CrossRef CAS.
- L. Wang and Y. Li, Nano Lett., 2006, 6, 1645–1649 CrossRef CAS PubMed.
- C. Homann, L. Krukewitt, F. Frenzel, B. Grauel, C. Würth, U. Resch-Genger and M. Haase, Angew. Chem., 2018, 130, 8901–8905 CrossRef.
- H. Guo, Z. Li, H. Qian, Y. Hu and I. N. Muhammad, Nanotechnology, 2008, 24, 12123–12125 Search PubMed.
- Y. Wang, L. Tu, J. Zhao, Y. Sun, X. Kong and H. Zhang, J. Phys. Chem. C, 2009, 113, 7164–7169 CrossRef CAS.
- H. Mai, Y. Zhang, L. Sun and C. Yan, J. Phys. Chem. C, 2007, 13721–13729 CrossRef CAS.
- Y. Wang, L. Tu, J. Zhao, Y. Sun, X. Kong and H. Zhang, J. Phys. Chem. C, 2009, 7164–7169 CrossRef CAS.
- N. J. J. Johnson, A. Korinek, C. Dong and F. C. J. M. van Veggel, J. Am. Chem. Soc., 2012, 134(27), 11068–11071 CrossRef CAS PubMed.
- D. Zhao, H. Chen, P. Zhang, H. Cui and W. Qin, Nanoscale Res. Lett., 2017, 12(1) DOI:10.1186/s11671-017-2306-3.
- H. Mai, Y. Zhang, L. Sun and C. Yan, J. Phys. Chem. C, 2007, 13730–13739 CrossRef CAS.
- R. A. Janjua, C. Gao, R. Dai, Z. Sui, M. A. Ahmad Raja, Z. Wang, X. Zhen and Z. Zhang, J. Phys. Chem. C, 2018, 122, 23242–23250 CrossRef CAS.
- L. Wang, X. Li, Z. Li, W. Chu, R. Li, K. Lin, H. Qian, Y. Wang, C. Wu, J. Li, D. Tu, Q. Zhang, L. Song, J. Jiang, X. Chen, Y. Luo, Y. Xie and Y. Xiong, Adv. Mater., 2015, 27, 5528–5533 CrossRef CAS PubMed.
- J. Park, K. An, Y. Hwang, J.-G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |