
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the sensitivity towards mercury via cooperative binding to D-fructose: dual fluorescent chemosensor based on 1,8-naphthyridine-boronic acid derivative†
Marina Rajadurai * and
E. Ramanjaneya Reddy
* and
E. Ramanjaneya Reddy
Center for Innovation in Molecular & Pharmaceuticals Sciences (CIMPS), Dr Reddy's Institute of Life Sciences, University of Hyderabad Campus, Gachibowli, Hyderabad 500046, Telangana, India. E-mail: marinasraj@gmail.com
First published on 21st April 2021
Abstract
A novel fluorescent chemosensor naphthyridine-boronic acid derivative (1.1) was synthesized and its ability to act as a selective chemosensor was examined for various metal ions. Compound 1.1 displayed highly selective fluorescence quenching upon interaction with Hg2+, possibly by means of photo induced electron transfer (PET) mechanism. The binding stoichiometry of the naphthyridine-boronic acid–Hg2+ complex and the association constant was determined. It was found that in the presence of D-fructose at physiological concentration, the sensitivity of chemosensor 1.1 towards Hg2+ improved by at least 7 times, perhaps as a result of the cooperative binding of both D-fructose and mercury ion to the sensor. Till now, the presented dual D-fructose–mercury chemosensor is the first example of utilizing boronic acid–diol complexation for enhancement of the sensor's sensitivity towards a toxic metal ion. The utility of compound 1.1 lays in applications in the food industry, e.g. for detection of mercury contamination of high fructose corn syrup, or in estimation of mercury in polluted biological samples and underground water.
1. Introduction
Mercury and mercuric salts are extensively circulated in atmospheric air, water and soil, and considered to be highly toxic and hazardous to humans and the environment.1 For humans, mercury contamination may cause a wide variety of symptoms, including neuro-disorders, neuromuscular changes, memory loss and carcinogenic diseases.2 Despite the danger mercury possesses, especially to unborn children, it is still used in industry. Its contaminants are often detected in various products, including food industry products, for example in high fructose corn syrup, which is cheaper and sweeter than regular sugar. Traces of mercury were found in high fructose corn syrup containing beverages/confectioneries, and the alarming amounts of up to 0.570 micrograms of mercury per gram of high fructose corn syrup were determined.3 Various methods have been employed for the detection of Hg2+, including cold vapor atomic absorption spectroscopy,4 high performance liquid chromatography5 and inductively coupled plasma atomic emission spectrometry.6 The above methods are extremely sensitive and able to detect Hg2+ in the nanomolar range. However, their drawbacks are: costly equipment, time-consuming and laborious procedures, and need for the trained professionals in order to perform the analysis. At the same time, mercury detection based on fluorescence changes allows rapid, convenient and inexpensive detection. Fluorescence chemosensors offer definite advantages, such as high selectivity, high sensitivity, accuracy and possibility to investigate molecule–molecule recognition in both environmental and biological samples.7 Till date, numerous fluorescent chemosensors for Hg2+ are developed based on rhodamine and BODIPY,8 various nanoparticles, for example silver,9 modified naphthalimide derivatives and other sensors.10 However, these sensors often suffer from such drawbacks, as laborious, complex or expensive synthesis, low aqueous solubility or limited selectivity as result of interference with other metal ions.1,8-Naphthyridine and its derivatives are extensively studied for molecular recognitions events, including nucleosides sensing (e.g. for guanine, cytosine and thymidine),11 monosaccharide sensing12 and heavy transition metal ions sensing13 (e.g. for Zn2+, Hg2+, Cd2+). In order to improve fluorescent properties, affinity, aqueous solubility and stability of 1,8-naphthyridine based chemosensors, we report design and synthesis of novel fluorescent chemosensor 1.1 [(2-((((5-(7-acetamido-1,8-naphthyridin-2-yl)thiophen-2-yl)methyl)(methyl)amino)methyl)phenyl)boronic acid]. In order to strengthen an interaction of mercury ion with the chemosensor, 1,8-naphthyridine was conjugated to thiophene moiety, keeping in mind high affinity of mercury to sulfur. At the same time, boronic acid group, which is known to easily form cyclic ester with monosaccharides, was additionally introduced in the same scaffold aiming cooperative action of boronic acid and metal chelate, which could possibly enhance sensitivity and selectivity either to metal ion or sugar. There are very few examples are known for using boronic acid containing compounds for direct detection of metal ions.14
Thus, we report the synthesis of naphthyridine-boronic acid based fluorescent chemosensor 1.1, its fluorescence properties and investigation of interaction with various metal ions and monosaccharides (D-glucose, D-fructose, D-mannose and D-galactose). The ability of chemosensor 1.1 to form reversible covalent bonds with D-fructose was employed for the first time to significantly enhance selectivity towards metal ion, namely Hg2+.
2. Materials and methods
2.1. General
The materials 2-thiophene carboxaldehyde, 2,6-diaminopyridine, n-butyl lithium, tributyltin chloride, bis(triphenylphosphine)palladium(II) dichloride, acetic anhydride, methyl amine (40% aqueous solution), 2-thiophenemethylamine, potassium bis(trimethylsilyl)amide, 2-(bromomethyl)phenylboronic acid and paraformaldehyde were purchased from Sigma Aldrich. N-Bromosuccinimide, phosphorus trichloride oxide, azobisisobutyronitrile, di-tert-butyl dicarbonate, trifluoroacetic acid, methyl iodide were purchased from Spectro Chem. Commercial reagents (DL-malic acid, ethane-1,2-diol, sodium borohydride, ammonium bicarbonate, mercury(I) chloride and HCl) were purchased from Merck and Rankem and were used as received. The solvents THF, DMF, toluene, 1,4-dioxane and DCM were distilled and dried before reactions and for extracting purposes. All reactions were carried out under an inert atmosphere with dry solvents, unless otherwise stated. Syringes and needles for the transfer of reagents were dried at 100 °C and allowed to cool in a desiccator over P2O5 before use. Reactions were monitored by thin layer chromatography (TLC) on silica gel plates (60 F254), using UV light detection. Merck silica gel (particle size 100–200 mesh) was used for column chromatography. For UV-vis and fluorescence measurements spectroscopic grade solvents were used.Buffers and samples preparations for fluorescence studies and synthesis of the compounds of routes 2–4 are given in ESI.†
2.2. Synthesis of target compound 1.1 using route I (Schemes 1 and 2)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) as an eluent to yield compound 1.4 in form of a golden needles (6.47 g, 60%). 1H NMR (DMSO-d6, 400 MHz) δ: 8.70 (1H, s), 8.56 (1H, d, J = 8.8 Hz), 8.20 (1H, d, J = 8.8 Hz), 8.18 (1H, d, J = 8.4 Hz), 7.40 (1H, d, J = 8.0 Hz), 2.30 (3H, S).:DCM (3 mL:3 mL) and stirred at RT for 2 h. After 2 h solvents were removed and the reaction mixture was treated with sodium bicarbonate solution and extracted with DCM. The organic solvent was removed under reduced pressure and the crude residue was then purified using column chromatography on silica gel using MeOH/DCM (2:8) as an eluent to yield compound 1.2 as a yellowish solid (0.189 g, 85%). Mp: 260–262 °C; TLC (20% MeOH in DCM): Rf = 0.21. 1H NMR (DMSO-d6, 400 MHz) δ: 11.08 (1H, brs), 8.35–8.28 (3H, m), 8.01 (1H, d, J = 8.4 Hz), 7.86 (1H, d, J = 4.4 Hz), 7.08 (1H, d, J = 3.6 Hz), 3.93 (2H, s), 2.35 (3H, s), 2.14 (3H, s). 13C NMR (DMSO-d6, 100 MHz) δ: 170.5, 155.2, 154.9, 154.8, 143.8, 139.6, 138.0, 127.9, 127.5, 119.2, 116.8, 114.2, 49.7, 35.2, 24.5. MS (EI, 70 eV): m/z [M]+ calcd for C16H16N4OS: 312.1, found 313.1 [M + 1]+. Anal. calcd for C16H16N4OS: C, 61.52; H, 5.16; N, 17.93; O, 5.12; S, 10.26, found: C, 61.42; H, 5.23; N, 17.85.
:9) as an eluent to yield compound 1.4 in form of a golden needles (6.47 g, 60%). 1H NMR (DMSO-d6, 400 MHz) δ: 8.70 (1H, s), 8.56 (1H, d, J = 8.8 Hz), 8.20 (1H, d, J = 8.8 Hz), 8.18 (1H, d, J = 8.4 Hz), 7.40 (1H, d, J = 8.0 Hz), 2.30 (3H, S).:DCM (3 mL:3 mL) and stirred at RT for 2 h. After 2 h solvents were removed and the reaction mixture was treated with sodium bicarbonate solution and extracted with DCM. The organic solvent was removed under reduced pressure and the crude residue was then purified using column chromatography on silica gel using MeOH/DCM (2:8) as an eluent to yield compound 1.2 as a yellowish solid (0.189 g, 85%). Mp: 260–262 °C; TLC (20% MeOH in DCM): Rf = 0.21. 1H NMR (DMSO-d6, 400 MHz) δ: 11.08 (1H, brs), 8.35–8.28 (3H, m), 8.01 (1H, d, J = 8.4 Hz), 7.86 (1H, d, J = 4.4 Hz), 7.08 (1H, d, J = 3.6 Hz), 3.93 (2H, s), 2.35 (3H, s), 2.14 (3H, s). 13C NMR (DMSO-d6, 100 MHz) δ: 170.5, 155.2, 154.9, 154.8, 143.8, 139.6, 138.0, 127.9, 127.5, 119.2, 116.8, 114.2, 49.7, 35.2, 24.5. MS (EI, 70 eV): m/z [M]+ calcd for C16H16N4OS: 312.1, found 313.1 [M + 1]+. Anal. calcd for C16H16N4OS: C, 61.52; H, 5.16; N, 17.93; O, 5.12; S, 10.26, found: C, 61.42; H, 5.23; N, 17.85.3. Results and discussion
3.1. Synthesis of naphthyridine-boronic acid (1.1)
The key intermediate in synthesis of the chemosensor 1.1 is compound 1.2, which in its turn could be synthesized through four different routes (Scheme 1). We selected the route I which allowed to successfully synthesizing key intermediate 1.2 and target compound 1.1 as described in Scheme 2. Thiophene methyl amine, which was protected with Boc-anhydride to give compound 2.1 served as the starting material for this synthetic route.15 Compound 2.1 was subjected to lithiation using n-BuLi followed by treatment with tributyl tin chloride resulting in compound 2.2 in excellent yield.15 Further, compound 2.2 was methylated using methyl iodide and potassium hexamethyldisilazane as a base, to give compound 1.3 in 96% yield. In order to build naphthyridine moiety, 2,6-diamino pyridine (2.3), was cyclised using DL-malic acid in the presence of conc. H2SO4, according to standard procedure,16 to give 7-amino-1,8-naphthyridin-2-ol (2.4) in 86% yield. Amino group of 7-amino-1,8-naphthyridin-2-ol was protected using acetic anhydride to obtain acetamide protected compound 2.5 in 89% yield.17 This step allowed subsequent selective conversion of hydroxyl group to the halogen via chlorination with phosphorus oxychloride18 to obtain 1.4 in 60% yield. In the next step, coupling of compound 1.3 with 1.4 in Stille coupling conditions in 1,4-dioxane using palladium(II) catalyst resulted in compound 2.6 in 67% yield. In order to introduce phenyl-boronic acid moiety in this scaffold, Boc-protection of the compound 2.6 was removed using trifluoroacetic acid to give key intermediate 1.2 in 82% yield, followed by N-alkylation with commercially available 2-bromomethylphenylboronic acid, using potassium carbonate as a base. Final compound was obtained in 70% yield and its structure was confirmed by different methods, including NMR spectroscopy and mass-spectrometry. Particularly, 1H NMR spectrum showed an appearance of characteristic N–CH2 peak at 3.72 ppm and B–(OH)2 peak at 9.03 ppm, additionally EI-MS spectrum showed predominant peak with m/z value of 447.1 corresponding to target molecule (ESI Fig. S1 and S2†). | ||
| Scheme 1 Plausible retrosynthesis of key intermediate 1.2 and target molecule 1.1. | ||
 | ||
| Scheme 2 Route I towards key intermediate 1.2 and target compound 1.1. | ||
Apart from the successful route described above, other attempts to synthesize target compound 1.1 were made as sketched in Scheme 1 (see experimental procedures and discussion in ESI, Schemes S1–S3†), including:
(a) Imine formation followed by reduction using intermediate 1.5, as presented in route II.
(b) Conversion of bromine to amine, via azide formation followed by N-alkylation using intermediate 1.6, as presented in route III.
(c) N-Alkylation using intermediate 1.7, as presented in route IV.
3.2. Photophysical properties of compound 1.1
To explore utility of the final fluorophore 1.1, we investigated the photophysical properties of 1.1 in MeOH and MeOH/H2O solutions. In both solutions, MeOH and MeOH/H2O fluorophore 1.1 displayed identical absorption maxima at 356 nm and a shoulder at 371 nm (Fig. 1 and Table S1 in ESI†). Fluorescent emission spectra (λex = 356 nm) in both solvents exhibited similar fluorescence bands, with a maximum at 401 nm for 1.1 in MeOH and 403 nm in MeOH/H2O. The solution state quantum yield of 1.1 in MeOH/H2O (Φf = 0.26) was slightly higher than that in MeOH (Φf = 0.20), Stokes shift of the new fluorophore 1.1 was 31 nm.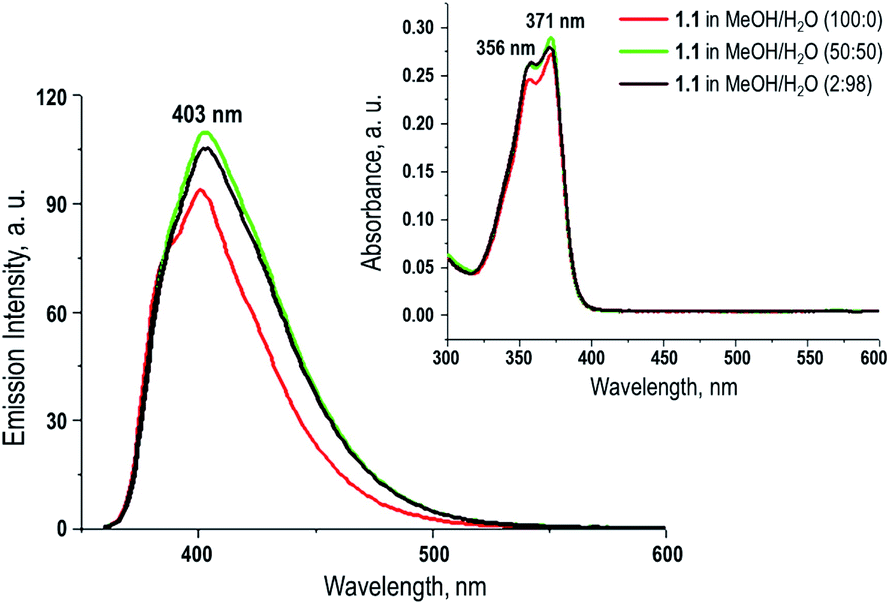 | ||
| Fig. 1 UV-vis absorption spectra of compound 1.1 in MeOH and MeOH/H2O, and its emission spectra in MeOH and MeOH/H2O, λex = 356 nm (insert). | ||
3.3. Examination of binding ability of 1.1 to metal ions
Fluorescence response of compound 1.1 in presence of various metal ions was investigated. The study was carried out in aqueous methanolic buffer at pH 7.5, and metal ions such as Ag1+, Zn2+, Cu1+, Mg2+, Pb2+, Cd2+, Hg2+, Li1+ and Fe3+ were tested. Among them, addition of Hg2+ to 1.1 showed a significant decrease of fluorescence intensity by 75% (Fig. 2A). This can be explained by the fact that after coordination to metal ion fluorophore becomes electron deficient, and as a result the PET from proximal tertiary nitrogen to adjacent chromophore increases and the fluorescence intensity of the fluorophore decreases. Apart from that, there could be possible contribution of strong spin–orbit coupling associated with Hg2+ to the luminescence quenching of 1.1. Fluorescence properties of compound 1.1 (10−5 M) were also studied upon increasing the concentration of Hg2+ (1–10 eq.), and it was observed that as the concentrations of Hg2+ increases, the fluorescent intensity of 1.1 gradually decreases as shown in Fig. 2B and C. In order to estimate the binding stoichiometry of naphthyridine-boronic acid–Hg2+ complex, Job plot experiments were employed.19 In Job method, equal concentrations of Hg2+ and 1.1 were prepared, and mixed in different proportions maintaining a total concentration of 10 μM. Using the obtained data, a graph was plotted between the emission intensity (fixed at 408 nm) and a mole fraction of compound 1.1 (Fig. 2D). The minimum emission intensity was reached when the mole fraction was 0.5. This result indicates the formation of naphthyridine-boronic acid–Hg2+ complex in a 1:1 ratio. The association constant (Ka) of naphthyridine-boronic acid–Hg2+ was calculated from the variation of fluorescence intensity as a function of the concentration of Hg2+ by using Benesi–Hildebrand equation20 (eqn (1)) and fitting the emission wavelength at 408 nm. The calculated Ka for a complex of 1.1 with Hg2+ was 2.842 × 105 M−1.| 1/(F − F0) = 1/(Ka(Fmax − F0)[Hg2+]) + 1/(Fmax − F0), | (1) |
 | ||
| Fig. 2 (A) Fluorescence spectra of compound 1.1 in MeOH/H2O in presence of various metal ions; (B) fluorescence spectra of compound 1.1 in MeOH/H2O in presence of Hg2+ at various concentrations (0–10 eq.); (C) Benesi–Hildebrand plot of the naphthyridine-boronic acid–Hg2+ complex in MeOH/H2O solutions; and (D) Job plot of naphthyridine-boronic acid–Hg2+ complex in MeOH/H2O. | ||
In order to understand the structure of naphthyridine-boronic acid–Hg2+ complex, proton NMR spectroscopy was employed.21 Addition of Hg2+ to the DMSO solution of 1.1 resulted in a downfield chemical shift of the Ha proton from 7.10 ppm to 7.55 ppm, additionally a moderate down field chemical shifts observed for He and Hh,i,j (Fig. 3). These changes indicate that Hg2+ interacts mainly with the sulphur atom, which was expected as high affinity of mercury for sulphur atoms is well known, and partly binds to the N1 and N8 of the naphthyridine moiety; at the same time there are no interaction with N-methyl nitrogen (no shift observed in NMR spectrum, not shown in Fig. 3). The decrease of fluorescence intensity or down field chemical shifts of Ha, He and Hh,i,j upon addition of Hg2+ are due to decrease of electron density after coordination of Hg2+ to the respective binding site as shown in Fig. 3. Therefore, these both observations support the theory of PET mechanism for presented here chemosensor.
 | ||
| Fig. 3 (A) Proposed coordination complex structure of Hg2+ ions with 1.1, and (B) 1H NMR spectra of compound 1.1 along and in the presence of Hg salt (1:3 eq.) in DMSO-d6. | ||
3.4. Examination of binding ability of 1.1 to monosaccharides
Fluorescence titration studies of compound 1.1 (10−5 M) in presence of various monosaccharides was carried out in aqueous methanolic phosphate buffer at pH 8.20. As expected, the fluorescence intensity of compound 1.1 increased upon increasing of saccharides concentration (D-glucose, D-mannose, D-fructose and D-galactose). As reported by Shinkai et al.22 such fluorescence response explained by the fact that boronic acids (such as compound 1.1) form cyclic esters with cis 1,2-diol or 1,3-diol of mono saccharides, which leads to increase of the Lewis acidity of the central boron atom. As result B–N (proximal tertiary nitrogen) interactions are strengthening23 and PET from proximal tertiary nitrogen to adjacent fluorophore is suppressed and the fluorescence of the fluorophore is enhancing.22,24 The fluorescence enhancement I/I0 obtained for D-glucose, D-galactose, D-mannose and D-fructose is in the order D-fructose > D-galactose > D-mannose > D-glucose as shown in Fig. 4. | ||
| Fig. 4 (A) Fluorescence intensity of compound 1.1 in MeOH/H2O (v/v = 1:1, 5 mM PBS, pH 8.20) in the presence of D-glucose, D-galactose, D-mannose and D-fructose; (B) fluorescence spectra of compound 1.1 (10−5 M) in presence of D-fructose at various concentrations (0–10 mM); (C) pH vs. fluorescent intensity of 1.1 (10−5 M) in presence and absence of D-fructose in MeOH/H2O (v/v = 1:1, 5 mM), at various pH solutions and (D) Benesi–Hildebrand plot of the naphthyridine-boronic acid–fructose complex in MeOH/H2O (v/v = 1:1, 5 mM PBS, pH 8.20) solutions, the emission wavelength was monitored at 403 nm. | ||
The binding selectivity of 1.1 towards D-fructose is around two and half fold higher compared to other monosaccharides (D-galactose, D-mannose, D-glucose) at lower concentrations (<10 mM). The association constant (Ka) of naphthyridine-boronic acid based fluorescence chemo sensor 1.1 with D-fructose, D-glucose, D-galactose and D-mannose were calculated from the variation of fluorescence intensity (Fig. 4 and S21–S23 in ESI†) as a function of the concentration of D-monosaccharides, using Benesi–Hildebrand equation20 (eqn (2)) and fitting the emission wavelength at 403, 409, 410 and 408 respectively. The highest affinity of 1.1 was observed towards fructose (the association constants (Ka) = 104.91 M−1), while to other sugars it was significantly lower (Ka = 14.42–39.46 M−1). The affinity for 1.1 were in the following order: D-fructose > D-galactose > D-mannose > D-glucose, and the calculated association constant are listed in Tables S2–S5 in ESI.†
| 1/(F − F0) = 1/(Ka(Fmax − F0)[sugar]) + 1/(Fmax − F0), | (2) |
Fluorescence response of the boronic acid based fluorescent chemosensors is usually pH dependent. Therefore, fluorescence response of naphthyridine-boronic acid fluorescent chemosensor 1.1 was measured at different pH, both in the presence and absence of D-fructose. As Fig. 4C displays, in both cases the fluorescent response was similar. Fluorescent intensity was increasing upon pH change from 2 to 5.5 due to protonation of tertiary amine, and was decreasing as pH raised from 5.5 to 12 due to deprotonation of tertiary amine and OH− adduct formation with boronic acid group.25
3.5. Competitive sensing of Hg2+ and D-monosaccharide
As compound 1.1 has incorporated phenyl boronic acid moiety which is expected to bind to mono-saccharides, its fluorescence response was examined in presence of Hg2+ and various D-monosaccharides. It was expected, that cooperative binding of both sugar and metal ion will result in increased sensitivity of the chemosensor to either of analytes, decreasing/eliminating at the same time interference by various factors, which are common for chemosensors acting through “turn-off” mechanism.25a The study was carried out in aqueous methanolic buffer at pH 7.5. Addition of Hg2+ to a solution containing both chemosensor 1.1 and D-fructose complex resulted in dramatic decrease of fluorescence intensity by seven folds (Fig. 5A). This fluorescent response was ∼2.8 times higher compared to one, when compound 1.1 was interacting with mercury without addition of D-fructose (Fig. 5B). At the same time, no any significant change was observed in the emission intensity of the probe 1.1 in the presence of other hexose sugars. | ||
| Fig. 5 Fluorescence spectra of 1.1 alone (10−5 M), 1.1 with Hg2+ and 1.1 with Hg2+ and D-monosaccharides in MeOH/H2O (v/v = 1:1, 5 mM, at various pH) solutions (A) and comparative bar graph of the emission intensity change for the same solutions (B). | ||
Till date, there are no reports, where the sensitivity of the chemosensor for metal ions was enhanced as result of interaction with fructose, although there are few investigations utilizing metal–sensor coordination in order to improve sensitivity towards sugar. For example, Seiji Shinkai et al. reported25a new saccharide receptor for uronic acids, based on two-point interactions of boronic acid and coordination with zinc(II), or another work14 employing cooperative binding of fructose and metal ion performed by group of Tony D. James. In contrary, we obtained an interesting result indicating that the sensitivity of the chemosensor 1.1 towards mercury ions increases significantly in presence of D-fructose moiety. Thus, presented here work is the first example of employing boronic acid–diol complexation to achieve significant enhancement of the sensor's sensitivity towards toxic metal ion.
4. Conclusions
In conclusion, we present the design and synthesis of novel fluorescent chemosensor 1.1 [(2-((((5-(7-acetamido-1,8-naphthyridin-2-yl)thiophen-2-yl)methyl)(methyl)amino)methyl)phenyl)boronic acid], its fluorescence properties and interaction with various metal ions and monosaccharides. Chemosensor 1.1 was designed to combine naphthyridine as a fluorescent metal chelating site and phenyl boronic acid as a saccharide recognition unit in one scaffold. The combination of these properties allowed investigation of the sensing abilities of 1.1 over various metal ions and D-monosaccharides. Fluorescence change, mediated by PET mechanism, was utilized as a reporting property of the chemosensor. Compound 1.1 found to have dramatic decrease of fluorescence intensity selectively upon binding to Hg2+, probably due to decrease of electron density after coordination of Hg2+ to the respective binding site. As a result, PET from proximal tertiary nitrogen to adjacent fluorophore enhanced and the fluorescence intensity changed by ∼2.5 folds. The binding stoichiometry of the naphthyridine-boronic acid–Hg2+ complex was found to be 1:1, according to Job plot experiments. The association constant for 1.1 with Hg2+ was found to be 2.842 × 105 M−1. Interestingly, the sensitivity of chemosensor 1.1 towards mercury ion greatly improved (7 fold) in the presence of D-fructose at physiological concentration, perhaps as result of the cooperative binding of both D-fructose and mercury ion to the sensor. This work is the first example of utilizing boronic acid–diol complexation to manipulate and enhance the sensor's sensitivity towards toxic metal ion. This finding is especially important for applications in food industry, e.g. for detection of mercury contamination of high fructose corn syrup, which often contains dangerous amounts of mercury residues,26 or for estimation of mercury in polluted biological samples and underground water.
Author contributions
E. Ramanjaneya Reddy: investigation, writing – original draft. Marina Rajadurai: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ERR acknowledges CSIR-New Delhi for a Senior Research Fellowship. This research was supported by grants from DST (SR/SI/CS-131/2009), CSIR (01(2411)/10/EMR-II) and DBT (BT/PR32386/MED/32/681/2019), New Delhi to MR.Notes and references
- P. D. Selid, H. Xu, E. M. Collins, M. S. Face-Collins and J. X. Zhao, Sensors, 2009, 9, 5446–5459 CrossRef CAS.
- F. M. M. Morel, A. M. L. Kraepiel and M. Amyot, Annu. Rev. Ecol. Syst., 1998, 29, 543–566 CrossRef.
- R. Dufault, B. LeBlanc, R. Schnoll, C. Cornett, L. Schweitzer, D. Wallinga, J. Hightower, L. Patrick and W. Lukiw, J. Environ. Health, 2009, 8, 2–8 CrossRef PubMed.
- A. F. Danet, M.-C. Bratu, M.-C. Radulescu and A. Bratu, Sens. Actuators, B, 2009, 137, 12–16 CrossRef.
- H. Hashemi-Moghaddam and M. J. Saber-Tehrani, J. AOAC Int., 2008, 91, 1453–1458 CrossRef CAS.
- O. T. Butler, J. M. Cook, C. F. Harrington, S. J. Hill, J. Rieuwerts and D. L. Miles, J. Anal. At. Spectrom., 2006, 21, 217–243 RSC.
- (a) W. Lin, L. Long and W. Tan, Chem. Commun., 2010, 46, 1503–1505 RSC; (b) D.-H. Wang, Y. Zhang, Z. Gong, R. Sun, D.-Z. Zhao and C.-L. Sun, RSC Adv., 2015, 5, 50540–50543 RSC.
- (a) J. Hu, Z. Hu, Y. Cui, X. Zhang, H.-W. Gao and K. Uvdal, Sens. Actuators, B, 2014, 203, 452–458 CrossRef CAS; (b) M. Vedamalai, D. Kedaria, R. Vasita, S. Mori and I. Gupta, Dalton Trans., 2016, 45, 2700–2708 RSC; (c) W. Sun, R. Chen, X. Cheng and L. Marin, New J. Chem., 2018, 42, 19224–19231 RSC; (d) A. N. Kursunlu, M. Oguz and M. Yilmaz, IEEE Sens. J., 2019, 19, 2009–2015 CAS.
- S. Balasurya, A. Syed, A. M. Thomas, N. Marraiki, A. M. Elgorban, L. L. Raju, A. Das and S. S. Khan, Spectrochim. Acta, Part A, 2020, 228, 117712–117719 CrossRef CAS PubMed.
- (a) P. Mahato, S. Saha, P. Das, H. Agarwalla and A. Das, RSC Adv., 2014, 4, 36140–36174 RSC; (b) J. Liu, K. Vellaisamy, G. Yang, C.-H. Leung and D.-L. Ma, Sci. Rep., 2017, 7, 3620, DOI:10.1038/s41598-017-03952-x; (c) M. Saleem, M. Rafiq and M. Hanif, J. Fluoresc., 2017, 27, 31–58 CrossRef CAS PubMed.
- (a) S. Djurdjevic, D. A. Leigh, H. McNab, S. Parsons, G. Teobaldi and F. Zerbetto, J. Am. Chem. Soc., 2007, 129, 476–477 CrossRef CAS PubMed; (b) H. Suda, A. Kobori, J. Zhang, G. Hayashi and K. Nakatani, Bioorg. Med. Chem., 2005, 13, 4507–4512 CrossRef CAS PubMed; (c) S.-H. Lu, S. Selvi and J.-M. Fang, J. Org. Chem., 2007, 72, 117–122 CrossRef CAS PubMed.
- J.-M. Fang, S. Selvi, J.-H. Liao, Z. Slanina, C.-T. Chen and P.-T. Chou, J. Am. Chem. Soc., 2004, 126, 3559–3566 CrossRef CAS PubMed.
- (a) M.-M. Yu, Z.-X. Li, L.-H. Wei, D.-H. Wei and M.-S. Tang, Org. Lett., 2008, 10, 5115–5118 CrossRef CAS PubMed; (b) M. K. Chahal and M. Sankar, Anal. Methods, 2015, 7, 4552–4559 RSC; (c) X. Liu, M. Chen, Z. Liu, M. Yu, L. Wei and Z. Li, Tetrahedron, 2014, 70, 658–663 CrossRef CAS.
- M. Li, H. Ge, R. L. Arrowsmith, V. Mirabello, S. W. Botchway, W. Zhu, S. I. Pascu and T. D. James, Chem. Commun., 2014, 50, 11806–11809 RSC.
- C. P. Harvey and J. D. Tovar, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4861–4874 CrossRef CAS.
- C. A. Anderson, P. G. Taylor, M. A. Zeller and S. C. Zimmerman, J. Org. Chem., 2010, 75, 4848–4851 CrossRef CAS PubMed.
- P. S. Corbin, S. C. Zimmerman, P. A. Thiessen, N. A. Hawryluk and T. J. Murray, J. Am. Chem. Soc., 2001, 123, 10475–10488 CrossRef CAS PubMed.
- J.-H. Jeon, K. Tanaka and Y. Chujo, Org. Biomol. Chem., 2014, 12, 6500–6506 RSC.
- J.-T. Yeh, W.-C. Chen, S.-R. Liu and S.-P. Wu, New J. Chem., 2014, 38, 4434–4439 RSC.
- (a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS; (b) R. B. Singh, S. Mahanta and N. Guchhait, J. Mol. Struct., 2010, 963, 92–97 CrossRef CAS.
- M. Vedamalai and S.-P. Wu, Org. Biomol. Chem., 2012, 10, 5410–5416 RSC.
- (a) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 4, 477–478 RSC; (b) T. D. James, K. R. A. S. Sandanayake and S. Shinkai, Angew. Chem., Int. Ed., 1994, 33, 2207–2209 CrossRef.
- (a) L. I. Bosch, T. M. Fyles and T. D. James, Tetrahedron, 2004, 60, 11175–11190 CrossRef CAS; (b) L. Zhu, S. H. Shabbir, M. Gray, V. M. Lynch, S. Sorey and E. V. Anslyn, J. Am. Chem. Soc., 2006, 128, 1222–1232 CrossRef CAS PubMed.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed.
- (a) M. Takeuchi, M. Yamamoto and S. Shinkai, Chem. Commun., 1997, 18, 1731–1732 RSC; (b) W. Yang, J. Yan, G. Springsteen, S. Deeter and B. Wang, Bioorg. Med. Chem. Lett., 2003, 13, 1019–1022 CrossRef CAS PubMed; (c) R. Hosseinzadeh, M. Mohadjerani and M. Pooryousef, Luminescence, 2015, 30, 549–555 CrossRef CAS PubMed.
- R. Dufault, B. LeBlanc and R. Schnoll, et al., Environ. Health, 2009, 8, 2 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02122b |
| This journal is © The Royal Society of Chemistry 2021 |