
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational structural modification of the isatin scaffold to develop new and potent antimicrobial agents targeting bacterial peptidoglycan glycosyltransferase†
Yong Wang‡
,
Zhiguang Liang‡,
Yuanyuan Zheng,
Alan Siu-Lun Leung,
Siu-Cheong Yan,
Pui-Kin So,
Yun-Chung Leung,
Wing-Leung Wong and
Kwok-Yin Wong *
*
State Key Laboratory of Chemical Biology and Drug Discovery, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hunghom, Kowloon, Hong Kong, P. R. China. E-mail: kwok-yin.wong@polyu.edu.hk
First published on 19th May 2021
Abstract
A series of isatin derivatives bearing three different substituent groups at the N-1, C-3 and C-5 positions of the isatin scaffold were systematically designed and synthesized to study the structure–activity relationship of their inhibition of bacterial peptidoglycan glycosyltransferase (PGT) activity and antimicrobial susceptibility against S. aureus, E. coli and methicillin-resistant Staphylococcus aureus (MRSA (BAA41)) strains. The substituents at these sites are pointing towards three different directions from the isatin scaffold to interact with the amino acid residues in the binding pocket of PGT. Comparative studies of their structure–activity relationship allow us to gain better understanding of the direction of the substituents that contribute critical interactions leading to inhibition activity against the bacterial enzyme. Our results indicate that the modification of these sites is able to maximize the antimicrobial potency and inhibitory action against the bacterial enzyme. Two compounds show good antimicrobial potency (MIC = 3 μg mL−1 against S. aureus and MRSA; 12–24 μg mL−1 against E. coli). Results of the inhibition study against the bacterial enzyme (E. coli PBP 1b) reveal that some compounds are able to achieve excellent in vitro inhibitions of bacterial enzymatic activity (up to 100%). The best half maximal inhibitory concentration (IC50) observed among the new compounds is 8.9 μM.
1. Introduction
Since the first discovery of the penicillin antibiotic in 1928, it has started the golden era of antibiotic discovery and development based on natural products.1 Over the past several decades, β-lactam antibiotics, such as penicillins, cephalosporins, monobactams, and carbacephems have been the most important and widely used clinical drugs to treat worldwide bacterial infections.2 The bacterial cell walls, of either Gram-positive or Gram-negative bacteria, play important roles to support bacteria to survive. Bacterial cell walls are constructed from peptidoglycans, which are cross-linked biopolymers.3 Most β-lactam antibiotics kill bacteria through the effective inhibition of the formation of peptidoglycan cross-links.4 Unfortunately, the overuse and sometimes abuse in various fields (e.g. ranging from clinical treatment to animal farming), bacteria evolve continuously into more powerful drug-resistant strains, and the ever evolution of drug resistance in many human pathogens has caused the emergence of worldwide ‘superbug crisis’, which seriously threaten global human health.5,6There are two generally accepted approaches to resolving the crisis of antimicrobial resistance: (i) development of synergistic combined therapies via the development of broad-spectrum inhibitors to restore the antibacterial activity of conventional antibiotics,7,8 and (ii) discovery of new antibiotics with a novel mode of antibacterial mechanism different from that of β-lactam antibiotics;9,10 to this end, scientists have to target at a new and highly conserved and critical bacterial enzyme for drug discovery. Over the last century, inhibition of bacterial cell wall syntheses is the most effective mean to inhibit bacterial growths by inducing their cell lysis.11 It is well known that both peptidoglycan glycosyltransferase (PGT) and transpeptidase (TP) activities of penicillin-binding proteins (PBPs) are essential for synthesizing bacterial cell walls. Although antibiotic resistance has emerged on the TP/PBP module, PGT is definitely another critical target for new drug discovery.12–16 However, compared to TP, the understanding and research findings about the structural and functional information of PGT are very limited, which largely restricts the rational design of new PGT inhibitors with high potency. The discovery of new inhibitors against PGT is therefore still very challenging.
Over the past twenty years, only a few potent inhibitors against PGT have been reported. Examples include disaccharide-modified molecules,17–20 monosaccharide-modified scaffolds,13 phosphoglycolipid AC326-α,21 coleophomones A and B,22 structurally modified moenomycin A derivative,23–26 modified peptidoglycan analogues,27 tryptamine-based compounds,28 and small molecules identified via high-throughput screening.29–33 Most inhibitors discovered so far are based on high-throughput screening, while inhibitors against the lipid-II binding site of PGT through molecular designs remain rare.34 Nonetheless, rational design of small molecule-based inhibitors with diverse structural scaffolds provides a good approach to understanding structure–activity relationship in depth. Moreover, this method enriches the molecular diversity for further improving the potency of PGT inhibitors. Our previous study via high-throughput screening has successfully identified a natural heterocyclic compound, isatin (1H indole-2,3-dione), which is found to be a potential PGT inhibitor after a simple modification on its N-1 and C-3 positions.32,35 The 2-site structural modifications, however, have not fully utilized the potential pharmacological properties of isatin moiety because its molecular scaffold allows multi-site three-directional structure modifications to create additional chemical features to fit for a specific purpose. Recent literature have shown that the diverse pharmacological properties of isatin-modified compounds have been identified in many other research areas,36,37 such as anti-cancer,38 anti-tubercular,39 antimalarial,40 anti-HIV41 and anti-bacterial42,43 treatments with good potency. In the present study, bacterial PGT inhibitors with three-directional structural modifications based on the isatin scaffold at the N-1, C-3 and C-5 positions (Scheme 1) was synthesized through structure-based design and then studied to gain better understanding of their structure–activity relationships towards the lipid-II binding pocket of bacterial peptidoglycan glycosyltransferase. The antimicrobial potency of the new inhibitors against Gram-positive (S. aureus (29213)), Gram-negative (E. coli (25922)) strains, and methicillin-resistant Staphylococcus aureus (MRSA (BAA41)) were also investigated.
 | ||
| Scheme 1 Three-site modified isatin derivatives for new antimicrobial studies on bacterial peptidoglycan glycosyltransferase (PGT). | ||
2. Results and discussion
Twelve isatin derivatives bearing three different substituent groups at the N-1, C-3 and C-5 positions of isatin scaffold (Scheme 1) were synthesized through two different synthetic approaches (Fig. S1†). The substituents at these positions of isatin are pointing to three different directions (Scheme 1), which allow maximizing the molecular interactions with the PGT binding pocket. More importantly, studying the activities of these compounds enable us to find out the direction of the substituent that provides critical interactions. The N-1 site was substituted with a flexible and lipophilic chain of various lengths (the carbon number of the aliphatic chain ranges from 4 to 14) as this substituent has been proposed to point towards the bacterial cell membrane region, where non-polar interactions are favorable. For the C-3 site, the functional ketone group of isatin was replaced with a polar guanidine group, which bears a positive charge (protonated form) and can therefore allow effective polar interactions with polar amino acid residues in the PGT binding pocket through hydrogen bonding and/or charge interactions. The PGT-inhibitory activity of the substituent effect at the C-5 site of isatin has not been investigated so far. This prompted us to synthesize four different substituents at this site so as to conduct comparative structure–activity relationship studies on the inhibitory activity against E. coli PBP 1b enzyme and the MIC effects against the Gram-positive and negative strains.The antimicrobial susceptibility tests of the new 3-site modified isatin derivatives were conducted with three strains: S. aureus, MRSA, and E. coli. The MIC results are summarized in Table 1. Compound 1, with no substituent at its C-5 site, show a MIC of 6 μg mL−1 (against S. aureus), 6 μg mL−1 (against MRSA), and 12 μg mL−1 (against E. coli), respectively. When a methyl group (–CH3) was introduced at the C-5 site (compound 2), its antimicrobial potency appears to increase against the strains (3 μg mL−1 for S. aureus and MRSA; 12 μg mL−1 for E. coli). These results indicate that the C-5 site of isatin scaffold is important for structural modifications. We therefore further modified the structure with an electron-donating methoxy group (–OCH3) and electron withdrawing groups (–F and –NO2) for comparison purposes. However, regarding the MIC values, a gradual reduction in the antimicrobial potency with these substituents appears (Table 1). Moreover, a MIC trend on the substituent groups at the C-5 site decreases in this order: –CH3 > –H ≈ –OCH3 > –F > –NO2 (Table 1). It seems that the nitro-group shows a significant decline in the antimicrobial potency (MIC > 192 μg mL−1), which is probably due to the strong electron-withdrawing character of –NO2 group that may reduce its ability to develop hydrogen bonding interactions in the PGT active site.44 To further verify these compounds can bind to PBP 1b enzyme and interfere with the lipid II polymerization, in vitro HPLC analyses using fluorescent dansyl-lipid II as the substrate45,46 were conducted against E. coli PBP 1b. The result indicates that compound 2 has the best inhibitory effect (the residual activity of the enzyme = 12%), while compound 5 with a nitro-substituent at the C-5 site shows no significant inhibitory effect. In this regard, the HPLC data are consistent with their MIC values. The results implies that the antimicrobial activity of the compounds is probably attributed to the inhibition of PBP 1b enzyme activity.
| Compounds | MIC (μg mL−1) | Inhibition of PBP 1b activity at 50 μM | Ki (μM) | ||
|---|---|---|---|---|---|
| S. aureus (29213) | MRSA (BAA41) | E. coli (25922) | |||
| a E. coli PBP 1b inhibitor.16 | |||||
| 1 | 6 | 6 | 12 | 17% | n.d. |
| 2 | 3 | 3 | 12 | 88% | 3.2 |
| 3 | 6 | 6 | 12 | 64% | n.d. |
| 4 | 12 | 12 | 48 | 14% | n.d. |
| 5 | >192 | >192 | >192 | 0% | n.d. |
| 6 | 96 | 96 | 96 | 4% | n.d. |
| 7 | 12 | 12 | 24 | 0% | n.d. |
| 8 | 6 | 6 | 12 | 71% | n.d. |
| 9 | 3 | 3 | 24 | 100% | 2.4 |
| 10 | 6 | 6 | 48 | 100% | n.d. |
| 11 | >192 | >192 | >192 | 100% | n.d. |
| 12 | >192 | >192 | >192 | 40% | n.d. |
| Moenomycin Aa | <0.75 | <0.75 | >96 | 100% | 1.9 × 10−3 |
We further modified the structure of compound 2 at the C-3 and N-1 sites for structure–activity comparison. At the N-1 site, a series of n-alkyl chains with various carbon atoms was synthesized, and the compounds (6–11) were also examined for their MIC values against three strains. Compound 6 has a short n-alkyl chain (butyl group) substituted at the N-1 site shows much poorer antimicrobial potency than compound 2 that bears a butylphenyl group at the same site. Interestingly, when the length of the n-alkyl chain increases from butyl to decyl (compound 7, 8, and 9), the antimicrobial activities of the compounds are improved gradually. This indicates that the N-1 site is sensitive to the hydrophobicity of the substituent, which probably interacts with the lipophilic region of the bacterial membrane. It seems that an aliphatic chain with 10 carbon atoms has optimal antimicrobial activity (MIC of compound 9 = 3 μg mL−1 against S. aureus and MRSA). Further increasing the chain length results in a significant reduction in antimicrobial potency; for example, compound 11 gave MIC >192 against S. aureus, MRSA, and E. coli. In general, the substitution at the N-1 site with a long aliphatic chain (8–11 carbon atoms) can enhance the inhibitory activity against PBP 1b but is unable to improve further the antibacterial potency, probably due to unfavorable factors, such as poor solubility and diffusion problem caused by the highly hydrophobic and bulky molecules. Based on the results of the compound series, compound 2 and 9 are the most potent antibacterial agents and their Ki values against E. coli PBP 1b are 3.2 and 2.4 μM, respectively (Fig. 1).
 | ||
| Fig. 1 Half maximal inhibitory concentration of compound 2 and 9 against E. coli PBP 1b. | ||
In order to demonstrate the importance of the guanidine moiety at the C-3 site of isatin, we replaced it with thiosemicarbazide (compound 12). Results of the enzyme inhibition assay and antibacterial ability test, the in vitro inhibitory enzymatic activity of compound 12 is significantly reduced as compared to compound 9. Inhibition assays with compound 12 at 50 μM, E. coli PBP 1b retains 60% activity while compound 9 achieves 100% inhibition (Table 1). Moreover, compound 12 reveals very weak antibacterial activity, as indicated by its relatively high MIC value > 192 μg mL−1 against the bacterial strains examined. These results suggest that the C-3 site requires a highly polar group (e.g. guanidine) capable of providing strong hydrogen bonding and/or charge interactions with the polar residues in the PGT binding pocket.
The cytotoxicity of compound 2, 9, and 12 were evaluated with MTT assays using a normal human cell line BJ (ATCC CRL-2522) for 24 h treatment. The results are shown in Fig. 2. Compound 2 and 9 show comparable cell proliferation profiles. At the concentration of 2 μg mL−1 or below, the cell viability is higher than 80%, indicating that both compounds are not toxic to the cells at these concentrations. The IC50 estimated for compound 2 and 9 against BJ cells are 6.6 μg mL−1 and 5.4 μg mL−1 (analyzed with Prism 6.0), which are higher than the MIC value (3 μg mL−1) of compound 2 and 9 against S. aureus and MRSA strains (Table 1). Moreover, it is noteworthy that compound 12 has a relatively low cytotoxicity, as revealed by about 80% cell viability after incubation at much higher concentration (18 μg mL−1). The results suggest that the cytotoxicity of the compounds is likely to be attributed from the guanidine moiety at the C-3 site of the isatin scaffold.
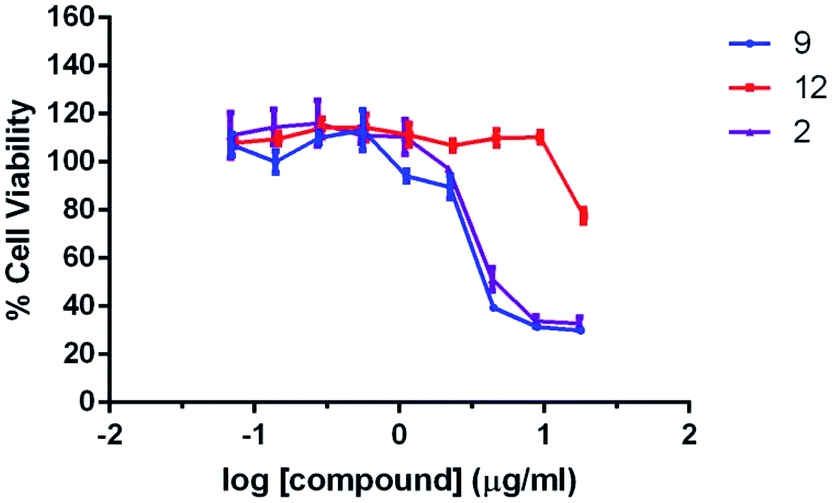 | ||
| Fig. 2 Cytotoxicity evaluation for compound 2, 9, and 12 in cell proliferation of a normal human cell line BJ (ATCC CRL-2522) in 24 h treatment. Data are presented in mean ± SD of three independence experiments, each performed in triplicate. | ||
Molecular docking study was then performed to understand the possible binding modes and locations of the molecules in the PGT binding pocket. The docking models between the selected compounds (compound 2, 6, 9 and 12) and S. aureus monofunctional glycosyltransferase (Mtg) were generated by AutoDock Vina. The most favorable interaction modes are shown in Fig. 3 and S5.† All these compounds are located at the same region, which is different from the natural macromolecular inhibitor moenomycin A. It is, however, interesting to note that both the non-polar aliphatic chains substituted at the N-1 site of the compounds (except compound 6 with a short butyl chain) and the long C25 isoprene tail of moenomycin A are pointing to the same direction to interact with the transmembrane region of the enzyme. This possibly causes compound 6 has much lower inhibitory enzymatic activity than compound 9. In addition, the decyl group of compound 9 also exhibits hydrophobic interactions with Phe110, Leu112, Phe157, and Leu158. The methyl group substituted at the C-5 site of the molecule shows a close contact with Thr115. Furthermore, its guanidine group at the C-3 site forms six hydrogen bonds with Gln100, Gln129, Gly130, Thr133, and Gln136, thus giving strong binding interactions. When the guanidine group is replaced by thiosemicarbazide, the binding between compound 12 and PGT is weakened due to loss of two hydrogen bonds. These docking results are consistent with their relatively low MIC values and the in vitro inhibition activities against the E. coli PBP 1b enzyme.
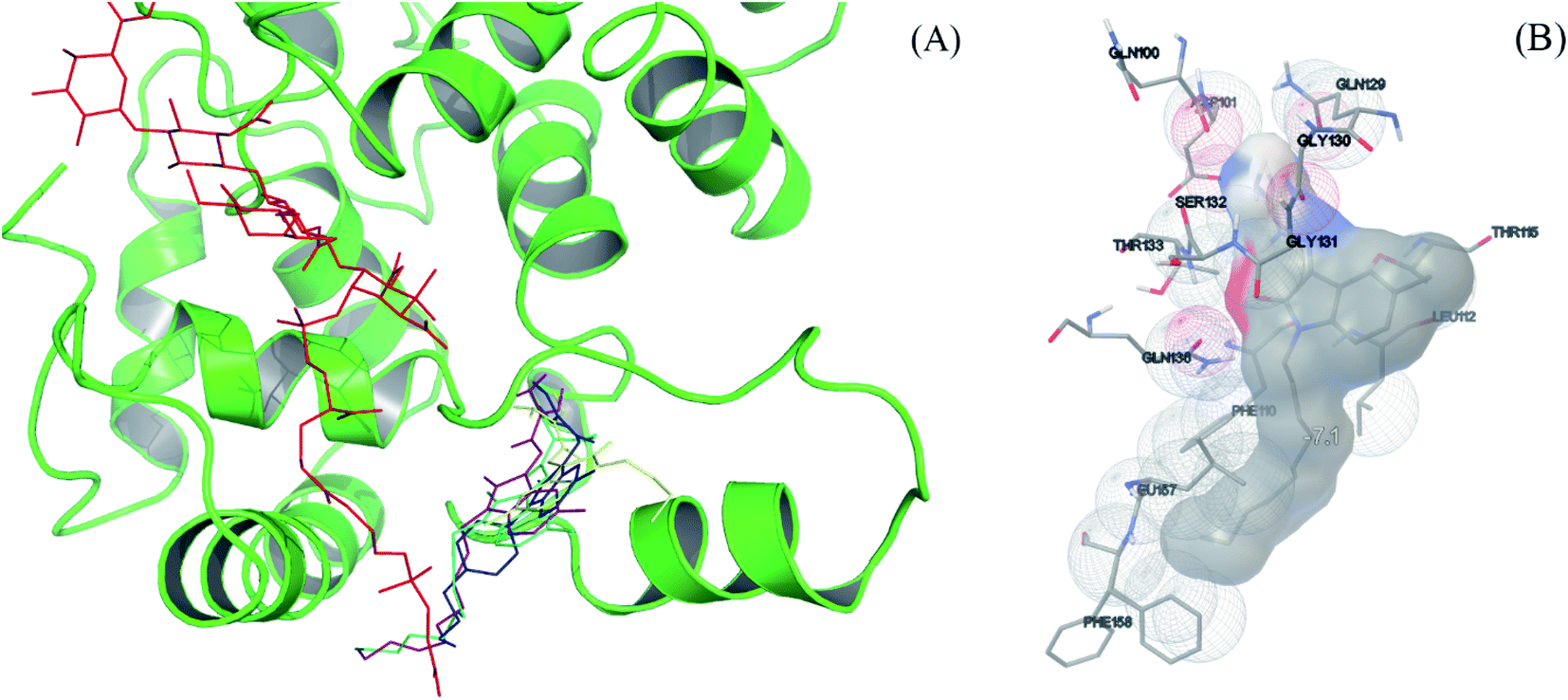 | ||
| Fig. 3 (A) Docking models between inhibitors (compound 2, 6, 9 and 12) and S. aureus monofunctional glycosyltransferase (protein ID: 3HZS):47 moenomycin A (red), compound 2 (blue), 6 (yellow), 9 (purple), and 12 (cyan). (B) The interactions between 9 and S. aureus monofunctional glycosyltransferase were generated from AutoDock tools.48,49 | ||
3. Experimental
3.1. General synthetic procedures of N-4-butylphenyl-5-substituent-isatin analogues (compound 1–5)
Compound 1 was synthesized and purified by following the reported procedures.35 For the synthesis of compound 2–5, to the mixture of 1 mM 5-substituent isatin, 1 mM 4-butylphenyl boronic acid, 3 mM triethylamine in 20 mL CH2Cl2 and activated 4 Å molecular sieve, 1 mM copper(II) acetate were added, and the mixture was kept stirring until reaction was completed. The solid was then removed and washed by CH2Cl2 (20 mL × 3). The combined organic layer was dried with anhydrous Na2SO4. After removal of the solvent, the product was purified by silica gel chromatography with ethyl acetate/petroleum ether (vol. ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) as the eluent. The final product was obtained by refluxing the mixture of 0.5 mM N-4-butylphenyl-5-substituent-isatin and 0.6 mM aminoguanidine hydrochloride in CH3COOH (10 mL) for 2 h. Yellow solid was obtained by introducing diethyl ether into the mixture. Finally, the product was purified by silica gel chromatography with methanol/dichloromethane (vol. ratio = 1:10) as the eluent.
:4) as the eluent. The final product was obtained by refluxing the mixture of 0.5 mM N-4-butylphenyl-5-substituent-isatin and 0.6 mM aminoguanidine hydrochloride in CH3COOH (10 mL) for 2 h. Yellow solid was obtained by introducing diethyl ether into the mixture. Finally, the product was purified by silica gel chromatography with methanol/dichloromethane (vol. ratio = 1:10) as the eluent.
3.2. General synthetic procedures of N-aliphatic chain-5-methyl-isatin analogues (compound 6–12)
To the mixture of 1 mM 5-methylisatin, 1 mM 1-bromo aliphatic alkane in 10 mL of DMF and 3 mM potassium carbonate were added, and the mixture was stirred at 80 °C for 5 h until starting compounds were consumed. The mixture was then poured into cold water and extracted by ethyl acetate (20 mL × 3) and washed by DI water (20 mL × 3), followed by drying with anhydrous Na2SO4. After removing the solvent, the product was purified by silica gel chromatography with ethyl acetate/petroleum ether (vol. ratio = 1:5) as the eluent. The final product was obtained as previously described.
3.3. Characterization of compounds
3.4. Antimicrobial susceptibility tests
The MIC values of the compounds (compound 1–12) was measured by antimicrobial susceptibility tests using the broth microdilution procedure according to the Clinical and Laboratory Standards Institute (CLSI) guidelines. Cation-adjusted Mueller Hinton broth (CA-MHB) for S. aureus strain ATCC 29213 and Mueller Hinton broth (MHB) for antibiotic-susceptible strains B. subtilis strain 168 and E. coli strain ATCC 25922 were used in the assays. Cells in exponential phase of growth were diluted to approximately 5 × 105 cfu mL−1. A stock solution of each compound were prepared in DMSO. Serial dilutions of each compound in DMSO and bacteria were mixed in a 96-well microplate. The final percentage of DMSO in the assay was 1% (v/v). Control experiments were performed with 1% (v/v) DMSO instead of the solution of the compounds. After incubation at 37 °C for 18 h, the OD600 value of the cells was measured on a microplate reader (Bio-Rad laboratory Ltd., UK), and the percentage of bacterial cell inhibition with respect to the controls calculated. The MIC value was defined as the lowest compound concentration at which the growth of bacteria was inhibited by ≥90%. Three independent assays were performed for each test.3.5. Lipid II-based in vitro transglycosylation assay
E. coli PBP 1b (13.2 nM) and the compounds (1–12) were pre-incubated at 30 °C for 5 min in reaction buffer (50 mM HEPES, pH 7.5 containing 200 mM NaCl and 10 mM CaCl2) with additional 0.08% (v/v) decyl PEG, 10% (v/v) DMSO, 15% (v/v) CH3OH and 2 U μL−1 muramidase. Dansyl-lipid II (working concentration = 5 μM) was then added into the reaction mixture, mixed well and incubated at 30 °C for another 5 min. MoeA (working concentration = 1 μM) was added and incubated for another 10 min to quench the reaction and completely cleave the produced glycan chain. The mixtures were stored at −80 °C before HPLC analysis. A 10 μL portion of samples was injected into an Agilent 1200 series equipped with a Supelco SAX1 anion-exchange column and eluted using a gradient elution method [NH4OAc (0.5 M in methanol) to MeOH (10% isopropanol) from 4%:96% to 100%:0%] in 30 min. All compounds were measured in duplicate, the half maximal inhibitory concentrations (IC50) of selected compounds were calculated by GraphPad Prism 8 with the model: Y = 1/(1 + 10((logIC50 − X) × h)) (Y, normalized response; X, log [inhibitor]; h, hillslope). Inhibition constants (Ki) were calculated according to the formula IC50 = Ki(1 + [s]/Km, Km = 1.8 μM) reported in literature.50,51
3.6. Cell proliferation assays
The normal human cell line BJ cells (ATCC CRL-2522) were cultured in a medium according to ATCC recommendations. All culture cell lines were maintained in a humidified culture chamber at 37 °C and 5% CO2. The cell lines were first seeded in a 96-well plate at a density of 5 × 103 cells per well. After incubation for one day, the cells were treated with the medium containing different concentrations of compounds (2, 9, 12). After 24 h of drug treatment, the culture medium was replaced with MTT solution (1 mg mL−1) (Invitrogen) and incubated at 37 °C for four hours. After incubation, the MTT solution was replaced with dimethyl sulfoxide (DMSO), and the absorbance at 570 nm measured with 650 nm as the reference. The cell viability was determined by dividing the absorbance of treated cells by the average absorbance of untreated cells. The IC50 values of the compounds (2, 9, and 12) on the normal human cell line BJ cells (at 95% confidence level) were analyzed by the software Prism 6.0. Three independent sets of experiments (n = 3) were performed for each cell line.3.7. Molecular modeling
Molecular models between the S. aureus monofunctional GT (protein PDB code: 3HZS) and isatin analogues was performed using AutoDock Vina.52 All the 3D coordinates of isatin analogues were generated by ChemDraw Ultra 8.0 and energy minimized by Avogadro with Merck Molecular Force Field 94 (MMFF94).53 The protein PDB and ligands MOL2 files were handled by reported procedures.54 The docking box contained the whole GT pocket.4. Conclusions
In conclusion, a series of new isatin derivatives was synthesized through three-direction modifications on the N-1, C-3 and C-5 positions of the isatin scaffold. The structure–activity relationship was investigated in antimicrobial susceptibility tests against Gram-positive (S. aureus (29213)), Gram-negative (E. coli (25922)) strains, and methicillin-resistant Staphylococcus aureus (MRSA (BAA41)) as well as the in vitro enzymatic inhibition assays with E. coli PBP 1b. It was found that the modifications on these 3 sites are able to maximize the antimicrobial potency and enzyme inhibitory activities. Two compounds (2 and 9) were identified as the potential candidates for new antimicrobial agent development against bacterial peptidoglycan glycosyltransferase. In addition, our cytotoxicity evaluation studies have shown that the toxicity of the compounds (2, 9, 12) may be primarily attributed to the guanidine substituent group at the C-3 site of isatin scaffold, which is also a crucial moiety of the molecule to achieve good antimicrobial potency against the three bacterial stains.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the General Research Fund of the Hong Kong Research Grants Council (PolyU 153338/16P), the Innovation and Technology Commission, the Ministry of Science and Technology of China to the State Key Laboratory of Chemical Biology and Drug Discovery, and The Hong Kong Polytechnic University. We also acknowledge the support of the University Research Facility on Chemical and Environmental Analysis (UCEA) of PolyU. KYW acknowledged the support from the Patrick S. C. Poon endowed professorship.References
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Antibiotics: past, present and future, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS PubMed.
- H. Cho, T. Uehara and T. G. Bernhardt, Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery, Cell, 2014, 159, 1300–1311 CrossRef CAS PubMed.
- K. Lazar and S. Walker, Substrate analogues to study cell-wall biosynthesis and its inhibition, Curr. Opin. Chem. Biol., 2002, 6, 786–793 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, beta-Lactams and beta-Lactamase Inhibitors: An Overview, Cold Spring Harbor Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- I. Roca, M. Akova, F. Baquero, J. Carlet, M. Cavaleri, S. Coenen, J. Cohen, D. Findlay, I. Gyssens, O. E. Heuer, G. Kahlmeter, H. Kruse, R. Laxminarayan, E. Liebana, L. Lopez-Cerero, A. MacGowan, M. Martins, J. Rodriguez-Bano, J. M. Rolain, C. Segovia, B. Sigauque, E. Tacconelli, E. Wellington and J. Vila, The global threat of antimicrobial resistance: science for intervention, New Microbes New Infect., 2015, 6, 22–29 CrossRef CAS PubMed.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Antibiotic resistance: a rundown of a global crisis, Infect. Drug Resist., 2018, 11, 1645–1658 CrossRef CAS PubMed.
- M. J. Cheesman, A. Ilanko, B. Blonk and I. E. Cock, Developing New Antimicrobial Therapies: Are Synergistic Combinations of Plant Extracts/Compounds with Conventional Antibiotics the Solution?, Pharmacogn. Rev., 2017, 11, 57–72 CrossRef CAS PubMed.
- A. R. M. Coates, Y. Hu, J. Holt and P. Yeh, Antibiotic combination therapy against resistant bacterial infections: synergy, rejuvenation and resistance reduction, Expert Rev. Anti-Infect. Ther., 2020, 18, 5–15 CrossRef CAS PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini and W. H. O. P. P. L. W. Group, Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef.
- G. Annunziato, Strategies to Overcome Antimicrobial Resistance (AMR) Making Use of Non-Essential Target Inhibitors: A Review, Int. J. Mol. Sci., 2019, 20, 5844 CrossRef CAS PubMed.
- A. Muller, A. Klockner and T. Schneider, Targeting a cell wall biosynthesis hot spot, Nat. Prod. Rep., 2017, 34, 909–932 RSC.
- E. Sauvage and M. Terrak, Glycosyltransferases and Transpeptidases/Penicillin-Binding Proteins: Valuable Targets for New Antibacterials, Antibiotics, 2016, 5, 12 CrossRef PubMed.
- J. Zuegg, C. Muldoon, G. Adamson, D. McKeveney, G. Le Thanh, R. Premraj, B. Becker, M. Cheng, A. G. Elliott, J. X. Huang, M. S. Butler, M. Bajaj, J. Seifert, L. Singh, N. F. Galley, D. I. Roper, A. J. Lloyd, C. G. Dowson, T. J. Cheng, W. C. Cheng, D. Demon, E. Meyer, W. Meutermans and M. A. Cooper, Carbohydrate scaffolds as glycosyltransferase inhibitors with in vivo antibacterial activity, Nat. Commun., 2015, 6, 7719 CrossRef CAS PubMed.
- Y. Yuan, S. Fuse, B. Ostash, P. Sliz, D. Kahne and S. Walker, Structural analysis of the contacts anchoring moenomycin to peptidoglycan glycosyltransferases and implications for antibiotic design, ACS Chem. Biol., 2008, 3, 429–436 CrossRef CAS PubMed.
- J. Halliday, D. McKeveney, C. Muldoon, P. Rajaratnam and W. Meutermans, Targeting the forgotten transglycosylases, Biochem. Pharmacol., 2006, 71, 957–967 CrossRef CAS.
- M. T. Sung, Y. T. Lai, C. Y. Huang, L. Y. Chou, H. W. Shih, W. C. Cheng, C. H. Wong and C. Ma, Crystal structure of the membrane-bound bifunctional transglycosylase PBP1b from Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8824–8829 CrossRef CAS.
- E. R. Baizman, A. A. Branstrom, C. B. Longley, N. Allanson, M. J. Sofia, D. Gange and R. C. Goldman, Antibacterial activity of synthetic analogues based on the disaccharide structure of moenomycin, an inhibitor of bacterial transglycosylase, Microbiology, 2000, 146 Pt 12, 3129–3140 CrossRef.
- S. Garneau, L. Qiao, L. Chen, S. Walker and J. C. Vederas, Synthesis of mono- and disaccharide analogs of moenomycin and lipid II for inhibition of transglycosylase activity of penicillin-binding protein 1b, Bioorg. Med. Chem., 2004, 12, 6473–6494 CrossRef CAS.
- M. F. Mesleh, P. Rajaratnam, M. Conrad, V. Chandrasekaran, C. M. Liu, B. A. Pandya, Y. S. Hwang, P. T. Rye, C. Muldoon, B. Becker, J. Zuegg, W. Meutermans and T. I. Moy, Targeting Bacterial Cell Wall Peptidoglycan Synthesis by Inhibition of Glycosyltransferase Activity, Chem. Biol. Drug Des., 2016, 87, 190–199 CrossRef CAS PubMed.
- M. J. Sofia, N. Allanson, N. T. Hatzenbuhler, R. Jain, R. Kakarla, N. Kogan, R. Liang, D. Liu, D. J. Silva, H. Wang, D. Gange, J. Anderson, A. Chen, F. Chi, R. Dulina, B. Huang, M. Kamau, C. Wang, E. Baizman, A. Branstrom, N. Bristol, R. Goldman, K. Han, C. Longley and H. R. Axelrod, et al., Discovery of novel disaccharide antibacterial agents using a combinatorial library approach, J. Med. Chem., 1999, 42, 3193–3198 CrossRef CAS PubMed.
- H. Y. He, B. Shen, J. Korshalla, M. M. Siegel and G. T. Carter, Isolation and structural elucidation of AC326-alpha, a new member of the moenomycin group, J. Antibiot., 2000, 53, 191–195 CrossRef CAS PubMed.
- K. E. Wilson, N. N. Tsou, Z. Q. Guan, C. L. Ruby, F. Pelaez, J. Gorrochategui, F. Vicente and H. R. Onishi, Isolation and structure elucidation of coleophomones A and B, novel inhibitors of bacterial cell wall transglycosylase, Tetrahedron Lett., 2000, 41, 8705–8709 CrossRef CAS.
- M. Adachi, Y. Zhang, C. Leimkuhler, B. Sun, J. V. LaTour and D. E. Kahne, Degradation and reconstruction of moenomycin A and derivatives: dissecting the function of the isoprenoid chain, J. Am. Chem. Soc., 2006, 128, 14012–14013 CrossRef CAS.
- S. Fuse, H. Tsukamoto, Y. Yuan, T. S. Wang, Y. Zhang, M. Bolla, S. Walker, P. Sliz and D. Kahne, Functional and structural analysis of a key region of the cell wall inhibitor moenomycin, ACS Chem. Biol., 2010, 5, 701–711 CrossRef CAS PubMed.
- C. M. Gampe, H. Tsukamoto, E. H. Doud, S. Walker and D. Kahne, Tuning the moenomycin pharmacophore to enable discovery of bacterial cell wall synthesis inhibitors, J. Am. Chem. Soc., 2013, 135, 3776–3779 CrossRef CAS PubMed.
- J. Y. Yu, H. J. Cheng, H. R. Wu, W. S. Wu, J. W. Lu, T. J. Cheng, Y. T. Wu and J. M. Fang, Structure-based design of bacterial transglycosylase inhibitors incorporating biphenyl, amine linker and 2-alkoxy-3-phosphorylpropanoate moieties, Eur. J. Med. Chem., 2018, 150, 729–741 CrossRef CAS PubMed.
- S. Dumbre, A. Derouaux, E. Lescrinier, A. Piette, B. Joris, M. Terrak and P. Herdewijn, Synthesis of modified peptidoglycan precursor analogues for the inhibition of glycosyltransferase, J. Am. Chem. Soc., 2012, 134, 9343–9351 CrossRef CAS.
- I. Sosic, M. Anderluh, M. Sova, M. Gobec, I. Mlinaric Rascan, A. Derouaux, A. Amoroso, M. Terrak, E. Breukink and S. Gobec, Structure-Activity Relationships of Novel Tryptamine-Based Inhibitors of Bacterial Transglycosylase, J. Med. Chem., 2015, 58, 9712–9721 CrossRef CAS PubMed.
- T. J. Cheng, M. T. Sung, H. Y. Liao, Y. F. Chang, C. W. Chen, C. Y. Huang, L. Y. Chou, Y. D. Wu, Y. H. Chen, Y. S. Cheng, C. H. Wong, C. Ma and W. C. Cheng, Domain requirement of moenomycin binding to bifunctional transglycosylases and development of high-throughput discovery of antibiotics, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 431–436 CrossRef CAS.
- T. J. Cheng, Y. T. Wu, S. T. Yang, K. H. Lo, S. K. Chen, Y. H. Chen, W. I. Huang, C. H. Yuan, C. W. Guo, L. Y. Huang, K. T. Chen, H. W. Shih, Y. S. Cheng, W. C. Cheng and C. H. Wong, High-throughput identification of antibacterials against methicillin-resistant Staphylococcus aureus (MRSA) and the transglycosylase, Bioorg. Med. Chem., 2010, 18, 8512–8529 CrossRef CAS PubMed.
- A. Derouaux, S. Turk, N. K. Olrichs, S. Gobec, E. Breukink, A. Amoroso, J. Offant, J. Bostock, K. Mariner, I. Chopra, T. Vernet, A. Zervosen, B. Joris, J. M. Frere, M. Nguyen-Disteche and M. Terrak, Small molecule inhibitors of peptidoglycan synthesis targeting the lipid II precursor, Biochem. Pharmacol., 2011, 81, 1098–1105 CrossRef CAS PubMed.
- Y. Wang, F. Y. Chan, N. Sun, H. K. Lui, P. K. So, S. C. Yan, K. F. Chan, J. Chiou, S. Chen, R. Abagyan, Y. C. Leung and K. Y. Wong, Structure-based Design, Synthesis, and Biological Evaluation of Isatin Derivatives as Potential Glycosyltransferase Inhibitors, Chem. Biol. Drug Des., 2014, 84, 685–696 CrossRef CAS PubMed.
- W. S. Wu, W. C. Cheng, T. J. R. Cheng and C. H. Wong, Affinity-Based Screen for Inhibitors of Bacterial Transglycosylase, J. Am. Chem. Soc., 2018, 140, 2752–2755 CrossRef CAS PubMed.
- A. Derouaux, E. Sauvage and M. Terrak, Peptidoglycan glycosyltransferase substrate mimics as templates for the design of new antibacterial drugs, Front. Immunol., 2013, 4, 1–6 CAS.
- Y. Wang, W. L. Cheong, Z. G. Liang, L. Y. So, K. F. Chan, P. K. So, Y. W. Chen, W. L. Wong and K. Y. Wong, Hydrophobic substituents on isatin derivatives enhance their inhibition against bacterial peptidoglycan glycosyltransferase activity, Bioorg. Chem., 2020, 97, 103710 CrossRef CAS PubMed.
- H. Guo, Isatin derivatives and their anti-bacterial activities, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- Z. Xu, S. J. Zhao, Z. S. Lv, F. Gao, Y. L. Wang, F. Zhang, L. Y. Bai and J. L. Deng, Fluoroquinolone-isatin hybrids and their biological activities, Eur. J. Med. Chem., 2019, 162, 396–406 CrossRef CAS PubMed.
- J. B. Wang, D. Yun, J. L. Yao, W. T. Fu, F. Y. Huang, L. P. Chen, T. Wei, C. J. Yu, H. N. Xu, X. O. Zhou, Y. Q. Huang, J. Z. Wu, P. H. Qiu and W. L. Li, Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds, Eur. J. Med. Chem., 2018, 144, 493–503 CrossRef CAS PubMed.
- D. Jiang, G. Q. Wang, X. F. Liu, Z. B. Zhang, L. S. Feng and M. L. Liu, Isatin Derivatives with Potential Antitubercular Activities, J. Heterocycl. Chem., 2018, 55, 1263–1279 CrossRef CAS.
- R. K. Thakur, P. Joshi, P. Baranwal, G. Sharma, S. K. Shukla, R. Tripathi and R. P. Tripathi, Synthesis and antiplasmodial activity of glyco-conjugate hybrids of phenylhydrazono-indolinones and glycosylated 1,2,3-triazolyl-methyl-indoline-2,3-diones, Eur. J. Med. Chem., 2018, 155, 764–771 CrossRef CAS PubMed.
- T. L. Devale, J. Parikh, P. Miniyar, P. Sharma, B. Shrivastava and P. Murumkar, Dihydropyrimidinone-isatin hybrids as novel non-nucleoside HIV-1 reverse transcriptase inhibitors, Bioorg. Chem., 2017, 70, 256–266 CrossRef CAS PubMed.
- G. F. Zhang, X. F. Liu, S. Zhang, B. F. Pan and M. L. Liu, Ciprofloxacin derivatives and their antibacterial activities, Eur. J. Med. Chem., 2018, 146, 599–612 CrossRef CAS PubMed.
- G. F. Zhang, S. Zhang, B. F. Pan, X. F. Liu and L. S. Feng, 4-Quinolone derivatives and their activities against Gram positive pathogens, Eur. J. Med. Chem., 2018, 143, 710–723 CrossRef CAS PubMed.
- W. F. Baitinger, P. v. R. Schleyer, T. S. S. R. Murty and L. Robinson, Nitro groups as proton acceptors in hydrogen bonding, Tetrahedron, 1964, 20, 1635–1647 CrossRef CAS.
- B. Schwartz, J. A. Markwalder, S. P. Seitz, Y. Wang and R. L. Stein, A kinetic characterization of the glycosyltransferase activity of Eschericia coli PBP1b and development of a continuous fluorescence assay, Biochemistry, 2002, 41, 12552–12561 CrossRef CAS PubMed.
- L. Y. Huang, S. H. Huang, Y. C. Chang, W. C. Cheng, T. J. Cheng and C. H. Wong, Enzymatic synthesis of lipid II and analogues, Angew Chem. Int. Ed. Engl., 2014, 53, 8060–8065 CrossRef CAS.
- H. Heaslet, B. Shaw, A. Mistry and A. A. Miller, Characterization of the active site of S-aureus monofunctional glycosyltransferase (Mtg) by site-directed mutation and structural analysis of the protein complexed with moenomycin, J. Struct. Biol., 2009, 167, 129–135 CrossRef CAS.
- M. F. Sanner, Python: A programming language for software integration and development, J. Mol. Graph. Model., 1999, 17, 57–61 CAS.
- R. Srinivasan and G. D. Rose, A physical basis for protein secondary structure, Proc. Natl. Acad. Sci. U.S.A., 1999, 96, 14258–14263 CrossRef CAS PubMed.
- S. Dumbre, A. Derouaux, E. Lescrinier, A. Piette, B. Joris, M. Terrak and P. Herdewijn, Synthesis of Modified Peptidoglycan Precursor Analogues for the Inhibition of Glycosyltransferase, J. Am. Chem. Soc., 2012, 134, 9343–9351 CrossRef CAS PubMed.
- M. Terrak, T. K. Ghosh, J. van Heijenoort, J. Van Beeumen, M. Lampilas, J. Aszodi, J. A. Ayala, J. M. Ghuysen and M. Nguyen-Disteche, The catalytic, glycosyl transferase and acyl transferase modules of the cell wall peptidoglycan-polymerizing penicillin-binding protein 1b of Escherichia coli, Mol. Microbiol., 1999, 34, 350–364 CrossRef CAS.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J. Cheminf., 2012, 4, 17 CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02119b |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |