
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro studies of maleidride-forming enzymes†
Sen Yin,
Steffen Friedrich,
Vjaceslavs Hrupins and
Russell J. Cox *
*
OCI, BMWZ, Leibniz University of Hannover, Schneiderberg 38, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
First published on 21st April 2021
Abstract
In vitro assays of enzymes involved in the biosynthesis of maleidrides from polyketides in fungi were performed. The results show that the enzymes are closely related to primary metabolism enzymes of the citric acid cycle in terms of stereochemical preferences, but with an expanded substrate selectivity. A key citrate synthase can react both saturated and unsaturated acyl CoA substrates to give solely anti substituted citrates. This undergoes anti-dehydration to afford an unsaturated precursor which is cyclised in vitro by ketosteroid-isomerase-like enzymes to give byssochlamic acid.
Introduction
Alkyl citrates are a class of fungal secondary metabolites derived from a polyketide or fatty acid component and oxaloacetic acid. They include relatively simple monomeric compounds such as 1 and 2, piliformic acid 3,1 oryzines A and B 4–5,2 sporothriolide 6,3 CJ-13,981 7 (ref. 4 and 5) and hexylitaconic acids6 known from Aspergillus niger (Fig. 1). More complex examples include compounds such as viridiofungin A 8 (ref. 7) and squalestatin S1 9 (ref. 8 and 9) which are potent inhibitors of squalene synthase. In many cases dehydration of the alkyl citrate affords a maleic anhydride as observed in 1 and 2, and dimerisation of these monomers leads to the formation of compounds with large alicyclic rings known as heptadrides, octadrides and nonadrides.10 Collectively such compounds are known as maleidrides.11 These include agnestadride A 10,11 zopfiellein 11 (ref. 12 and 13) byssochlamic acid 12 (ref. 14) and the selective herbicide cornexistin 13.15,16 These types of compounds are wide-spread in fungi (Fig. 1).17 | ||
| Fig. 1 Structures mentioned in the text. Bold bonds indicate atoms derived from oxaloacetate. | ||
To-date the main evidence for the biochemical reactions involved in the biosynthesis of alkyl citrates and maleidrides has come indirectly from in vivo genetic knockout or heterologous expression experiments. Thus, in the case of byssochlamic acid 12, for example, we showed that co-expression in the host Aspergillus oryzae of genes encoding: a fungal highly-reducing polyketide synthase (hr-PKS); an αβ-hydrolase; a citrate synthase-like protein (CS); and a homolog of 2-methylcitrate dehydratase (2MCDH) results in production of 1 and 2. Likewise, Oikawa and coworkers expressed homologous genes from the phomoidride BGC from unidentified fungus ATCC 74256 and related genes from Talaromyces stipitatus and showed production of anhydride monomers related to 1 and 2.18
These experiments suggest a series of events in which a polyketide synthase produces a linear acyl group attached to its acyl carrier protein (ACP) 14. This must be released (e.g. 15) and reacted with oxaloacetate to form a vinyl citrate 16 by the citrate synthase enzyme (Scheme 1). Dehydration by the 2MCDH enzyme would then provide the observed substituted maleic acids such as 17 and its equilibraiting anhydride from 1. Anhydride 1 decarboxylates spontaneously to give 2, which is also in equilibrium with a diacid form 18.19 We also showed that 1 appears to be a substrate for a pair of enzymes with homologies to ketosteroid isomerase-like (KI) and phosphatidyl-ethanolamine-binding protein (PEBP) which then produce the alicylic rings of dimeric maleidrides 10, 12 and 13 by an as-yet unknown mechanism. In order to probe these reactions and enzymes in more detail we chose to study them in vitro with the aim of determining their selectivities. Since the byssochlamic acid 12 and cornexistin 13 pathways were previously described in our hands, we focussed our efforts on the study of enzymes from these systems.
Results
Gene cloning, expression and protein purification
Initial attempts to clone codon optimised fungal whole-length ORFs in E. coli expression systems (pRSETA, Rosetta, IPTG induction) with an N-terminal his6-tag were successful for BfL1 (hydrolase), BfL2 (citrate synthase) and BfL9 (PEBP) only (Table 1), giving soluble protein. In other cases either insoluble protein was detected, or no significant expressed protein was detected. In the cases of BfL5, BfL6 and BfL10 (PEBP and KI) putative signal peptides sequences were detected at their N-termini (SignalP 5.0 server).20 These were removed by cloning N-truncated versions into pET28a and expression trials were set up in E. coli BL21(DE3). This resulted observable production of protein, but in the insoluble fraction in each case. The truncated bfL5, bfL6 and BfL10 genes were then individually co expressed in the presence of the pG-KJE8 chaperone system in E. coli, resulting in observation of soluble protein in each case. The final genes, bfL3 and pvL2, encoding putative dehydratases could not be expressed in E. coli under any of the previously tried conditions. These genes were cloned into pESC-URA vectors and expressed in Saccharomyces cerevisiae strain W303-1b, induced by the addition of galactose. Soluble protein was detected in each case by SDS-PAGE analysis. |
The ACP region of the B. fulva PKS was synthesised in E. coli optimised form (S2511 – E2618) in pET28a by a commercial supplier. The fragment was expressed in E. coli BL21 DE3 and soluble apo-protein was purified by nickel affinity chromatography and converted to holo-ACP using the Sfp phosphopantetheine transferase in vitro.21 Both apo and holo-proteins were confirmed by mass spectrometry (see ESI† for details).
Hydrolase
Previous in vivo results indicate that BfPKS builds the hex-2-enoyl intermediate required for construction of monomeric maleic anhydrides (Scheme 1). We assumed that release of the hexenoyl unit from the PKS is achieved by the BfL1 hydrolase, and observations from coexpression studies were in agreement with this hypothesis.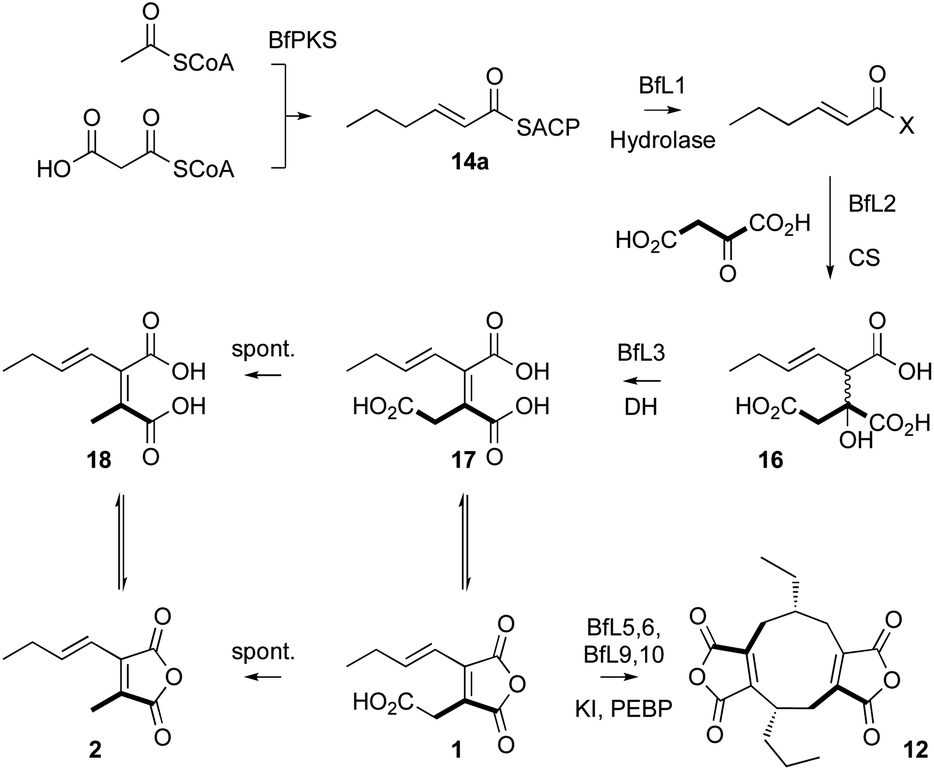 | ||
| Scheme 1 Proposed pathway to the maleidride byssochlamic acid 12. | ||
In order to probe the selectivity of the BfL1 hydrolase we synthesised SNAC, pantetheine, CoA and ACP-bound potential substrates (Scheme 2).22,23 In our previous work we assumed that the acyl group should be unsaturated (e.g. an E-hex-2-enoyl substrate) but this was unproven,10 so we made both E-hex-2-enoyl 14a–d and hexanoyl 15a–d substrates. Initial experiments showed that direct formation of thiolesters of α,β-unsaturated species (e.g. in carbodiimide type couplings) was accompanied by unwanted Michael addition, the products of which could not be easily removed. This was overcome by formation of the ylides 19c and 20c from the corresponding thiols 19a and 20a. These reacted smoothly with aldehydes to give the desired 14b and 14c after deprotection (Scheme 2). CoA thiolesters were formed more easily by direct coupling of mixed anyhydrides with CoASH itself, and in turn these were loaded onto the isolated apo-ACP of the B. fulva PKS using the phospho-pantetheinyl transferase (PPTase) enzyme Sfp.24,25 Finally, saturated SNACs and panthetheins 15b and 15c were made by standard EDCI coupling reactions.
 | ||
| Scheme 2 Synthesis of hex(en/an)oyl substrates. Reagents and conditions: (a) BrCH2CO2H, EDCI, DMAP, CH2Cl2, RT; (b) PPh3, toluene, 80 °C, then aq. NaOH; (c) 19c, CH2Cl2, RT, 72 h; (d) 20c, CH2Cl2, 40 °C, 3 d then THF/CF3CO2H, RT, 60 min; (e) ClCO2Et, CH2Cl2, Et3N, 0 °C; (f) CoASH, aq. NaHCO3; (g) 19a, EDCI, DMAP, CH2Cl2; (h) 20a, EDCI, DMAP, CH2Cl2, then THF/CF3CO2H, RT, 60 min. | ||
In order to assess the substrate selectivity of the BfL1 hydrolase we incubated it with hex-2-enoyl and hexanoyl thiolester species 14b–d and 15b–d and acyl-BfACP 14a and 15a. Assays were set up in vitro and contained substrate and purified BfL1 in buffer only. Reactions were incubated at 30 °C in Tris buffer containing MgCl2, NaCl and the non-nucleophilic reducing agent tris(2-carboxyethyl)phosphine (TCEP, 1 mM), then quenched by addition of 1 volume of CH3CN. Protein was precipitated by centrifugation and the residue analysed directly by LCMS.
In the case of the ACP species 14a and 15a, the acyl ACP was directly monitored by ESIMS. In all cases 14a–d and 15a–d we observed very rapid hydrolysis to the respective carboxylic acids, and by direct observation of the formation of holo-BfACP (Scheme 3A, see ESI† for bfACP mass data).
 | ||
| Scheme 3 In vitro assays of substrate analogs. A, results of incubation with the BfL1 hydrolase; B, results of incubation with the BfL2 citrate synthase and oxaloacetic acid. | ||
Citrate synthase
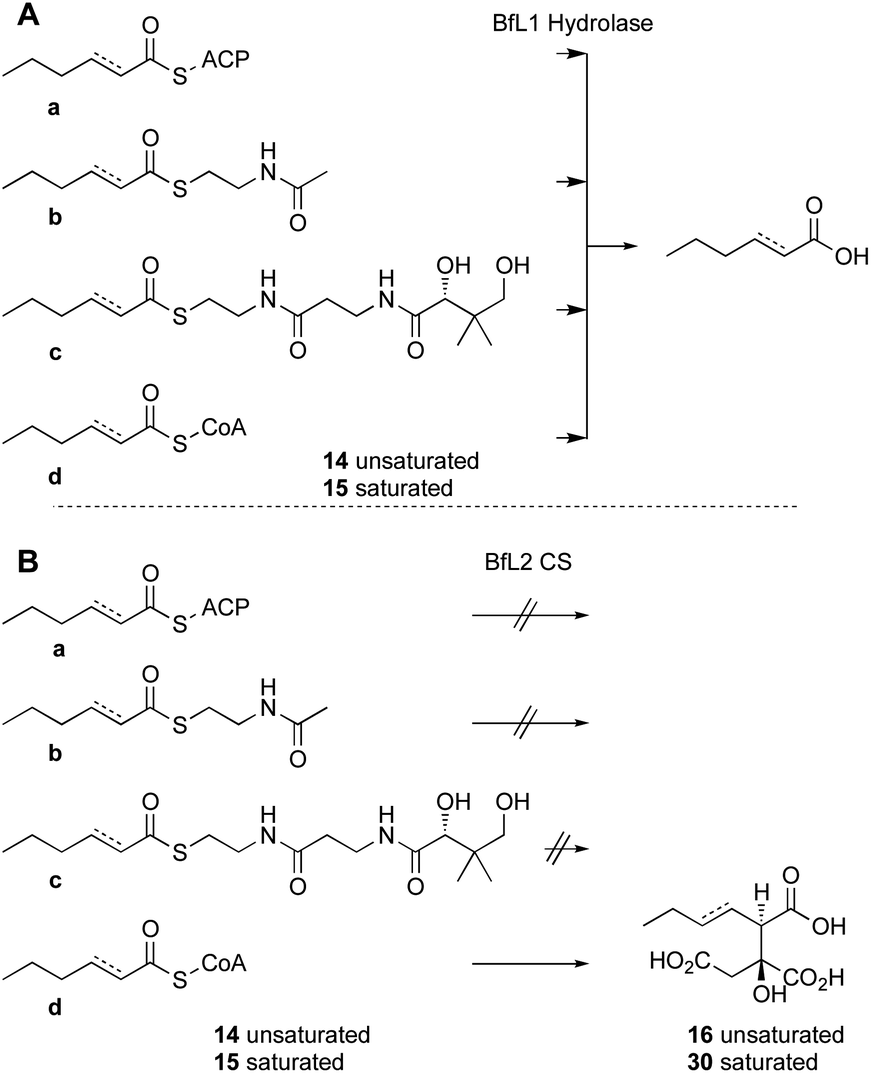
Citrate synthase [EC 2.3.3.1] is a key enzyme of primary metabolism and is known to react acetyl CoA with oxaloacetic acid to produce citric acid. The enzyme performs both the condensation reaction and the hydrolytic-release of CoA. The gene bfL2 from the byssochlamic acid BGC in B. fulva encodes a citrate synthase homolog which is 20% identical to the mitochondrial (presumed primary metabolism) CS of B. fulva.10 In vitro assays were set up containing purified BfL2, oxaloacetic acid and substrates 14a–d and 15a–d. The reactions were followed by LCMS. ACPs, SNACs and pantetheines were not substrates for BfL3, but both CoA thiolesters 14d and 15d were converted to the corresponding citrates 16 and 30 (Scheme 3, and Fig. 2).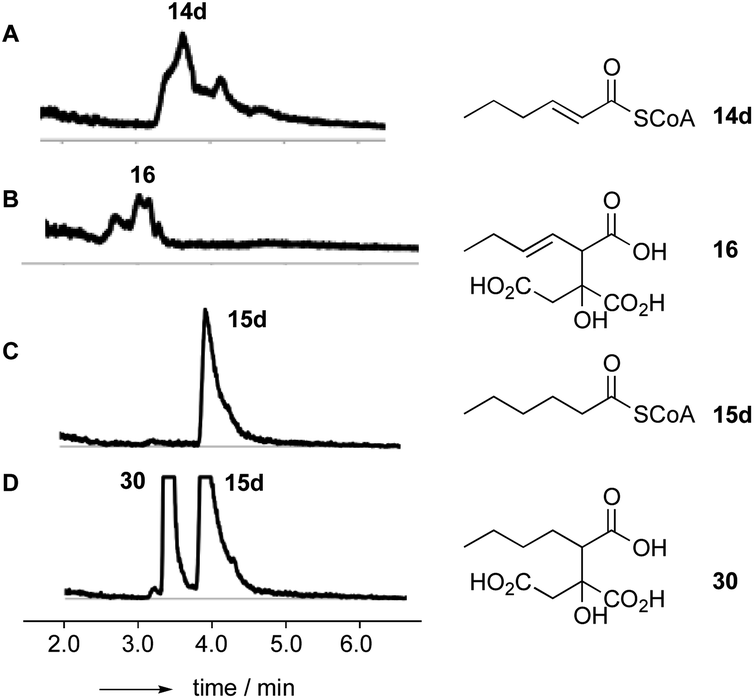 | ||
| Fig. 2 LCMS results for incubation of saturated and unsaturated CoAs with oxaloacetic acid and BfL2 (CS), ELSD traces. A, hex-2-enoyl CoA 14d; B, 14d + BfL2 + oxaloacetic acid; C, hexanoyl CoA 15d; D, 15d + BfL2 + oxaloacetic acid. | ||
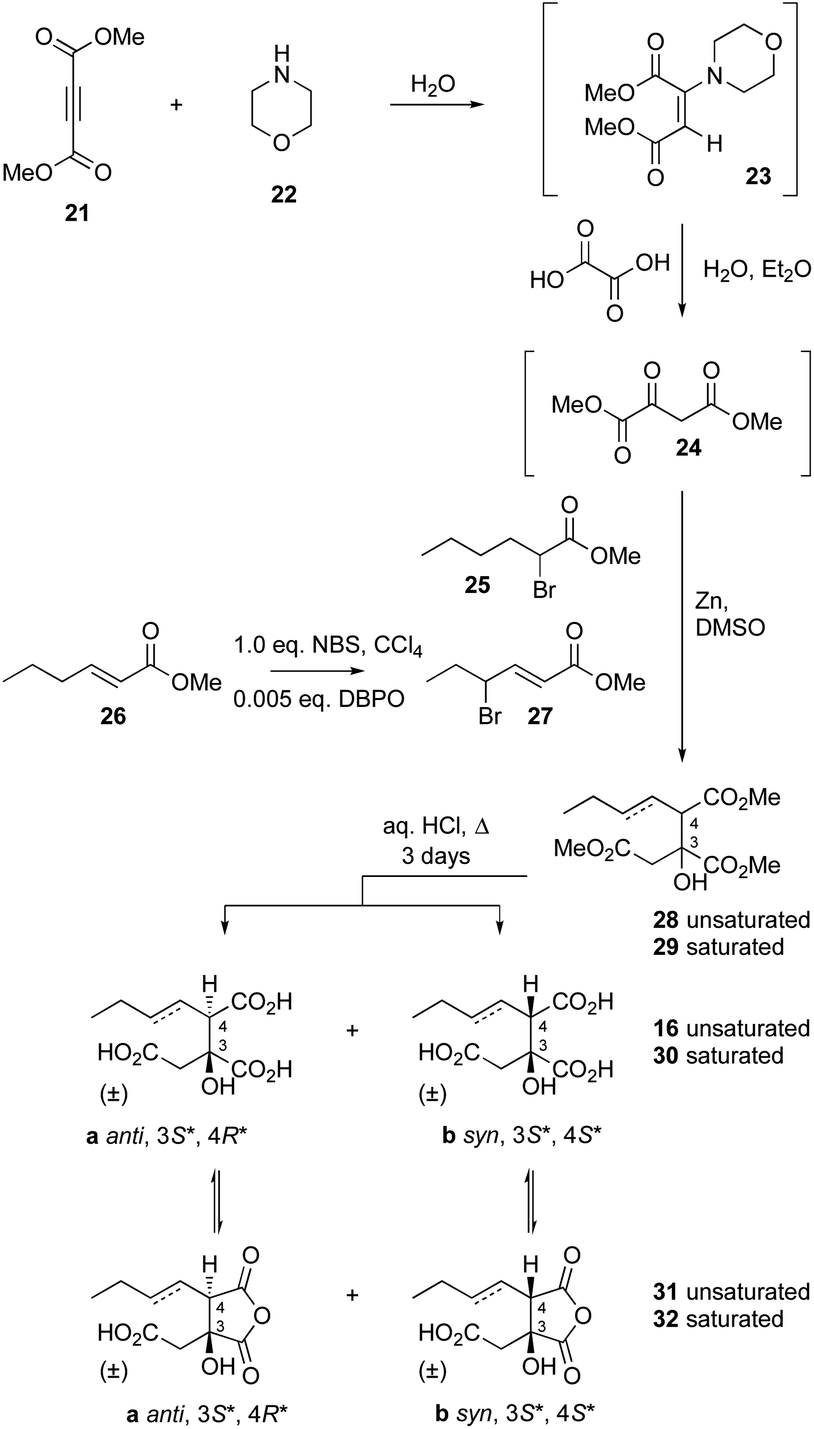
A synthesis was developed for the production of the proposed substituted-citrate products of the CS (Scheme 4). The synthesis involves a Reformatsky reaction with an oxalate diester. Diethy oxaloacetate is commercially available, and it reacts satisfactorily to give citrate products, however the ethyl citrates could not be hydrolysed in our hands. We therefore required dimethyl oxalate 24. This innocuous compound is not commercially available and attempts to produce it by esterification of oxaloacetic acid, or transesterification of diethyloxalate were unsuccessful. However, starting from dimethylacetylene dicarboxylate (DMAD) 21, selective hydration of the alkyne was achieved in two steps by the controlled addition of morpholine 22 to give the intermediate 23, followed by hydrolysis in the presence of oxalic acid. This yielded 24 as yellow crystals.
 | ||
| Scheme 4 Synthesis of citrates. Abbreviations: DBPO, dibenzoylperoxide; NBS, N-bromosuccinimide; DMSO, dimethylsulfoxide. | ||
Although this material did not give satisfactory spectroscopic analysis, it did react smoothly and as expected in the following step in which it was reacted with either methyl-α-bromohexanoate 25 or its 3-unsaturated congener 27 (itself derived from 26) in a Reformatsky reaction in DMF.26 This afforded a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers of the required trimethyl esters 28 (unsaturated) and 29 (saturated). These were hydrolysed by extended reflux in aqueous HCl (3 days) to afford good yields of the required triacids 16 (unsaturated) and 30 (saturated) – again as 2:1 mixtures of diastereomers.
:1 mixture of diastereomers of the required trimethyl esters 28 (unsaturated) and 29 (saturated). These were hydrolysed by extended reflux in aqueous HCl (3 days) to afford good yields of the required triacids 16 (unsaturated) and 30 (saturated) – again as 2:1 mixtures of diastereomers.
Previous work by Barrett and coworkers showed that similar reactions are anti-selective27,28 and this was confirmed by 1H NMR analysis of the mixture of diastereomers of 16 which showed that the major compound is the 2,3-anti (3S*, 4R*) diastereomer 16a. The 1H chemical shifts of H-4 are particularly diagnostic, with H-4syn consistently resonating at lower field than H-4anti (see ESI† for details). HPLC analysis showed that the major anti diastereomer 16a elutes slightly before the minor syn diastereomer 16b.
The individual diastereomers of synthetic 16 were separated by HPLC to yield the pure racemic stereoisomers. NMR and LCMS analysis also revealed the presence of equilibrating mixtures of the corresponding cyclic anhydrides 31, again with a prevalence of the 3,4 anti diastereomer (3S*, 4R*, ESI Fig S5.1†). Chromatographic comparison of the pure synthetic diastereomers with the products of the in vitro assay of BfL2 showed that the enzyme product is exclusively the anti diastereomer 16a (ESI Fig S5.3†).
Structures of many primary metabolism citrate synthases have been determined at high resolution. In particular the structures of CS from Escherichia coli,29 Thermus thermophilus,30 Pyrococcus furiosus31 and Sus scrofa32 are understood in significant detail. Multiple sequence alignment between BfL2 and CS from these organisms showed that the known active site residues D377, H284 and R332 are fully conserved (BfL2 numbering, see ESI†).
Other residues known to be involved in binding oxaloacetate (e.g. G322, H323, R402 and R422*) and acyl CoA (e.g. K/R317, L/I318, G320, R324, R/K370 and N375) are also conserved. N249 is replaced by histidine in the other organisms, but modelling (vide infra) shows that this residue forms an auxiliary hydrogen bond to the C-4 carboxylate of oxaloacetate which can be achieved by either histidine or asparagine.
We built a 3D model of BfL2 using the SwissModel server33 and CS from Acetobacter aceti34 as a template (pdb 2H12, Scheme 5B, see ESI† for details). The A. aceti structure was obtained in complex with oxaloacetate and an acetyl CoA mimic which allows the determination of residues which bind these substrates.33 Overlay of the BfL2 model and the A. aceti structure shows that all of the residues involved in catalysis and binding oxaloacetate and acyl CoA are structurally highly conserved (Scheme 5B). The model shows that the expected Si face of the oxaloacetate ketone is within 3.7 Å of the nucleophilic carbon, consistent with the formation of an S-configured tertiary alcohol upon carbon–carbon bond formation.
 | ||
| Scheme 5 Conserved residues in citrate synthase: A, mechanism of citrate synthase; B, model of BfL2 (cyan) overlayed with crystal structure data from A. aceti citrate synthase 2H12 (green). Residue numbering from BfL2 throughout. Oxaloacetate and an acetyl CoA mimic bound to 2H12 are shown in grey. * = residue from other dimer subunit. | ||
Since the oxaloacetate-binding and catalytic residues of the BfL2 model and the structure of 2H12 are identical, and overlay to within 1.15 Å RMSD over 13 residues, it is highly likely that the BfL2 alkyl citrate synthase also creates a 2S-stereocentre. Since we already know that BfL2 synthesises exclusively the anti diastereomer, then we conclude that the product of BfL2 possesses 2S,3R configuration.
Dehydratase
Dehydratase assays were also set up in vitro and monitored by analytical LCMS. In initial reactions, the synthetic mixture of E-1-butenyl-citrate diastereomers 16ab was incubated with the BfL3 and PvL2 dehydratase enzymes in separate reactions. When observed by diode array detector (200–300 nm), an increase in the area of the later-running peak corresponding to the syn citrate was noted. The citrates themselves have no significant UV absorbtion above 220 nm, but the new peak has a maximum UV absorbtion at 270 nm consistent with the formation of the expected unsaturated product 17 which co-elutes with 16b (Fig. 3). | ||
| Fig. 3 LCMS analysis of PvL2 and BfL3 reactions in vitro. A, 16 alone; B, 16 + BfL3; C, 16 + PvL2; D, 16 alone; E, 16 + BfL3; F, 16 + PvL2. | ||
Observation of the reaction at 270 nm more clearly showed the increase in this species. Mass analysis also indicated a loss of 18 mass units, consistent with the expected dehydration. PvL2 was more active that BfL3 and was therefore used for all further assays.
We next tested the anti 16a and syn 16b diastereomers of the citrate substrate separately. The anti diastereomer 16a was converted to the new peak, while the syn diastereomer 16b was unreactive. The new compound 17 elutes with the same retention time as the syn diastereomer of the citrate 16b. Extended reaction never reduced the amount of (±) starting material below 50% suggesting that the enzyme is selective for a single enantiomer, presumably the 2S,3R stereoisomer. Although the UV and MS analysis of the product of the reaction were consistent with formation of the expected 2,3-unsaturated system, the in vitro assays did not yield enough material for full NMR characterisation (Fig. 4).
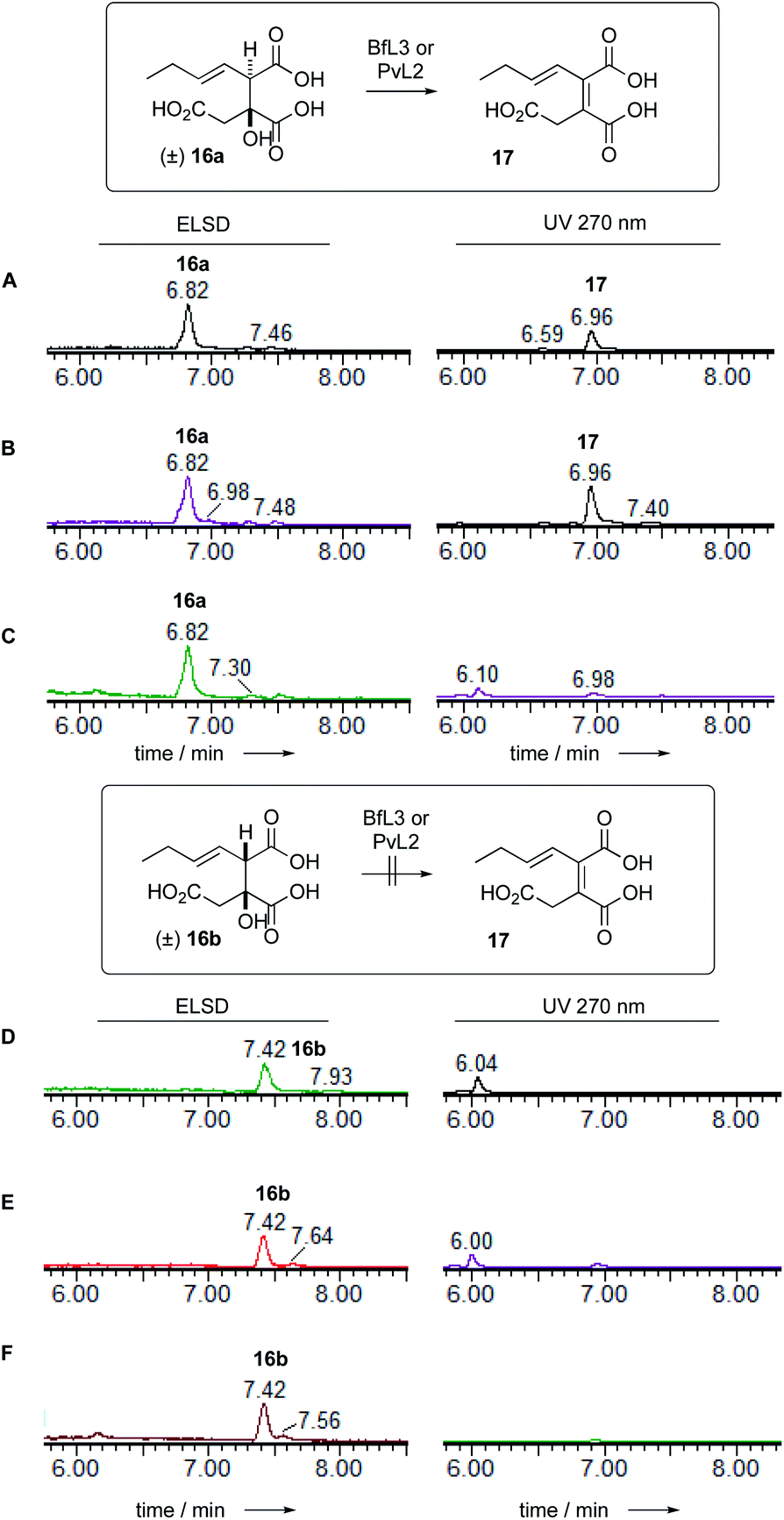 | ||
| Fig. 4 LCMS analysis of the indicated DH reactions. Top (2R,3S/2S,3R) anti substrate 16a reacted with enzyme: A, BfL3; B, PvL2; and C, empty vector control respectively. Bottom (2R,3R/2S,3S) syn substrate 16b reacted with enzyme: D, BfL3; E, PvL2; and F, empty vector control. | ||
In order to fully characterise 1 it was re-isolated from a fermentation of A. oryzae expressing bfPKS, bfL1 (hydrolase), bfL2 (CS) and bfl3 (dehydratase). A time course experiment determined that 1 reaches maximum concentration after 4 days of fermentation. After this time cells were collected, and extracted with EtOAc. Extended contact with organic solvents such as CHCl3 and DMSO causes 1 to decarboxylate to give 2. Similarly, freeze-drying procedures also decarboxylated 1. However, rapid purification by preparative HPLC in CH3CN/H2O mixtures, followed by removal of CH3CN under mild vacuum led to stable aqueous solutions of 1 which could be analysed by NMR and used for further in vitro assays (vide infra). The product of the in vitro reaction of PvL2 and BfL3 was thus proven to be identical to 1 isolated from fermentation (Scheme 6).
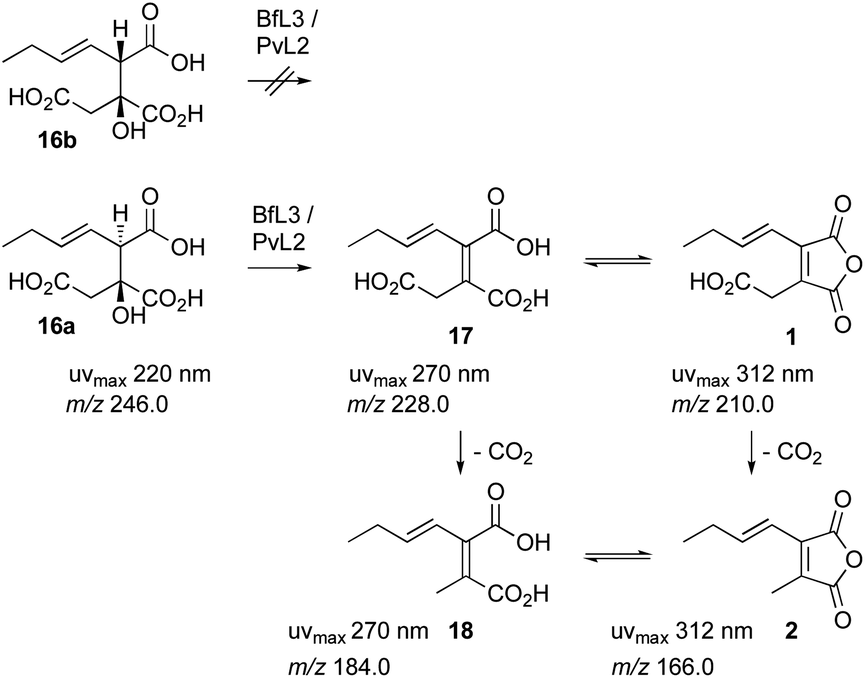 | ||
| Scheme 6 Reactions catalysed by BfL3 and Pvl2 dehydratases, and facile decarboxylation. | ||
LCMS analysis also showed that the maleic acid 17 (UV max 268 nm) and maleic anhydride 1 (UV max 313 nm) forms are in equilibrium. The diacid 17b elutes significantly earlier than the anhydride 1. Attempts to purify either compound always led to recovery of mixtures.
KI and PEBP
Our previous in vitro work showed that expression of KI genes with the earlier pathway genes led to dimerisation of 1 and formation of byssochlamic acid, while addition of the PEBP genes increased the titre. However, we were unable to conclude whether the PEBP proteins played a role in the catalysis or, alternatively, provided an uncharacterised resistance mechanism to byssochlamic acid 12 which is known to be toxic to fungi.35 Here we tested the effects of the isolated proteins with the proposed substrate 1 in vitro, once again following the reactions by analytical LCMS.Initial work focussed on attempts to detect conversion of maleic anhydride monomer 1 to bysochlamic acid 12 using BfL5, BfL6, BfL9 and BfL10 obtained by E. coli expression of the genes (lacking signal peptides and containing N-terminal his6 tags) either singly or in combination. However despite numerous attempts and varied conditions we could never observe any activity. We then attempted to express the full-length genes in yeast using the pESC system already described. In these cases we were not able to observe significant amounts of new soluble proteins by SDS-PAGE, or purify the proteins because they were cloned without his6 tags. However, we tested cell-free extracts of the induced yeast strains and compared the effects vs. control strains lacking the expression plasmids. In these cases we could clearly observe the conversion of 1 to byssochlamic acid 12 by a combination of all 4 protein extracts. The same results were observed for the combination of the two KI components, and for the KI components tested singly. The PEBP proteins appeared to be inactive in all conditions tested. They did not form 12 either alone or in combination, and their addition to the KI proteins did not seem to significantly increase the amount of 12 formed (Scheme 7).
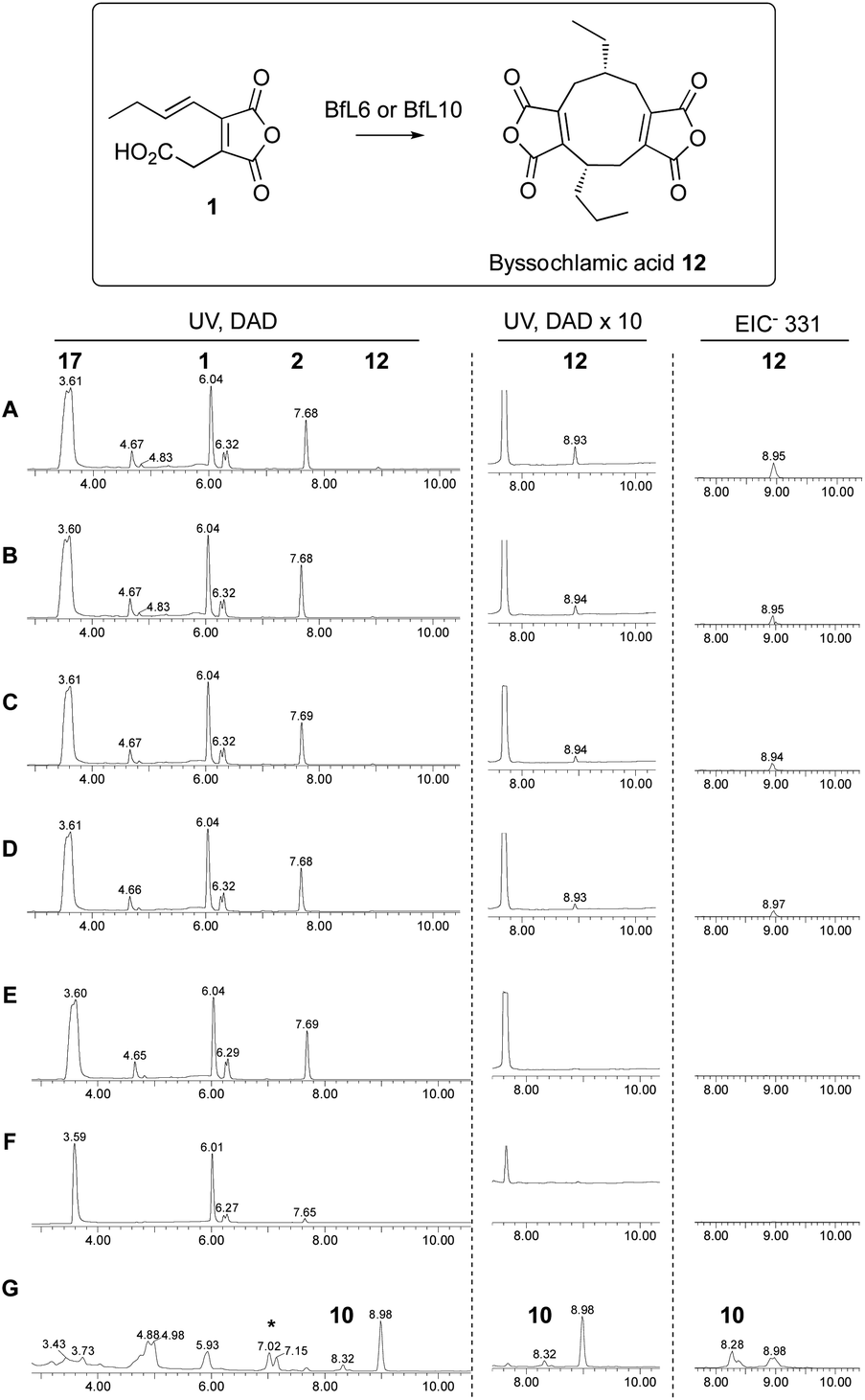 | ||
| Scheme 7 LCMS analysis of reactions catalysed by KI enzymes BfL6 and BfL10, and PEBP proteins BfL5 and BfL9: A, two KSI and two PEBP; B, two KI; C, BfL6; D, BfL10; E, empty vector control; F, only substrate; G, producing A. orzyae transformant. * mixture of diacid forms of 12. Note that data is qualitative rather than quantitative. | ||
Finally, decarboxylated maleic anhydride 2 was also tested as a substrate in the dimerisation assay. However no conversion to byssochlamic acid 12 was observed, and 2 and its hydrolysed congener 18 were the only compounds observed by LCMS analysis (see ESI† for details).
Discussion
Alkyl citrates are a class of fungal secondary metabolites which are closely related to primary metabolites. In the case of the maleidrides byssochlamic acid 12 and cornexistin 13 the pathway starts with a highly-reducing polyketide synthase (hr-PKS),36 but in many other cases such as the sporothriolides3 the pathway begins with a typical fungal fatty acid synthase (FAS). The function of these synthases at the start of the pathways is subtly different.hr-PKS such as BfPKS1 can control chain-length and functionalisation of the acyl chain, but these hr-PKS usually lack an integrated release mechanism. Thus BfPKS1 appears to synthesise a hex-2-enoyl product which is released by a dedicated trans-acting hydrolase BfL1 as a carboxylic acid. This is demonstrated in vitro here where we showed that BfL1 can hydrolyse acyl-BfPKS-ACP species. Similar hydrolytic release mechanisms are known in other fungal PKS systems such as phaeospelide A.37 However, BfL1 also appears to rapidly hydrolyse other thiolesters, so how its selectivity is controlled in vivo is not yet known. This release mechanism differs from that used by fungal FAS (e.g. SpoFas)3 where the malonyl-palmitoyl transferase (MPT) domain is known to both load malonyl CoA extenders, and release products directly as CoA thiolesters.38
The next step of the pathway is also closely related to primary metabolism. We showed that the citrate synthase BfL2 selectively reacts acyl CoAs 14d and 15d with oxaloacetate. This differs from the suggestion by Tang and coworkers that similar CS enzymes react directly with ACP-bound acyl groups, for example during the biosynthesis of squalestatins.39 This reaction does not occur in the case of the byssochlamic acid pathway where we showed that acyl-ACPs (i.e. 14a and 15a) are not substrates for BfL2.
The requirement for CoA substrates by the CS raises an interesting question. In the case of FAS-based pathways acyl CoAs are released directly by the FAS, but for the PKS + hydrolase pathways the released carboxylic acids must be activated to CoA thiolesters, however the required CoA synthetase does not appear to be encoded within the Bf BGC. Presumably such short-chain thiolesters could be synthesised by endogenous primary metabolism enzymes.
The BfL2 CS can react both saturated and E-unsaturated substrates. In the case of E-unsaturated substrates the alkene migrates during the reaction. Since it is known that the active site base of CS is a highly conserved aspartate (D377, Scheme 8), we speculate that this residue could be involved in both processes, forming a dienol intermediate during the synthesis of 16a (Scheme 8B). Thus-far we have not experimented with Z-alkenes as substrates, but this would be an interesting future goal.
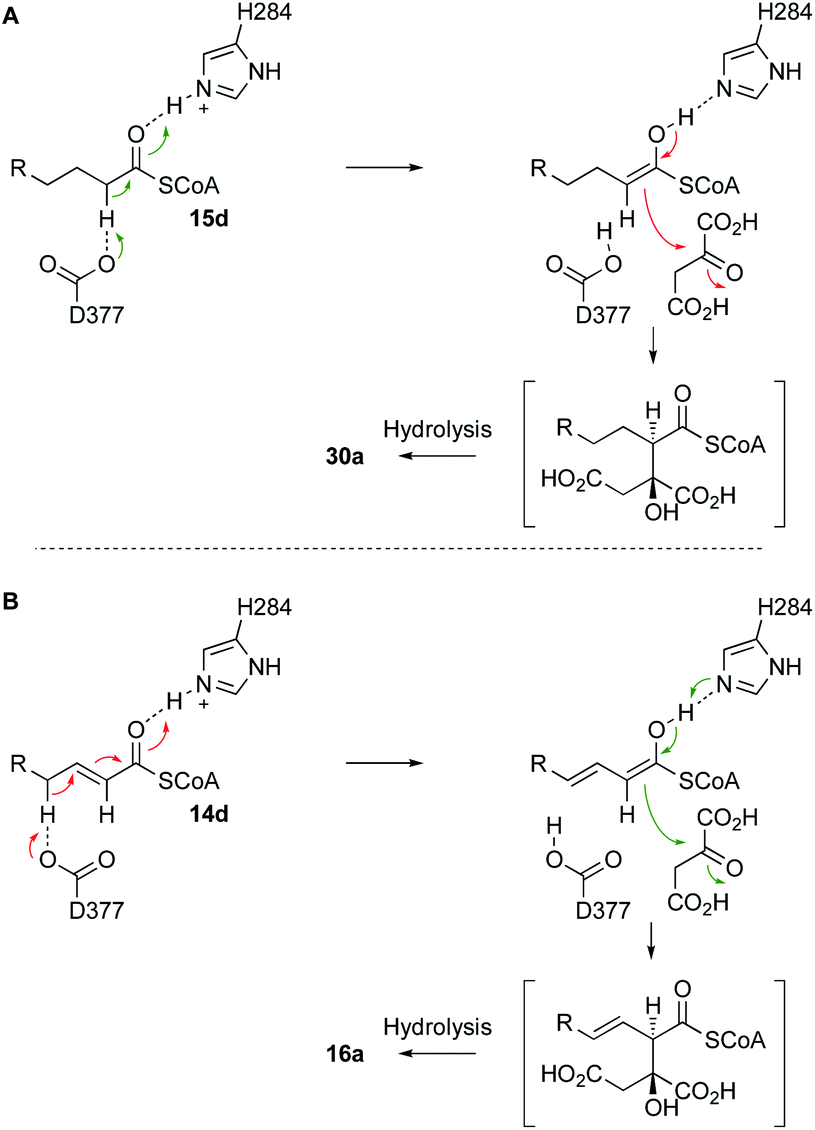 | ||
| Scheme 8 A, Likely mechanism of BfL2 during the synthesis of alkyl citrates; B, adapted mechanism for the synthesis of vinyl citrates via a dienol intermediate. | ||
We showed that the BfL2 CS produces exclusively 3,4-anti products, and structural considerations strongly suggest this is the 3S,4R stereoisomer. In the cases of other known citrate-derived metabolites (and in primary metabolism) the 3S configuration is also observed, but in the known cases of squalestatin S1 9, viridiofungin 8 and CJ-13,981 7, for example, the CS products are 3S,4S.7,40,41 Differences in configuration at the 4-position must be controlled by the geometry of the enoyl CoA intermediate, and in-turn this may be related to the ability of the BfL2 CS to react 2,3-unsaturated substrates. Detailed structural work will be required to probe this question further. The family of secondary metabolite alkyl citrate synthases resemble methylcitrate synthases involved in propionate metabolism. Recent work by Reddick and coworkers has reassigned the product of these enzymes as 3S*,4R* in agreement with our stereochemical assignment.42
In the next step the dehydratase BfL3 removes a water molecule. This step is closely related to the primary metabolism enzyme 2-methylcitrate dehydratase (2MCDH). Again the stereoselectivity of this step by the primary metabolism 2-methylcitrate dehydratase has been debated, but Reddick's results41 conclusively show that the enzyme takes 3S*,4R* isomer to give Z-methyl aconitate – i.e. anti dehydration.43 Again, the BfL3 and PvL2 dehydratases follow exactly the same stereochemical course, emphasising the similarity of the primary and secondary metabolism pathways. The dehydrated products spontaneously equilibrate between diacid 17 and anhydride 1 forms. Reddick showed that the primary metabolism 2MCDH enzymes can dehydrate both the anti and syn diastereomers of 2-methylcitrate, but the BfL3 enzyme cannot dehydrate the syn substrate diastereomer 16b.
Finally, we showed that ketosteroid-isomerase-like (KI-like) enzymes BfL6 and BfL10 catalyse the dimerisation of 1 to the nonadride byssochlamic acid 12. They appear to act alone and both are individually active. Interestingly, decarboxylated congener 2 is not a substrate for the enzyme catalysed reaction. In previous in vitro work Baldwin44 and others45 have shown that related decarboxylated compounds can give low yields of dimeric products in organic solvents in the presence of medium to strong bases which are likely to be able to form low concentrations of anionic intermediates. However our results suggest that while such intermediates could be involved in dimerisation, they are likely to be generated by enzyme-catalysed decarboxylation of 1.
We could not obtain high yields of the KI-like enzymes in soluble form, and only cell-free extracts produced in yeast showed any tangible activity. We were therefore unable to perform more detailed studies of the mechanisms of these intriguing enzymes, but never-the-less this is the first report of their activity in vitro and sets the scene for future more detailed studies. Also unexpected is the observation that the KI proteins appear to make a single nonadride product in vitro: formation of the heptadride 10 was not observed in vitro, raising the question of how this is formed in vivo.
The PEBP proteins studied here appeared catalytically inactive. The B. fulva 12 BGC encodes two pairs of KI and PEBP enzymes, but most other maleidride clusters encode only a single pair, so it seems likely that the B. fulva system has undergone gene duplication. Lack of catalytic activity of PEBP proteins supports previous suggestions that these may be part of resistance systems as they are encoded by most maleidride BGC, however another function cannot be ruled out.
Thus the in vitro studies of the function and stereoselectivity of enzymes involved in fungal alkyl citrate biosynthesis show that the pathway is very closely related to primary metabolism. Indeed simple compounds like CJ-13981 7 should require only a fatty acid synthase and an alkylcitrate synthase, both presumably duplicated from primary metabolism, to produce a compound with useful activity as an inhibitor of squalene synthase.4,5 Exchange of the FAS for an hr-PKS (and hydrolase) allows more complex chains to be built – for example squalestatin precursors which are also squalene synthase inhibitors.46–48 Finally, gain of a dehydratase (again almost directly from primary metabolism) and a KI-like protein then allows the pathway to make maleidrides. Other pathways like those involved in squalestatin or sporothriolide diversify by gain of more obviously secondary metabolism steps involving C–H activation by oxidation. The close relationship of such secondary metabolism pathways to key primary metabolic enzymes shows how complex secondary metabolic pathways have likely evolved. However, for the same reasons, this makes it difficult for bioinformatic systems trained to find secondary metabolism genes and pathways to recognise these pathways.
Experimental details
All experimental details and characterisations of individual compounds and proteins is contained in the ESI.†Author contributions
VH cloned the BfPKS ACP, prepared acylated ACPs and conducted assays with these species, and worked with Sfp. SY cloned all other genes, expressed all other proteins and performed all other in vitro assays. SF synthesised all small molecules and performed hydrolase assays. RJC devised the study, gained funding and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Ben Schleibaum for assistance with the synthetic chemistry. The China Scholarship Council is thanked for funding (SY, CSC201606210136). DFG is thanked for analytical instrumentation (INST 187/686-1) and funding for VH (CO 1328/3-1). Prof. Jianqiang Kong is thanked for sending us yeast plasmids and strains. The publication of this article was funded by the Open Access Fund of the Leibniz Universität Hannover.Notes and references
- N. Chesters and D. O'Hagan, J. Chem. Soc., Perkin Trans. 1, 1997, 827–834 RSC; H. Culceth, J. Fuchser, S. J. Moss, J. Nieschalk and D. O'Hagan, Tetrahedron Lett., 1998, 39, 1949–1952 CrossRef CAS.
- Z. Wasil, E. Kuhnert, T. J. Simpson and R. J. Cox, J. Fungi, 2018, 4, 96 CrossRef CAS PubMed.
- D.-S. Tian, E. Kuhnert, J. Ouazzani, D. Wibberg, J. Kalinowski and R. J. Cox, Chem. Sci., 2020, 11, 12477–12484 RSC.
- F. Calo, A. Bondke, J. Richardson, A. J. P. White and A. G. M. Barrett, Tetrahedron Lett., 2009, 50, 3388–3390 CrossRef CAS.
- S. Watanabe, H. Hirai, T. Kambara, Y. Kojima, H. Nishida, A. Sugiura, Y. Yamauchi, N. Yoshikawa, H. J. Harwood, L. H. Huang and N. Kojima, J. Antibiot., 2001, 54, 1025–1030 CrossRef CAS PubMed.
- S. Palys, T. T. M. Pham and A. Tsang, Front. Microbiol., 2020, 11, 1378 CrossRef PubMed.
- G. H. Harris, E. T. T. Jones, M. S. Meinz, M. Nallin-Omstead, G. L. Helms, G. F. Bills, D. Zink and K. E. Wilson, Tetrahedron Lett., 1993, 34, 5235–5238 CrossRef CAS.
- A. Baxter, B. J. Fitzgerald, J. L. Hutson, A. D. McCarthy, J. M. Motteram, B. C. Ross, M. Sapra, M. A. Snowden, N. S. Watson and R. J. Williams, J. Biol. Chem., 1992, 267, 11705–11708 CrossRef CAS; C. A. Jones, P. J. Sidebottom, R. J. P. Cannell, D. Noble and B. A. M. Rudd, J. Antibiot., 1992, 45, 1492–1498 CrossRef PubMed.
- B. Bonsch, V. Belt, C. Bartel, N. Duensing, M. Koziol, C. Lazarus, A. Bailey, T. Simpson and R. Cox, Chem. Commun., 2016, 52, 6777–6780 RSC; K. E. Lebe and R. J. Cox, Chem. Sci., 2019, 10, 1227–1231 RSC.
- K. Williams, A. J. Szwalbe, N. P. Mulholland, J. L. Vincent, A. M. Bailey, C. L. Willis, T. J. Simpson and R. J. Cox, Angew. Chem. Int. Ed., 2016, 55, 6784–6788 CrossRef CAS PubMed.
- A. J. Szwalbe, K. Williams, D. E. O'Flynn, A. M. Bailey, N. P. Mulholland, J. L. Vincent, C. L. Willis, R. J. Cox and T. J. Simpson, Chem. Commun., 2015, 51, 17088–17091 RSC.
- M. Futagawa, D. E. Wedge and F. E. Dayan, Pestic. Biochem. Physiol., 2002, 73, 87–93 CrossRef CAS.
- T. Shiina, T. Ozaki, Y. Matsu, S. Nagamine, C. Liu, M. Hashimoto, A. Minami and H. Oikawa, Org. Lett., 2020, 22, 1997–2001 CrossRef CAS PubMed; K. M. J. de Mattos-Shipley, C. E. Spencer, C. Greco, D. M. Heard, D. E. O'Flynn, T. T. Dao, Z. Song, N. P. Mulholland, J. L. Vincent, T. J. Simpson, R. J. Cox, A. M. Bailey and C. L. Willis, Chem. Sci., 2020, 11, 11570–11578 RSC.
- T. A. Hamor, I. C. Paul, J. M. Robertson and G. A. Sim, Experientia, 1962, 18, 352–354 CrossRef CAS PubMed; J. E. Baldwin, D. H. R. Barton and J. K. Sutherland, J. Chem. Soc., 1965, 1787–1798 RSC.
- M. Nakajima, K. Itoi, Y. Takamatsu, S. Sato, Y. Furukawa, K. Furuya, T. Honma, J. Kadotani, M. Kozasa and T. Haneishi, J. Antibiot., 1991, 44, 1065–1072 CrossRef CAS PubMed.
- K. Williams, A. J. Szwalbe, C. Dickson, T. R. Desson, N. P. Mulholland, J. L. Vincent, J. M. Clough, A. M. Bailey, C. P. Butts, C. L. Willis, T. J. Simpson and R. J. Cox, Chem. Commun., 2017, 53, 7965–7968 RSC.
- R. Schor and R. Cox, Nat. Prod. Rep., 2018, 35, 230–256 RSC.
- R. Fujii, Y. Matsu, A. Minami, S. Nagamine, I. Takeuchi, K. Gomi and H. Oikawa, Org. Lett., 2015, 17, 5658–5661 CrossRef CAS PubMed.
- A. J. Szwalbe, PhD thesis, University of Bristol, 2016.
- J. J. A. Armenteros, K. D. Tsirigos, C. K. Sønderby, T. N. Petersen, O. Winther, S. Brunak, G. von Heijne and H. Nielsen, Nat. Biotechnol., 2019, 37, 420–423 CrossRef PubMed.
- K. Reuter, M. R. Mofid, M. A. Marahiel and R. Ficner, EMBO J., 1999, 18, 6823–6831 CrossRef CAS PubMed.
- D. M. Roberts, C. Bartel, A. Scott, D. Ivison, T. J. Simpson and R. J. Cox, Chem. Sci., 2016, 8, 1116–1126 RSC.
- J. Wunderlich, T. Roß, M. Schröder and F. Hahn, Org. Lett., 2020, 22, 4955–4959 CrossRef CAS PubMed.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef CAS PubMed.
- R. J. Cox, T. S. Hitchman, K. J. Byrom, I. S. C. Findlow, J. A. Tanner, J. Crosby and T. J. Simpson, FEBS Lett., 1997, 405, 267–272 CrossRef CAS PubMed.
- D. J. Ritson, R. J. Cox and J. Berge, Org. Biomol. Chem., 2004, 2, 1921–1933 RSC.
- S. M. Goldup, C. J. Pilkington, A. J. P. White, A. Burton and A. G. M. Barrett, J. Org. Chem., 2006, 71, 6185–6191 CrossRef CAS PubMed.
- Note that Barrett and coworkers use a different definition of syn and anti. We prefer to use consistent nomenclature with the cyclic anhydrides 1 and 2..
- D. J. Stokell, L. J. Donald, R. Maurus, N. T. Nguyen, G. Sadler, K. Choudhary, P. G. Hultin, G. D. Brayer and H. W. Duckworth, J. Biol. Chem., 2003, 278, 35435–35443 CrossRef CAS PubMed.
- E. Kanamori, S. Kawaguchi, S. Kuramitsu, T. Kouyama and M. Murakami, Biophys. Physicobiol., 2015, 12, 47–56 CrossRef CAS PubMed.
- R. J. M. Russell, J. M. C. Ferguson, D. W. Hough, M. J. Danson and G. L. Taylor, Biochemistry, 1997, 36, 9983–9994 CrossRef CAS PubMed.
- M. Karpusas, B. Branchaud and S. J. Remington, Biochemistry, 1990, 29, 2213–2219 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303, DOI:10.1093/nar/gky427.
- J. A. Francois, C. M. Starks, S. Sivanuntakorn, H. Jiang, A. E. Ransome, J.-W. Nam, C. Z. Constantine and T. J. Kappock, Biochemistry, 2006, 45, 13487–13499 CrossRef CAS PubMed.
- X. Chen, Y. Zheng and Y. Shen, Chem. Rev., 2007, 107, 1777–1830 CrossRef CAS PubMed.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 RSC.
- Y. Morishita, H. Zhang, T. Taniguchi, K. Mori and T. Asai, Org. Lett., 2019, 21, 4788–4792 CrossRef CAS PubMed.
- Z. Zhu, Y. J. Zhou, A. Krivoruchko, M. Grininger, Z. K. Zhao and J. Nielsen, Nat. Chem. Biol., 2017, 13, 360–362 CrossRef CAS PubMed.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS PubMed.
- A. Baxter, B. J. Fitzgerald, J. L. Hutson, A. D. McCarthy, J. M. Motteram, B. C. Ross, M. Sapra, M. A. Snowden, N. S. Watson and R. J. Williams, J. Biol. Chem., 1992, 267, 11705–11708 CrossRef CAS.
- D. Sturgess, Z. Chen, J. M. White and M. A. Rizzacasa, J. Antibiot., 2018, 71, 234–239 CrossRef CAS PubMed.
- J. J. Reddick, S. Sirkisoon, R. A. Dahal, G. Hardesty, N. E. Hage, W. T. Booth, A. L. Quattlebaum, S. N. Mills, V. G. Meadows, S. L. H. Adams, J. S. Doyle and B. E. Kiel, Biochemistry, 2017, 56, 5698–5711 CrossRef CAS PubMed.
- J. R. Mohrig, K. A. Moerke, D. L. Cloutier, B. D. Lane, E. C. Person and T. B. Onasch, Science, 1995, 269, 527–529 CrossRef CAS PubMed.
- J. E. Baldwin, A. Beyeler, R. J. Cox, C. Keats, G. J. Pritchard, R. M. Adlington and D. J. Watkin, Tetrahedron, 1999, 55, 7363–7374 CrossRef CAS.
- R. K. Huff, C. E. Moppett and J. K. Sutherland, J. Chem. Soc., Perkin Trans. 1, 1972, 2584–2590 RSC.
- G. H. Harris, C. Dufresne, H. Joshua, L. A. Koch, D. L. Zink, P. M. Salmon, K. E. Göklen, M. M. Kurtz, D. J. Rew, J. D. Bergstrom and K. E. Wilson, Bioorg. Med. Chem. Lett., 1995, 5, 2403–2408 CrossRef CAS.
- K. E. Lebe and R. J. Cox, Chem. Sci., 2019, 10, 1227–1231 RSC.
- N. Liu, Y.-S. Hung, S.-S. Gao, L. Hang, Y. Zou, Y.-H. Chooi and Y. Tang, Org. Lett., 2017, 19, 3560–3563 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02118d |
| This journal is © The Royal Society of Chemistry 2021 |