
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing protein dissociation from gold nanoparticles and the influence of temperature from the protein corona formation mechanism†
Meifeng Lia,
Xiaoning Zhang *b,
Sining Lic,
Xiaoqing Shaob,
Huixian Chenb,
Lei Lv*b and
Xiaowen Huang*d
*b,
Sining Lic,
Xiaoqing Shaob,
Huixian Chenb,
Lei Lv*b and
Xiaowen Huang*d
aSchool of Public Health, Chengdu University of Traditional Chinese Medicine, Chengdu 610075, China
bSchool of Food Science & Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China. E-mail: xiaoningzhang@126.com; lvlei831005@163.com
cCollege of Food Science and Technology, Southwest Minzu University, Chengdu 610041, China
dSchool of Bioengineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, China. E-mail: huangxiaowen2013@gmail.com
First published on 19th May 2021
Abstract
Gold nanoparticles (AuNPs) provide a novel approach for protein enrichment and analysis due to their protein adsorption properties, forming a so called protein corona. This corona can significantly influence the protein's structure and characteristics, hindering their identification in situ. Dissociation is an important solution to analyze and identify the composition of protein coronas. However, a comprehensive picture of adsorbed protein dissociation is lacking. In this study, the protein dissociation from the protein corona and influencing factors were investigated on the basis of the formation mechanism and time evolution. Temperature and cysteine are the key factors influencing protein dissociation by altering the protein's binding ability. The results showed that half Au–S formation time is an important time point for thio-protein dissociation by the method of high speed centrifugation. When incubated for longer than that time, the thio-protein located in the hard corona could only be separated by β-mercaptoethanol replacement under analytical ultracentrifugation. However, Fourier-transform infrared spectroscopy (FTIR) revealed significant changes that occurred in βlg's secondary structure after ultracentrifugation. The Au–S bond formation time offers the potential to define the protein enrichment time of AuNPs.
Introduction
Nanotechnology and nanoparticles (NPs) have gained great developments in the biomedicine and biodetection fields.1–3 This is especially true for protein enrichment and analysis in gold nanoparticle (AuNP)-based microfluidic chips4,5 due to their high specific surface area and unique physicochemical properties.6 This technology has led to creating novel strategies for rapidly examining food composition and diagnosing early-stage cancers.7,8 These applications mainly rely on the adsorption of biomacromolecules, including proteins, to AuNPs.9 As a result of this adsorption, a “protein corona” is formed on the NPs surface.10,11 However, this corona could alter the adsorbed protein structure and biological characteristics,12,13 subsequently hindering their analysis and identification in situ.14,15 Our previous work revealed that AuNPs can be explored as a novel strategy to desensitize βlg since the formation of protein corona.16 While, the application in practical samples is limited, such as milk or whey protein isolate (WPI) because of the identification of corona composition is the precondition of bioapplication. Therefore, it is crucial to probe the adsorbed protein dissociation and influence factors from the aspect of protein corona formation mechanism, which may potentially define practical guidelines for AuNPs' bioapplications.Protein corona formation is a complex process and is kinetically divided into two phases.17,18 In phase one, proteins are absorbed on the NP surface in seconds or minutes by fast electrostatic or hydrophobic forces. For example, more than 300 plasma protein species rapidly adsorb onto AuNPs within 0.5 min.19 In phase two, thio-protein sulfhydryl groups, accessible to Au+, form Au–thiol covalent bonds (Au–S bonds) on the order of hours.13 The protein corona contains a two-component system that the proteins direct/indirect interact with NPs. Au–S bonds form a tightly bound layer, called the “hard” corona. Other layers are then surrounded by a “protein cloud,” referred to as the “soft” corona. In general, the hard corona is an irreversible process after the formation of Au–S bonds,20 which is a critical factor for understanding the protein dissociation mechanism.
Thio-proteins and non-thio-proteins exhibit distinct binding mechanisms to AuNPs. The binding mechanism, including the formation of Au–S bond and formation time, is crucial for understanding the adsorbed protein dissociation from the AuNP surface. In practical applications, protein enrichment occurs in the order of minutes to hours, depending on the protein sample and specific application.21,22 The main strategies for improving the enrichment process are decreasing the enrichment time, enhancing the detection efficiency, or lengthening the Au–S formation time. Numerous studies demonstrate that one can decrease the enrichment time or increase the detection efficiency by modifying the AuNPs with probes or combining the technology with a resonance energy transfer technique.4,23 while, the system research for lengthening the Au–S formation time and temperature influences remain elusive.
Here, we characterized the protein adsorption to AuNPs using two model proteins, β-lactoglobulin (βlg, thio-protein) and myoglobin (MB, non-thio-protein), which have similar molecular weights and global conformation.24,25 The binding parameters, including the binding affinities (Ka), binding numbers, and Au–S bond formation time as a function of temperature (4 °C to 44 °C), were detected to assess the centrifugation efficiency method for dissociating adsorbed proteins from the AuNP surface. Fourier transform infrared spectroscopy (FTIR) was used to examine the secondary structure changes before and after dissociation. Our results provide a guideline for the design of AuNPs as a protein enrichment material. Fig. 1 shows the adsorbed protein binding mechanism on AuNPs for the thio- and non-thio-proteins.
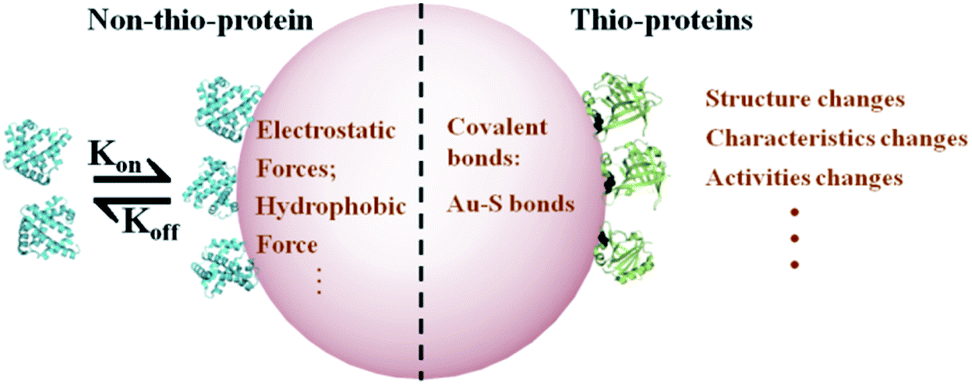 | ||
| Fig. 1 A schematic representation of proteins adsorbed on AuNPs and the consequences for the thio- and non-thio-proteins. | ||
Results and discussion
The corona thickness
The protein corona thickness related to the protein binding number is a crucial criterion for understanding the protein binding to NPs.26 Dynamic light scattering (DLS) is the commonly used tool for measuring NPs and protein coronas' hydrodynamic diameters.6,27 Typical AuNPs–protein (βlg and MB) corona DLS spectrum that were prepared at different temperatures are shown in Fig. 2.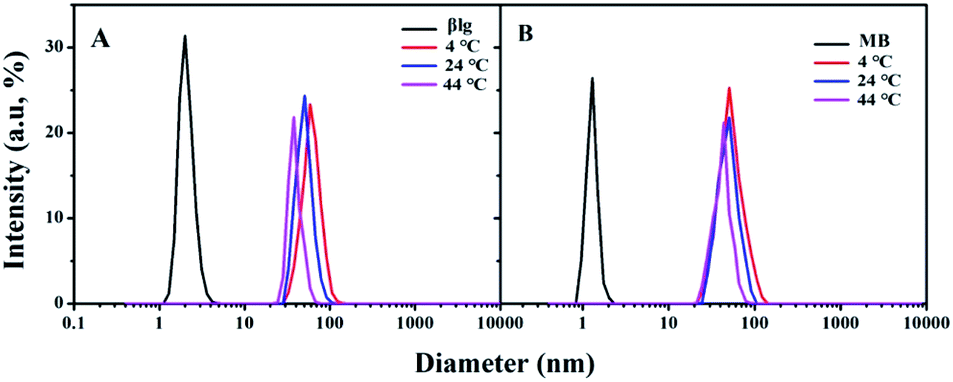 | ||
| Fig. 2 Typical DLS spectrum of AuNPs–protein coronas incubated at different temperature. (A) AuNPs–βlg corona; (B) AuNPs–MB corona. | ||
The results showed that the AuNPs–βlg corona diameters decreased from 60 ± 2.6 nm to 44 ± 3.1 nm with an increase in temperature from 4 °C to 44 °C (Fig. 2A). In contrast, the AuNPs–MB coronas exhibited a smaller decrease in diameter (ΔD = 10 nm) with temperature increasing. The AuNPs–βlg protein corona thickness was several times larger than the βlg protein diameter (2.1 ± 0.5 nm) at all experimental temperatures, indicating that it consisted of both “hard” and “soft” coronas.16 The hard corona proteins form Au–S bonds with the surface of the AuNPs, representing approximately irreversible bound proteins.19 Thereby, the hard corona defined the centrifugation dissociation efficiency. Cysteine residues and temperature played key roles in the protein corona formation. Temperature influences the degree of protein coverage and the composition of adsorbed proteins on AuNPs surface at which the protein corona is formed.28,29 The higher temperature means the smaller proteins were adsorbed by the AuNPs.
Binding parameters detection
Isothermal titration calorimetry (ITC) is a straightforward method for quantitatively determining crucial thermodynamic parameters of the protein–NP interaction by assessing the associated heat change in real-time with a high signal-to-noise ratio.30 The results of this assay shed light on the interaction mechanisms by revealing the stoichiometry (Ka) and enthalpy (ΔH), allowing for a comparative analysis of influencing factors (e.g., temperature, pH, and NP properties) important for NP bio-application.31 The free energy change (ΔG) and entropy change (ΔS) were calculated from the fundamental equations:6| ΔG = −RT ln Ka | (1) |
| ΔG = ΔH − TΔS | (2) |
The raw data for βlg titration into the AuNP solutions at different temperatures are shown in Fig. 3. Each heat exchange curve corresponds to a single injection. The negative signals (ΔH < 0) indicate that the protein binding to the AuNPs is a strongly exothermic process.30 The signal intensities gradually decreased with an increasing number of injections and reached equilibrium at distinct molar ratios at different temperatures. Additionally, the equilibrium molar ratio decreased from ∼7500 to ∼5000 at ∼20 min when the temperature increased from 4 °C to 44 °C, which indicated that the decrease of the binding number and the fast electrostatic forces, including van der Waals interactions, electrostatics and hydrogen bond dominated the formation of protein corona during the ITC detection. The MB titration into AuNPs exhibited a smaller heat exchange, albeit with the similar behaviour (Fig. S1†).
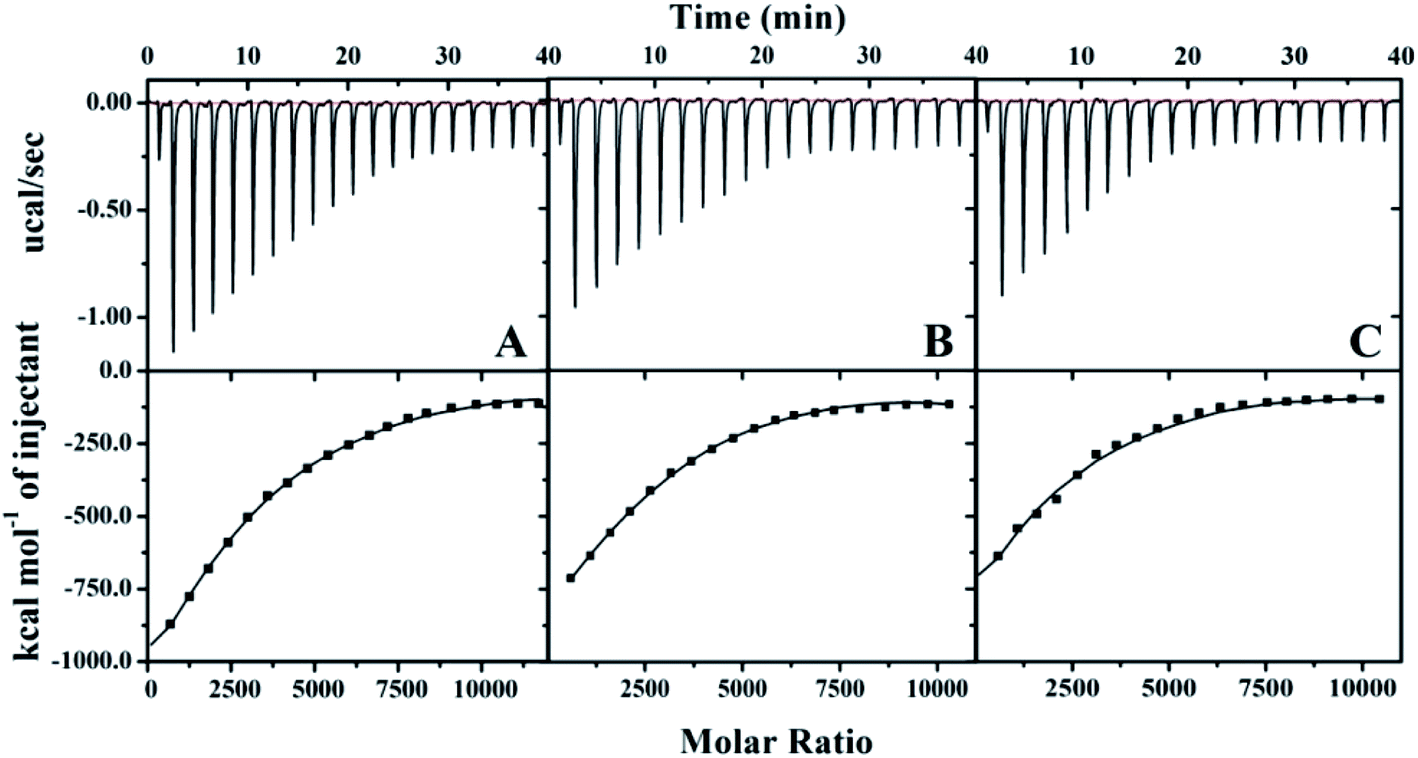 | ||
| Fig. 3 The Raw ITC data for βlg (30 μM) titrated into the AuNP solution (4 × 10−4 μM) with continuous stirring at 1000 rpm at different temperatures. (A) 4 °C; (B) 24 °C; (C) 44 °C. | ||
The stoichiometry thermodynamic parameters, Ka, ΔH, ΔS, and ΔG for proteins titrated into the AuNP solutions were derived using a nonlinear least-squares model and are summarized in Table 1. The results indicate that protein association with AuNPs is exothermic and therefore enthalpically favoured. Ka is important for describing the interaction between proteins and NPs, and it can give a general idea of the binding force and strength. A higher incubation temperature resulted in a lower binding affinity and stoichiometry of the protein adsorbed to AuNPs. The βlg's Ka decreased from 34 ± 10 × 105 M−1 to 25 ± 6 × 105 M−1, and the stoichiometry decreased from 3900 to 2600 when the temperature increased from 4 °C to 44 °C. Likewise, the MB's Ka decreased from 2.6 ± 0.4 × 105 M−1 to 2.1 ± 0.5 × 105 M−1, and the stoichiometry decreased from 3600 to 2600. Based on these decrease of stoichiometry data, inconsistent with the DLS, temperature exerts a greater influence on the adsorption of thio-proteins than non-thio-proteins in thermodynamics.
| Protein | Temperature (°C) | Stoichiometry (103) | Kaa (105 M−1) | ΔH (105 J M−1) | ΔSb (103 J M−1 deg.) | ΔGb (104 J) |
|---|---|---|---|---|---|---|
| a Derived from one site binding model.b Calculated according to the eqn (1) and (2). | ||||||
| βlg | 4 | 3.9 | 34 ± 10 | −39 ± 12 | −13.9 | −3.5 |
| 24 | 3.1 | 29 ± 7 | −32 ± 9 | −10.6 | −3.7 | |
| 44 | 2.6 | 25 ± 6 | −29 ± 7 | −9.0 | −3.9 | |
| MB | 4 | 3.6 | 2.6 ± 0.7 | −4.9 ± 0.7 | −1.6 | −2.9 |
| 24 | 3.0 | 2.3 ± 0.4 | −4.6 ± 0.8 | −1.4 | −3.0 | |
| 44 | 2.6 | 2.1 ± 0.5 | −4.3 ± 0.5 | −1.2 | −3.2 | |
Au–S covalent bond detection
Surface-enhanced Raman spectroscopy (SERS) has been extensively applied in various ultrasensitive chemical detection applications in a wide variety of fields and can be applied even at the single-molecule level.32 SERS can provide information about the structure, composition, and chemical bonds of molecules interacting with AuNPs, enabling the study of adsorbed proteins by the protein corona in stiu.33 βlg contains five cysteine residues; four are involved in two disulfide bridges and one free thiol group. The thiol groups are accessible to Au+ and can form the Au–S covalent bond rapidly, which can be observed at 296 cm−1 in Raman spectroscopy.16To investigate how temperature influences the protein adsorption speed to AuNPs, we examined the changes in the AuNPs–βlg corona SERS intensity prepared at different temperatures as a function of incubation time; these results are shown in Fig. 4. The intensity increased as time went on until reaching a plateau, indicating the formation time of Au–S bond. The Au–S bond formation for AuNPs–βlg coronas took ∼12 h at 4 °C, whereas it decreased to ∼9 h and ∼6 h at 24 °C and 44 °C, respectively. Notably, this result (about 9 h at 24 °C) was approximate to our previous results that detected at 20 °C because of the experimental error.16 In contrast, there was no Raman shift in the AuNPs–MB corona spectroscopy (Fig. S2†). No Au–S bonds were formed in AuNPs–MB corona, indicating weak forces, such as electrostatic or hydrophobic forces, were the main binding force for MB adsorbed on AuNPs. Higher temperatures promoted a faster AuNP–protein interaction and a shorter Au–S formation time. Au–S bonds are the main force involved in attaching thio-proteins to the surface of AuNPs, resulting in these proteins secondary structure and characteristics changes.
 | ||
| Fig. 4 AuNPs–βlg corona SERS intensity change, prepared at different temperatures as a function of incubation time. Insert: the Au–S formation time at different temperatures. | ||
Centrifugation
Centrifugation is an efficient method for separating adsorbed proteins from the NP surface onto microfluidic chips, which requires properly controlled centrifugation speed and time.34,35 Fluorescence emission is an inherent property of proteins containing tryptophan, tyrosine, and phenylalanine residues, quenched by association with AuNPs.36,37 For the same protein and specific AuNP sizes, the quenching rate is the same. Therefore, a change in protein emission intensity can be attributed to protein dissociation from the AuNPs, which can investigate protein corona changes after centrifugation.The AuNPs–protein corona fluorescence emission intensities prepared at 4 °C as the function of time after centrifugation are shown in Fig. 5. Centrifugation resulted in different dissociation efficiencies for thio-proteins and non-thio-proteins. The MB amount dissociated was temperature-independent, as most of the adsorbed MB in both “hard” and “soft” coronas were separated by high-speed centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min, for all incubation times (Fig. 5A). Whereas, the βlg amount dissociated was strongly related to the incubation time. The longer incubation time shows the higher emission intensities, which indicated that βlg was weakly dissociated. In general, most of the adsorbed βlg could be separated from the corona by high-speed centrifugation when incubated shorter than 4 h (half the Au–S bond formation time). However, there were no significant changes after incubated longer than 9 h (the Au–S bond formation time), maintaining a high level of fluorescence intensity due to the residual βlg located in the “hard” corona (Fig. 5B). The fluorescence intensities of protein corona prepared at 24 °C and 44 °C showed similar phenomenon (data not show).
000 rpm for 15 min, for all incubation times (Fig. 5A). Whereas, the βlg amount dissociated was strongly related to the incubation time. The longer incubation time shows the higher emission intensities, which indicated that βlg was weakly dissociated. In general, most of the adsorbed βlg could be separated from the corona by high-speed centrifugation when incubated shorter than 4 h (half the Au–S bond formation time). However, there were no significant changes after incubated longer than 9 h (the Au–S bond formation time), maintaining a high level of fluorescence intensity due to the residual βlg located in the “hard” corona (Fig. 5B). The fluorescence intensities of protein corona prepared at 24 °C and 44 °C showed similar phenomenon (data not show).
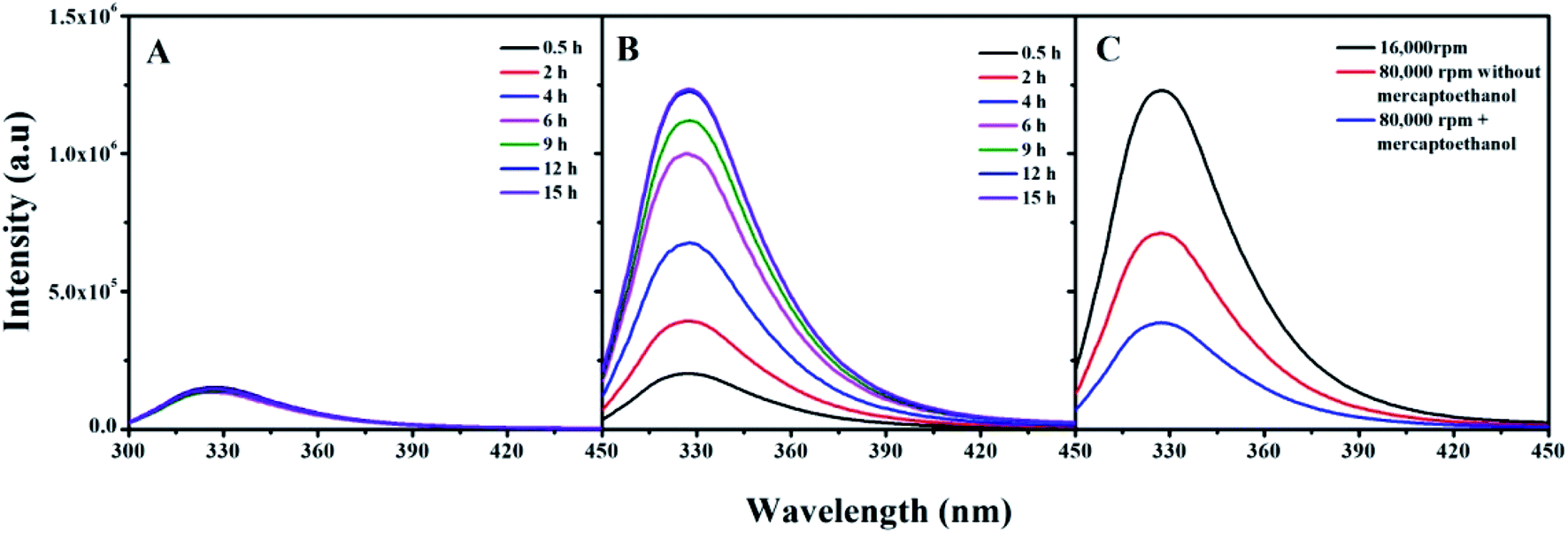 | ||
| Fig. 5 The protein emission intensities in protein coronas as a function of time after centrifugation. (A) AuNPs–MB corona centrifuged at 15000 rpm for 15 min, (B) AuNPs–βlg corona centrifuged at 15000 rpm for 15 min, (C) AuNPs–βlg corona incubated for 15 h and ultracentrifuged at 80000 rpm for 20 min in the absence and presence of β-mercaptoethanol. | ||
In order to separate those remaining βlg in hard corona, β-mercaptoethanol added in AuNPs–βlg corona solution and analytical ultracentrifugation at 80000 rpm for 20 min were performed. The result shows that most of these remaining βlg in “hard” corona can be separated by this synergistic action (Fig. 5C). While this action, especially of the presence of β-mercaptoethanol may bring new challenges for the proteins, such as altering the structure and functional properties. The bind force, in terms of Au–S bonds or weak force, and incubation time are the key criteria for choosing the dissociation methods.
Secondary structure detection
Fourier-transform infrared spectroscopy (FTIR) is a versatile tool for studying protein and polypeptide conformation in H2O-based solutions.24,38 Proteins exhibit nine characteristic infrared spectroscopy (IR) absorption bands. Of those bands, the amide I in the second-derivative spectrum (1700–1600 cm−1) is the most sensitive spectral region. It is closely correlated with the protein secondary structure, which is almost entirely due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch vibrations in the peptide linkages (∼80%).39 The fitted inverted second-derivative amide I curves of βlg, AuNPs–βlg coronas, and dissociated βlg by centrifugation and ultracentrifugation with β-mercaptoethanol were shown in Fig. 6. The resulting secondary structural components were summarized in Table 2. The β-sheet (1628 and 1637 cm−1) is the most common secondary structure component in native βlg and in separated βlg by high-speed centrifugation (approximately 80%, Fig. 6A and C). The β-sheet and 310-helix (1663 cm−1) decreased after being adsorbed on AuNPs, thereby, the bands assigned to the α-helix (1658 cm−1), β-turn (1667 and 1675 cm−1), and random coil (1648 cm−1) increased (Fig. 6B). Most cysteine residues located in β-sheet conformation according to the three-dimensional model,23 which would form Au–S bond with AuNPs, resulting in destroying the β-sheet conformation.40 Although most of the adsorbed βlg can be separated by analytical ultracentrifugation, the secondary structure occurs significantly changes due to the presence of β-mercaptoethanol that β-sheet decreased, while other structure components increased (Fig. 6D).
O stretch vibrations in the peptide linkages (∼80%).39 The fitted inverted second-derivative amide I curves of βlg, AuNPs–βlg coronas, and dissociated βlg by centrifugation and ultracentrifugation with β-mercaptoethanol were shown in Fig. 6. The resulting secondary structural components were summarized in Table 2. The β-sheet (1628 and 1637 cm−1) is the most common secondary structure component in native βlg and in separated βlg by high-speed centrifugation (approximately 80%, Fig. 6A and C). The β-sheet and 310-helix (1663 cm−1) decreased after being adsorbed on AuNPs, thereby, the bands assigned to the α-helix (1658 cm−1), β-turn (1667 and 1675 cm−1), and random coil (1648 cm−1) increased (Fig. 6B). Most cysteine residues located in β-sheet conformation according to the three-dimensional model,23 which would form Au–S bond with AuNPs, resulting in destroying the β-sheet conformation.40 Although most of the adsorbed βlg can be separated by analytical ultracentrifugation, the secondary structure occurs significantly changes due to the presence of β-mercaptoethanol that β-sheet decreased, while other structure components increased (Fig. 6D).
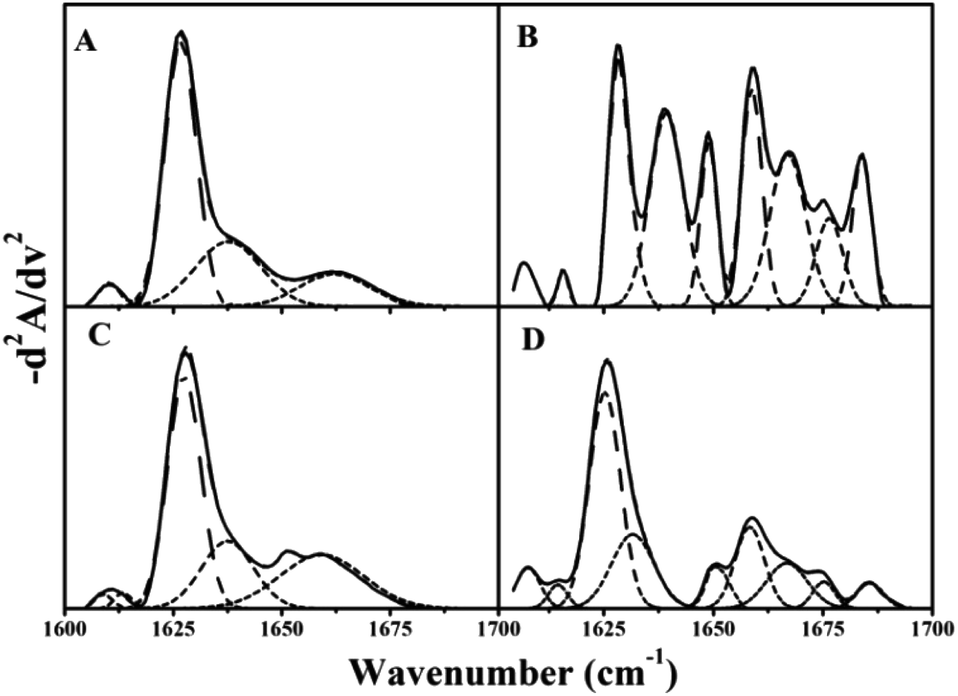 | ||
| Fig. 6 The fitted curve for the second-derivative amide I (1600–1700 cm−1) spectrum of βlg in different states. (A) Native βlg; (B) βlg in AuNPs–βlg corona prepared at 4 °C; (C) βlg after common centrifugation; (D) βlg after β-mercaptoethanol replacement and ultracentrifugation. | ||
| 310-Helixa | α-Helixb | β-Sheetc | β-Turnd | Random coilse | |
|---|---|---|---|---|---|
| a 310-Helix is 1663 cm−1.b α-Helix is 1658 cm−1.c β-Sheet is 1628 and 1637 cm−1.d β-Turn is 1667 and 1675 cm−1.e Random coils is 1648 cm−1 in the FTIR spectrum. | |||||
| Native | 15% | 0 | 81% | 0 | 4% |
| Corona | 0 | 26% | 31% | 24% | 19% |
| Centrifugation | 19% | 0 | 76% | 0 | 5% |
| Ultra-centrifugation | 0 | 15% | 56% | 18% | 11% |
MB exhibited an α-helix content above 70%, representing the most common secondary structure in the protein (Fig. S3†). Table S1† shows no changes in secondary structure components after MB was adsorbed on and dissociated from AuNPs. MB likely adsorbed to the AuNP surface because of a weak force that did not destroy the forces stabilizing the protein spatial structure.
Cysteine residues and Au–S bonds are the key factors influencing the proteins' dissociation efficiency from protein corona. Half the Au–S formation time has significant meanings to the AuNPs bioapplications. When the enrichment time is shorter than that time, most of the adsorbed proteins can be separated by high speed centrifugation without structure changes, conversely, when the enrichment is longer than that time, the dissociation is more difficult and complex, even changing the secondary structures after separation.
Experimental
Materials
Tetrachloroaurate (HAuCl4·3H2O), βlg from bovine milk, and MB from the equine heart were purchased from Sigma Aldrich (USA). AuNPs, with a theoretical diameter of 40 nm, were fabricated using 100 mL of HAuCl4 (10−2%, w/v) reduced by 1 mL of sodium citrate (1%, w/v).41 The actual size and concentration were determined by DLS and UV spectra analysis.42 The stock protein solutions were prepared in 10 mM phosphate buffer solution (PBS, pH 7.2).Dynamic light scattering (DLS)
AuNPs–protein (βlg or MB) coronas were prepared by incubating AuNPs (3 mL, 5 × 10−4 μM) with proteins (0.3 mL, 50 μM) for 2 h at 4 °C, 24 °C and 44 °C, and then centrifuged at 10000 rpm for 5 min. 50 μL PBS (10 mM, pH 7.2) was used to re-suspend the sediment. The protein solutions of AuNPs and protein coronas formed at different temperatures, and were filtered through a PTFE 0.45 μm filter. Their hydrometers were measured by DLS (Nicomp 380, PSS, USA) to estimate the protein corona thickness change with increasing temperature. The data collection time was set to 90 s for temperature equilibrium and 5 s during the kinetic series acquisition.
Isothermal titration calorimetry (ITC)
All ITC experiments were conducted on a MicroCal ITC-200 system (GE Healthcare, USA) at 4 °C, 24 °C, or 44 °C. Proteins and AuNP solutions were thoroughly degassed by gently stirring under a vacuum for 5 min at the corresponding temperature before titration to avoid the formation of bubbles. An injection syringe was filled with 40 μl (30 μM) of protein, and 200 μl-AuNPs (4 × 10−4 μM) were placed in the sample cell. The reference cell was filled with 10 mM PBS (pH 7.2). The proteins were titrated into the AuNP solution. The titrations consisted of 20 injections; 0.5 μl for the first injection and 2 μl for each subsequent injection, under continuous stirring at 1000 rpm. A blank protein titration was also performed to detect the heat dilution, which was later subtracted during calculations. The titration data were analyzed with MicroCal origin V7.0 software.Surface-enhanced Raman spectroscopy (SERS)
To monitor the time evolution of the Au–S bonds between AuNPs and proteins (βlg or MB), SERS experiments at different temperatures were performed on a Jobin Yvon XploRA (France). The coronas were obtained by incubating proteins (0.1 mL, 50 μM) with AuNPs (1 mL, 4 × 10−4 μM) for 1, 4, 6, 8, 10, 12, 15, and 20 h at temperatures from 4 °C to 44 °C. The corona spectra were measured at 532 nm (10 mW) excitation, and the Rayleigh scattering light was removed by an edge filter. The collection time was set to 30 s, and the emission signal was collected from 200 to 800 cm−1. All SERS spectra were the result of a single 1 s accumulation.Centrifugation
The AuNPs–protein coronas were centrifuged at 15000 rpm for 15 min to separate the adsorbed MB and βlg located on the soft corona. AuNPs–βlg corona solution (0.3 mL) was incubated for 15 h and interacted with 0.5 mL of β-mercaptoethanol for 2 h at 44 °C, then centrifuged at 80000 rpm for 20 min. All the precipitates were re-suspended in 1 mL of PBS to detect the fluorescence emission intensity (Varian, Sweden). Excitation experiments were performed at 280 nm with a slit width of 5 nm, and the emission signal was collected from 300 to 450 nm. The values were calculated as an average of triplicate measurements.
Fourier-transform infrared spectroscopy (FTIR)
The dissociated proteins were prepared following the centrifugation protocols. FTIR spectra of the proteins, coronas, and dissociated proteins were recorded at 20 °C on a Bomen MB series FITR Spectrometer (Quebec, Canada) equipped with a dTGS detector, and constantly purged with dry air, as described in our previous study.23 A 128-scan interferogram was collected in single-beam mode with 4 cm−1 resolution. The relative secondary structure content was determined from a curve-fitting analysis of the amide I band in the range of 1600–1700 cm−1.Conclusions
AuNPs are commonly used for protein enrichment in microfluidic chips due to their fast protein adsorption, leading to the formation of a protein corona. The protein corona is mainly stabilized by non-thio-protein electrostatic forces or thio-protein Au–S covalent bonds. We found that the corona formation altered the adsorbed protein structure and characteristics, especially for thio-proteins, hindering subsequent in situ macromolecule detection and identification. Thus, NPs dissociation is an important supplemental measurement in protein analysis and identification. The binding mechanism and factors influencing binding are key to understanding the method of dissociation, including the centrifugation dissociation efficiency.In summary, temperature plays a key role in the protein adsorption on the AuNP surface, especially for thio-proteins. The binding force decreased with increasing temperatures, resulting in lower Ka and a reduced adsorbed protein number. And the Au–S bond formation speed was faster in the AuNPs–βlg corona. The Au–S bond formation time for AuNPs–βlg corona was ∼12 h at 4 °C. Centrifugation at 16000 rpm for 15 min separated most of the adsorbed βlg; the incubation time was shorter than half the Au–S bond formation time. However, βlg was only be separated by β-mercaptoethanol replacement and ultracentrifugation. Moreover, FTIR revealed significant changes that occurred in βlg's secondary structure after ultracentrifugation. Contrasting, most of the MB was be separated from the AuNPs in a short time because of the weak binding forces. An enrichment time that is shorter than half the Au–S formation time provides a guide for AuNP bio-application and can help avoid the need for more complex dissociation methods.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Xinglin Scholar Research Premotion Project of Chengdu University of TCM (no. QNXZ202012) and Sichuan Provincial Major Science and Technology Program (no. 2020YFN0153).Notes and references
- J. Kaufman, R. Ottman, G. Tao, S. Shabahang, E. Banaei, X. Liang, S. Johnson, Y. Fink, R. Chakrabarti and A. Abouraddy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15549–15554 CrossRef CAS PubMed.
- D. Li, Y. Zhang, M. Yu, J. Guo, D. Chaudhary and C. Wang, Biomaterials, 2013, 34, 7913–7922 CrossRef CAS PubMed.
- J. Liu, Z. Ni, P. Nandi, U. Mirsaidov and Z. Huang, Nano Lett., 2019, 19, 7427–7433 CrossRef CAS PubMed.
- C. Wang, Y. Shi, J. Wang, J. Pang and X. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 6835–6841 CrossRef CAS PubMed.
- Y. Kim, D. Kim, S. Lee, D. Kim, S. Park and S. Kim, Small, 2019, 15, 1905076 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, F. Zhang and S. Yu, Nanoscale, 2017, 9, 4787–4792 RSC.
- K. Haume, S. Rosa, S. Grellet, A. Śmiałek, T. Butterworth, V. Solov'yov, M. Prise, J. Golding and J. Mason, Cancer Nanotechnol., 2016, 7, 8–28 CrossRef PubMed.
- Q. Ma, Y. Wang, J. Jia and Y. Xiang, Food Chem., 2018, 249, 98–103 CrossRef CAS PubMed.
- D. Docter, U. Distler, W. Storck, J. Kuharev, D. Wünsch, A. Hahlbrock, S. Knauer, S. Tenzer and R. Stauber, Nat. Protoc., 2014, 9, 2030–2044 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, K. Knauer and H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- M. Wang, C. Fu, X. Liu, Z. Lin, N. Yang and S. Yu, Nanoscale, 2015, 7, 15191–15196 RSC.
- C. Fu, H. Yang, M. Wang, H. Xiong and S. Yu, Chem. Commun., 2015, 51, 3634–3636 RSC.
- K. Schey, D. Anderson and K. Rose, Anal. Chem., 2013, 85, 6767–6774 CrossRef CAS PubMed.
- D. Shteynberg, E. Deutsch, H. Lam, J. Eng, Z. Sun, N. Tasman and A. Nesvizhskii, Mol. Cell. Proteomics, 2011, 10, 7690–7705 CrossRef PubMed.
- X. Zhang, M. Li, Y. Lv, X. Sun, Y. Han, B. Liu, X. Zhao and X. Huang, Food Chem., 2021, 342, 128329 CrossRef CAS PubMed.
- X. Zhang, H. Shi, R. Zhang, J. Zhang, F. Xu, L. Qiao and S. Yu, Part. Part. Syst. Charact., 2019, 36, 1800257–1800263 CrossRef.
- W. Liu, J. Rose, S. Plantevin, M. Auffan, J. Bottero and C. Vidaud, Nanoscale, 2013, 5, 1658–1668 RSC.
- T. Göppert and R. Müller, Eur. J. Pharm. Biopharm., 2005, 60, 361–372 CrossRef PubMed.
- S. Milani, B. Baldelli, A. Pitek, K. Dawson and J. Radler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS PubMed.
- Y. Jiang, M. Shi, Y. Liu, S. Wan, C. Cui, L. Zhang and W. Tan, Angew. Chem., Int. Ed., 2017, 56, 11916–11920 CrossRef CAS PubMed.
- J. Lin and W. Tseng, Rev. Anal. Chem., 2012, 31, 153–162 CAS.
- W. Chang and W. Tseng, Anal. Chem., 2010, 82, 2696–2702 CrossRef PubMed.
- X. Zhang, H. Hemar, L. Lv, T. Zhao, Y. Yang, Z. Han, M. Li and J. He, Int. J. Biol. Macromol., 2019, 140, 377–383 CrossRef CAS PubMed.
- B. Debipreeta and S. Kumar, J. Biomol. Struct. Dyn., 2016, 35, 1260–1271 Search PubMed.
- L. Marichal, G. Giraudon-Colas, F. Cousin, A. Thill and J. Renault, Langmuir, 2019, 35, 10831–10837 CrossRef CAS PubMed.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- M. Mahmoudi, A. Abdelmonem, S. Behzadi, J. Clement, S. Dutz, M. Ejtehadi, R. Hartmann, K. Kantner, U. Linne, P. Maffre, S. Metzler, M. Moghadam, C. Pfeiffer, M. Rezaei, P. Ruiz-Lozano, V. Serpooshan, M. Shokrgozar, G. Nienhaus and W. Parak, ACS Nano, 2013, 7, 6555–6562 CrossRef CAS PubMed.
- A. Lesniak, A. Campbell, M. P. Monopoli, I. Lynch, A. Salvati and K. A. Dawson, Biomaterials, 2010, 31, 9511–9518 CrossRef CAS PubMed.
- A. Velazquez-Campoy and E. Freire, Nat. Protoc., 2006, 47, 186–191 CrossRef PubMed.
- R. Huang, R. Carney, K. Ikuma, F. Stellacci and B. Lau, ACS Nano, 2014, 8, 5402–5412 CrossRef CAS PubMed.
- J. Zhang, X. Zhang, F. Zhang and S. Yu, Anal. Bioanal. Chem., 2017, 409, 4459–4465 CrossRef CAS PubMed.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- E. Casals, T. Pfaller, A. Duschl, G. Oostingh and V. Puntes, ACS Nano, 2010, 4, 3623–3632 CrossRef CAS PubMed.
- C. Weber, S. Morsbach and K. Landfester, Angew. Chem., Int. Ed., 2019, 58, 12787–12794 CrossRef CAS PubMed.
- S. Lacerda, J. Park, C. Meuse, D. Pristinski, M. Becker, A. Karim and J. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- C. Carrillo-Carrion, M. Carril and W. Parak, Curr. Opin. Biotechnol., 2017, 46, 106–113 CrossRef CAS PubMed.
- S. Yang, X. Dai, B. Stogin and T. Wong, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 268–273 CrossRef CAS PubMed.
- X. Han, B. Zhao and Y. Ozaki, Anal. Bioanal. Chem., 2009, 394, 1719–1727 CrossRef CAS PubMed.
- D. Zhang, O. Neumann, H. Wang, V. Yuwono, A. Barhoumi, M. Perham, D. Hartgerink, P. Wittung-Stafshede and N. Halas, Nano Lett., 2009, 9, 666–671 CrossRef CAS PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- W. Haiss, N. Thanh, J. Aveyard and D. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02116h |
| This journal is © The Royal Society of Chemistry 2021 |