
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile fabrication of hollow tubular covalent organic frameworks using decomposable monomer as building block†
Ya Liab,
Chang Wanga,
Shujuan Maa,
Junwen Xuac,
Xiaowei Liac,
Yinmao Wei b and
Junjie Ou*ac
b and
Junjie Ou*ac
aCAS Key Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: junjieou@dicp.ac.cn; Fax: +86-411-84379620; Tel: +86-411-84379576
bKey Laboratory of Synthetic and Natural Function Molecule Chemistry of Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi’an 710127, China
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 14th June 2021
Abstract
In this study, a commercial and low-toxicity hydrazide-containing building block has been used to construct azine-linked covalent organic frameworks (COFs). New style COFs were constructed between flexible formic hydrazide (FH) and 1,3,5-triformylphloroglucinal (Tp) or 1,3,5-triformylbenzene (TFB). The two resulting COFs (TpFH and TFBFH) exhibited uniform hollow tubular morphology (20–50 nm for TpFH, 50–100 nm for TFBFH). Compared to hydrazine, FH has low-toxicity and is a flexible monomer, consisting of amine and aldehyde groups. The decomposition of FH slows down the reaction rate and the as-synthesized FH-series COFs (708 m2 g−1 for TpFH and 888 m2 g−1 for TFBFH) had higher specific surface area than hydrazine-series COFs (617 m2 g−1 for TpAzine and 472 m2 g−1 for TFBAzine). A detailed time-dependent investigation was carried out to interpret the mechanism of hollow structure formation, and Ostwald ripening possibly happens during the formation of hollow COF microstructures. Considering the porous and high density N, O elements of these materials, preliminary applications of the metal ions removal from aqueous solution and gas storage were implemented.
1. Introduction
Since the seminal work published by Yaghi in 2005, covalent organic frameworks (COFs) have attracted a great deal of attention.1 Being analogous to metal–organic frameworks (MOFs), COFs are an emerging class of crystalline porous materials which are composed entirely of covalent bonds between light elements.2 In particular, two-dimensional COFs have well-defined crystalline structures with regular slow channels. Over the past decade, various potential applications of these materials have been explored, including gas storage and separation,3 heterogeneous catalysis,4 optoelectronics,5 sensing,6 etc. It is essential to maintain the reversibility of covalent bonds in the synthesis of COFs. Based on the widely implemented four reversible organic reactions7,8 – Schiff base reactions, trimerization of nitriles, boronic acid trimerization and boronate ester formation – the obtained 2D COFs were characterized with limited long-range crystal growth and morphological definition. Although several researchers have also reported on the defined morphology of COFs such as films,9 belts,10 spheres11 and fibers,12 so far, there are only a few detailed structure and mechanism investigations for hollow tubular COFs.13,14 The majority of the reported COFs have been constructed from rigid building blocks, which are beneficial for the formation of crystalline products,15,16 conversely, they limit the variety and properties of COFs. In addition, new functionalized monomers synthesis requires a high expenditure of energy, long reaction time and the use of hazardous organic solvents. Thus, it is essential to look for new commercial flexible monomers to synthesize economic COFs with new functions.Hollow-structured porous materials have attracted great attention because of their potential for extraordinary properties arising from their hierarchically porous structures.17 To synthesize the functionalized hollow COFs with specific surface chemistry, besides the template-assisted method, the self-templated method was also adopted to fabricate hollow materials, such as polymer-based hollow materials and metal oxide/sulfide-based nanotubes.18–21 The latter method is simple and facile which not only maintains the surface properties, but avoids the need to remove templates.22 Banerjee et al. synthesized two hydroxyl-decorated COFs via a Schiff base reaction of 2,3-dihydroxyterethaldehyde with 1,3,5-tris(4-aminophenyl)benzene and 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline.13 They also fabricated a hollow spherical COF using the self-templated method via Schiff base reaction of 2,5-dihydroxyterephthaldehyde and 1,3,5-tris(4-aminophenyl)benzene.14 Other building blocks, including aldehyde and carboxyl group decorated monomers, have been explored less to synthesize the COFs with hollow tubular structures. State-of-the-art approaches to stabilize COFs are based on the introduction of enol–keto tautomerization23,24 or hydrogen bonding interactions25 in the COF skeletons. In the formation process of COFs, the different molecular conformations of the building block are known depending on the hydrogen bonding and electron resonance, etc. Considering these matters, a variety of untested building blocks with specific properties can be explored to enrich the function and variety of COFs.
Recently, green synthesis, under mild conditions and using harmless or eco-friendly solvents or monomers has drawn extensive attention from academia and industry. Here, we propose a facile and relatively green strategy for constructing two highly crystalline COFs. Formic hydrazide (FH) is a commercial and low-toxic flexible hydrazide-containing monomer, which owns intramolecular hydrogen bonding between the hydrogen atoms attached to the nitrogen atoms and the oxygen atom. Although significant progress on the synthesis of well-defined and customizable COFs has recently been achieved, research on the precise construction of template-free nano- and microstructure hollow tubular COFs are rare, especially with flexible monomers as the building block. The hydrogen bonding between the amine and aldehyde in FH improves the planarity, but the flexibility increases the potential for crystallographic defects and undermines the process of dynamic error correction required for growth of crystalline COF.26 It should be highlighted that the chemical reactivity of azine amine is higher than FH, and the aldehydes of FH are inclined to decompose in harsh conditions. Herein, we took full advantage of the unique characteristic of FH and high efficiency of Schiff base condensation reaction with 1,3,5-triformylphloroglucinal (Tp) or 1,3,5-triformylbenzene (TFB), to synthesize two new 2D COFs: TpFH and TFBFH. In view of the hollow microtubular structures and abundant of N, O elements on the skeleton, the hollow-shape COFs were applied to metal ion adsorption and gas storage.
2. Experimental section
2.1 Preparation of COFs: TpFH and TFBFH
An ampoule bottle measuring 10 × 8 mm (o.d. × i.d.) was charged with FH (9 mg, 0.15 mmol) and Tp (21 mg, 0.10 mmol), 500 μL of anhydrous dioxane and 500 μL of mesitylene, and then immersed in an ultrasonic bath for 15 min. After sonication for 30 min, 0.3 mL of aqueous HAc (6 M) was added. The bottle was rapidly frozen at 77 K (liquid N2 bath), evacuated to an internal pressure of below 5 Pa and sealed by flame. The length of the tube sealed was reduced to 10–15 cm. The reaction was heated to 120 °C for 72 h yielding a red solid, which was isolated by filtration and washed with anhydrous DMF and THF, the resulting powder was immersed in anhydrous THF for 24 h and dried under vacuum at 100 °C overnight. Yield: 18 mg, 60% for C7H6N2O4. Elemental analysis calculated: C, 43.65%; H, 4.38%; N, 16.83%. Found: C, 46.15%; H, 3.29%; N, 15.38%.Similarly, TFBFH was synthesized by TFB (0.1 mmol) and FH (0.15 mmol) in the same style of ampoule bottle. Yield: 14 mg, 56% for C7H6N2O2. Elemental analysis calculated: C, 52.37%; H, 5.76%; N, 19.61%. Found C, 56.00%; H, 4.00%; N, 18.67%.
2.2 Preparation of COFs of TpAzine and TFBAzine
An ampoule bottle measuring 10 × 8 mm (o.d. × i.d.) was charged with hydrazine hydrate (7.5 mg, 0.15 mmol) and Tp (21 mg, 0.10 mmol), 500 μL of anhydrous dioxane and 500 μL of mesitylene, and then immersed in an ultrasonic bath for 15 min. After sonication for 30 min, 0.3 mL of aqueous HAc (6 M) was added. The bottle was rapidly frozen at 77 K (liquid N2 bath), evacuated to an internal pressure of below 5 Pa and sealed by flame. The length of the tube sealed was reduced to 10–15 cm. The reaction was heated to 120 °C for 72 h yielding a red solid, which was isolated by filtration and washed with anhydrous DMF and THF. The resulting powder was immersed in anhydrous THF for 24 h and dried under vacuum at 100 °C overnight. Yield: 14 mg, 49% for C9H6N3O3.Similarly, TFBAzine was synthesized by TFB (0.1 mmol) and hydrazine hydrate (0.15 mmol) in the same style of ampoule bottle. Yield: 10 mg, 43% for C9H6N3.
2.3 Stability experiment of TpFH and TFBFH
The stability of TpFH and TFBFH was investigated by dissolution experiments. Three portions of the materials (10 mg for each) were immersed in 10 mL of 0.1 M HCl, boiling water and 0.1 M NaOH solutions, respectively, for 7 days. Then, the mixture was centrifuged and washed with anhydrous THF overnight. The product was dried at 100 °C for PXRD analysis.2.4 Characterization
Powder X-ray diffraction (XRD) patterns were recorded at room temperature with a PANalytical X’Pert Pro multipurpose diffractometer using Cu Kα (α = 1.54) radiation at 40 kV and 40 mA, with a scan speed of 1° min−1 and a step size of 0.02°.Fourier-transform infrared spectroscopy (FT-IR) characterization was carried out using a FT-IR spectrometer using KBr pellets (Nicolet, USA).
Solid-state 13C NMR was recorded on an Advance III-500 MHz spectrometer (Bruker, Switzerland). Carbon chemical shifts are expressed in parts per million (δ scale).
Thermogravimetric analysis from 20–800 °C was carried out on a STA449F3. Thermogravimetric Analyzer (Netzsch, Germany) in a nitrogen atmosphere with a heating rate of 10 °C min−1.
Helium ion microscopy (HIM) images were taken using a helium microscope (Zeiss, Germany), equipped with helium gas field as the ion source. The samples were prepared simply by putting COFs on a clean piece of silicon wafer.
Transmission electron microscopy (TEM) images were obtained by JEM-2000 EX (JEOL, Japan) at an accelerating voltage of 200 kV. The samples were prepared for TEM analysis by drop casting the samples (dispersed in ethanol) on copper grids (TED PELLA, INC.).
Nitrogen adsorption/desorption measurements were performed using a liquid nitrogen bath (77 K) on a Micrometrics surface area analyzer (Micrometrics, USA). The samples were outgassed under vacuum for at least 8 h at 120 °C before measurement. Surface area was calculated using the Brunauer–Emmett–Teller (BET) model. The pore size distributions were calculated using the non-localized density functional theory (NLDFT) method.
Molecular modeling of the COF was carried out using Materials Studio (8.0) (Accelrys Inc.). The initial structure was geometry optimized using the MS Forcite molecular dynamics module (Universal force fields, Ewald Summations).
We performed Pawley refinement to optimize the lattice parameters iteratively until the Rwp value converges. The pseudo-Voigt profile function was used for whole profile fitting and Berrar–Baldinozzi function was used for asymmetry correction during the refinement processes.
2.5 Batch adsorption experiments
The adsorption properties of the COF for metal ions were tested under non-competitive conditions by immersing COF into a solution containing one type of metal ion.Adsorption isotherms were conducted at 298 K. A 10 mg sample of dried COF was placed in a series of flasks containing different initial concentrations of each metal ion. Each flask was agitated at 150 rpm for 24 h. The filtrate was collected for the determination of the metal ion concentrations. The amount of metal ion adsorbed was calculated according to eqn (1). The adsorption isotherm of the metal ion was obtained by plotting Qe against Ce:
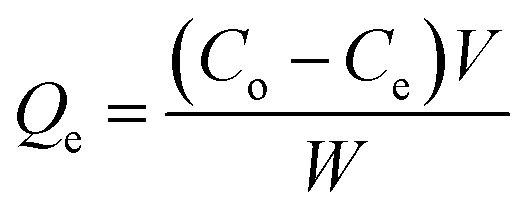 | (1) |
A 10 mg sample of dried TpFH was placed in a series of flasks containing 50 mL of each metal ion solution (2 mmol L−1). Each flask was collected for the determination of the metal ion concentration. The kinetic curve was obtained by plotting the adsorption capacities of the metal ions vs. the adsorption time. The experimental results were fitted with the pseudo-second-order kinetic model using the equation:
| t/qt = (k2qe2)−1 + t/qe | (2) |
A 5 mg sample of dried TpFH was placed in a flask containing 50 mL aqueous solution of Cu(II), Pb(II), Cd(II), Cr(III) and Ni(II) (10 mg L−1). This flask was agitated at 150 rpm for 24 h. The filtrate was collected for the determination of the metal ion concentrations.
One measure of a sorbent’s affinity for a target metal ion is the distribution coefficient (Kd). The equation is defined as:
| Kd = (Ci − Cf)V/Cfm | (3) |
2.6 Gas adsorption experiments
CO2 adsorption measurements were performed at 289 K using a IGA-100 volumetric adsorption analyzer (Micrometrics, Britain). The samples of 80 mg COFs were degassed under vacuum for a minimum 8 h at 120 °C before measurement. Ultrahigh purity gas was used at 298 K/1.0 bar for all measurements and the free volume was measured using helium.3. Results and discussion
3.1 Synthesis, characterization and stability analysis of COFs
In an effort to build flexible linkage covalent materials, our strategy is to choose the building block with a specific functional group which can produce strong interaction forces in the 2D COFs. FH, as a prototype of these building blocks, was selected to certify our hypothesis. Synthesis of TpFH and TFBFH was carried out via Schiff base reaction between FH and Tp or TFB using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. For comparison, we also fabricated two azine-based COFs, TpAzine and TFBAzine, via Schiff base reaction between hydrazine and Tp or TFB according to previous reports with a little modification.27,28
:2 molar ratio. For comparison, we also fabricated two azine-based COFs, TpAzine and TFBAzine, via Schiff base reaction between hydrazine and Tp or TFB according to previous reports with a little modification.27,28
Fig. 1 shows the scheme for construction of 2D FH-based COFs which have the same structure skeleton as azine-based COFs, TpAzine and TFBAzine. Fluffy powders, red TpFH and yellow TFBFH, were isolated from the reaction mixture by centrifugation and washed with dioxane, N,N-dimethylformamide (DMF) and tetrahydrofuran (THF) in sequence.
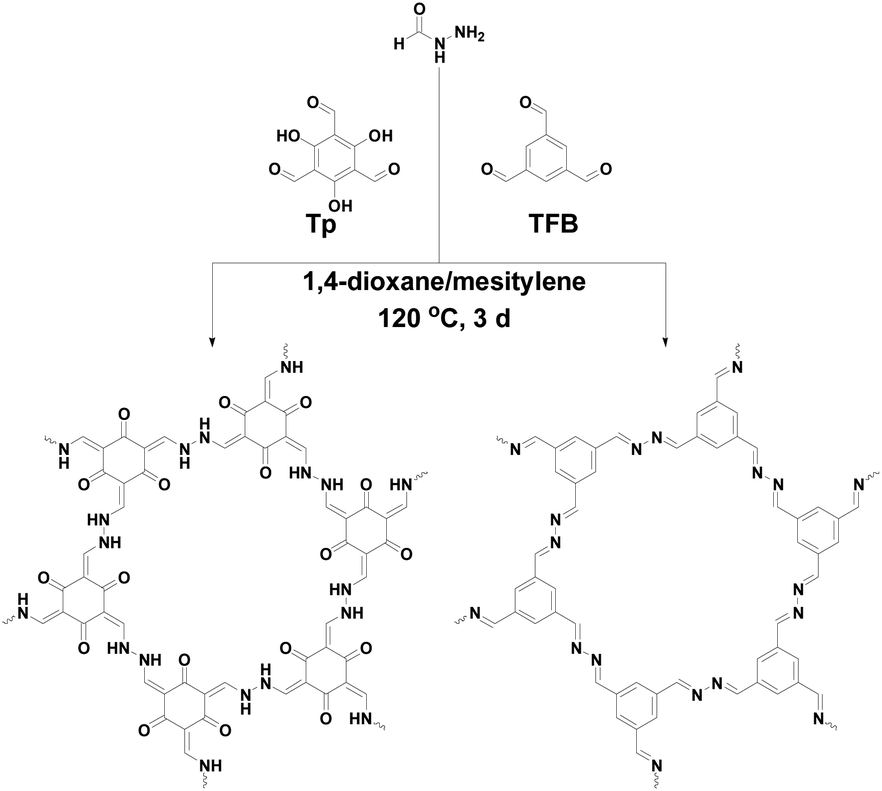 | ||
| Fig. 1 Schematic of the construction of COFs. | ||
The atomic-level construction of two FH-based COFs was ascertained by FT-IR (Fig. S1 and S2†). FT-IR spectra indicated the successful synthesis of COFs, as the characteristic –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands (1640, 2891 cm−1 for Tp, and 1696, 2868 cm−1 for TFB) of the two aldehyde monomers, and –N–H stretching band (∼3320 cm−1) of FH were absent. Two peaks at 1593 and 1285 cm−1 (Fig. S1a†) affirmed the presence of –CC (nonaromatic) and –C–N bonds of TpFH. Similarly, Fig. S2a† shows the imine bond (–CN) stretching at 1628 cm−1, indicating the successful construction of TFBFH. The broad peak at ∼3500 cm−1 emerges in Fig. S1a and S2a,† indicating the presence of hydrogen bonding in both TpFH and TFBFH. Four reference compounds – N,N′-hydrazinebis(salicylideneimine) (salicylaldehyde reacts with hydrazine hydrate, SAAzine), 1,2-di(benzylidene)hydrazine (benzaldehyde reacts with hydrazine hydrate, BAAzine), N′-(2-hydroxybenzylidene)formohydrazide (salicylaldehyde reacts with formic hydrazide, SAFH) and 2-(benzylidene)-1-methylhydrazide (benzaldehyde reacts with formic hydrazide, BAFH) – were also synthesized. The FT-IR spectra of SAAzine and SAFH are shown in Fig. S3a and c,† both of them exhibited almost the same characteristic peaks, indicating that FH was decomposed into azine in the process of TpFH synthesis. An imine bond (–CN) stretching peak could be observed in both Fig. S3b and d,† while the –CO characteristic stretching band of BAFH at 1680 cm−1 indicated that aldehyde groups still existed in the model reference of TFBFH.
O stretching bands (1640, 2891 cm−1 for Tp, and 1696, 2868 cm−1 for TFB) of the two aldehyde monomers, and –N–H stretching band (∼3320 cm−1) of FH were absent. Two peaks at 1593 and 1285 cm−1 (Fig. S1a†) affirmed the presence of –CC (nonaromatic) and –C–N bonds of TpFH. Similarly, Fig. S2a† shows the imine bond (–CN) stretching at 1628 cm−1, indicating the successful construction of TFBFH. The broad peak at ∼3500 cm−1 emerges in Fig. S1a and S2a,† indicating the presence of hydrogen bonding in both TpFH and TFBFH. Four reference compounds – N,N′-hydrazinebis(salicylideneimine) (salicylaldehyde reacts with hydrazine hydrate, SAAzine), 1,2-di(benzylidene)hydrazine (benzaldehyde reacts with hydrazine hydrate, BAAzine), N′-(2-hydroxybenzylidene)formohydrazide (salicylaldehyde reacts with formic hydrazide, SAFH) and 2-(benzylidene)-1-methylhydrazide (benzaldehyde reacts with formic hydrazide, BAFH) – were also synthesized. The FT-IR spectra of SAAzine and SAFH are shown in Fig. S3a and c,† both of them exhibited almost the same characteristic peaks, indicating that FH was decomposed into azine in the process of TpFH synthesis. An imine bond (–CN) stretching peak could be observed in both Fig. S3b and d,† while the –CO characteristic stretching band of BAFH at 1680 cm−1 indicated that aldehyde groups still existed in the model reference of TFBFH.
Two COFs were further characterized by solid-state 13C NMR. As shown in Fig. 2a, the resonances at 104 and 99 ppm were assigned to the exocyclic carbons (CC) in the keto-form and carbons on the benzene ring in the enol-form, respectively, and both of them belong to the structure of TpFH. In addition, the resonance signal at 181 and 164 ppm were assigned to the carbonyl carbons (CO) of the quaternary carbons (ring) and carbons of C–OH, respectively. Correspondingly, the signal at 163 ppm in Fig. 2b indicates the formation of –CN– in TFBFH, which overlaps with the peak of the CO carbons of aldehyde in FH uninvolved in the COF synthesis, and the signals at 134 and 125 ppm could correspond to the carbon atoms of the benzene ring on the framework.
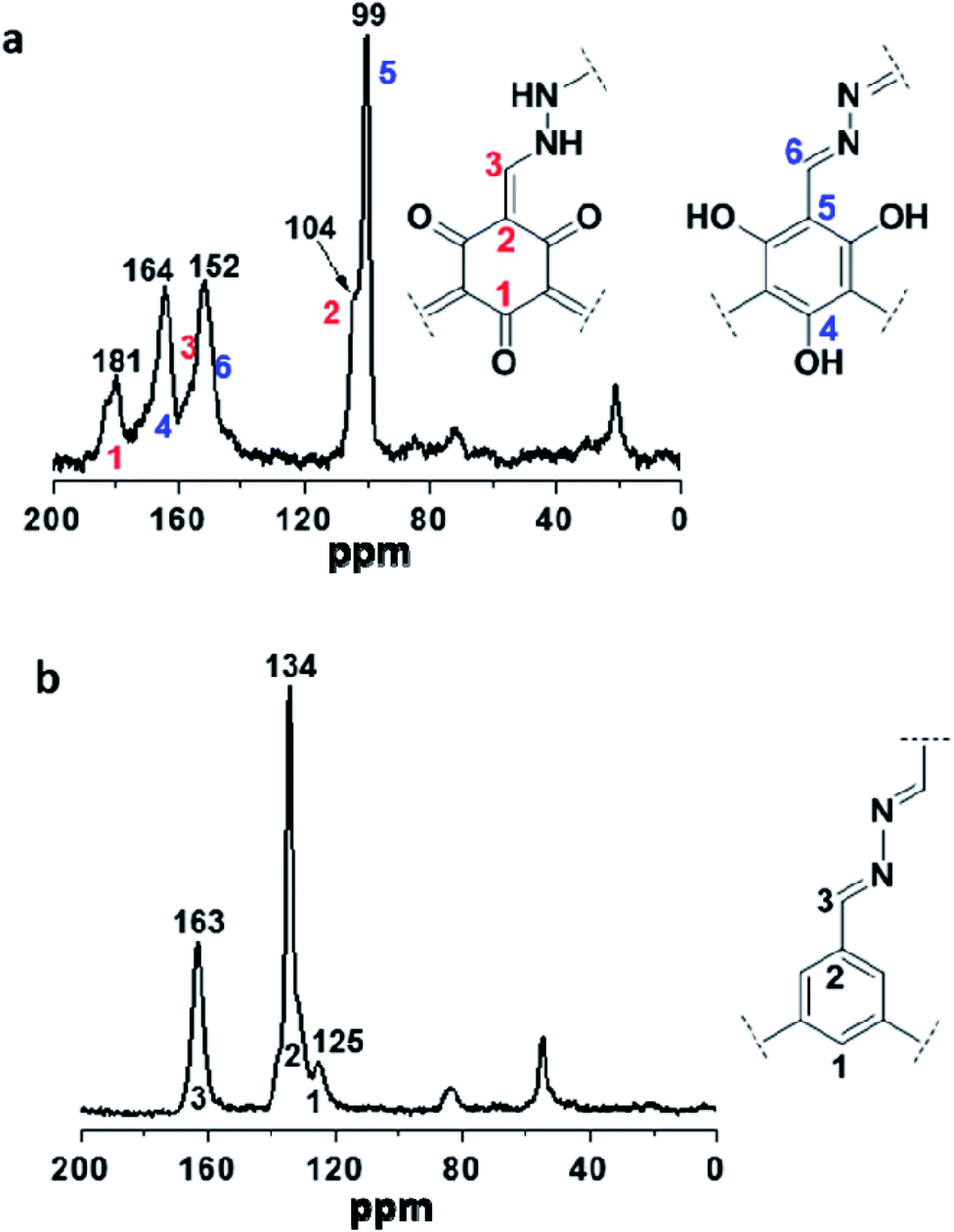 | ||
| Fig. 2 13C NMR spectra of (a) TpFH and (b) TFBFH. | ||
The liquid-state NMR spectra of the reference compounds are shown in Fig. S4† (1H spectra) and Fig. S5† (13C spectra). The absence of the peaks at 10 ppm in Fig. S4d–g,† and at 192 ppm in Fig. S5d–g† show the complete consumption of aromatic aldehyde. In detail, except for the characteristic peaks of the benzene ring in the 1H spectra of SAAzine (Fig. S4d†) and BAAzine (Fig. S4e†), two peaks (Fig. S4d†) at 9.0 and 11.1 ppm corresponded to the hydrogen of the Schiff-base and phenolic hydroxyl of SAAzine respectively, and the peak (Fig. S4e†) at 8.7 ppm corresponded to the hydrogen of the Schiff-base of BAAzine. Similarly, except for the characteristic peaks of the benzene ring in the 13C spectra of SAAzine (Fig. S5d†) and BAAzine (Fig. S5e†), two peaks (Fig. S5d†) at 159 and 163 ppm belonged to the carbons of the Schiff-base and phenolic hydroxyl of SAAzine, respectively, and the peak (Fig. S5e†) at 161 ppm belonged to the carbons of the Schiff-base of BAAzine.
It is obvious that the number and chemical shift of the characteristic peaks in SAAzine (Fig. S4d and S5d†) and SAFH (Fig. S4f and S5f†) were almost the same. The 1H and 13C spectra similarity of SAFH and SAAzine further illustrated the complete decomposition of FH in the conditions of COFs synthesis. Compared with the 1H and 13C spectra of BAAzine (Fig. S4e and S5e†) and BAFH (Fig. S4g and S5g†), the hydrogen signal at 8.0 ppm (Fig. S5e†) and carbon signal at 165 ppm (Fig. S5g†) indicated the retained existence of the aldehyde group of FH, which is the same as the result of FT-IR. In addition, in Fig. S5g,† the presence of the carbon signal peak of the Schiff-base at 145 ppm indicated there were two kinds of compounds in the BA and FH condensation reaction. As a result, it was deduced that the FH decomposed in the formation of COFs. The reaction mechanism was proposed, and the possible routes are shown in Fig. S6.† First, FH was decomposed into hydrazine and further reacted with Tp or TFB to form COFs. Second, FH reacted with Tp or TFB to form intermediates, which were consumed to form COFs. According to the above-mentioned results, the latter was more credible to interpret the formation of FH-based COFs.
Helium ion microscopy (HIM) and transmission electron microscopy (TEM) images clearly show the new style COFs predominantly assemble into well-defined structures with a hollow nature and smooth surface. As shown in Fig. 3a and c, the HIM images showed that TpFH and TFBFH comprise a large number of interconnected hollow microtubes with a relatively uniform size. In general, most microtubes have flower-like aggregation, and the hollow interior could be clearly observed through partially broken walls. TEM images showed the main feature of hollow tubular COFs with a light regime at the center of each tube and a well-defined dark area contrast at the tube wall (Fig. 3b and d). The inner diameter of the microtubes was found to be 20 nm, and the outer diameter was 50 nm for TpFH (Fig. S7a†). In the case of TFBFH, the hollow was much broader, about 50 nm inner diameter and 100 nm outer diameter (Fig. S7b†). Additionally, there are individual tubes and fragments, possibly caused by mechanical separation in the course of sample preparation. As shown in Fig. S8,† both hydrazine-series COFs (TpAzine and TFBAzine) had flower-like morphology, without hollow tube-like characteristics, which was obviously different from the morphology of the FH-series COFs.
 | ||
| Fig. 3 (a and c) HIM and (b and d) TEM images of (a and b) TpFH and (c and d) TFBFH showing a hollow nanorod morphology. | ||
In order to explore the effect of reaction conditions on the evolution of the crystallinity and surface area, various solvents systems including N,N-dimethylacetamide (DMAc), 1,2-dichlorobenzene (o-DCB), DMF and dimethyl sulfoxide (DMSO), as well as binary dioxane/mesitylene and DMAc/o-DCB, were used to synthesize the COFs. Through observing the PXRD patterns of TpFH and TFBFH, the peak intensities were obviously distinct (Fig. S9†). When DMAc, o-DCB or binary DMAc/o-DCB were selected as the solvents for synthesis of TpFH, the peak intensity of PXRD almost remained constant (Fig. S9a†), and the specific surface area was 568, 435 and 633 m2 g−1, respectively (Fig. S10†). The resulting TpFH material from using DMSO possessed poor crystallinity and a small specific surface area of 16 m2 g−1, however, using DMF, the TpFH exhibited a PXRD pattern with higher peak intensity and specific surface area of 350 m2 g−1. When o-DCB or DMF, and other solvents, were used to fabricate TFBFH, the resulting materials demonstrated a strong PXRD pattern (Fig. S9b†), but their specific surface areas were less than 84 m2 g−1 (Fig. S11†). As shown in Fig. S12,† HIM images of the synthesized COFs using DMF and DMSO indicated a tree-like morphology without a hollow tubular structure. As a result, these solvents were not well suited to the synthesis of TpFH and TFBFH. The possible reason for this phenomenon was the distinction of crystal growth rate in various solvents.29 In detail, based on the different polarity of various organic solvents, it is certain that the solubility of building blocks in different solvents is quite different. With good solubility solvent, the crystal growth rate is faster than with poor solubility solvents, which leads to a lack of the repeat assembly and repair cycles of the microcrystal.
After optimizing the fabrication conditions, the PXRD patterns of TpFH and TFBFH were measured and are shown in Fig. 4. Meanwhile, the experimental PXRD patterns of three precursors (Tp, TFB and FH) are given in Fig. S13,† and their peaks were remarkably different from those of the synthesized COFs. Good crystallinity with a highly intense peak at ∼6.90° (±0.2, 2θ) with d spacing of 12.80 Å for as-synthesized TpFH and TFBFH appear owing to the strong reflection from the 100 plane diffraction. Other peaks at 12.08, 20.77, 27.26° (TpFH) and 12.06, 18.35, 26.67° (TFBFH) correspond to 110120 and 001 plane diffractions. The d-spacing value at the 001 plane is mainly due to π–π stacking between vertically stacked successive layers. A value of 3.32 ± ∼0.04 Å was obtained for both COFs, which is in good agreement with the simulated slipped-AA stacking model. In addition, enol–keto tautomerism equilibrium of TpFH was beneficial for the decomposition of aldehydes and construction of AA eclipsed structures. Nevertheless, based on the relatively slow decomposition and reaction rate of FH in the reaction system of TFB and FH, TFBFH COF was inclined to adopt an AA and ABC mixture stacking model, which can be observed from the relatively higher signal intensity ratio of 18.35/6.90° and 12.06/6.90° in Fig. 4e. In addition, the PXRD pattern characteristic peaks of TpFH and TFBFH were almost the same as those of TpAzine (Fig. S13d†) and TFBAzine (Fig. S13e†), respectively.
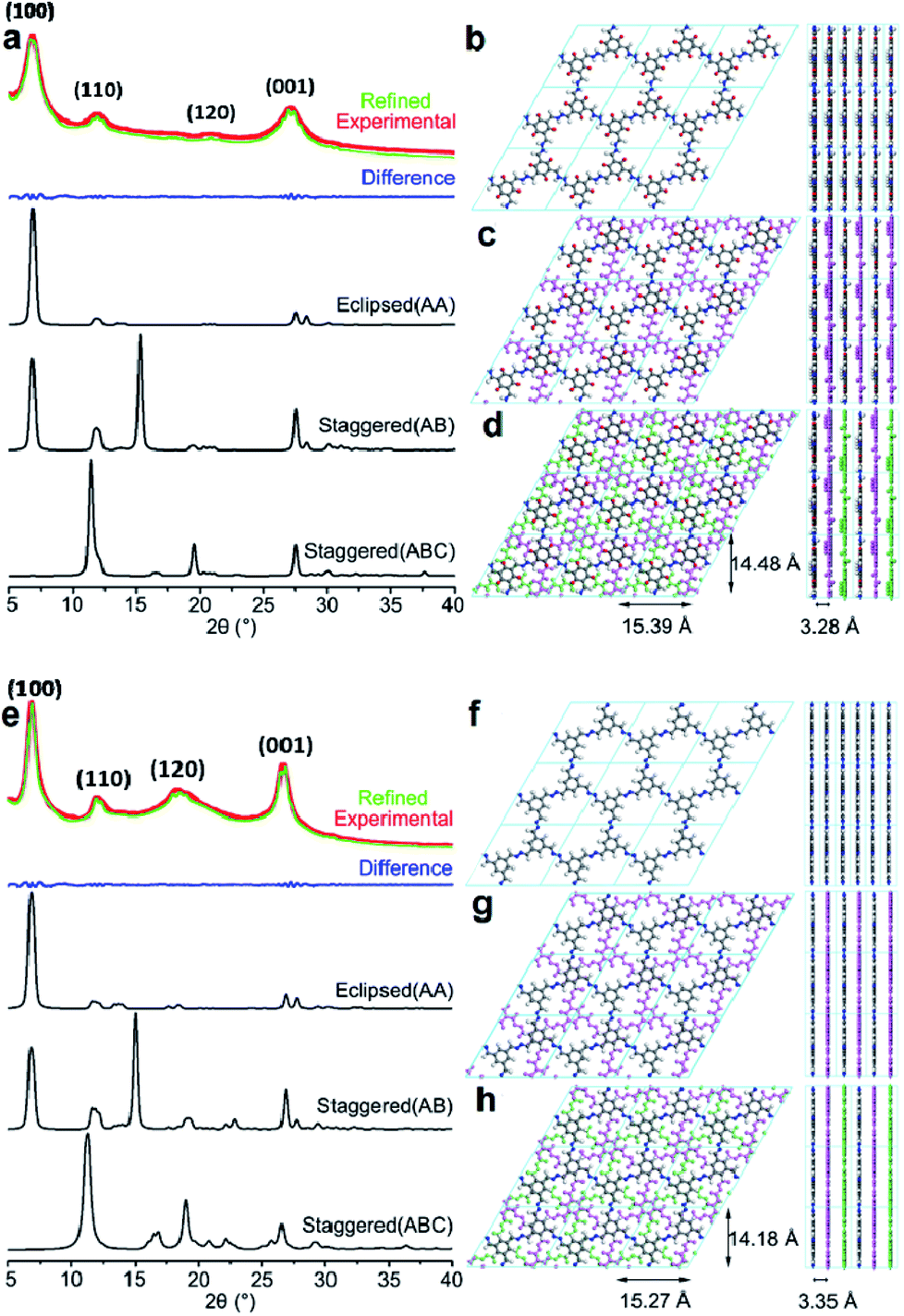 | ||
| Fig. 4 The experimental PXRD patterns compared with the simulated patterns of (a) TpFH and (e) TFBFH. Space-filling eclipsed stacking model of (b) TpFH and (f) TFBFH. Space-filling staggered stacking model of (c and d) TpFH and (g and h) TFBFH. Carbon (black), nitrogen (blue), oxygen (red) and hydrogen (white). | ||
The strong peak at ∼6.90° emerged, indicating that two kinds of FH-series and hydrazine-series COFs possessed the same size of imine linker in the skeleton. These results strongly demonstrated that the FH-series COFs were constructed via the Schiff-base condensation between FH and Tp (or TFB). Particularly, the relative peak intensities of 26.0/6.90° in the FH-series COFs were higher than those of the hydrazine-series COFs, which was possibly attributed to the aldehyde group decomposition of FH. The decomposition process would be influential in the direction of the phenyl rings in the COF framework, and reduce the reaction rates and increase stacking interactions between adjacent COF layers. The Pawley-refined PXRD profiles matched the experimental PXRD patterns very well (Rwp = 3.60%, Rp = 2.84% for TpFH and Rwp = 3.26%, Rp = 2.53% for TFBFH). Comparison of the experimental and the calculated PXRD patterns indicated that the stacking structures of these two COFs were mainly eclipsed arrangements. The unit cell parameters using the P1 space group for TpFH (Table S1†) with a = 15.39 Å, b = 3.28 Å, c = 14.48 Å, α = γ = 90°, β = 120° and TFBFH (Table S2†) with a = 15.27 Å, b = 3.35 Å c = 14.18 Å, α = γ = 90°, β = 120°, respectively. The possible eclipsed (AA model, Fig. 4b for TpFH and Fig. 4f for TFBFH) and staggered structures (including two different models, AB and ABC, Fig. 4c and d for TpFH and Fig. 4g and h for TFBFH) of COFs are given.
Nitrogen sorption isotherms were measured after activation of samples at 77 K under high vacuum for 12 h to estimate the permanent porosity of as-synthesized COFs (Fig. 5a and b). The specific surface areas of the two materials were calculated to be 708 (TpFH, Fig. S14a†) and 888 m2 g−1 (TFBFH, Fig. S14c†) using the Brunauer–Emmett–Teller (BET) model, whereas using the Langmuir model, they were 953 m2 g−1 for TpFH (Fig. S14b†) and 1150 m2 g−1 for TFBFH (Fig. S14d†). Using the common BET model, the specific surface areas of the hydrazine-series COFs were calculated to be 617 (TpAzine, Fig. S15a†) and 472 m2 g−1 (TFBAzine, Fig. S15b†). The presence and decomposition of aldehyde groups on the edge of FH reduces the chemical reaction rate and dominates the direction and location of imine linkers in the skeleton, enhancing the COF specific surface area. In addition, the intramolecular hydrogen bonds formed between the H atom of primary amine and the oxygen atom is relatively stabilized.30 At P/P0 = 0.99, the total pore volumes of the two materials were evaluated to be 0.87 and 0.99 cm3 g−1, respectively. A micropore size distribution (0.54–1.16 nm) for TpFH (Fig. 5c) was obtained using the nonlocal density functional theory (NLDFT) model. The same model was fitted to the isotherm of TFBFH to estimate its pore size with a narrow distribution in 0.93–1.18 nm (Fig. 5d).
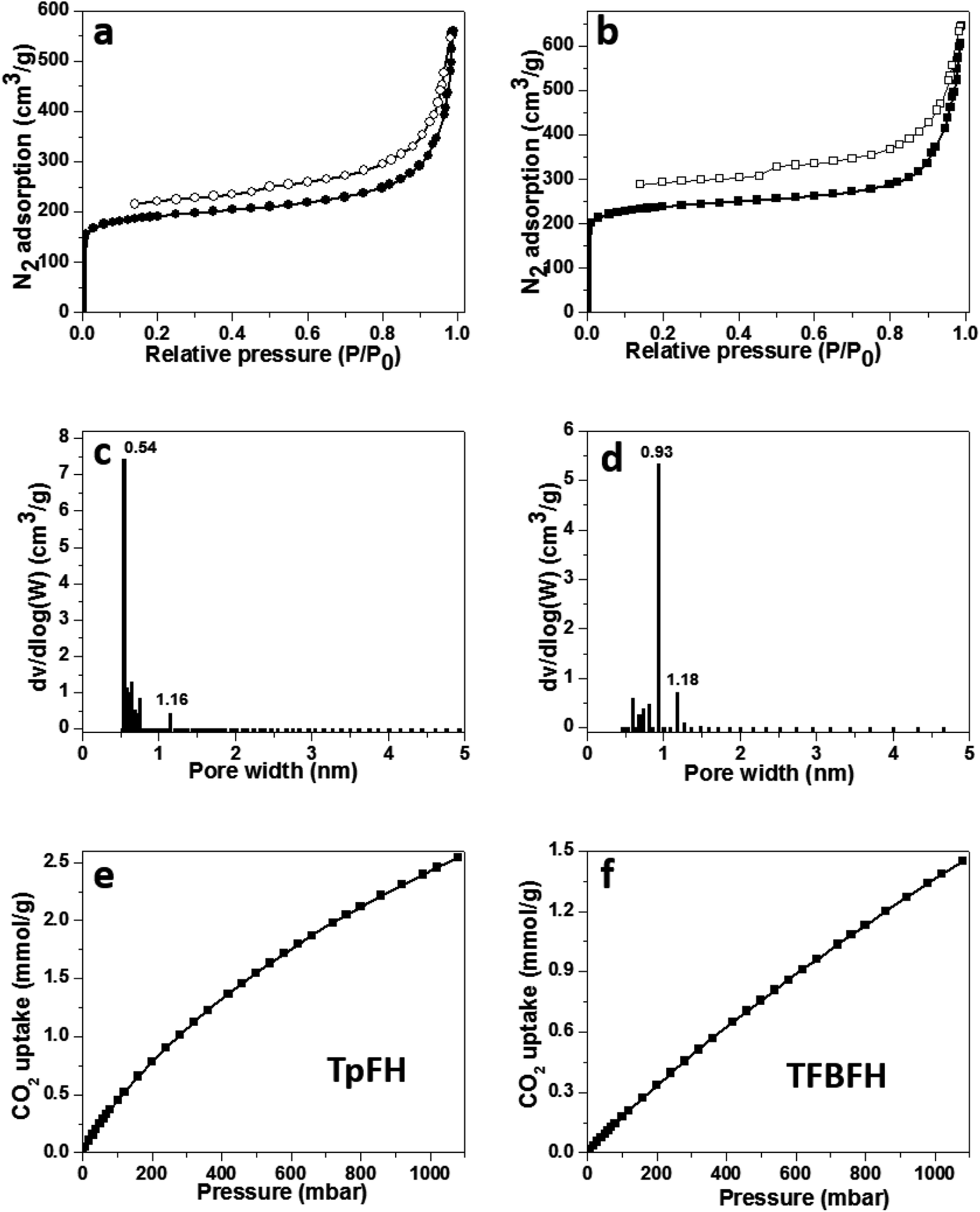 | ||
| Fig. 5 (a and b) N2 adsorption isotherm (77 K), (c and d) pore size distribution profiles calculated according to the NLDFT model of (a and c) TpFH and (b and d) TFBFH. Carbon dioxide isotherms of (e) TpFH and (f) TFBFH. The isotherms were collected at 298 K. | ||
The micropore size was directly correlated to the dimensions and geometry of the precursor molecules.31 Owing to the combustion of fossil fuels, CO2 emissions are known to be primarily responsible for abnormal climate change, rising sea levels, and an irreversible increase in the acidity of the oceans, resulting in adverse impacts on the ecology and environment. These issues have inspired the pursuit of state-of-the-art CO2 capture technology and materials.32 There are many reports indicating that microporous materials have excellent potential for storing gas molecules.33 Thus, the microporous nature and high surface area of the as-synthesized COFs prompted us to study their CO2 sorption properties. A more relevant measurement for post-combustion CO2 capture is the sorption isotherm collected at a temperature of 298 K, since this is close to the conditions required for post-combustion capture.34 Low pressure CO2 uptake of COFs was measured. TpFH could uptake 2.5 mmol g−1 CO2 (Fig. 5e), while TFBFH could uptake 1.6 mmol g−1 (Fig. 5f) at 298 K/1.0 bar. The capture capacities of two COFs surpass those of the published hydrazine-series COFs, TpAzine (2.4 mmol g−1 at 298 K) and TFBAzine (1.5 mmol g−1 at 298 K), but are lower than those of other porous polymers such as COF TpPa-1 (ref. 9) (3.57 mmol g−1), COF-6 (ref. 35) (3.84 mmol g−1) and a pyrene-based benzimidazole-linked nanofiber BILP-10 (ref. 36) (4.02 mmol g−1). The capture ability of CO2 should be related to the specific functional groups and uniform pore size distribution in materials.
Stability represents an important parameter to evaluate the COFs for widespread application. The resulting hollow-shape COFs were treated at different pH environments for 7 days, including boiling water, HCl (pH = 1) and NaOH (pH = 13) aqueous solutions. TpFH was stable in boiling water and HCl, as evident from the PXRD patterns not showing a distinct difference in the peak position (Fig. S16a†). Furthermore, it was persuasive that the crystallinity remained under different harsh conditions, even after immersing in strongly alkaline solution, three peaks were also retained, compared with Fig. 4a. Due to the backward keto-to-enol conversion, the crystallinity of TpFH shows a small distinction under alkaline conditions.37 As shown in Fig. S16b,† the PXRD of TFBFH was changed only a small amount when compared to the pristine TFBFH in Fig. 4e, demonstrating the retention of the framework structure of TFBFH. Because these COFs are composed of carbon–carbon and carbon–hydrogen bonds, they are thermally robust and chemically stable.38 It should be highlighted that TpFH after treatment with alkaline solution shows a weight loss of 30%, whereas the mass of TFBFH is almost retained. Fig. S17† shows the results of thermogravimetric analysis. Both TpFH and TFBFH in N2 atmosphere exhibited almost an identical decomposition temperature up to ∼300 °C, showing the high thermal stability of the two COFs; the initial weight loss continued up to 86 °C and was attributed to the loss of physically adsorbed water molecules in TpFH.
3.2 The mechanism of hollow tube formation
To investigate the mechanism of hollow tube formation, TpFH or TFBFH were synthesized with the same reaction mixture in several ampoule tubes and quenched at different time intervals 3, 12, 36 and 72 h. As shown in Fig. 6, the crystallites of COFs initially emerged at 3 h as random agglomerates, and the nanorods with hollow structures can be observed after 12 h, indicating the growth of these microcrystals. The hollow microtubes became longer and aggregated together with relatively uniform size and dimensions within 36 h, but they were not remarkably changed until 72 h (Fig. 3). The Kirkendall effect39,40 and Ostwald ripening41 are the popular theories of self-templated hollow structure formation, while another mechanism that the microtubes originate from rolled-up crystallite sheets42 is uncommon and has rarely been documented. In this research, self-assembly and Ostwald ripening processes were simultaneous during the formation of the hollow microstructure, which is supported by the PXRD of TpFH and TFBFH synthesized at different time intervals (Fig. S18†). There were only a few crystals formed at the beginning of 3 h. After 12 h, the peak intensities of PXRD obviously increased. Interestingly, comparing the PXRD patterns of crystals formed at 36 h and 72 h (Fig. 4), the peak intensity ratio between the 100 to 001 planes of the sample were increased, demonstrating that each individual layer utilizes the strong π–π stacking interaction to interact with other layers to further extend along the Z direction compared to X and Y directions. As mentioned above, the hollow structure of COFs was obtained within a shorter time (12 h).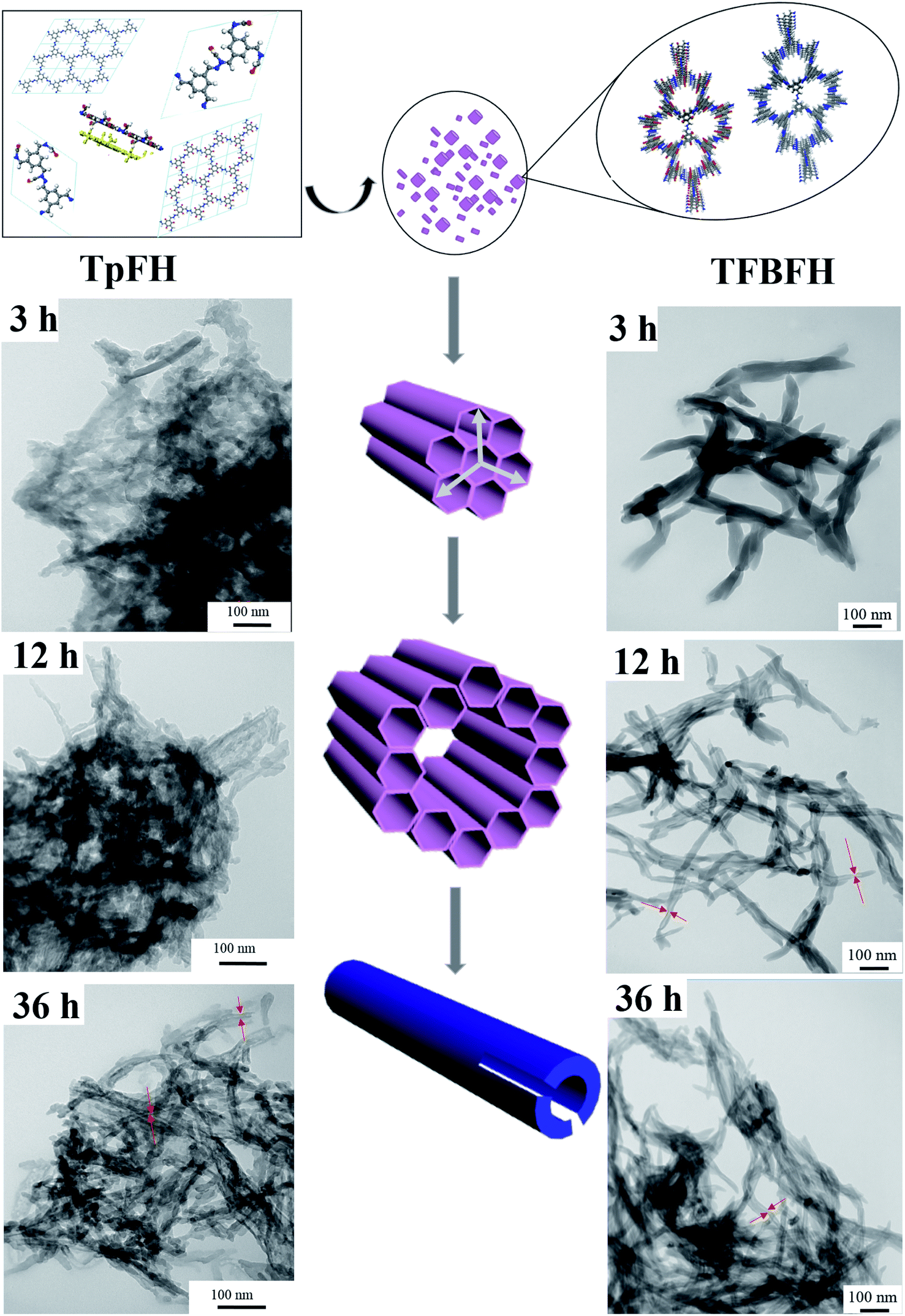 | ||
| Fig. 6 Proposed mechanism for hollow tubular formation and SEM images of TpFH and TFBFH at different time intervals. | ||
As a result, it is assumed that self-assembly of the crystallites happened at the beginning of the crystal formation, which is presumably induced by van der Waals interactions through intramolecular hydrogen bonds. These small crystallites further redissolve and migrate from the inner core to the outer walls of the boundary in order to reduce the surface energy of the materials, as shown in reported supramolecular nanotubes43 and microporous polymers.44 To assess the adsorption ability of these materials, the adsorption isotherm for copper was plotted after measuring the metal ions in aqueous solutions with different concentrations. As shown in Fig. 7a, a maximal adsorption capacity of Cu(II) was calculated to be 162 mg g−1 for TpFH, while another was only 8 mg g−1 for TFBFH (Fig. 7b). The outstanding capacity should be ascribed to the plentiful nitrogen and oxygen covering the skeleton of TpFH, acting to trap Cu(II). Then, a Langmuir adsorption model was selected to investigate the adsorption mechanism of copper on TpFH. It could be clearly observed that the equilibrium adsorption isotherm well matched the Langmuir model with a high correlation coefficient (RL > 0.998). This result demonstrates that the adsorption process would be monolayer. As presented in Fig. 7c, a pseudo-second-order kinetic model was used to fit the kinetic data of copper and interpret the mechanism of the adsorption process. It can be proved by fitting the experimental data with the pseudo-second-order kinetic model using eqn (2) (ESI).† A high correlation coefficient (0.999) indicated that a pseudo-second-order kinetic model was fitted for the adsorption of Cu(II) onto the surface of TpFH. As a result, the adsorption process was chemical adsorption between COF and the metal ion. In order to assess the ability of the COF material to remove the Cu(II) under realistic conditions, the adsorption selectivity experiment was carried out with mixed metal ions aqueous solution including Cu(II), Pb(II), Cd(II), Cr(III) and Ni(II) ions. Fig. 7d shows the capture efficiency of different heavy metal ions, in detail, the adsorbent exhibited more than 50% removal efficiency for Cu(II) but less than 10% for Pb(II), Cd(II), Cr(III) and Ni(II) ions. The relatively higher removal ability of the adsorbent to Cu(II) ions might be attributed to the fact that the electron donor is N and O in the adsorption process. In addition, the distribution coefficient (Kd) was used to evaluate the affinity of adsorbent to adsorbate, and a Kd value of 1.0 × 104 mL g−1 is generally regarded as showing excellent affinity for copper.45 A Kd value of 4.16 × 106 mL g−1 was acquired under conditions of 10 ppm copper after reaching equilibrium at room temperature (Fig. S19†), indicating the strong affinity of copper with TpFH.
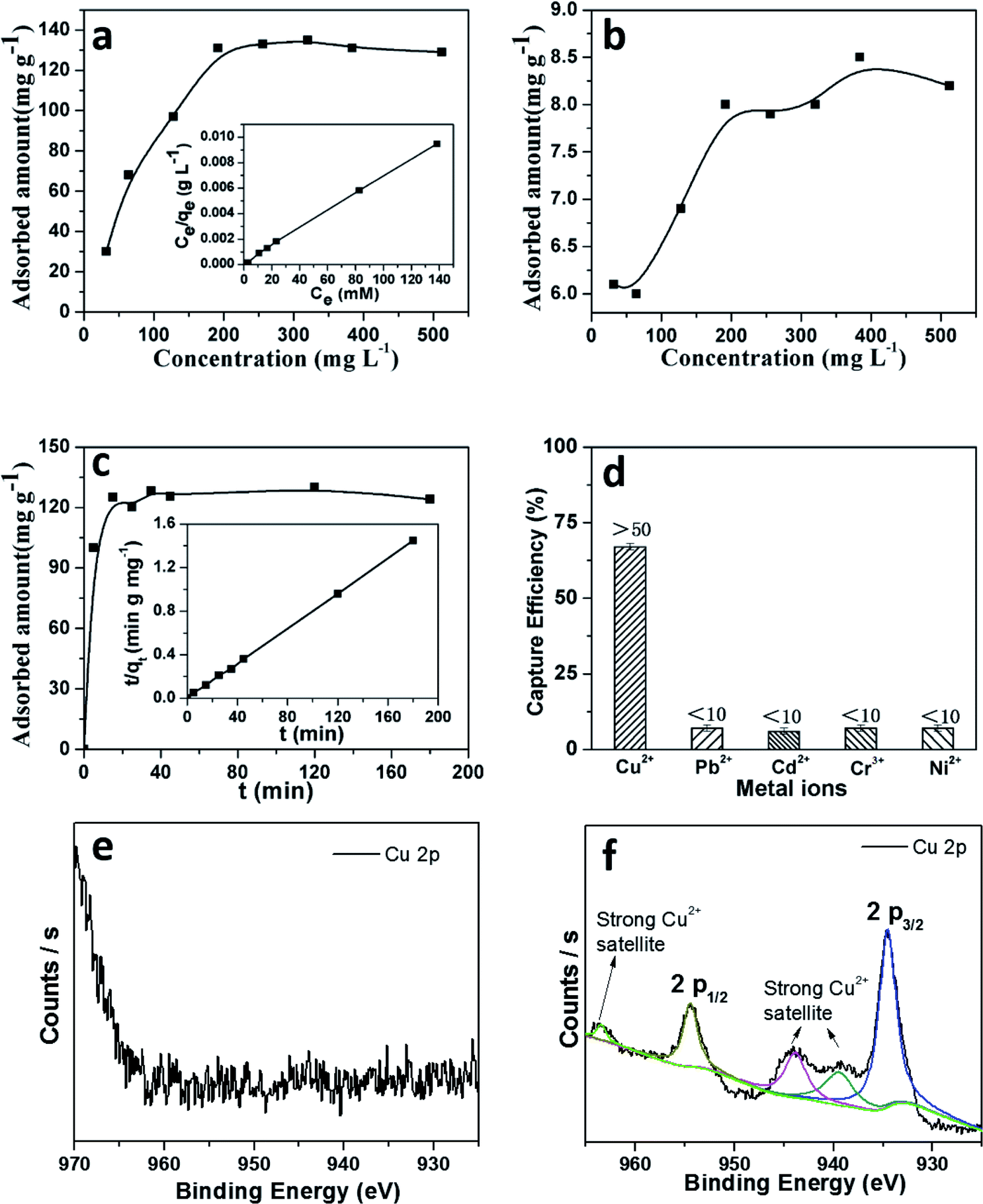 | ||
| Fig. 7 (a) Cu(II) adsorption isotherm of TpFH at room temperature. Inset shows the linear regression with the Langmuir adsorption model by fitting the isotherm. (b) Cu(II) adsorption isotherm of TFBFH at room temperature. (c) Adsorption kinetics of Cu(II). Inset presents the pseudo-second-order kinetic plot for the adsorption process of TpFH to Cu(II). (d) Capture efficiency in removing different metal ions. High-resolution XPS spectra of (e) TpFH and (f) TpFH–Cu. | ||
3.3 Adsorption of metal ions
Two-dimensional materials with abundant nitrogen and π–π conjugation may provide more possibility for interaction with copper ions.The adsorption mechanism of heavy metal ions using the TpFH adsorbent relied on a synergistic combination of electrostatic (between metal ion and functional groups such as amide, amino and carbonyl groups) and coordination interactions. On the basis of the hard and soft acids and bases (HSAB) theory, the adsorption may be dominated by coordination interactions between the adsorbate and adsorbent. XPS survey wide scan spectra of TpFH and TpFH–Cu were obtained. As displayed in Fig. S20a,† there were three characteristics peaks for TpFH – C 1s (285 eV), N 1s (399 eV) and O 1s (532 eV) – while Cu 2p (934 eV) and Cl 2p (198 eV) peaks for TpFH–Cu are clearly observed in Fig. S20b† besides the three above-mentioned peaks, indicating that the copper ions were adsorbed on the TpFH via coordination interactions.46 Furthermore, the data for elemental content and composition of the TpFH and TpFH–Cu samples based upon analysis of the survey spectra are shown in Table S3.† High-resolution XPS spectra of TpFH and TpFH–Cu were also given. There were no characteristic peaks for copper ions in Fig. 7e, whereas in Fig. 7f, the binding energies in 932.5 and 952.2 eV correspond to satellite peaks of Cu 2p3/2 and Cu 2p1/2, regarded as the Cu 2p peak, indicating the presence of Cu(II).47 The satellite peaks were related to the interactions between Cu(II) and functional groups (–NH, –CO).48 Furthermore, Cu LMM spectrum of characteristic TpFH–Cu XPS band of Cu(II) can be clearly seen in Fig. S20c and d.† A more detailed chemical analysis on XPS was obtained from the C 1s spectrum illustrated in Fig. S21a and b.† There are slight shifts of three characteristic peaks, manifesting that the electron cloud density of different C atoms was changed along with the adsorption process. N 1 s (Fig. S21c and d†) and O 1 s (Fig. S21e and f†) XPS spectra of TpFH and TpFH–Cu further confirmed that the metallic ions could be adsorbed on TpFH via electrostatic interactions and coordination interactions. In addition, after saturation adsorption of Cu(II) with TpFH, the pH of Cu(II) solution was measured. We found that the acidity was increased from the initial pH 4.5 to the final pH 1.5, further indicating the adsorbent chelation with metal along with deprotonation.49
4. Conclusions
In summary, we have developed an efficient and green strategy to synthesize chemically stable COFs, TpFH and TFBFH, with a hollow microtubular structure using aldehyde-containing monomer, FH, as the building block. The aldehyde group of FH would decompose during the formation of FH-based COFs, which possess the same structure as the hydrazine-series COFs. The formation process of the hollow structure was investigated, the FH-series COFs synthesized at different reaction times displayed various morphologies. Furthermore, the morphologies of COFs strictly depended on the reaction solvents. All of the COFs were resistant to moisture and highly stable in acidic or basic aqueous solutions. Remarkable stability and high density of N, O elements aligned in periodic arrays on the walls of TpFH, facilitating their interactions and high affinities toward copper ions. The decomposition of FH in the process of COF synthesis was the basis of the high specific surface area and crystallinity of the as-synthesized COFs. Therefore, it is expected that such a simple method could be explored for the construction of various novel COFs in the future.Author contributions
Ya Li: methodology, investigation, writing-original draft, formal analysis. Chang Wang: investigation, formal analysis. Shujuan Ma: investigation. Junwen Xu: investigation. Xiaowei Li: validation. Yinmao Wei: investigation, formal analysis. Junjie Ou: conceptualization, resources, funding acquisition, supervision, writing-review and editingConflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support is gratefully acknowledged from the National Natural Sciences Foundation of China (No. 21974137), the CAS-Weigao Research & Development Program ([2017]-009) and the Innovation Program (DICP I202005) of Science and Research from the DICP, CAS to J. Ou.References
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O’Keeffe, A. J. Matzger and O. M. Yaghi, Porous, crystalline, covalent organic frameworks, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Function-led design of new porous materials, Science, 2015, 348, 8075 CrossRef PubMed.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, Metalation of a mesoporous three-dimensional covalent organic framework, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed.
- H. Xu, J. Gao and D. Jiang, Stable, crystalline, porous, covalent organic frameworks as a platform for chiral organocatalysts, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- Y. Du, H. Yang, J. M. Whiteley, S. Wan, Y. Jin, S. H. Lee and W. Zhang, Ionic covalent organic frameworks with spiroborate linkage, Angew. Chem., Int. Ed., 2016, 55, 1737–1741 CrossRef CAS PubMed.
- M. R. Rao, Y. Fang, S. De Feyter and D. F. Perepichka, Conjugated covalent organic frameworks via michael addition-elimination, J. Am. Chem. Soc., 2017, 139, 2421–2427 CrossRef CAS PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Porous, covalent triazine-based frameworks prepared by ionothermal synthesis, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS PubMed.
- F. J. Uribe-Romo, C. J. Doonan, H. Furukawa, K. Oisaki and O. M. Yaghi, Crystalline covalent organic frameworks with hydrazone linkages, J. Am. Chem. Soc., 2011, 133, 11478–11481 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, Chemically stable multilayered covalent organic nanosheets from covalent organic frameworks via mechanical delamination, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, A belt-shaped, blue luminescent, and semiconducting covalent organic framework, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- Q. Fang, Z. Zhuang, S. Gu, R. B. Kaspar, J. Zheng, J. Wang, S. Qiu and Y. Yan, Designed synthesis of large-pore crystalline polyimide covalent organic frameworks, Nat. Commun., 2014, 5, 4503 CrossRef PubMed.
- W. Huang, Y. Jiang, X. Li, X. Li, J. Wang, Q. Wu and X. Liu, Solvothermal synthesis of microporous, crystalline covalent organic framework nanofibers and their colorimetric nanohybrid structures, ACS Appl. Mater. Interfaces, 2013, 5, 8845–8849 CrossRef CAS PubMed.
- A. Halder, S. Kandambeth, B. P. Biswal, G. Kaur, N. C. Roy, M. Addicoat, J. K. Salunke, S. Banerjee, K. Vanka, T. Heine, S. Verma and R. Banerjee, Decoding the morphological diversity in two dimensional crystalline porous polymers by core planarity modulation, Angew. Chem., Int. Ed., 2016, 55, 7806–7810 CrossRef CAS PubMed.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Self-templated chemically stable hollow spherical covalent organic framework, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. Mar Ramos, Post-synthetic modification of covalent organic frameworks, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- P. J. Waller, S. J. Lyle, T. M. Osborn Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, Chemical conversion of linkages in covalent organic frameworks, J. Am. Chem. Soc., 2016, 138, 15519–15522 CrossRef CAS PubMed.
- X. W. Lou, L. A. Archer and Z. Yang, Hollow micro-/nanostructures: synthesis and applications, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- P. Peng, X. M. Lin, Y. Liu, A. S. Filatov, D. Li, V. R. Stamenkovic, D. Yang, V. B. Prakapenka, A. Lei and E. V. Shevchenko, Binary transition-metal oxide hollow nanoparticles for oxygen evolution reaction, ACS Appl. Mater. Interfaces, 2018, 10, 24715–24724 CrossRef CAS PubMed.
- J. G. Railsback, A. C. Johnston-Peck, J. W. Wang and J. B. Tracy, Size-dependent nanoscale Kirkendall effect during the oxidation of nickel nanoparticles, ACS Nano, 2010, 4, 1913–1920 CrossRef CAS PubMed.
- G. D. Fu, J. P. Zhao, Y. M. Sun, E. T. Kang and K. G. Neoh, Conductive hollow nanospheres of polyaniline via surface-initiated atom transfer radical polymerization of 4-vinylaniline and oxidative graft copolymerization of aniline, Macromolecules, 2007, 40, 2271–2275 CrossRef CAS.
- C. Wang, H. Su, Y. Ma, D. Yang, Y. Dong, D. Li, L. Wang, Y. Liu and J. Zhang, Coordination polymers-derived three-dimensional hierarchical CoFe2O4 hollow spheres as high-performance lithium ion storage, ACS Appl. Mater. Interfaces, 2018, 10, 28679–28685 CrossRef CAS PubMed.
- J. Hu, M. Chen, X. Fang and L. Wu, Fabrication and application of inorganic hollow spheres, Chem. Soc. Rev., 2011, 40, 5472–5491 RSC.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Diaz Diaz and R. Banerjee, Targeted drug delivery in covalent organic nanosheets (CONs) via sequential postsynthetic modification, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef CAS PubMed.
- C. R. DeBlase, K. E. Silberstein, T. T. Truong, H. D. Abruna and W. R. Dichtel, Beta-ketoenamine-linked covalent organic frameworks capable of pseudocapacitive energy storage, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Enhancement of chemical stability and crystallinity in porphyrin-containing covalent organic frameworks by intramolecular hydrogen bonds, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS PubMed.
- C. Zhao, C. S. Diercks, C. Zhu, N. Hanikel, X. Pei and O. M. Yaghi, Urea-linked covalent organic frameworks, J. Am. Chem. Soc., 2018, 140, 16438–16441 CrossRef CAS PubMed.
- L. Stegbauer, M. W. Hahn, A. Jentys, G. Savasci, C. Ochsenfeld, J. A. Lercher and B. V. Lotsch, Tunable water and CO2 sorption properties in isostructural azine-based covalent organic frameworks through polarity engineering, Chem. Mater., 2015, 27, 7874–7881 CrossRef CAS.
- Z. Li, Y. Zhi, X. Feng, X. Ding, Y. Zou, X. Liu and Y. Mu, An azine-linked covalent organic framework: synthesis, characterization and efficient gas storage, Chem.-Eur. J., 2015, 21, 12079–12084 CrossRef CAS PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gandara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J. C. Olsen and A. Trabolsi, Viologen-based conjugated covalent organic networks via zincke reaction, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- S. Samdal and H. Mollendal, The structural and conformational properties of formic hydrazide (formylhydrazine) studied by microwave spectroscopy and quantum chemical calculations, J. Phys. Chem. A, 2003, 107, 8845–8850 CrossRef CAS.
- S. Zhang, Q. Yang, C. Wang, X. Luo, J. Kim, Z. Wang and Y. Yamauchi, Porous organic frameworks: advanced materials in analytical chemistry, Adv. Sci., 2018, 5, 1801116 CrossRef PubMed.
- J. Mahmood, S. J. Kim, H. J. Noh, S. M. Jung, I. Ahmad, F. Li, J. M. Seo and J. B. Baek, A robust 3D cage-like ultramicroporous network structure with high gas-uptake capacity, Angew. Chem., Int. Ed., 2018, 57, 3415–3420 CrossRef CAS PubMed.
- Y. Jin, B. A. Voss, R. D. Noble and W. Zhang, A shape-persistent organic molecular cage with high selectivity for the adsorption of CO2 over N2, Angew. Chem., Int. Ed., 2010, 49, 6348–6351 CrossRef CAS PubMed.
- R. Dawson, E. Stöckel, J. R. A. D. J. Holst and A. I. Cooper, Microporous organic polymers for carbon dioxide capture, Energy Environ. Sci., 2011, 4, 4239–4245 RSC.
- H. Furukawa and O. M. Yaghi, Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- M. G. Rabbani, A. K. Sekizkardes, O. M. El-Kadri, B. R. Kaafarani and H. M. El-Kaderi, Pyrene-directed growth of nanoporous benzimidazole-linked nanofibers and their application to selective CO2 capture and separation, J. Mater. Chem., 2012, 22, 25409–25417 RSC.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharu and R. Banerjee, Self-exfoliated guanidinium-based ionic covalent organic nanosheets (iCONs), J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, S. Kandambeth, R. Babarao, Y. Marathe, S. M. Kunjir and R. Banerjee, Phosphoric acid loaded azo (-N=N-) based covalent organic framework for proton conduction, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- A. Cabot, V. F. Puntes, E. Shevchenko, Y. Yin, L. Balcells, M. A. Marcus, S. M. Hughes and A. P. Alivisatos, Vacancy coalescence during oxidation of iron nanoparticles, J. Am. Chem. Soc., 2007, 129, 10358–10360 CrossRef CAS PubMed.
- Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Formation of hollow nanocrystals through the nanoscale Kirkendall Effect, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- J. Huo, L. Wang, E. Irran, H. Yu, J. Gao, D. Fan, B. Li, J. Wang, W. Ding, A. M. Amin, C. Li and L. Ma, Hollow ferrocenyl coordination polymer microspheres with micropores in shells prepared by Ostwald ripening, Angew. Chem., Int. Ed., 2010, 49, 9237–9241 CrossRef CAS PubMed.
- B. Gole, V. Stepanenko, S. Rager, M. Grune, D. D. Medina, T. Bein, F. Wurthner and F. Beuerle, Microtubular self-assembly of covalent organic frameworks, Angew. Chem., Int. Ed., 2018, 57, 846–850 CrossRef CAS PubMed.
- N. Kameta, H. Minamikawa and M. Masuda, Supramolecular organic nanotubes: how to utilize the inner nanospace and the outer space, Soft Matter, 2011, 7, 4539–4561 RSC.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Conjugated microporous poly(aryleneethynylene) networks, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Removal of heavy metals from aqueous systems with thiol functionalized superparamagnetic nanoparticles, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS PubMed.
- S. S. Zhang, Q. F. Niu, L. X. Lan and T. D. Li, Novel oligothiophene-phenylamine based Schiff base as a fluorescent chemosensor for the dual-channel detection of Hg2+ and Cu2+ with high sensitivity and selectivity, Sens. Actuators, B, 2017, 240, 793–800 CrossRef CAS.
- H. Mai, D. Zhang, L. Shi, T. Yan and H. Li, Highly active Ce1−xCuxO2 nanocomposite catalysts for the low temperature oxidation of CO, Appl. Surf. Sci., 2011, 257, 7551–7559 CrossRef CAS.
- L. Merí-Bofí, S. Royuela, F. Zamora, M. L. Ruiz-González, J. L. Segura, R. Muñoz-Olivas and M. J. Mancheño, Thiol grafted imine-based covalent organic frameworks for water remediation through selective removal of Hg(II), J. Mater. Chem. A, 2017, 5, 17973–17981 RSC.
- J. Li, S. Chen, G. Sheng, J. Hu, X. Tan and X. Wang, Effect of surfactants on Pb(II) adsorption from aqueous solutions using oxidized multiwall carbon nanotubes, Chem. Eng. J., 2011, 166, 551–558 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02104d |
| This journal is © The Royal Society of Chemistry 2021 |