
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreener and facile synthesis of Cu/ZnO catalysts for CO2 hydrogenation to methanol by urea hydrolysis of acetates†
Nat Phongprueksathat ab,
Atul Bansodeab,
Takashi Toyaocd and
Atsushi Urakawa*ab
ab,
Atul Bansodeab,
Takashi Toyaocd and
Atsushi Urakawa*ab
aCatalysis Engineering, Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, Netherlands. E-mail: A.Urakawa@tudelft.nl
bInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain
cInstitute for Catalysis, Hokkaido University, N-21, W-10, Sapporo 001-0021, Japan
dElements Strategy Initiative for Catalysis and Batteries, Kyoto University, Katsura, Kyoto 615-8520, Japan
First published on 20th April 2021
Abstract
Cu/ZnO-based catalysts for methanol synthesis by COx hydrogenation are widely prepared via co-precipitation of sodium carbonates and nitrate salts, which eventually produces a large amount of wastewater from the washing step to remove sodium (Na+) and/or nitrate (NO3−) residues. The step is inevitable since the remaining Na+ acts as a catalyst poison whereas leftover NO3− induces metal agglomeration during the calcination. In this study, sodium- and nitrate-free hydroxy-carbonate precursors were prepared via urea hydrolysis co-precipitation of acetate salt and compared with the case using nitrate salts. The Cu/ZnO catalysts derived from calcination of the washed and unwashed precursors show catalytic performance comparable to the commercial Cu/ZnO/Al2O3 catalyst in CO2 hydrogenation at 240–280 °C and 331 bar. By the combination of urea hydrolysis and the nitrate-free precipitants, the catalyst preparation is simpler with fewer steps, even without the need for a washing step and pH control, rendering the synthesis more sustainable.
Introduction
One of the strategies to reduce greenhouse gas emission and alleviate the impacts of the escalating global warming is carbon dioxide (CO2) conversion with green H2 (e.g. produced through water electrolysis sourced by renewable energies) into chemicals such as methane, formic acid, methanol, dimethyl ether, and methyl formate. Among those chemicals, methanol is positioned as the most versatile chemical feedstock and energy carrier towards a fossil-fuel-free economy, known as the “methanol economy”.1 The green methanol production from CO2 hydrogenation has been demonstrated successfully on a relatively large scale at the “George Olah Carbon Dioxide to Renewable Methanol Plant” in 2012 and it paves the way for sustainable recycling of CO2.2 On the other hand, the current synthesis processes for methanol synthesis catalysts are still far from eco-friendly. Most traditional synthesis processes inevitably produce contaminated wastewater, which requires extensive treatment before its release to the environment.3 This harmful effluent must be minimized and not released according to green chemistry principles as a key path for sustainable chemical synthesis in the 21st century.4Conventionally, the majority of industrial methanol synthesis catalysts (e.g. Cu/ZnO/Al2O3) have been prepared by co-precipitation of metal nitrate salts and NaCO3 precipitant,5 that contributes to a considerable amount of nitrate-containing wastewater from the washing process of the as-precipitated precursors (approximately 500 L kg−1 of catalyst (ESI†)). Washing off nitrate and sodium residues is crucial to prevent agglomeration of the active metal (Cu), its poisoning, and thus catalyst deactivation. Concerning residual nitrate anions, replacing Cu and Zn nitrates with other soluble inorganic salts such as respective chlorides and sulfates are detrimental to catalytic activity since Cl and S residues could act as poisons.6,7 An effective approach is the use of organic salts such as formates or acetates, avoiding the generation of nitrate-contaminated wastewater while forming active catalysts.7,8 Concerning the residual sodium cations, employing salts containing thermally decomposable cation, such as (NH4)HCO3 and (NH4)2CO3, as a precipitant allows eliminating the washing step of sodium cations, although an effective removal of anion, typically nitrate, by calcination in the gas stream is still required to achieve the maximum activity.9 Supercritical antisolvent process is a recent approach to avoid the use precipitant completely although a special equipment for high volume of supercritical CO2 required.10,11
Typically, a precipitant is added together with the metal nitrate precursor(s) dropwise and in a controlled manner to precisely regulate the pH of the solution and control the growth of catalyst precursor crystals. In this regard, hydrolysis of urea (NH2CONH2) is interesting and potentially advantageous because the precipitant, (NH4)2CO3, can be produced in situ in solution (eqn (1)).12
| NH2CONH2 + 2H2O → (NH4)2CO3 | (1) |
Similar to the co-precipitation using (NH4)2CO3 precipitant, the homogeneous alkalinization via urea hydrolysis of metal nitrate or chloride salts can yield sodium-free hydroxycarbonates, such as copper hydrozincite ((CuxZn1−x)5(OH)6(CO3)2, when x < 0.1), aurichalcite ((CuxZn1−x)5(OH)6(CO3)2, when x < 0.5), rosasite ((CuxZn1−x)2(OH)2(CO3), when 0.5 < x < 0.7), and zincian malachite ((CuxZn1−x)2(OH)2(CO3) when x > 0.7),12–14 in which Cu and Zn are closely located in the same crystalline structure.15,16 These hydroxycarbonates are essential for the formation of CuO–ZnO inter-dispersion during calcination, improving Cu–ZnO contact after reduction, and eventually producing more active catalysts than those obtained by the impregnation method.17,18 On the contrary, the catalysts prepared by urea hydrolysis usually possess higher crystallinity, smaller particle size, and more uniform size distribution than co-precipitation using conventional precipitants because of the gradient-free nature and less-fluctuating pH during the precipitation process thanks to the in situ precipitant (NH4)2CO3 formation (eqn (1)) whose concentration is regulated by the rate of hydrolysis influenced by the consumption of the precipitant in the solution. These features are beneficial to enhance the reproducibility of the complex and highly sensitive synthesis process, where precise semi-automatic synthesis equipment is generally required in the case of conventional co-precipitation.19 Moreover, its application can be readily transferred to industrial-scale process employing batch reactor.
In the past, Cu-based catalysts (e.g. Cu/ZnO and Cu/ZnO/Al2O3) prepared by urea hydrolysis of nitrate salts have been reported for steam reforming of methanol,20–28 water–gas shift reaction,28–32 selective hydrogenation,33–36 and liquid phase methanol synthesis from syngas.37–39 Most studies have shown higher copper surface area, stronger metal–support interaction, and better catalytic performance for CO2-related reaction than conventional co-precipitated catalysts. These properties should be highly beneficial for methanol synthesis catalysts.40,41 The major parameters reported to influence the synthesis are temperature, aging time, urea content, and precursors (metal salts) type,12 and they have been optimized in case of urea hydrolysis of nitrate salts. Such parameters, however, cannot be applied directly for urea hydrolysis of acetates due to the formation of different meta-stable/stable phases.8 Moreover, the washing remains crucial for nitrates-derived catalysts, and the influence of such a step has never been investigated in urea hydrolysis of both nitrates and acetates.
In this study, we aim at simplifying the synthesis procedure of Cu/ZnO catalyst and improving the quality of resulting material as methanol synthesis catalyst by urea hydrolysis of metal acetates to (i) better control the precipitation process, (ii) skip washing step of cation like Na+ and (iii) avoid the use of nitrates in the precipitated precursor to prevent agglomeration of active Cu species upon calcination. The focus of this work is given to optimize the precipitation temperature, urea-to-metal salt ratio, and Cu-to-Zn ratio using acetate salts.
Results and discussion
Influence of precipitation temperature
Temperature is one of the most critical parameters in solid synthesis by precipitation. Here, the optimum precipitation temperature was determined experimentally by correlating with the catalytic activity of the resulting catalyst. In literature, the optimum temperature for co-precipitation of the precursor yielding Cu/ZnO/Al2O3 catalyst for methanol synthesis is reported to be 60–70 °C.42 In the case of urea hydrolysis, however, the rate of urea hydrolysis (eqn (1)) is associated with precipitation temperature, and eventually determines the rate of alkilination (the rate OH− generation and consequently increasing pH) in the solution. The promoted nucleation rate from a rapid pH increase is beneficial for the formation of small particle size and high crystallinity of the as-precipitated precursor.14 The smaller Cu and ZnO particle sizes after calcination have been reported as increasing precipitating temperature and the optimal temperature is reported at 95 °C.14,23,43 The catalysts prepared at the same temperature using nitrate salts in this study possess comparable textural properties as reported in the aforementioned literature, as shown in Table 2. However, the temperature of 95 °C is not suitable with acetate salt since the catalytic activity obtained is inferior to that of 80 °C (Table 1). It should be noted that mostly methanol and carbon monoxide are detected under all conditions with only a trace amount (<1% selectivity) of other products (e.g. methane, methyl formate, and diethyl ether).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn = 1:1) prepared by urea hydrolysis of acetate at various U/M ratio at 70, 80, and 95 °C
:Zn = 1:1) prepared by urea hydrolysis of acetate at various U/M ratio at 70, 80, and 95 °C
Clearly, incomplete precipitation of Zn2+ is observed at 70 °C after 24 h of synthesis since the rate constant of urea hydrolysis is 4 times lower than at 80 °C and results in insufficient alkalization of the solution.44 As described in the phase diagrams of the Cu2+ + Zn2+ system, the Cu2+ would precipitate first due to the larger energy requirement for dehydration of aqueous Zn2+,14 which is directly related to the higher solubility of zinc acetate (0.43 g mL−1) than copper acetate (0.072 g mL−1). Therefore, it is still challenging to carry out urea hydrolysis at even lower temperatures e.g. 40 °C to obtain a superior zincian georgeite phase reported recently.8
As shown in the X-ray diffraction (XRD) patterns of the as-precipitated precursors (Fig. 1), the major component/phase obtained at 70, 80, and 95 °C is the aurichalcite phase. However, a large amount of CuO is precipitating at a temperature of 70 °C. The XRD patterns of calcined catalysts are analyzed using Rietveld refinement to estimate crystallite size and approximate phase composition (Table 1). The lower CuO content in the catalyst obtained at the synthesis temperature of 95 °C is likely associated with Cu leaching.31 On the other hand, higher CuO content in the catalyst obtained at 70 °C can be associated with the formation and decomposition of thermally unstable Cu2(OH)3(CH3COO)·H2O intermediate.45 It is likely that the incomplete precipitation of Zn2+ could limit the formation of the aurichalcite phase and allows the firstly precipitated copper intermediate to decompose. Based on these observations and also catalytic activity (Table 1), the synthesis temperature of 80 °C is concluded to be optimal and is used throughout this work.
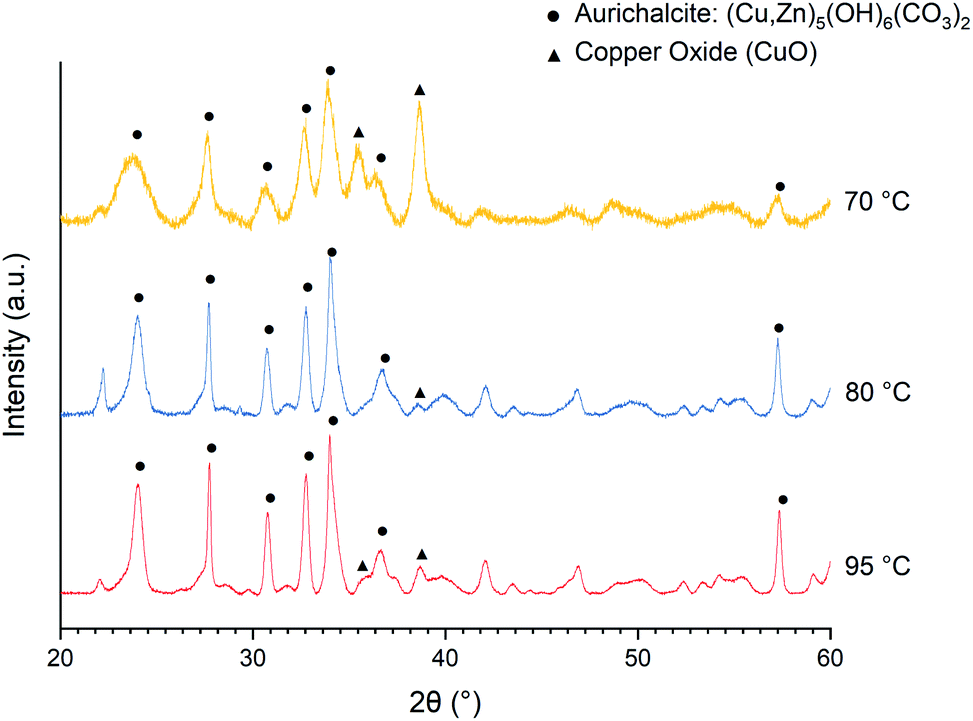 | ||
| Fig. 1 XRD patterns of the as-precipitated precursors for Cu/ZnO catalysts (Cu:Zn = 1:1) prepared by urea hydrolysis of acetate salts with urea to metal cations molar ratio (U/M) of 10 at 70, 80, and 95 °C. | ||
Influence of urea to metal ratio
The amount of urea used in co-precipitation is one of the key factors determining the alkalinity of the solution in urea hydrolysis (eqn (1)), thus impacting the precipitating time, the structure of as-precipitated precursors, and physical properties of the final catalyst. In early studies, an extremely excessive amount of urea had been used together with a diluted metal salts solution to obtain the aurichalcite structure.14,31,46 However, the excess of urea promotes the formation of copper ammonia complexes ([Cu(NH3)4(H2O)2]2+) that easily aggregate and in turn produce larger Cu particle size.9,37 The optimal urea concentration should be identified to yield a precursor containing both Cu and Zn at the optimal molar ratio with atomic dispersion and resulting in a highly active catalyst upon calcination.To study the influences of urea concentration, the relationship between catalytic activity and the urea to metal cations molar ratio (U/M ratio) was studied using both nitrate and acetate salts (Fig. 2). CO2 conversion and CH3OH selectivity at 260 °C and 331 bar increase significantly at a higher U/M ratio and reach a constant value for both nitrate- and acetate-derived catalysts. The catalysts synthesized at a higher U/M ratio exhibit a comparable catalytic activity and higher intrinsic activity than the highly active and optimized commercial Cu/ZnO/Al2O3 catalyst (also containing MgO promoter).
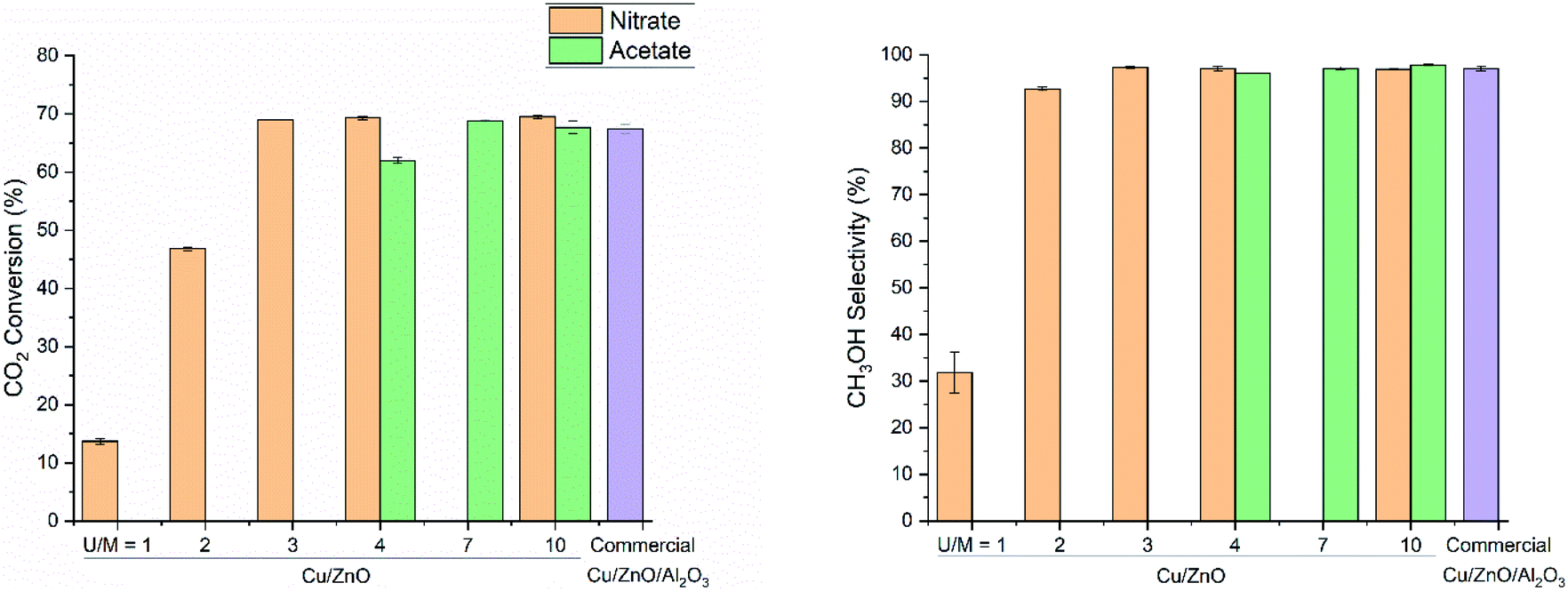 | ||
| Fig. 2 Influence of urea-to-metal molar ratio on the catalytic activity of Cu/ZnO catalyst (Cu:Zn = 1:1) derived from nitrate and acetate salts, and commercial Cu/ZnO/Al2O3 catalyst for CO2 hydrogenation to methanol. H2/CO2 = 3, T = 260 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | ||
Moreover, extraordinary catalytic performances are achieved by high pressure where the CO2 conversion and CH3OH selectivity are boosted by the enhanced reaction rate, thermodynamically favorable conditions, and surpassed chemical equilibrium due to in situ condensation of methanol and water.40,41,47,48 CO2 conversion and CH3OH selectivity obtained with Cu/ZnO catalysts under such conditions are by far the state-of-the-art.49 Nevertheless, the values of intrinsic activity are probably not representing a true intrinsic activity since the specific Cu surface area determined by N2O titration may not be identical to the surface area during the reaction (severe deactivation).
The inferior activity of catalysts prepared at low U/M is attributed to poorer physical properties of calcined catalysts such as lower BET surface area and larger crystallite size of CuO and ZnO (Table 2). The XRD patterns of as-precipitated precursors using the nitrate salts (Fig. S1†) indicate the formation of the gerhardtite phase (Cu2(OH)3(NO3)) at low U/M as the main phase. This phase was reported as an intermediate for aurichalcite and rosasite phases which were observed during precipitation at low pH.50 The presence of such a crystal phase containing Cu as only metal element should be avoided to obtain finely mixed CuO–ZnO after calcination. Too low alkalinity due to the little amount of urea likely induced incomplete precipitation of Zn2+ since it requires a higher pH value (pH 10.1) than Cu2+ (pH 8.1) due to significantly higher solubility of zinc nitrate (184 g mL−1) compare to copper nitrate (0.419 g mL−1).46
:Zn = 1:1) prepared by urea hydrolysis of nitrate and acetate salts at various U/M ratios
| Catalysts | Precipitation temperature (°C) | U/M ratio | BET surface area (m2 g−1) | Cu surface areaa (m2 g−1) | Average crystallite sizeb (nm) | Intrinsic activityc (mmolCH3OH mCu−2 h−1) | |
|---|---|---|---|---|---|---|---|
| CuO | ZnO | ||||||
| a Determined by N2O chemisorption.b Estimated by Rietveld refinement.c Based on methanol productivity at H2/CO2 = 3, T = 260 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | |||||||
| Cu/ZnO (nitrate) | 95 | 1 | 9 | — | 40.5 | 42.7 | — |
| 95 | 2 | 28 | — | 27.5 | 18.7 | — | |
| 95 | 3 | 74 | — | 6.2 | 7.8 | — | |
| 95 | 4 | 57 | — | 6.9 | 6.9 | — | |
| 95 | 10 | 64 | — | 5.1 | 7.1 | — | |
| 80 | 10 | 53 | 7 | 6.9 | 8.5 | 5.0 | |
| Cu/ZnO (acetate) | 80 | 4 | 56 | 11 | 8.5 | 5.1 | 2.8 |
| 80 | 7 | 54 | 19 | 8.7 | 8.8 | 1.8 | |
| 80 | 10 | 70 | 13 | 4.1 | 6.7 | 2.6 | |
| Commercial Cu/ZnO/Al2O3 | — | — | 102 | 19 | 6.3 | 4.2 | 1.8 |
When acetate salts are used, the XRD patterns of as-precipitated precursors (Fig. 3) show the mixture of aurichalcite and CuO phase at a low U/M ratio of 4. The surprising formation of CuO without calcination treatment is explained by the formation and decomposition of thermally unstable Cu2(OH)3(CH3COO)·H2O intermediate as reported by Jia et al.45 On the other hand, the undesired copper ammonia complex ([Cu(NH3)4(H2O)2]2+) is not observed at a high U/M ratio of 7 and 10 (Fig. 3). Therefore, the optimal U/M ratio for urea hydrolysis of acetate is found to be at least 7 up to 10.
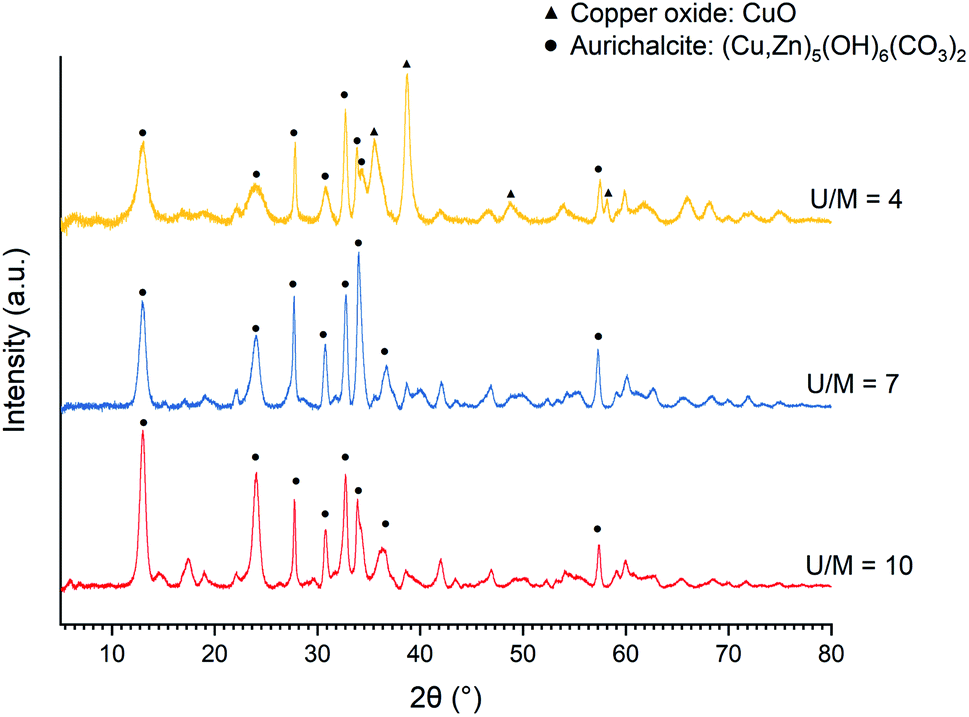 | ||
| Fig. 3 XRD patterns of the as-precipitated precursors of Cu/ZnO catalysts (Cu:Zn = 1:1) prepared by urea hydrolysis of acetate salts with various urea to metal molar ratios (U/M) of 4–7 at 80 °C. | ||
Influence of metal salts and washing step
To compare the influence of metal salts on as-precipitated precursors, the urea hydrolysis of nitrate and acetate salts is carried out at the same temperature (80 °C) and U/M ratio of 10. The pH evolution of the suspension of nitrate and acetate salts is measured as shown in Fig. S2 (ESI†). The nitrate and acetate solutions have different initial pH and progression suggesting the formation of different meta-stable phases. The overall pH of both nitrate and acetate suspension increases during urea hydrolysis and eventually reaches the same value of 6.5 after 24 h. It should be noted that the pH of 6.5–7 is commonly used for conventional co-precipitation where the alkaline solution is constantly added to maintain the pH value.8,9 A gradual increase in pH and simultaneous aging allows precipitation and active phase transformation to take place slowly, which improves crystallinity. However, there are sudden drops in pH during 2–8 h, which may indicate the crystallization of rosasite ((Cu,Zn)2(OH)2CO3) and aurichalcite ((Cu,Zn)5(OH)6(CO3)2).50,51 As shown by XRD patterns of as-precipitated precursors (Fig. S3, ESI†), only the aurichalcite phase is observed in the acetate-derived precursors, while the rosasite phase is abundant in the nitrate-derived precursors. It should be noted that the precursor structure is sensitive to the precipitation temperature; only aurichalcite phase is observed in the nitrate-derived precursors if prepared at 95 °C.Moreover, the platelet shape of aurichalcite phase and the needle-like shape of rosasite are confirmed by scanning electron microscope (SEM) (Fig. 4). The needle-like structure of rosasite has been proposed to give a superior catalyst after calcination than an unfavorably larger platelet structure of aurichalcite.16 However, it is noticeable that the crystallite sizes of the platelet aurichalcite derived from acetate salts (Fig. 4B) are apparently much smaller than needle-like rosasite derived from nitrate salts (Fig. 4A). The smaller crystallite size of the aurichalcite phase is evident from the XRD patterns (Fig. S3†). The less thickness (smaller size) of the crystallite needle is reported to be one of the critical properties that affect Cu particle size since it makes Cu more accessible to reactant gas upon decomposition.7,52 Therefore, the smaller crystallite size of aurichalcite may explain the favorable textural properties of the acetate-derived catalysts after calcination in terms of metallic copper and total surface area than those of the nitrate-derived one (Table 3).
 | ||
| Fig. 4 Representative SEM images of washed as-precipitated precursors for CuO/ZnO (Cu:Zn = 1:1) derived from urea hydrolysis of (A) nitrate and (B) acetate salts. | ||
:Zn = 1:1) derived from urea hydrolysis of nitrate and acetate salts at 80 °C for 24 h with the urea-to-metal molar ratio of 10 and commercial Cu/ZnO/Al2O3 catalyst
| Catalyst | BET surface area (m2 g−1) | Cu surface areaa (m2 g−1) | Crystallite sizeb (nm) | Intrinsic activityd (mmolCH3OH/mCu−2 h−1) | |
|---|---|---|---|---|---|
| CuO | ZnO | ||||
| a Determined by N2O chemisorption.b Estimated by Rietveld refinement.c Determined by ICP elemental analysis.40d Based on methanol productivity at H2/CO2 = 3, T = 280 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | |||||
| Nitrate-unwashed Cu/ZnO | 25 | 4 | 19.2 | 47.9 | 3.9 |
| Nitrate-washed Cu/ZnO | 53 | 7 | 6.9 | 8.5 | 5.1 |
| Acetate-unwashed Cu/ZnO | 70 | 16 | 4.8 | 6.6 | 2.3 |
| Acetate-washed Cu/ZnO | 70 | 13 | 4.1 | 6.7 | 2.9 |
| Commercial Cu/ZnO/Al2O3 (64/25/10 wt%)c | 102 | 19 | 6.3 | 4.2 | 1.9 |
Scanning transmission electron microscopy (STEM) with high-angle annular dark field (HAADF) and X-ray energy dispersive spectroscopy (EDS) mapping images of the catalyst obtained after calcination of the washed acetate-derived precursor are shown in Fig. 5. The images confirm the inter-dispersion of ca. 8–10 nm size CuO and ZnO comparable to crystallite size as obtained from the Rietveld refinement (Table 3). Similar homogeneous inter-dispersion of CuO, ZnO, and Al2O3 particles is observed in the EDS mapping from commercial Cu/ZnO/Al2O3 (Fig. 6). On the other hand, the EDS mapping of the washed nitrate-derived catalyst (Fig. 7) shows inter-dispersion Cu and Zn components but with larger CuO and ZnO particle sizes of 30–100 nm, which results in a relatively lower copper surface area than the acetate-derived catalyst (Table 3).
 | ||
| Fig. 5 Representative HAADF-STEM images (A–C) and EDS analysis (D–F) of fresh CuO/ZnO (Cu:Zn = 1:1) catalyst derived from urea hydrolysis of acetate salts (washed). | ||
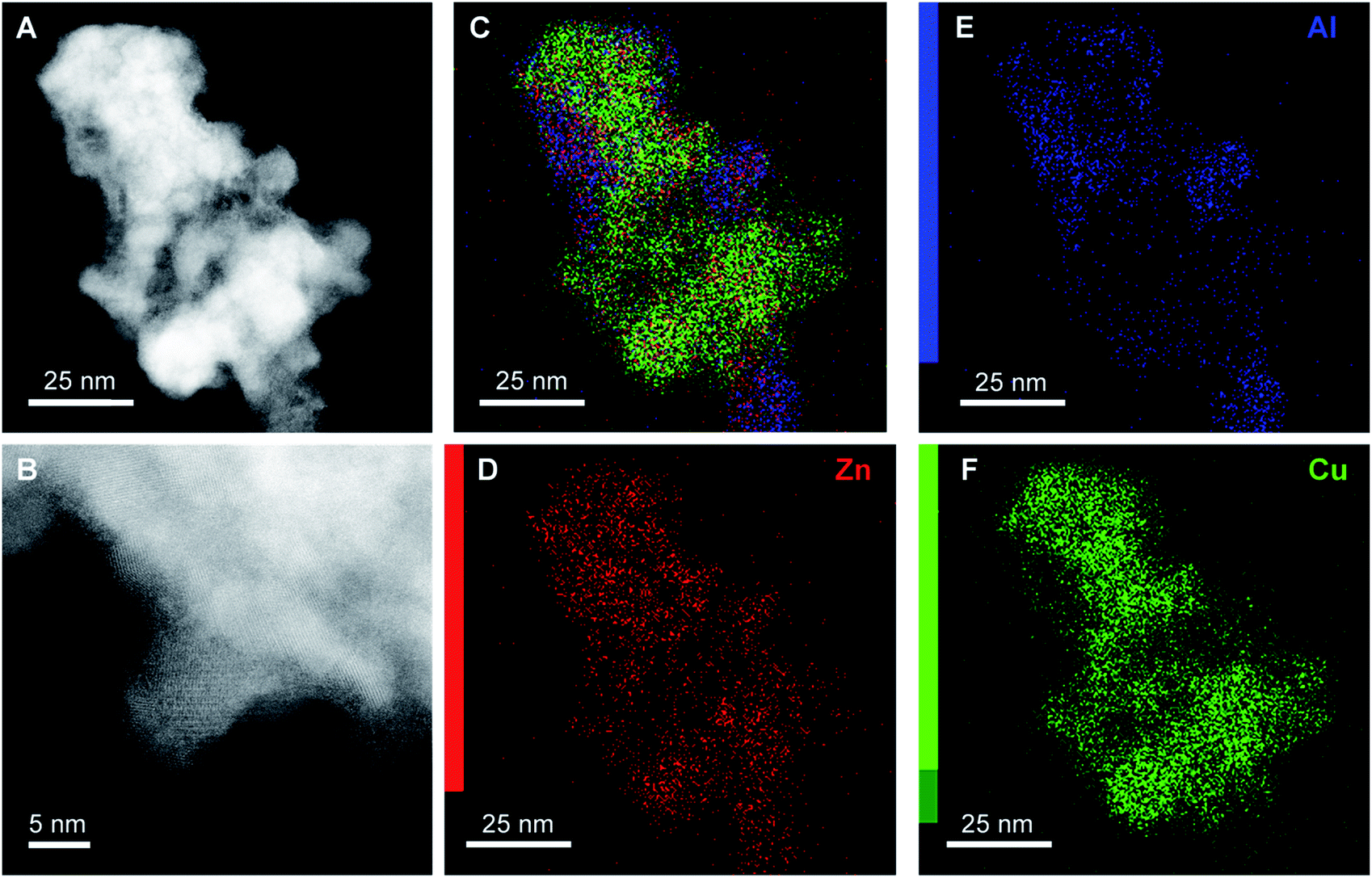 | ||
| Fig. 6 Representative HAADF-STEM images (A and B) and EDS analysis (C–F) of fresh commercial CuO/ZnO/Al2O3 catalyst. | ||
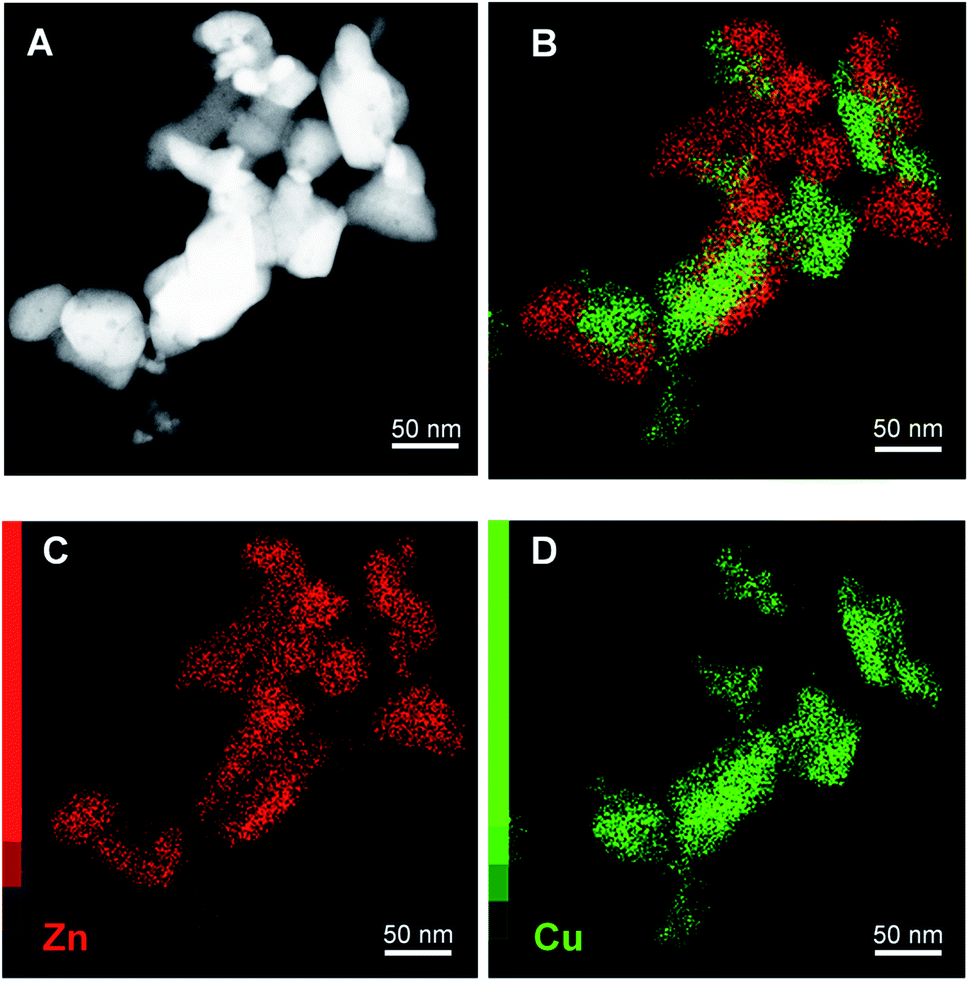 | ||
| Fig. 7 Representative HAADF-STEM images (A) and EDS analysis (B–D) of fresh CuO/ZnO (Cu:Zn = 1:1) catalyst derived from urea hydrolysis of nitrate salts (washed). | ||
The washing procedure did not affect XRD patterns (Fig. S3, ESI†) and those of the washed and unwashed precursors are identical. On the other hand, the presence of unwashed residues is evident from the thermogravimetric analysis with derivative thermogravimetry (TGA/DTG) results (Fig. S4, ESI†). The total mass loss of as-precipitated precursors is 28–30% for the nitrate-derived catalysts and 26–28% for the acetate-derived catalysts. The unwashed residues account for ca. 2.5% of extra mass loss. The major mass losses at 350 and 400 °C are attributed to the decomposition of aurichalcite and rosasite, respectively,15 which correspond to the phases identified by XRD (Fig. S3, ESI†). From the TGA/DTG results (Fig. S4, ESI†), a high-temperature carbonate phase (HT-CO3) is found only in the case of acetate-derived precursors at ca. 480 °C,15 similar to previously reported zincian georgeite precursor.8 The existence of HT-CO3 after calcination provides a positive effect on Cu dispersion by suppressing sintering Cu during exothermic reduction pretreatment.53,54 However, low-temperature mass losses at 225 and 260 °C are detected only in the case of unwashed nitrate and acetate precursors, which are attributed to the decomposition of NH4(NO3) and CH3COONH4, respectively.8,55 The decomposition of such ammonium residues during calcination is exothermic and promotes not only the metal agglomeration but also the removal of the high-temperature carbonate phase.8 Table 3 shows that the washing step is essential for nitrate-derived catalysts to exhibit more favorable textural properties as an active catalyst. The activity loss during calcination of nitrate is due to the formation of NOx that promotes metal agglomeration,56 and can be avoided by decomposition under the gas flow (N2, NO, or air), which was found to effectively remove remaining nitrate from the unwashed Na-free precursors.9 On the other hand, the textural properties of the catalyst derived from unwashed acetate precursors are not penalized, even upon calcination in a stagnant air of the muffle furnace, suggesting that the washing step could be skipped completely.
The catalytic performance of the materials listed in Table 3 in comparison to the commercial Cu/ZnO/Al2O3 catalyst in high-pressure CO2 hydrogenation to methanol at 240–280 °C and 331 bar is shown in Fig. 8. The commercial Cu/ZnO/Al2O3 catalyst shows increasing CO2 conversion and CH3OH selectivity with increasing temperature from 240 to 280 °C. The improvement in the catalytic performance is small above 260 °C due to the shift from kinetically-controlled to the thermodynamically-controlled regime which can be achieved under very high-pressure conditions.40 The optimal temperature is found to be 280 °C and higher temperatures will in turn decrease both CO2 conversion and CH3OH selectivity due to the thermodynamic equilibrium limitation.41 The catalysts derived from washed acetate and nitrate precursors exhibit similar trends and achieve comparable CO2 conversion and CH3OH selectivity as commercial Cu/ZnO/Al2O3 catalyst, although the performance of the acetate-derived catalyst is superior. Importantly, the washing step did not affect the catalytic performance of the acetate-derived catalysts. In contrast, the unwashed nitrate-derived catalyst shows much lower activity than the washed one, as expected from the inferior textural properties (Table 3). The results clearly show that the urea hydrolysis using acetate salts can produce a very active catalyst and render the catalyst preparation simpler with less amount of wastewater.
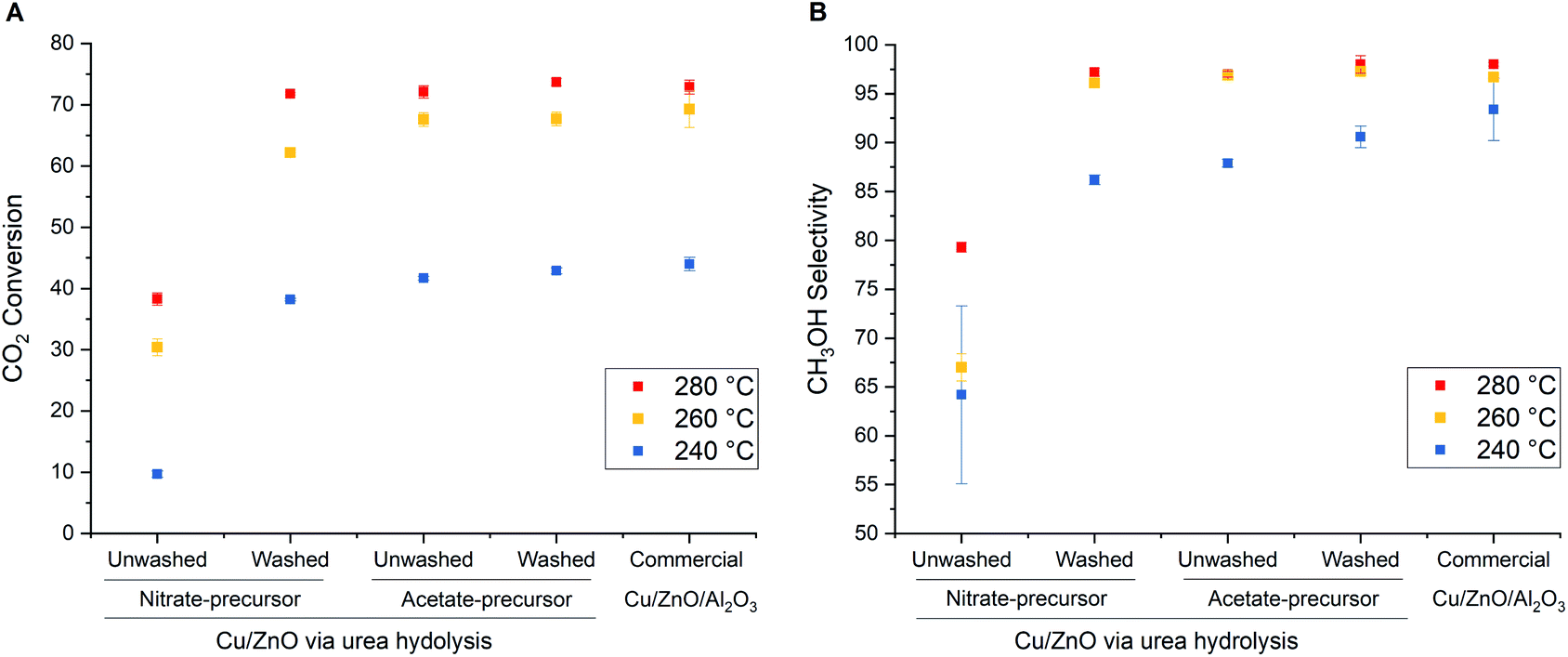 | ||
| Fig. 8 Influences of washing of as-precipitated precursors derived from nitrate and acetate salts on (A) CO2 conversion (B) CH3OH selectivity of Cu/ZnO catalyst (Cu:Zn = 1:1) for CO2 hydrogenation to methanol. H2/CO2 = 3, T = 240, 260 and 280 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | ||
It should be noted that Cu content in Cu/ZnO (1:1) catalysts (50 wt% CuO) is lower than the commercial Cu/ZnO/Al2O3 catalyst (64 wt% CuO) by 14 wt% so is the Cu surface area (Table 3). This indicates that the intrinsic activity is higher, which agrees with the study done by Behrens et al.; the intrinsic activity of Cu0 significantly decreases with increasing Al2O3 content and higher Cu surface area.57 However, the Cu surface area alone cannot reflect the catalytic activity of the catalyst since Cu in the vicinity to ZnO has been reported to be particularly active for methanol production (e.g. Cu steps decorated with Zn atoms or strong metal–support interaction).58,59 The Cu content up to 80 wt% is employed for the preparation of Cu/ZnO or Cu/ZnO/Al2O3 catalysts via conventional co-precipitation since it is favorable for the formation of zincian malachite phase forming a highly active structure after calcination.15,42 With urea hydrolysis of acetate salts, an active catalyst can be prepared with lower active Cu metal usage and less water treatment, which likely, in turn, reduces the cost of catalyst production. Therefore, the effect of Cu content (Cu:Zn ratio) will be further investigated in the following section.
Influence of Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :Zn ratio
:Zn ratio
In addition to the studied parameters reported above, the ratio between Cu2+ and Zn2+ during co-precipitation plays also a decisive role in determining the structure of precipitated hydroxylcarbonate precursors that finally determine the Cu–ZnO inter-dispersion and the catalytic activity of final Cu/ZnO catalysts. The common reported structures are copper hydrozincite ((CuxZn1−x)5(OH)6(CO3)2, when x < 0.1), aurichalcite ((CuxZn1−x)5(OH)6(CO3)2, when x < 0.5), rosasite ((CuxZn1−x)2(OH)2(CO3), when 0.5 < x < 0.7), and zincian malachite ((CuxZn1−x)2(OH)2(CO3) when x > 0.7).15,16 Industrially, Cu:Zn ratios in the range of 7:3 to 2:1 are chosen to yield a desired zincian malachite precursor.42 In this study, catalysts derived from acetate salts (washed) having Cu:Zn ratios of 1:3, 1:1, 3:1 are tested. The XRD patterns of as-precipitated precursors confirm the formation of different phases upon changing from the Cu-rich to the Zn-rich solution. Aurichalcite phase is the main phase at Cu:Zn ratio of 1:3 and 1:1, while zincian malachite phase is the main one at 3:1 (Fig. S5, ESI†). Calcination of these precursors produces CuO/ZnO (or Cu/ZnO after reduction) with textural properties shown in Table 4. As expected, the increasing relative amount of Cu (i.e. higher Cu/Zn ratio) results in decreased Cu dispersion, while increasing the specific Cu surface area with a maximum at the Cu:Zn ratio of 1:1 (Table 4). In literature, the Cu:Zn ratio of 1:1 is hypothetically favorable for a highly active catalyst. The hydroxylcarbonate precursors with the Cu:Zn ratio of 1:1 should yield the smallest CuO and ZnO particles and highest inter-dispersion upon decomposition (nano-structuring).42,52 The excess amount of Cu leads to lower specific Cu surface area and larger crystallite size mainly due to CuO agglomeration and insufficient ZnO stabilizer functioning as a spacer to prevent sintering (3:1, Table 4). As a reflection of the highest Cu surface area, the highest CO2 conversion and CH3OH selectivity are obtained with Cu/ZnO (1:1) catalyst (Fig. 9).
| Cu:Zn molar ratio of Cu/ZnO catalysts |
BET surface area (m2 g−1) | Cu surface areaa (m2 g−1) | Cu dispersion (%) | Average crystallite sizeb (nm) | Intrinsic activity (mmolCH3OH mCu−2 h−1)d | |
|---|---|---|---|---|---|---|
| CuO | ZnO | |||||
| a Determined by N2O chemisorption.b Estimated by Rietveld refinement.c Determined by ICP elemental analysis.40d Based on methanol productivity at H2/CO2 = 3, T = 280 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | ||||||
| 1:3 |
65 | 9 | 18 | 2.8 | 7.1 | 3.8 |
| 1:1 |
70 | 13 | 13 | 4.1 | 6.7 | 2.9 |
| 3:1 |
71 | 12 | 8 | 4.8 | 3.8 | 3.0 |
| Commercial Cu/ZnO/Al2O3 (64/25/10 wt%)c | 102 | 19 | 16 | 6.3 | 4.2 | 1.9 |
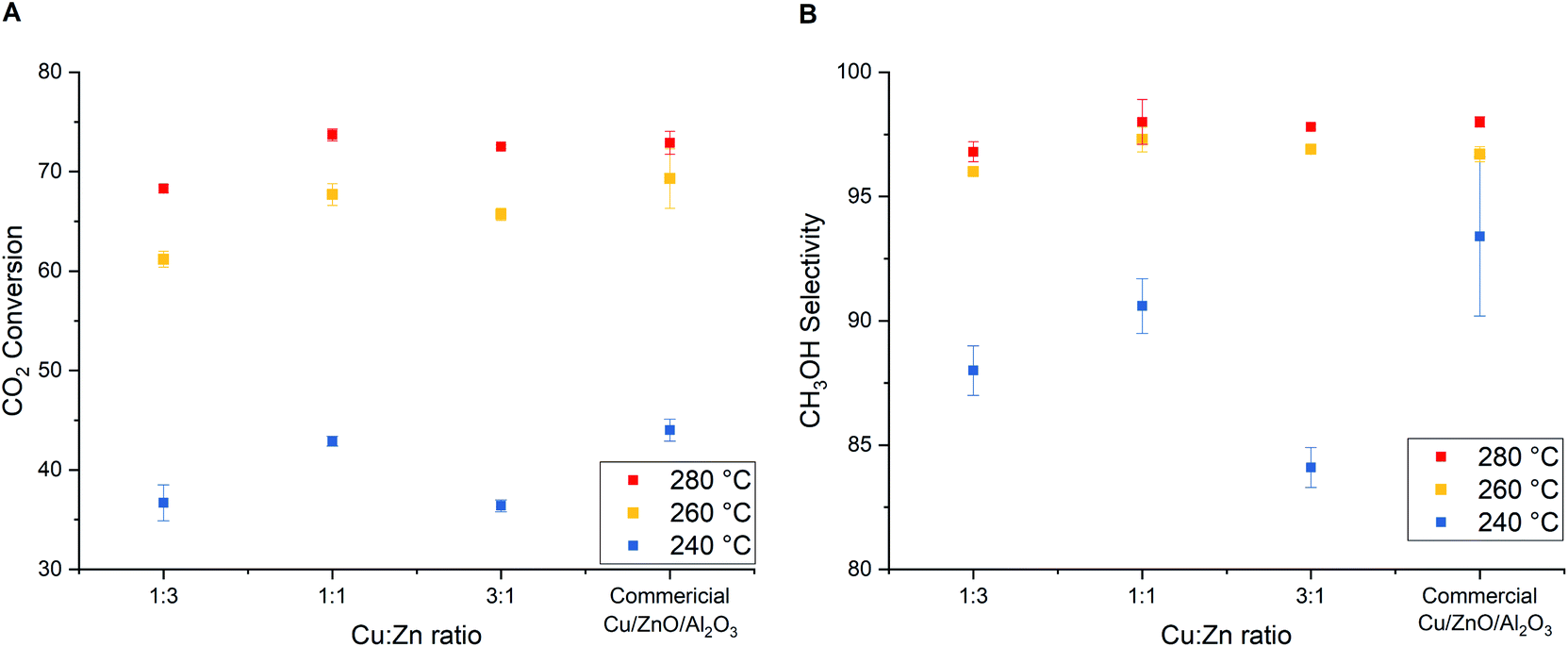 | ||
| Fig. 9 Influences of Cu:Zn molar ratio in Cu/ZnO catalyst derived from acetate salts (washed) on (A) CO2 conversion and (B) CH3OH selectivity from CO2 hydrogenation to methanol. H2/CO2 = 3, T = 260 °C, P = 331 bar, GHSV = 8500 h−1, and TOS = 6 h. | ||
Even though the catalytic activity of Cu/ZnO (1:3) is slightly lower than the commercial Cu/ZnO/Al2O3 and other Cu/ZnO catalysts reported in Fig. 9, Cu utilization (weight basis) towards methanol formation is the highest thanks to the high copper dispersion. The weight–time–yields of methanol (WTYCH3OH) per amount of Cu at 280 °C and 331 bar are found to be Cu/ZnO (1:3) > Cu/ZnO (1:1) > Cu/ZnO/Al2O3 > Cu/ZnO (3:1) for 5177 > 2780 > 1887 > 1656 mg gCu−1 h−1, respectively. This suggests that the amount of Cu metal could be reduced with a slight compromise of catalytic activity.
Stability test
The stability of the Cu/ZnO derived from the unwashed acetate precursors was compared with a commercial Cu/ZnO/Al2O3 catalyst at 280 °C and at a relatively high space velocity to induce faster deactivation (Fig. 10). Both catalysts have shown similar trends of activity with time-on-stream (TOS) followed by a gradual decrease in CO2 conversion. The Cu/ZnO/Al2O3 catalyst (Fig. 10A) has reached the maximum conversion of 76% and CH3OH selectivity of 96% within 7 h, then these values started to decrease by 9% and 1.5% after 94 h, respectively. On the other hand, the Cu/ZnO catalyst (Fig. 10B) has reached the same value of maximum conversion and selectivity within 5 h of TOS. However, the activity started to decline more rapidly, and the catalyst has lost up to 24% lower CO2 conversion and 3% lower methanol selectivity after 147 h. This comparably poor stability of the Cu/ZnO catalyst is expected since Al2O3 is known to function as a structural promoter that provides resistance against sintering.60 Incorporation of such promoters using Al(CH3COO)2(OH) via this synthesis route is possible without precursor washing; however, this is out of the scope of this work and the influences of Al amount on catalyst structures and activity need to be investigated.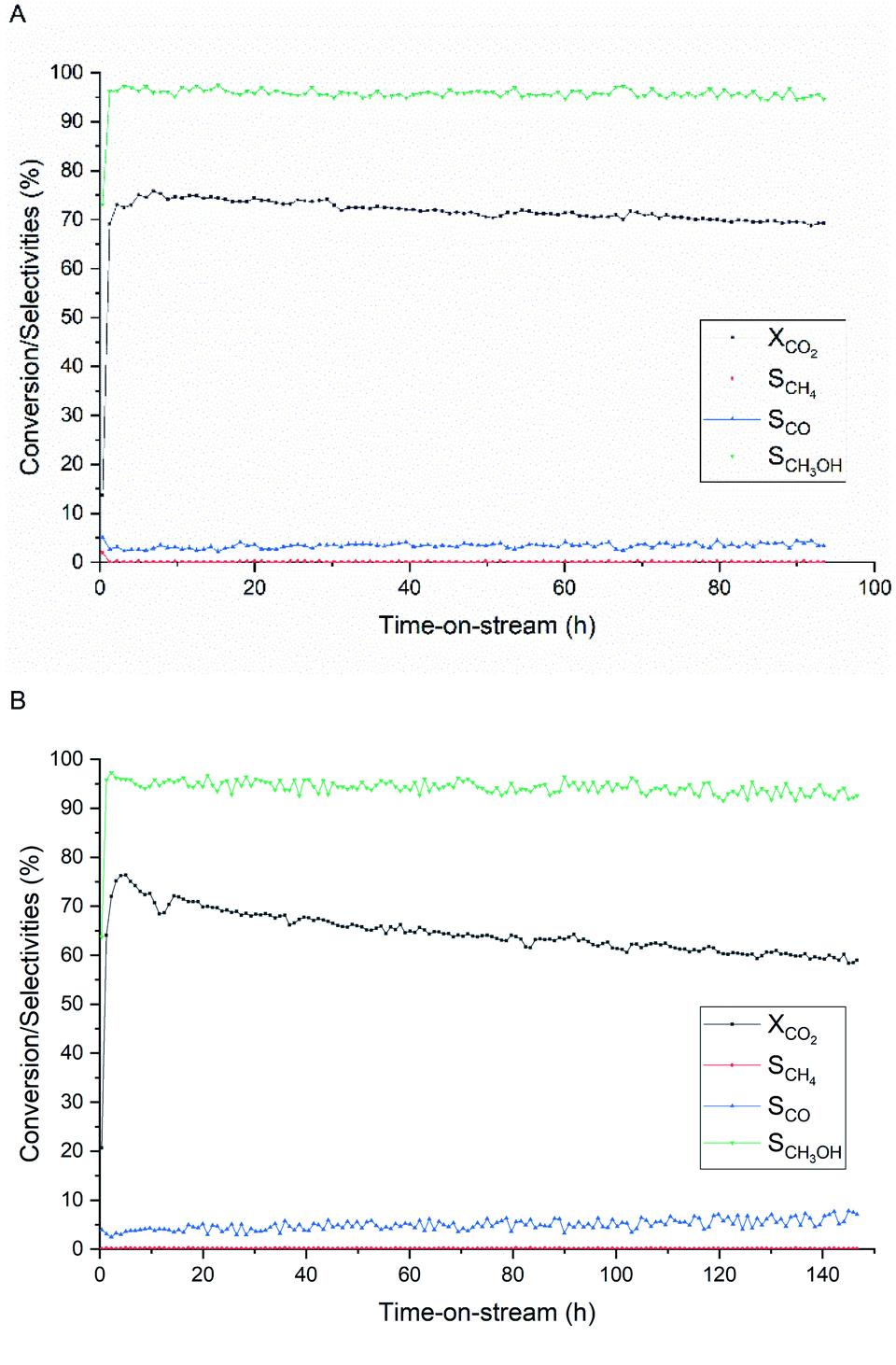 | ||
| Fig. 10 Catalytic stability during CO2 hydrogenation to methanol of (A) commercial Cu/ZnO/Al2O3 and (B) Cu/ZnO catalyst (Cu:Zn = 1:1) derived from acetate salts (unwashed, U/M = 7). H2/CO2 = 3, T = 280 °C, P = 331 bar, and GHSV 17000 h−1. | ||
Characterization of the spent catalysts by XRD (Fig. S6†) confirms that Cu sintering is the main cause of deactivation; the crystallite size of metallic Cu increases on average from 11.2 nm to 20.7 nm toward the end of stability testing. The increase in Cu size over time is related to a slight increase in CO selectivity, which reflects the structure–activity of Cu/ZnO catalyst.61 The Cu crystallite growth over the Cu/ZnO/Al2O3 catalyst due to the presence of the water partial pressure is reported.48 Under a very high-pressure condition of 331 bar, CO2 can react with ZnO into a more stable phase ZnCO3.62 The formation of rigid ZnCO3 helps slower growth of Cu crystallite size.63 Moreover, weaker interaction between water and ZnCO3 than ZnO seems to improve the activity of the Cu-based catalyst under liquid phase methanol synthesis.62 The understanding of the involved mechanisms and electronic properties of both ZnO or ZnCO3 is still limited, and the role of ZnCO3 on catalytic activity and stability will be investigated in future work.
Experimental
Raw materials
The following raw materials were used for co-precipitation: copper(II) nitrate trihydrate (p.a. 99–104%, Sigma-Aldrich), zinc(II) nitrate hexahydrate (98%, Sigma-Aldrich), copper(II) acetate hydrate (98%, Sigma-Aldrich), zinc(II) acetate dihydrate (≥97%, Alfa Aesar), urea (99%), deionized water was used from a Millipore system. The commercial methanol synthesis catalyst (Cu/ZnO/Al2O3) was purchased from Alfa Aesar (Product ID: 45776).Preparation of Cu/ZnO via urea hydrolysis method
The Cu/ZnO catalysts were prepared batchwise in a 500 mL round-bottom flask with a reflux condensation and internal temperature control. An aqueous solution containing Cu(CH3COO)2, Zn(CH3COO)2, and urea were freshly prepared and mixed at room temperature. The total concentration of Cu2+ and Zn2+ in the solution was kept constant at 0.25 M. The Cu:Zn ratio in the solution was varied from 1:3 to 3:1 to adjust the Cu content of the CuO/ZnO catalyst, while the molar ratio of [urea]/[Cu2+ + Zn2+] was varied from 1–10. The mixed solution was added to the flask and heated to 70–95 °C with 10 °C min−1 using a heating mantle and while stirring vigorously at 1000 rpm using a magnetic stirrer. After 24 hours of precipitation process, the as-precipitate precursor was filtrated, optionally washed with adequate deionized water, and dried in an oven at 80 °C overnight. The dried precursor was calcined at 300 °C in a muffle furnace for 1 hour at a heating rate of 2 °C min−1. The calcined catalyst powder was pelletized with a pressing die, crushed in a mortar, and sieved to the size of 100–300 μm. The palletization pressure was 370 kg cm−2 or ca. 363 bar, similar to the reaction pressure, to ensure no deformation of the catalyst pellets during gas pressurization. The same synthesis procedure was used to prepared catalysts from aqueous Cu(NO3)2, and Zn(NO3)2 solution.
Catalyst characterizations
The fresh and spent catalysts were characterized by various methods. The thermal decomposition of catalyst precursors is measured by a Mettler Toledo DSC822 thermogravimetric analyzer (TGA). The BET surface area of the catalyst was analyzed using a Quantachrome Autosorb 1-MP surface area analyzer. The reducibility of catalyst is studied by temperature-programmed reduction (H2-TPR) on Thermo TPDRO 1100 equipped with a TCD detector. The copper surface areas were measured by N2O pulse chemisorption at 90 °C, in which samples were reduced in 5% H2 in He stream at 300 °C before analysis.40 A relatively harsh reduction condition is used to ensure the reduction of the catalysts as shown (Fig. S7†). Powder X-ray diffraction (XRD) patterns were attained using a Bruker AXS D8 Advance diffractometer equipped with a Cu tube. The Rietveld refinement ware calculated using X'Pert HighScore Plus. Scanning electron microscope (SEM) images were obtained by Hitachi HD-2000. High-angle annular dark-field imaging (HAADF) was performed using a JEM-ARM200F scanning transmission electron microscope (STEM) equipped with a JEOL JED-2300 X-ray energy dispersive spectrometer (EDS). Samples were prepared by dropping an ethanol solution containing the catalyst on carbon-supported Mo grids.Catalyst testing
The catalytic tests were carried out in a high-pressure setup as reported elsewhere.40 In a typical test, 200 mg catalyst was packed between quartz wool inside a 1/4 inch fixed-bed continuous flow reactor (ID 2.79 mm). The catalyst was reduced in situ at 260 °C with 90% H2/Ar (25 mL min−1) for 2 h under atmospheric pressure. A relatively harsh reduction condition was used to ensure the reduction of the catalysts and to reach a stable state of the catalyst shortly. The reduction temperature had negligible effects on the catalytic activity (Fig. S8†). After cooling down to 30 °C, the H2/CO2/Ar mixture with vol% of 69%/23%/8% was fed into the reactor and pressurized to 360 bar (the reactant pressure is 331 bar). The total flow rate of the gas mixture is kept at 16.7 mL min−1 to achieve a gas-hourly space velocity of 8500 h−1 equivalents to 5 N L gcat−1 h−1. The products were analyzed by an online gas chromatograph (Bruker, GC-450) equipped with a flame ionization detector for methanol, methyl formate, diethyl ether, and other hydrocarbons, and a thermal conductivity detector for permanent gases e.g. CO2, H2, Ar, CO, CH4.Conclusions
The sodium- and nitrate-free precursors of the Cu/ZnO catalysts were successfully prepared via the facile urea hydrolysis method using acetate and nitrate salts. At the optimal preparation conditions, the small aurichalcite structure is obtained leading to superior catalytic activity from the nitrate-derived catalyst. Moreover, the washing step was completely omitted from the acetate-derived catalyst while retaining high catalytic activity. This unwashed acetate-derived catalyst exhibited excellent time-on-stream stability even in an absence of Al2O3 which is normally present as a structural promoter in the commercial Cu/ZnO/Al2O3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Swiss National Science Foundation (Sinergia grant no. CRSII5-183495) and MINECO, Spain (CTQ2016-75499-R (FEDER-UE)). We also thank the technical staffs of the Open Facility of Hokkaido University for their help with STEM analysis.References
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley, 2009, vol. 44 Search PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 2013, 52, 104–107 CrossRef CAS.
- K. Klier, Adv. Catal., 1982, 31, 243–313 CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- S. Schimpf and M. Muhler, in Synthesis of Solid Catalysts, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 329–351 Search PubMed.
- Y. Tang, Y. Liu, P. Zhu, Q. Xue, L. Chen and Y. Lu, AIChE J., 2009, 55, 1217–1228 CrossRef CAS.
- M. Behrens, S. Kißner, F. Girsgdies, I. Kasatkin, F. Hermerschmidt, K. Mette, H. Ruland, M. Muhler and R. Schlögl, Chem. Commun., 2011, 47, 1701 RSC.
- P. J. Smith, S. A. Kondrat, P. A. Chater, B. R. Yeo, G. M. Shaw, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park and G. J. Hutchings, Chem. Sci., 2017, 8, 2436–2447 RSC.
- G. Prieto, K. P. de Jong and P. E. de Jongh, Catal. Today, 2013, 215, 142–151 CrossRef CAS.
- S. A. Kondrat, P. J. Smith, P. P. Wells, P. A. Chater, J. H. Carter, D. J. Morgan, E. M. Fiordaliso, J. B. Wagner, T. E. Davies, L. Lu, J. K. Bartley, S. H. Taylor, M. S. Spencer, C. J. Kiely, G. J. Kelly, C. W. Park, M. J. Rosseinsky and G. J. Hutchings, Nature, 2016, 531, 83–87 CrossRef CAS PubMed.
- S. A. Kondrat, P. J. Smith, J. H. Carter, J. S. Hayward, G. J. Pudge, G. Shaw, M. S. Spencer, J. K. Bartley, S. H. Taylor and G. J. Hutchings, Faraday Discuss., 2017, 197, 287–307 RSC.
- U. Costantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1998, 1439–1446 CrossRef.
- R. J. Candal, A. E. Regazzoni and M. A. Blesa, J. Mater. Chem., 1992, 2, 657–661 RSC.
- G. J. d. A. A. Soler-Illia, R. J. Candal, A. E. Regazzoni and M. A. Blesa, Chem. Mater., 1997, 9, 184–191 CrossRef.
- M. Behrens, F. Girgsdies, A. Trunschke and R. Schlögl, Eur. J. Inorg. Chem., 2009, 1347–1357 CrossRef CAS.
- M. Behrens and F. Girgsdies, Z. Anorg. Allg. Chem., 2010, 636, 919–927 CrossRef CAS.
- A. Álvarez, A. Bansode, A. Urakawa, A. V. Bavykina, T. A. Wezendonk, M. Makkee, J. Gascon and F. Kapteijn, Chem. Rev., 2017, 117, 9804–9838 CrossRef PubMed.
- X.-M. Liu, G. Q. Lu, Z.-F. Yan and J. Beltramini, Ind. Eng. Chem. Res., 2003, 42, 6518–6530 CrossRef CAS.
- J. Schumann, T. Lunkenbein, A. Tarasov, N. Thomas, R. Schlögl and M. Behrens, ChemCatChem, 2014, 6, 2889–2897 CrossRef CAS.
- S. Murcia-Mascarós, R. M. Navarro, L. Gómez-Sainero, U. Costantino, M. Nocchetti and J. L. G. Fierro, J. Catal., 2001, 198, 338–347 CrossRef.
- M. Turco, G. Bagnasco, U. Costantino, F. Marmottini, T. Montanari, G. Ramis and G. Busca, J. Catal., 2004, 228, 43–55 CAS.
- M. Turco, G. Bagnasco, U. Costantino, F. Marmottini, T. Montanari, G. Ramis and G. Busca, J. Catal., 2004, 228, 56–65 CAS.
- T. Shishido, Y. Yamamoto, H. Morioka, K. Takaki and K. Takehira, Appl. Catal., A, 2004, 263, 249–253 CrossRef CAS.
- U. Costantino, F. Marmottini, M. Sisani, T. Montanari, G. Ramis, G. Busca, M. Turco and G. Bagnasco, Solid State Ionics, 2005, 176, 2917–2922 CrossRef CAS.
- T. Shishido, Y. Yamamoto, H. Morioka and K. Takehira, J. Mol. Catal. A: Chem., 2007, 268, 185–194 CrossRef CAS.
- S. G. Sanches, J. H. Flores, R. R. De Avillez and M. I. Pais Da Silva, Int. J. Hydrogen Energy, 2012, 37, 6572–6579 CrossRef CAS.
- J. Baneshi, M. Haghighi, N. Jodeiri, M. Abdollahifar and H. Ajamein, Energy Convers. Manage., 2014, 87, 928–937 CrossRef CAS.
- S. G. Sanches, J. Huertas Flores and M. I. P. da Silva, React. Kinet., Mech. Catal., 2017, 121, 473–485 CrossRef CAS.
- T. Shishido, M. Yamamoto, I. Atake, D. Li, Y. Tian, H. Morioka, M. Honda, T. Sano and K. Takehira, J. Mol. Catal. A: Chem., 2006, 253, 270–278 CrossRef CAS.
- T. Shishido, M. Yamamoto, D. Li, Y. Tian, H. Morioka, M. Honda, T. Sano and K. Takehira, Appl. Catal., A, 2006, 303, 62–71 CrossRef CAS.
- I. Atake, K. Nishida, D. Li, T. Shishido, Y. Oumi, T. Sano and K. Takehira, J. Mol. Catal. A: Chem., 2007, 275, 130–138 CrossRef CAS.
- F. Meshkani, M. Rezaei and M. Jafarbegloo, Mater. Res. Bull., 2015, 64, 418–424 CrossRef CAS.
- S. Wang, X. Li, Q. Yin, L. Zhu and Z. Luo, Catal. Commun., 2011, 12, 1246–1250 CrossRef CAS.
- S. Mondal, R. Janardhan, M. L. Meena and P. Biswas, J. Environ. Chem. Eng., 2017, 5, 5695–5706 CrossRef CAS.
- S. Wang, Y. Zhang and H. Liu, Chem. - Asian J., 2010, 5, 1100–1111 CrossRef CAS PubMed.
- S. Wang and H. Liu, Chin. J. Catal., 2014, 35, 631–643 CrossRef CAS.
- R. Fan, M. Kyodo, L. Tan, X. Peng, G. Yang, Y. Yoneyama, R. Yang, Q. Zhang and N. Tsubaki, Fuel Process. Technol., 2017, 167, 69–77 CrossRef CAS.
- H. Liu, T. Chen and G. Wang, Catal. Lett., 2018, 148, 1462–1471 CrossRef CAS.
- P. Zhang, Y. Araki, X. Feng, H. Li, Y. Fang, F. Chen, L. Shi, X. Peng, Y. Yoneyama, G. Yang and N. Tsubaki, Fuel, 2020, 268, 117213 CrossRef CAS.
- A. Bansode and A. Urakawa, J. Catal., 2014, 309, 66–70 CrossRef CAS.
- R. Gaikwad, A. Bansode and A. Urakawa, J. Catal., 2016, 343, 127–132 CrossRef CAS.
- M. Behrens and R. Schlögl, Z. Anorg. Allg. Chem., 2013, 639, 2683–2695 CrossRef CAS.
- F. Zhang, Y. Zhang, L. Yuan, K. A. M. Gasem, J. Chen, F. Chiang, Y. Wang and M. Fan, Mol. Catal., 2017, 441, 190–198 CrossRef CAS.
- W. H. R. Shaw and J. J. Bordeaux, J. Am. Chem. Soc., 1955, 77, 4729–4733 CrossRef CAS.
- W. Jia, E. Reitz, P. Shimpi, E. G. Rodriguez, P. X. Gao and Y. Lei, Mater. Res. Bull., 2009, 44, 1681–1686 CrossRef CAS.
- Y. Fernández, J. A. Menéndez, A. Arenillas, E. Fuente, J. H. Peng, Z. B. Zhang, W. Li and Z. Y. Zhang, Solid State Ionics, 2009, 180, 1372–1378 CrossRef.
- J. G. van Bennekom, R. H. Venderbosch, J. G. M. Winkelman, E. Wilbers, D. Assink, K. P. J. Lemmens and H. J. Heeres, Chem. Eng. Sci., 2013, 87, 204–208 CrossRef CAS.
- R. Gaikwad, H. Reymond, N. Phongprueksathat, P. Rudolf von Rohr and A. Urakawa, Catal. Sci. Technol., 2020, 10, 2763–2768 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem. - Eur. J., 2003, 9, 2039–2052 CrossRef CAS PubMed.
- C. Baltes, S. Vukojević and F. Schüth, J. Catal., 2008, 258, 334–344 CrossRef CAS.
- M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef CAS.
- M. Schur, B. Bems, A. Dassenoy, I. Kassatkine, J. Urban, H. Wilmes, O. Hinrichsen, M. Muhler and R. Schlögl, Angew. Chem., Int. Ed., 2003, 42, 3815–3817 CrossRef CAS PubMed.
- P. Kowalik, M. Konkol, K. Antoniak, W. Próchniak and P. Wiercioch, J. Mol. Catal. A: Chem., 2014, 392, 127–133 CrossRef CAS.
- M. Olszak-Humienik, Thermochim. Acta, 2001, 378, 107–112 CrossRef CAS.
- J. R. A. Sietsma, H. Friedrich, A. Broersma, M. Versluijs-Helder, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, J. Catal., 2008, 260, 227–235 CrossRef CAS.
- M. Behrens, S. Zander, P. Kurr, N. Jacobsen, J. Senker, G. Koch, T. Ressler, R. W. Fischer and R. Schlögl, J. Am. Chem. Soc., 2013, 135, 6061–6068 CrossRef CAS PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kuhl, M. Havecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Norskov and R. Schlogl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- S. Zander, E. L. Kunkes, M. E. Schuster, J. Schumann, G. Weinberg, D. Teschner, N. Jacobsen, R. Schlögl and M. Behrens, Angew. Chem., Int. Ed., 2013, 52, 6536–6540 CrossRef CAS PubMed.
- M. Kurtz, H. Wilmer, T. Genger, O. Hinrichsen and M. Muhler, Catal. Lett., 2003, 86, 77–80 CrossRef CAS.
- R. Van Den Berg, G. Prieto, G. Korpershoek, L. I. Van Der Wal, A. J. Van Bunningen, S. Lægsgaard-Jørgensen, P. E. De Jongh and K. P. De Jong, Nat. Commun., 2016, 7, 13057 CrossRef CAS PubMed.
- S. Lee, B. G. Lee and C. J. Kulik, Fuel Sci. Technol. Int., 1991, 9, 977–998 CrossRef CAS.
- S. Lee and A. Sardesai, Top. Catal., 2005, 32, 197–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02103f |
| This journal is © The Royal Society of Chemistry 2021 |