
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light driven photocatalytic degradation of aqueous acetamiprid over nitrogen and graphene oxide doped ZnO composites
Carolina Sayury Miyashiro and
Safia Hamoudi *
*
Department of Soil Sciences & Agri-Food Engineering, Centre in Green Chemistry & Catalysis, Centr'Eau, Université Laval, Québec, G1V 0A6, Canada. E-mail: safia.hamoudi@fsaa.ulaval.ca; Fax: + 418 656 3723; Tel: +1 418 656 2131 ext. 408460
First published on 24th June 2021
Abstract
The present investigation focused on the photocatalytic degradation of acetamiprid in aqueous solutions under visible light over bare ZnO as well as N- and N-GO-doped photocatalysts. The synthesised materials were characterised using SEM, TEM, XRD, nitrogen sorption, photoluminescence, UV-Vis, FTIR and electrochemical impedance spectroscopy techniques. The obtained results pointed out the high photocatalytic performances of the N-GO-ZnO allowing complete degradation of the acetamiprid after 5 hours of reaction at ambient temperature. Under otherwise the same operating conditions, 12, 38 and 68% conversion were reached in the absence of any photocatalyst, over the bare ZnO and N-doped ZnO materials, respectively.
Introduction
Neonicotinoids are the most widely and extensively used insecticides in the world, for crop protection, pest management or parasite control.1,2 They can be applied flexibly in a variety of ways, such as sprays, injections and drenches.3 Along with the properties of relatively long half-life in soil and high solubility in water, neonicotinoids have the potential to accumulate in soil and leach into surface water and groundwater, which could represent a direct threat to non-target organisms, especially honeybees.2Acetamiprid, a widely used neonicotinoid in crop protection, was considered to be a favourable choice for controlling those pests that are severely resistant to organophosphorus pesticides, owing to its broad insecticidal spectrum and relatively low acute and chronic mammalian toxicity.4,5 Because of the widespread use of acetamiprid in many areas and its potential toxicity to humans,1 the residues present in the environment have received considerable attention, and methods for the degradation of neonicotinoids are being actively investigated.6
The advanced oxidative processes (AOPs) are potential candidates for the treatment of water polluted from industry and agriculture.7 Among the AOPs, photocatalytic degradation under visible light represents a potential process to get rid of numerous organic pollutants in water and wastewaters. The oxides, sulfides, and halides of Ti, Zn, Cd, Ag and so forth are the most explored photocatalysts.8 To enhance their photocatalytic activity, features like adsorption, utilisation of visible light or sunlight and charge separation can be tuned by doping with transition metals, rare earth metals as well as non-metals such as N and S.9,10
The use of semiconductors as photocatalysts is common and well known in advanced oxidation. ZnO material proved to be effective when compared to other semiconductors and has been studied extensively due to its interesting features like weak toxicity, high electron mobility, and strong oxidizing power.11 However, using ZnO as a photocatalyst exhibits some drawbacks which include its wide band gap energy, the low surface area of the bulk form as well as the high rate at which the photogenerated electron and hole recombine. In addition, ZnO has the intrinsic disadvantage of the photocorrosion effect which limits its practical applications, particularly in water treatment.12,13 To overcome these challenges and drawbacks, several modifications have been made on ZnO to extend its light absorption to the visible light region and thus enhance its photocatalytic capability and applicability. These modifications included the preparation of nanocomposites with organic materials, co-doping, and coupling with other semiconductors.14
The nitrogen atoms can easily substitute oxygen atoms and dope into ZnO.15 Nitrogen doping has been widely used to increase visible light absorption by modifying the band gap energy. Indeed, while the band gap energy was generally evaluated to be around 3.21 eV for pristine ZnO, nitrogen doped ZnO materials exhibited lower values within the range [3.08–2.95 eV].16–19 The graphene oxide (GO) is also a promising material having an atomic thick nanosheet of covalently organised two-dimensional lattice of carbon atoms.20 Moreover, the surface properties of GO can be adjusted through chemical modification, which favours its perfect solubility in solvents and provides efficient opportunities for the synthesis of GO based nanocomposites.21 The advantages of doping with graphene oxide are related to the reduction of the bandgap energy of ZnO and suppression of the rapid recombination of electrons and holes. Both effects have been proven to enhance the photocatalytic performances of ZnO.22 For instance, band gap energy values comprised between 3.06 and 3.17 eV were reported for GO-doped ZnO materials.23,24 Moreover, doping with a combination of nitrogen and graphene oxide also offers the advantage of lowering the band gap energy of ZnO. In this connection, the band gap energy was reported to be close to 2.83–2.90 for the N-GO doped ZnO.19
So far, various methods of ZnO nanoparticles doping with nitrogen and graphene oxide have been explored, such as thermal nitridation,25 in situ synthesis,26 chemical vapor deposition (CVD)27 and so forth. However, these methods exhibited disadvantages such as severe synthesis conditions and random distribution of the doping species. Thus, finding a facile doping method is still challenging.28,29
The present work is devoted to the investigation of the visible light-driven photocatalytic degradation for acetamiprid aqueous solutions over ZnO, N-doped ZnO and N-GO- doped ZnO photocatalysts. The specific objectives are: (1) catalysts synthesis and characterisation and (2) investigation of the effect of different operating conditions on the photocatalytic degradation reaction.
Experimental
Materials
All reagents were of the highest purity and were used without further purification. Acetamiprid (N-(6-chloro-3-pyridylmethyl)-N′-cyano-acetamidine) was purchased from Sigma-Aldrich. Zinc nitrate (Zn(NO3)2·6H2O) and sodium hydroxide were acquired form J. T. Baker. Urea was purchased from BDH. For the synthesis of graphene oxide, natural graphite flakes (Alfa Aesar, 325 mesh, 99.8%), potassium persulfate (K2S2O8, Anachemia, ≥99%), phosphorus pentoxide (P2O5, J. T. Baker, ≥99%), sulfuric acid (H2SO4, Fisher, 98%), potassium permanganate (KMnO4, Fisher, ≥99%), hydrogen peroxide solution (H2O2, EMD Millipore, 30 wt% in water), hydrochloric acid (HCl, Fisher, 36%), and ethylene glycol (BDH, ≥99%) were used.Synthesis of ZnO and N-ZnO materials
ZnO photocatalyst was synthesised according to a modified methodology reported by Perillo et al.30 To this purpose, solutions of Zn (NO3)2 (0.3 M) and NaOH (5 M) were prepared. Precipitation was performed at 65 °C ±3 by adding NaOH solution to the zinc solution dropwise. The mixture was left under intense agitation for 2 hours. The obtained white precipitate was filtered, washed, and dried overnight at 150 °C, then calcined at 420 °C for 4 hours. N-ZnO material was synthesised following the same procedure except that urea was added to the Zn precursor solution (0.5/1 molar ratio) before precipitation.Synthesis of the nanocomposite N-ZnO mixed with GO
The synthesis of graphene oxide was carried out as reported by Andrade et al.31 The nanocomposites N-GO-ZnO were prepared according to a modified method reported by Yousaf, et al.32 Therefore, 90, 92.5 and 95 mg of N-ZnO and correspondingly 10, 7.5 and 5 mg of GO were suspended in deionised water in separate beakers and sonicated for one hour. Then, the contents of both bakers were mixed and sonicated for 2 hours. The suspensions were then dried at 100 °C overnight and grinded to a fine powder. The obtained materials were correspondingly denoted as N-GO-ZnO-5, N-GO-ZnO-7.5 and N-GO-ZnO-10.Characterisation
The morphology and elemental composition of the photocatalysts were examined by scanning electron microscopy (SEM) using a JEOL model JSM-840A microscope equipped with an energy dispersive X-ray spectrometry (EDX). Transmission electron microscopy (TEM) was performed using JEOL JEM-1230 electron microscope, at an accelerating voltage of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kV. Fourier transform infrared (FTIR) spectra were collected between 300 and 4000 cm−1 on a Varian 1000 (Scimitar series) spectrometer with a resolution of 2 cm−1 using KBr pellets at room temperature. The textural properties of the samples were investigated using nitrogen adsorption/desorption analyses at 77 K using a volumetric adsorption analyser (Model Autosorb-1, Quantachrome Instruments). The specific surface area was determined from the linear part of the Brunauer–Emmett–Teller (BET) plot (P/P0 = 0.05–0.30). The pore size distribution curves were calculated from the desorption branch, using the Barrett–Joyner–Hallenda (BJH) method. The total pore volume was evaluated from the adsorbed amount at a relative pressure P/P0 = 0.99 single point. The crystal structures of the samples were determined by X-ray powder diffraction (XRD) using a Rigaku D-Max-Ultima III diffractometer with nickel-filtered Cu Kα radiation of wavelength 1.5406 Å with a step size of 0.04° for 2θ angles ranging from 10° to 80°. The optical properties were recorded on a UV-visible HP UV-8453 spectrophotometer. The photoluminescence (PL) spectra were obtained using a fluorescence spectrometer Horiba QuantaMaster 8000. The photoelectrochemical measurements were conducted in a standard three-electrode system, using Pt wire and Ag/AgCl as working and reference electrodes, respectively. The working electrode was prepared on fluorine-doped tin oxide (FTO) glass cells (2.5 cm × 2.5 cm) by spray-coating with 10 mL of a suspension containing the photocatalyst (10 mg) and methanol (2.5 mL) under sonication for 3 h. The side part of the FTO slide was previously protected with scotch tape. The transient photocurrent response was performed on an Autolab PGSTAT204 electrochemical workstation with a 0.5 M aqueous solution of Na2SO4 as the electrolyte. The photocurrent was measured under solar-light irradiation (150 W xenon lamp) with a 20 s light on–off cycle with a reference voltage of 0.8 V. The surface chemical composition of the photocatalysts was investigated by X-ray Photoelectron Spectroscopy, using a PHI 5600-ci spectrometer (Physical Electronics, Eden Prairie, MN). The main XPS chamber was maintained at a base pressure <8 × 10−9 torr. A standard aluminium X-ray source was used to record survey and high-resolution spectra without charge neutralisation. The binding energies (B.E.) of core levels in the observed XPS spectra were referenced to C1s core (B.E. at 285 eV).
kV. Fourier transform infrared (FTIR) spectra were collected between 300 and 4000 cm−1 on a Varian 1000 (Scimitar series) spectrometer with a resolution of 2 cm−1 using KBr pellets at room temperature. The textural properties of the samples were investigated using nitrogen adsorption/desorption analyses at 77 K using a volumetric adsorption analyser (Model Autosorb-1, Quantachrome Instruments). The specific surface area was determined from the linear part of the Brunauer–Emmett–Teller (BET) plot (P/P0 = 0.05–0.30). The pore size distribution curves were calculated from the desorption branch, using the Barrett–Joyner–Hallenda (BJH) method. The total pore volume was evaluated from the adsorbed amount at a relative pressure P/P0 = 0.99 single point. The crystal structures of the samples were determined by X-ray powder diffraction (XRD) using a Rigaku D-Max-Ultima III diffractometer with nickel-filtered Cu Kα radiation of wavelength 1.5406 Å with a step size of 0.04° for 2θ angles ranging from 10° to 80°. The optical properties were recorded on a UV-visible HP UV-8453 spectrophotometer. The photoluminescence (PL) spectra were obtained using a fluorescence spectrometer Horiba QuantaMaster 8000. The photoelectrochemical measurements were conducted in a standard three-electrode system, using Pt wire and Ag/AgCl as working and reference electrodes, respectively. The working electrode was prepared on fluorine-doped tin oxide (FTO) glass cells (2.5 cm × 2.5 cm) by spray-coating with 10 mL of a suspension containing the photocatalyst (10 mg) and methanol (2.5 mL) under sonication for 3 h. The side part of the FTO slide was previously protected with scotch tape. The transient photocurrent response was performed on an Autolab PGSTAT204 electrochemical workstation with a 0.5 M aqueous solution of Na2SO4 as the electrolyte. The photocurrent was measured under solar-light irradiation (150 W xenon lamp) with a 20 s light on–off cycle with a reference voltage of 0.8 V. The surface chemical composition of the photocatalysts was investigated by X-ray Photoelectron Spectroscopy, using a PHI 5600-ci spectrometer (Physical Electronics, Eden Prairie, MN). The main XPS chamber was maintained at a base pressure <8 × 10−9 torr. A standard aluminium X-ray source was used to record survey and high-resolution spectra without charge neutralisation. The binding energies (B.E.) of core levels in the observed XPS spectra were referenced to C1s core (B.E. at 285 eV).
Photocatalytic activity
The acetamiprid was photo-oxidised in a home-made photocatalytic reactor operating under visible light at a wavelength λ > 420 nm. The tests were operated batchwise under the following ranges of initial acetamiprid concentration [5–20 mg L−1], photocatalyst loading [0.1–0.4 g L−1], temperature [25–45 °C] and time [0–300 min]. The reacting volume was 250 mL for all the tests performed under agitation (600 rpm). Furthermore, tests were performed without catalyst to assess the photolysis. At pre-set reaction times, aliquots of the aqueous solution were withdrawn, filtered with a microsyringe (0.2 μm) and analysed for acetamiprid content using an HP 6890 Series gas chromatograph equipped with a flame ionisation detector (Agilent ChemStation). The column used was an HP-5 (30 m × 0.25 mm ID × 0.25 μm film thickness). The injector and detector temperatures were set to 250 °C and 1 μL sample was injected with a split ratio of 50:1. The oven temperature profile was: initial temperature 100 °C, ramp at 10 °C min−1 to 250 °C, hold for 5 min; ramp at 30 °C min−1 to 300 °C, hold for 5 min. The conversion of acetamiprid during experiment was calculated by the following equation:33where, C0 and Ct are the initial concentration of the acetamiprid solution and the concentration after different intervals of the irradiation time, respectively. All the tests reported in the present investigation were performed in duplicate and analysed twice.
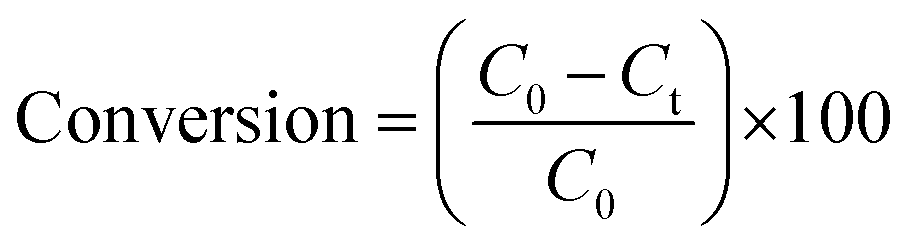
Results and discussion
Characterisation
The SEM images of ZnO, N-ZnO and N-GO-ZnO-5 are depicted in Fig. 1A–C respectively, while Fig. 1D is relative to the EDX profile of the N-GO-ZnO-5 material. The predominant morphology of the samples is almost homogeneous spherical nanoparticles sized between ca. 110 and 130 nm in the form of aggregates for ZnO and N-ZnO samples (see Fig. 1A and B). The N-GO-ZnO-5 exhibited nanoparticles of relatively uniform size densely packed on the surface of GO nanosheets (see Fig. 1C). The coupling of N-ZnO onto the surface of GO nanosheets may be attributed to the presence of epoxide together with hydroxide groups on the GO sheets which act as attachment sites for N-ZnO nanoparticles.34 Moreover, the EDX analysis of the N-GO-ZnO-5 (see Fig. 1D) confirmed the composition of this composite material demonstrating the occurrence of C, N, Zn and O. The C peak is attributed to the basal plane of graphene oxide nanosheets, while O is assigned to both ZnO and GO. | ||
| Fig. 1 SEM images for: (A) ZnO; (B) N-ZnO; (C) N-GO-ZnO-5 and (D) N-GO-ZnO-5 EDX analysis. | ||
The TEM images (Fig. 2A–C) of the different materials revealed the occurrence of nanoparticles aggregated in hexagonal-like structures having sizes around 100 nm. This behaviour was especially marked in the case of the N-doped ZnO-5 sample (Fig. 2B). The unit particles embedded in the hexagonal-like structures exhibited much lower sizes around 25 nm for all the samples. As evidenced in Fig. 2C, N-ZnO aggregates were successfully linked on the of GO nanosheets surface (Fig. 2).
 | ||
| Fig. 2 TEM images for: (A) ZnO; (B) N-ZnO and (C) N-GO-ZnO-5. | ||
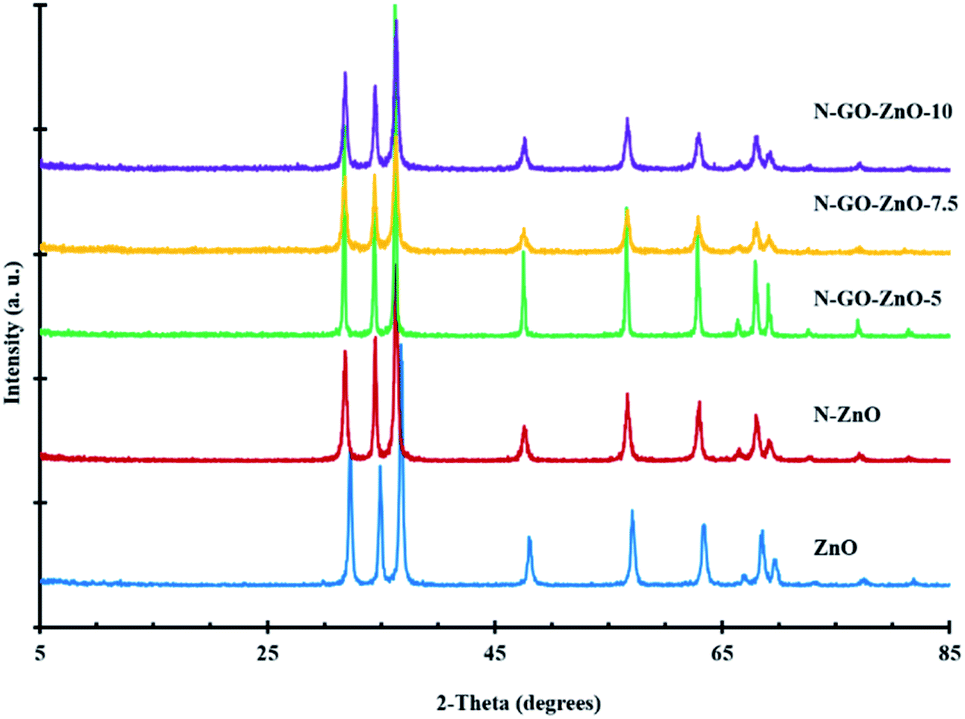 | ||
| Fig. 3 XRD patterns of the synthesised materials. | ||
The X-ray diffractograms of the ZnO, N-ZnO and N-GO-ZnO materials are depicted in Fig. 3. As seen, the diffractograms exhibited prominent XRD lines at 31.6°, 34.3°, 36.1°, 47.4°, 56.4°, 62.7°, 66.4° 67.8° and 69.1°. These lines, indexed respectively as (100), (002), (101), (102), (110), (103), (200), (112) and (201), were assigned to the hexagonal wurtzite phase of ZnO.35 Both doped materials exhibited similar XRD patterns to bare ZnO and no other crystallite phases were detected, indicating that the presence of GO did not result in the growth of new crystals or changes in preferential orientation of ZnO. The average crystallite sizes evaluated using Scherrer's equation were 90, 110 and around 170 nm for ZnO, N-ZnO and N-GO-ZnO materials, respectively.
FTIR spectra registered in the range [300–4000 cm−1] for ZnO, N-ZnO and N-GO-ZnO materials are presented in Fig. 4. All the spectra revealed absorption peaks at 3426 cm−1 corresponding to characteristic O–H stretching and bonding vibration of the water molecule. The peak at 1502 cm−1 for N-doped ZnO sample may correspond to the vibration of the Zn–N bond.35 The other peaks at 1180 cm−1 and 1565 cm−1 were assigned to C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The spectra exhibited also an absorption peak at 500 cm−1, usually attributed to the characteristic absorption of Zn–O bond in zinc oxide.36,37
C bond. The spectra exhibited also an absorption peak at 500 cm−1, usually attributed to the characteristic absorption of Zn–O bond in zinc oxide.36,37
 | ||
| Fig. 4 FTIR spectra of the synthesised materials. | ||
The optical absorption behaviour of the pure ZnO as well as N-ZnO and N-GO-ZnO samples was investigated using UV-visible spectroscopy. As presented in Fig. 5A, the absorbance of the N-GO composite increased even in the visible light region due to the increase of surface electric charge of the oxides, and the modification of the fundamental process of electron–hole pair formation during irradiation.38 Therefore, the presence of N-GO in ZnO can increase the light absorption intensity and range, which is beneficial to the photocatalytic performance.39,40 The band gap energy (Eg) values of the investigated photocatalysts were estimated from the UV-vis data (Fig. 5B) using the following equation:
| (αhν)2 = A(hν − Eg) |
 | ||
| Fig. 5 (A) UV-vis spectra and (B) estimated band gap energies from the Tauc plot. | ||
After doping with N-GO, the obtained spectra exhibited an additional tail-like absorption in the visible region. The band gap energies of the ZnO and N-ZnO materials were very close to 3.20 eV. The N-GO-ZnO-5 material exhibited slightly lower band gap energy estimated to be 3.17 eV. The contribution of nitrogen and graphene oxide to the top of the valence band (VB) of ZnO played a major role in extending the absorption of doped ZnO to the visible region. Interestingly, the band gap energy was evaluated to be ca. 3.05 and 3.02 eV for the N-GO-ZnO-7.5 and N-GO-ZnO-10 materials, respectively. A scrutiny of the literature allowed to note that band gap energy was generally evaluated to be around 3.21 eV for pristine ZnO, while nitrogen doped ZnO exhibited lower values in the vicinity of 3.08 to 2.95 eV.16–19 As for the N-GO doped ZnO, the band gap energy was reported to be close to 2.83–2.90 eV.19
Photoluminescence (PL) spectra were recorded for the five photocatalysts investigated, i.e., ZnO, N-ZnO and N-GO-ZnO-5, 7.5 and 10. The obtained spectra are depicted in Fig. 6. Such characterisation is usually used to assess the effect of nitrogen and GO on the electron–hole recombination rate of ZnO. As seen, the ZnO photocatalyst exhibited the highest photoluminescence emission intensity, thus indicating a faster electron–hole recombination rate. The photoluminescence emission was lowered for the nitrogen doped ZnO. It is worth to notice that the photoluminescence emission was remarkably lowered for the three N-GO-ZnO photocatalysts, indicating a slower electron–hole recombination rate. This behaviour reveals an enhancement in the transfer charges occurring at the interface which eliminates the electron–hole recombination arising at the defect sites. As a result, the generation of photo-induced electrons in the conduction band of N-ZnO nanoparticles is transferred speedily to GO sheets.19 As to be demonstrated later, the three N-GO-ZnO photocatalysts exhibited the best performances in the photodegradation of aqueous acetamiprid.
 | ||
| Fig. 6 Photoluminescence spectra of the synthesised materials. | ||
The surface elemental composition of the different synthesised photocatalysts was analysed by XPS. Fig. 7A depicts the wide survey spectra of bare ZnO, N-ZnO and N-GO-ZnO-5 confirming the occurrence of the different constituting elements present on the materials surfaces, i.e., Zn, O, N and C. In the case of pure ZnO, the detected carbon is attributed to the adsorption of carbonaceous species upon exposure to the ambient atmosphere. The Zn 2p3/2 high resolution spectra indicated a clear shift in the binding energies which were evaluated to 1022.52, 1023.30 and 1022.70 eV for the ZnO, N-ZnO and N-GO-ZnO-5 samples, respectively (see Fig. 7 (inset)). Such variation in the binding energies indicates that the chemical environment of the Zn surface atoms was modified upon incorporation of nitrogen and graphene oxide on the ZnO material. Moreover, the occurrence of nitrogen in the prepared nanocomposites N-ZnO and N-GO-ZnO-5 with the N 1s peak was detected at ca. 399.96 eV (Fig. 7). Indeed, the nitrogen atomic surface concentration was 2.7% for the N-ZnO material, while it oscillated between 0.9 and 2.5% for the N-GO-ZnO materials.
 | ||
| Fig. 7 (A) XPS survey spectra; (B) high-resolution Zn 2p3/2 core level spectra of ZnO, N-ZnO and N-GO-ZnO-5 materials. | ||
The electrochemical impedance spectroscopy and photocurrent density measurements were conducted to evaluate the internal resistance and photocurrent for the charge transfer process of the synthesised N-ZnO and N-GO-ZnO-5 materials. To understand the improved performance of these two materials, the photoelectrochemical behaviour from the photocurrent was evaluated. Nyquist plots were measured as shown in Fig. 8A. It is well known that the smaller arc of Nyquist plots represents the higher separation efficiency of the photo-generated charges. Clearly, the arc of N-ZnO material decreased dramatically after incorporating GO on the N-ZnO, thus indicating the high separation of photo-induced charges on the N-GO-ZnO-5 photocatalyst. This reveals that the prepared nanocomposite could facilitate charge carrier's separation leading to the improved photocatalytic performances of the GO containing photocatalyst as previously reported for graphene–ZnO nanocomposites.41 In Fig. 8B, it is evident that the short-circuit current of N-GO-ZnO-5 was largely higher than that of N-ZnO. Such a remarkable enhancement of the photocurrent is attributed to the higher photo-absorption and better carrier transport in N-GO-ZnO-5 material.42
 | ||
| Fig. 8 (A) Electrochemical impedance spectroscopy; (B) photocurrent of N-ZnO and N-GO-ZnO-5 materials. | ||
Textural properties of the ZnO, N-ZnO and N-GO-ZnO materials were investigated using N2 adsorption–desorption volumetric analysis. As depicted in Fig. 9, the obtained isotherms were attributed to the type IV with H3 hysteresis loop according to the IUPAC classification. The BET specific surface area varied from 17.22 to 47.60 m2 g−1. The highest value was observed for the N-GO-ZnO-10 material. This greater surface area along with highly porous structure provide more active sites and abundant transport pathways for the adsorption of the acetamiprid molecules, leading to superior photocatalytic performance as reported in the next section (Table 1).
 | ||
| Fig. 9 Nitrogen physisorption isotherms of ZnO, N-ZnO and N-GO-ZnO materials. | ||
| Material | Surface area (m2 g−1) | Pore volume (mL g−1) | Average pore size (nm) |
|---|---|---|---|
| ZnO | 20.85 | 0.16 | 5.14 |
| N-ZnO | 17.22 | 0.17 | 3.92 |
| N-GO-ZnO-5 | 29.69 | 0.13 | 1.92 |
| N-GO-ZnO-7.5 | 41.19 | 0.14 | 4.81 |
| N-GO-ZnO-10 | 47.60 | 0.12 | 7.71 |
Photocatalytic efficiency on the degradation of acetamiprid
Acetamiprid degradation reactions were carried out under two different conditions, i.e., photolysis (no catalyst) and heterogeneous photocatalysis over ZnO catalyst (see Fig. 10). | ||
| Fig. 10 Acetamiprid photodegradation time profiles in the absence and over bare ZnO, N-ZnO and N-GO-ZnO materials. Catalyst loading = 0.2 g L−1; temperature = 25 °C; C0 = 15 ppm; error bars indicate standard deviation. | ||
In the absence of a catalyst, the photodegradation of acetamiprid under visible light occurred at a lower rate than that observed in the photocatalytic reaction, and only 12% conversion of the initial concentration of acetamiprid was reached after 5 hours of reaction. Under otherwise the same operating conditions, the presence of ZnO catalyst allowed reaching 38% acetamiprid conversion after 5 hours of reaction. Although the uncatalysed photolysis reaction did occur, clearly ZnO photocatalyst enhanced appreciably the acetamiprid degradation.
The photodegradation of acetamiprid was investigated comparatively in the presence of bare ZnO as well as N-ZnO and N-GO-ZnO under otherwise the same operating conditions (see Fig. 10). As seen, after 5 hours of reaction under visible light, while only 38% acetamiprid degradation was reached in the presence of bare ZnO, 68% degradation was obtained in the presence of N-doped ZnO. More interestingly, almost complete (99.4%) acetamiprid degradation was achieved in the presence of the three N-GO-ZnO photocatalysts with no major difference between them. The high degradation efficiency under visible light radiation of the N-GO doped ZnO can be explained by the presence of N surface species, such as CN, rich in electrons providing a great potential to activate the catalyst. Moreover, N functional groups conjugating with the long-pair electrons can activate the effective sites of the sp2 carbon of graphene oxide with abundant free-flowing π-electrons of nitrogen species.43 Furthermore, charge transport by graphene oxide leads to increased lifetimes of photogenerated electrons and holes, and thus promotes the formation of hydroxyl and superoxide radicals, which are responsible for the oxidation and degradation of acetamiprid molecules.44
Effect of catalyst loading
The influence of N-GO-ZnO-5 loading, within the range of 0.1–0.4 g L−1 is presented in Fig. 11A. As seen, conversion of acetamiprid reached ca. 50% with the lowest catalyst loading (0.1 g L−1) within 300 min of reaction. A two-fold increase of the catalyst loading (from 0.1 to 0.2 g L−1) allowed reaching almost 100% acetamiprid conversion after 300 min of reaction. However, a further increase of the catalyst loading to 0.4 g L−1 translated into acetamiprid conversion of ca. 85%. This behaviour may be attributed to the catalyst particles agglomeration, thus making the active sites unavailable due to the opacity induced in the reaction medium (suspension). Hence, the catalyst becomes less illuminated which decreases the photocatalytic conversion due to a lower production rate of hydroxyl radicals.45 | ||
| Fig. 11 (A) Effect of N-GO-ZnO-5 photocatalyst loading; temperature = 25 °C; C0 = 15 ppm; (B) effect of acetamiprid initial concentration. N-GO-ZnO-5 catalyst loading = 0.2 g L−1; temperature = 25 °C. Error bars indicate standard deviation. | ||
Effect of acetamiprid initial concentration
The effect of the initial pesticide concentration was investigated in the range 5–20 ppm at ambient temperature over N-GO-ZnO-5 at a loading of 0.2 g L−1. As evidenced in Fig. 11B, pesticide was efficiently degraded (more than 85%) when the initial concentration was below 15 ppm. However, with further increase in pesticide concentration, the degradation was less efficient reaching a maximum conversion of 68% after 5 h of reaction. This behaviour can be due to the lack of available surface-bound reactive oxygen species on the photocatalyst surface when the pesticide initial concentration is higher in agreement with previous investigations.46 As presented in Table 2, comparison of the acetamiprid degradation data obtained in this work with those reported in the recent literature under comparable operating conditions, reveals the remarkable performances of the N-GO-ZnO photocatalyst. Furthermore, it is worth to mention that acetamiprid is known to be recalcitrant to oxidative degradation.52| Photocatalysta | C0b (mg L−1) | Conversionc (%) | Ref. |
|---|---|---|---|
| a Values in parentheses are for photocatalyst loading (g L−1).b C0 is the acetamiprid initial concentration.c Values in parentheses are for the reaction time (min). | |||
| C3N4/HPW (0.7) | 10 | 58 (180) | 47 |
| g-C3N4 (1) | 5 | 82 (300) | 48 |
| SO4-Ag3PO4 (0.8) | 5 | 50 (60) | 49 |
| C3N4 (0.33) | 50 | 55 (180) | 50 |
| TiO2 (1) | 22 | 38 (80) | 51 |
| N-GO-ZnO-5 (0.2) | 15 | 98 (300) | This work |
Effect of reaction temperature
The effect of temperature on acetamiprid photodegradation over N-GO-ZnO-5 in the range [25–45 °C] under constant agitation (600 rpm), 15 ppm initial pesticide concentration, 0.2 g L−1 catalyst loading, is depicted in Fig. 12A as pesticide conversion time profiles. As seen, while complete removal of the pollutant was achieved within 120 min at 45 °C, almost 300 min were required at 25 °C to get rid of the pesticide under otherwise the same operating conditions. Moreover, when acetamiprid solutions were photodegraded at very short reaction times (2 min) at the three temperatures investigated, initial rates could be evaluated. Considering that the reaction can be assumed to be first order with respect to acetamiprid at very short reaction times, rate constants were estimated. The corresponding Arrhenius plots (Fig. 12B) allowed to determine the activation energies which were 34.45 and 25.04 kJ mol−1 for the uncatalysed and photocatalysed (over N-GO-ZnO-5) reactions, respectively. | ||
| Fig. 12 (A) Photodegradation profiles of the acetamiprid over N-GO-ZnO-5 under visible light at different temperatures. C0 = 15 ppm; catalyst loading = 0.2 g L−1. (B) Arrhenius plots for the catalysed and uncatalysed photoreaction. Error bars indicate standard deviation. | ||
Photocatalyst reusability
The reusability was verified by conducting successive trials of the repeatedly used N-GO-ZnO-5 catalyst (Table 3). Therefore, the used N-GO-ZnO-5 photocatalyst was washed several times with distilled water and alcohol and dried in the oven at 60 °C. The recovered N-GO-ZnO-5 was then reused for a new photodegradation batch, without further treatment. As evidenced, 90% degradation of acetamiprid was still achieved even after five cycles of photodegradation. Clearly, this illustrates the photocatalyst stability and long lasting as it keeps its high photo-degradative performances towards acetamiprid making it suitable for a large-scale use.| Reaction cycle | Acetamiprid conversion (%) |
|---|---|
| 1 | 99.41 |
| 2 | 99.16 |
| 3 | 98.75 |
| 4 | 94.12 |
| 5 | 90.85 |
Conclusions
The photocatalytic degradation of acetamiprid in aqueous solutions was investigated in the presence of ZnO, N-ZnO and N-GO-ZnO photocatalysts under visible light. Both photocatalysts were characterised using powerful and appropriate techniques. The effects of doping as well as different operating conditions such as the photocatalyst loading, acetamiprid initial concentration and temperature were investigated. In the absence of any photocatalyst, only 12% acetamiprid conversion was reached after 5 h of photolysis. The different N-GO-ZnO photocatalysts exhibited the highest activities in comparison to the N-ZnO and bare ZnO materials. Indeed, acetamiprid conversion after 5 h of reaction was almost 100% over N-GO-ZnO-(5, 7.5 and 10), while it levelled off at 68 and 38% over N-ZnO and ZnO photocatalysts, respectively, thus demonstrating the high potential of N-GO-ZnO. This remarkable improvement in the photocatalytic performances was attributed to the synergetic effect of N-doping and the occurrence of oxygen vacancies ascribed to the increased electrical conductivity of graphene oxide.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
Funding provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canada Foundation for Innovation (CFI) are gratefully acknowledged. The authors would like to thank Prof. T. O. Do for the use of the UV-Vis spectrometer, ElS and Autolab PGSTAT204 electrochemical workstation. Also, the authors are grateful for M. J. Becerra for his kind help in the photoelectrochemical measurements.References
- A. M. Cimino, A. L. Boyles, K. A. Thayer and M. J. Perry, Environ. Health Perspect., 2017, 125, 155–162 CrossRef CAS PubMed.
- T. J. Wood and D. Goulson, Environ. Sci. Pollut. Res., 2017, 24, 17285–17325 CrossRef CAS PubMed.
- P. Jeschke, R. Nauen, M. Schindler and A. Elbert, J. Agric. Food Chem., 2011, 59, 2897–2908 CrossRef CAS PubMed.
- D. Fu, S. Zhang, M. Wang, X. Liang, Y. Xie, Y. Zhang and C. Zhang, J. Sci. Food Agric., 2020, 100, 4540–4548 CrossRef CAS PubMed.
- E. Watanabe, T. Yamasaki, Y. Hirakawa, A. Harada, S. Iwasa and S. Miyake, Anal. Methods, 2018, 10, 3162–3169 RSC.
- T. Kanjilal, C. Bhattacharjee and S. Datta, J. Water Process. Eng., 2015, 6, 21–31 CrossRef.
- B. Bethi, S. H. Sonawane, B. A. Bhanvase and S. P. Gumfekar, Chem. Eng. Process., 2016, 109, 178–189 CrossRef CAS.
- F. Zhou, C. Yan, T. Liang, Q. Sun and H. Wang, Chem. Eng. Sci., 2018, 183, 231–239 CrossRef CAS.
- A. Abdelhaleem, W. Chu and X. Liang, Appl. Catal., B, 2019, 244, 823–835 CrossRef CAS.
- K. Wetchakun, N. Wetchakun and S. Sakulsermsuk, J. Ind. Eng. Chem., 2019, 71, 19–49 CrossRef CAS.
- B. M. Rajbongshi, A. Ramchiary and S. Samdarshi, Mater. Lett., 2014, 134, 111–114 CrossRef CAS.
- T. T. Guaraldo, J. Wenk and D. Mattia, Adv. Sustainable Syst., 2021, 5, 2000208 CrossRef.
- K. Sridharan, E. Jang, Y. M. Park and T. Park, Chem.–Eur. J., 2015, 21, 19136–19141 CrossRef CAS PubMed.
- M. Mzoughi, W. W. Anku, S. O. B. Oppong, S. K. Shukla, E. S. Agorku and P. P. Govender, Adv. Mater. Lett., 2016, 7, 946–950 CrossRef CAS.
- S. Lautenschlaeger, M. Hofmann, S. Eisermann, G. Haas, M. Pinnisch, A. Laufer and B. K. Meyer, Phys. Status Solidi B, 2011, 248, 1217–1221 CrossRef CAS.
- P. K. Baitha and J. Manam, J. Rare Earths, 2015, 33, 805–813 CrossRef CAS.
- S. Y. Sawant and M. H. Cho, RSC Adv., 2015, 5, 97788–97797 RSC.
- H. Sudrajat and S. Hartuti, Mater. Res. Bull., 2018, 102, 319–323 CrossRef CAS.
- C. N. Peter, W. W. Anku, R. Sharma, G. M. Joshi, S. K. Shukla and P. P. Govender, Ionics, 2019, 25, 327–339 CrossRef CAS.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- E. Kusiak-Nejman, A. Wanag, J. Kapica-Kozar, Ł. Kowalczyk, M. Zgrzebnicki, B. Tryba, J. Przepiórski and A. W. Morawski, Catal. Today, 2020, 357, 630–637 CrossRef CAS.
- Z. Durmus, B. Z. Kurt and A. Durmus, ChemistrySelect, 2019, 4, 271–278 CrossRef CAS.
- M. Suresh and A. Sivasamy, J. Mol. Liq., 2020, 317, 114112 CrossRef CAS.
- K. Dai, L. Lu, C. Liang, J. Dai, G. Zhu, Z. Liu, Q. Liu and Y. Zhang, Mater. Chem. Phys., 2014, 143, 1410–1416 CrossRef CAS.
- A. Valour, F. Cheviré, F. Tessier, F. Grasset, B. Dierre, T. Jiang, E. Faulques, L. Cario and S. Jobic, Solid State Sci., 2016, 54, 30–36 CrossRef CAS.
- Q. Gan, K. Zhao, S. Liu and Z. He, Electrochim. Acta, 2017, 250, 292–301 CrossRef CAS.
- S. Li, W. Wang, X. Zeng and X. Ma, Desalin. Water Treat., 2015, 54, 1925–1938 CrossRef CAS.
- M. Kaur, M. Kaur and V. K. Sharma, Adv. Colloid Interface Sci., 2018, 259, 44–64 CrossRef CAS PubMed.
- H. Lee, K. Paeng and I. S. Kim, Synth. Met., 2018, 244, 36–47 CrossRef CAS.
- P. M. Perillo, M. N. Atia and D. F. Rodríguez, Phys. E, 2017, 85, 185–192 CrossRef CAS.
- M. B. Andrade, T. R. T. Santos, M. F. Silva, M. F. Vieira, R. Bergamasco and S. Hamoudi, Sep. Sci. Technol., 2019, 54, 2653–2670 CrossRef CAS.
- S. Yousaf, T. Kousar, M. B. Taj, P. O. Agboola, I. Shakir and M. F. Warsi, Ceram. Int., 2019, 45, 17806–17817 CrossRef CAS.
- S. Danwittayakul, M. Jaisai and J. Dutta, Appl. Catal., B, 2015, 163, 1–8 CrossRef CAS.
- S. Park, K.-S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- A. Sahai and N. Goswami, AIP Conf. Proc., 2014, 40, 14569–14578 CAS.
- A. B. Lavand and Y. S. Malghe, J. Australas. Ceram. Soc., 2015, 3, 305–310 CrossRef.
- P. Singh, S. Gautam, P. Shandilya, B. Priya, V. P. Singh and P. Raizada, Adv. Mater. Lett., 2017, 8, 229–238 CrossRef CAS.
- N. Chandel, K. Sharma, A. Sudhaik, P. Raizada, A. Hosseini-Bandegharaei, V. K. Thakur and P. Singh, Arabian J. Chem., 2020, 13, 4324–4340 CrossRef CAS.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- Y. L. Min, K. Zhang, W. Zhao, F. C. Zheng, Y. C. Chen and Y. G. Zhang, Chem. Eng. J., 2012, 193–194, 203–210 CrossRef CAS.
- M. Salem, S. Akir, I. Massoudi, Y. Litaiem, M. Gaidi and K. Khirouni, J. Alloys Compd., 2018, 767, 982–987 CrossRef CAS.
- D. Neena, K. K. Kondamareddy, M. Humayun, V. B. Mohan, D. Lu and W. Gao, Appl. Surf. Sci., 2019, 488, 611–619 CrossRef CAS.
- L. Wei, P. Wang, Y. Yang, R. Luo, J. Li, X. Gu, Z. Zhan, Y. Dong, W. Song and R. Fan, J. Nanopart. Res., 2018, 20, 110 CrossRef.
- X. Liu, L. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Sun, Catal. Sci. Technol., 2011, 1, 1189–1193 RSC.
- Y. Qiu, M. Yang, H. Fan, Y. Xu, Y. Shao, X. Yang and S. Yang, Mater. Lett., 2013, 99, 105–107 CrossRef CAS.
- A. Tabasum, M. Zahid, H. N. Bhatti and M. Asghar, Mater. Res. Express, 2019, 6, 015608 CrossRef.
- Y. Sun and X. Liu, Appl. Surf. Sci., 2019, 485, 423–431 CrossRef CAS.
- X. Liu, C. Li, B. Zhang, M. Yuan, Y. Ma and F. Kong, Chemosphere, 2020, 253, 126672 CrossRef CAS PubMed.
- Y. Lee, J. Kang, S. Park, C. Lee, J. Moon and P. J. J. Alvarez, Chem. Eng. J., 2020, 402, 126183 CrossRef CAS.
- M. Padervand, S. Ghasemi, S. Hajiahmadi and C. Wang, Appl. Surf. Sci., 2021, 544, 148939 CrossRef CAS.
- A. Khan, M. M. Haque, N. A. Mir, M. Muneer and C. Boxall, Desalination, 2010, 261, 169–174 CrossRef CAS.
- T. Gonzalez, J. R. Dominguez and S. Correia, J. Environ. Manage., 2020, 261, 110156 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |