
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: The anchoring of a Cu(II)–salophen complex on magnetic mesoporous cellulose nanofibers: green synthesis and an investigation of its catalytic role in tetrazole reactions through a facile one-pot route†
Pouya Ghamari kargar a and
Ghodsieh Bagherzade*b
a and
Ghodsieh Bagherzade*b
aDepartment of Chemistry, Faculty of Sciences, University of Birjand, Birjand, 97175-615, Iran. E-mail: P.ghamari71@gmail.com
bDepartment of Chemistry, Faculty of Sciences, University of Birjand, Birjand, 97175-615, Iran. E-mail: gbagherzade@gmail.com; bagherzade@birjand.ac.ir; Fax: +98 56 32345192; Tel: +98 56 32345192
First published on 27th May 2021
Abstract
Today, most synthetic methods are aimed at carrying out reactions under more efficient conditions and the realization of the twelve principles of green chemistry. Due to the importance and widespread applications of tetrazoles in various industries, especially in the field of pharmaceutical chemistry, and the expansion of the use of nanocatalysts in the preparation of valuable chemical reaction products, we decided to use an (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst in this project. In this study, the synthesis of the nanocatalyst (Fe3O4@NFC@NSalophCu)CO2H was explained in a step-by-step manner. Confirmation of the structure was obtained based on FT-IR, EDX, FE-SEM, TEM, XRD, VSM, DLS, TGA, H-NMR, and CHNO analyses. The catalyst was applied to the synthesis of 5-substituted-1H-tetrazole and 1-substituted-1H-tetrazole derivatives through multi-component reactions (MCRs), and the performance was assessed. With advances in science and technology and increasing environmental pollution, the use of reagents and methods that are less dangerous for the environment has received much attention. Therefore, following green chemistry principles, with the help of the (Fe3O4@NFC@NSalophCu)CO2H salen complex as a nanocatalyst that is recyclable, cheap, safe, and available, the use of water as a green solvent, and reduced reaction times, the synthesis of tetrazoles can be achieved.
Introduction
Multi-component reactions are reactions that start by combining several reagents at the same time and create the target molecule in the same pot; they reduce energy consumption and solvent loss, and also show reduced reaction times, ultimately having the advantages of efficiency, simplicity, and atom economy.1 Nitrogen-containing heterocyclic and aromatic compounds are necessary and important structural components in chemical and biochemical compounds, and these heterocyclic compounds have acquired immense importance recently.2,3 Tetrazoles are a group of nitrogen-rich heterocyclic compounds that have a high specific heat density due to the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–N bonds. These compounds are highly regarded as high-energy green materials for use in engines, gas production, and explosive propellants due to their high potential energies and low impact and friction sensitivities. Tetrazoles are used in coordination chemistry,4 explosives, photography, information-recording systems,5 in materials engineering as anti-corrosion agents, and many other areas. In addition, they are widely used in the fields of medicinal and agricultural chemistry.6 1-Substituted-1-H-tetrazoles and 5-substituted-1H-tetrazoles, which represent significant classes of heterocycles, have attracted considerable interest in recent years because of their wide utility7 and because of their hopeful biological properties, particularly their antifungal,8 antiulcer,9 antinociceptive,10 anti-HIV,11 antiallergic,12 anti-inflammatory,13 antiproliferative,14 hormonal,15 carboxylic acid isostere,16 herbicidal,17 diuretic,18 and anticancer19 potential (Scheme 1). Due to the many benefits that these compounds have shown, various synthesis approaches for tetrazole derivatives have been well-documented in the literature. In general, the easiest and most versatile method for the synthesis of 5-substituted-1H-tetrazoles is the [3 + 2] cyclization of nitriles and azide in the presence of appropriate catalysts and various solvents. Several new catalysts have been investigated, such as zinc salts,20 β-cyclodextrin,21 Fe(OAc)2,22 CdCl2,23 AgNO3,24 SiO2–H3BO3,25 Cu–MCM-41,26 PCl5,27 Zn/Al hydrotalcite,28 Pd(OAc)2/ZnBr,29 Fe3O4@MCM-41–SB–Cu,30 Fe3O4@L-lysine–Pd(0),31 and copper triflates.32
N and C–N bonds. These compounds are highly regarded as high-energy green materials for use in engines, gas production, and explosive propellants due to their high potential energies and low impact and friction sensitivities. Tetrazoles are used in coordination chemistry,4 explosives, photography, information-recording systems,5 in materials engineering as anti-corrosion agents, and many other areas. In addition, they are widely used in the fields of medicinal and agricultural chemistry.6 1-Substituted-1-H-tetrazoles and 5-substituted-1H-tetrazoles, which represent significant classes of heterocycles, have attracted considerable interest in recent years because of their wide utility7 and because of their hopeful biological properties, particularly their antifungal,8 antiulcer,9 antinociceptive,10 anti-HIV,11 antiallergic,12 anti-inflammatory,13 antiproliferative,14 hormonal,15 carboxylic acid isostere,16 herbicidal,17 diuretic,18 and anticancer19 potential (Scheme 1). Due to the many benefits that these compounds have shown, various synthesis approaches for tetrazole derivatives have been well-documented in the literature. In general, the easiest and most versatile method for the synthesis of 5-substituted-1H-tetrazoles is the [3 + 2] cyclization of nitriles and azide in the presence of appropriate catalysts and various solvents. Several new catalysts have been investigated, such as zinc salts,20 β-cyclodextrin,21 Fe(OAc)2,22 CdCl2,23 AgNO3,24 SiO2–H3BO3,25 Cu–MCM-41,26 PCl5,27 Zn/Al hydrotalcite,28 Pd(OAc)2/ZnBr,29 Fe3O4@MCM-41–SB–Cu,30 Fe3O4@L-lysine–Pd(0),31 and copper triflates.32
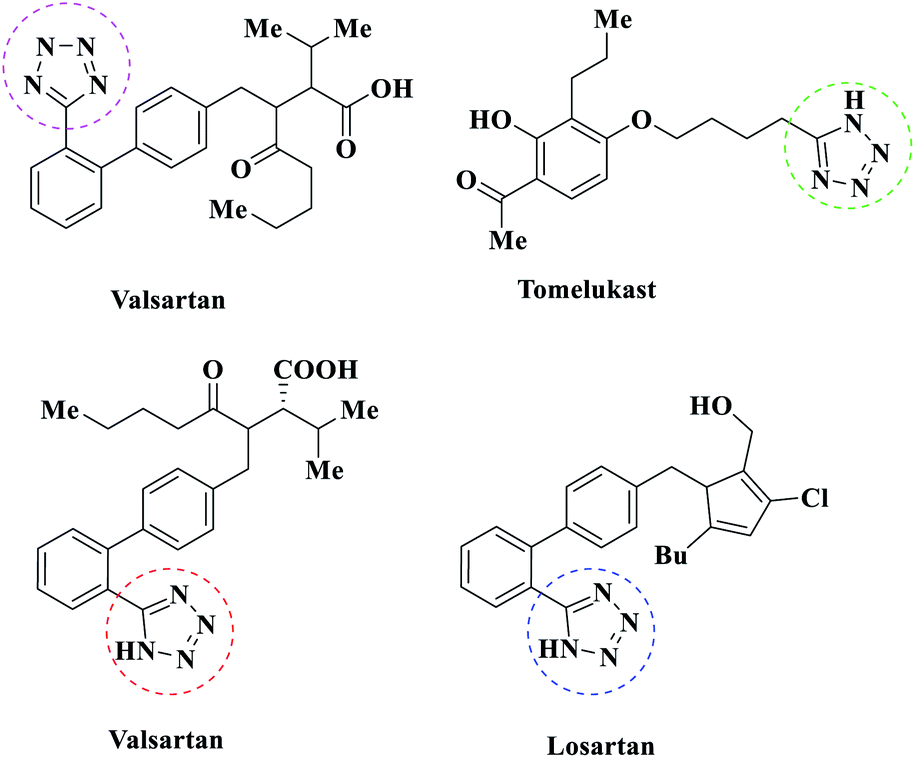 | ||
| Scheme 1 Drugs on the market with a tetrazole moiety. | ||
Although these methods have good advantages, some of them also have drawbacks, including the use of dimethylformamide as a solvent, the need for harsh reaction conditions and long reaction times, the use of toxic and corrosive reagents, difficulties in the separation and recovery of catalysts, low yields, the need for tedious work-up procedures and expensive moisture-sensitive reaction conditions, and the presence of hydrazoic acid, which can be highly toxic and explosive, as well as volatile. Thus, the development of a simple, convenient, and environmentally benign method for synthesizing tetrazoles remains necessary. However, to achieve good results in the reaction process, it is crucial to choose a suitable heterogeneous catalyst and an environmentally benign method that can overcome all these problems. For minimizing chemical waste, increasing energy efficiency, and obtaining better economy, the use of nanocatalysts that comply with the principles of green chemistry is recommended. These heterogeneous catalysts can be easily recovered from the reaction medium, but the activity and selectivity of the catalysts can decrease due to the existence of limited active sites on the surfaces. Nanoscale supports can be used to solve this problem. Due to the non-toxicity of Fe3O4, it is more often used as a magnetic substance in catalyst supports than other metallic counterparts.33,34 It is notable that nanomagnetic compounds have had major effects on the development of other areas, such as medications, drug delivery, and chemical modifications.35 A clear path in organic chemistry synthesis has been created due to the modifications of the surfaces of magnetic nanoparticles. Also, their physical properties, comprising high surface areas, a superparamagnetic nature, low toxicity, give them potential applications in various fields, such as biotechnology, medicine, drug delivery, and environmental science.36 However, pure magnetic particles oxidate in the presence of air and the magnetic properties are altered. Therefore, proper coverage is important to prevent such an issue.37,38 Thus, much consideration has been paid to the synthesis of magnetic core–shell structures via the use of a cellulose layer around the synthesized nanoparticles. Nanofiber cellulose (NFC) coatings can give magnetic nanoparticles an easily adjustable surface, which may be used to graft various desirable functional groups. Cellulose is abundantly available from diverse natural sources, such as plants, bacteria, tunicates, and algae, making it a green polymer suitable for use in sustainable materials engineering.39–41
From a “green chemistry” perspective, substantial attempts to find alternatives to organic solvents, such as solvent-free methods or those carried out in the presence of water, have been made, due to the reduced pollution, low cost, safety, and ease of processing and use. They are of industrial importance. The first 11 principles of green chemistry state that preventing the generation of waste is better than refining or cleaning-up waste after generation. Here, our research team has presented a simple and efficient method for the preparation of 1-H-tetrazole and 5-H-tetrazole derivatives, considering the importance of green chemistry and the issues mentioned above. We have illustrated it via forming successive C–N and N–N bonds catalyzed by a new heterogeneous (Fe3O4@NFC@NSalophCu)CO2H catalyst. The interesting thing is that (Fe3O4@NFC@NSalophCu)CO2H has shown good catalytic activity. We reacted a wide range of aromatic amines and aromatic aldehydes with electron-donating and electron-withdrawing groups to obtain the desired products. We also reported the use of recyclable heterogeneous catalysts with an aqueous solvent as a green solvent for the synthesis of 5H-tetrazoles and under neat conditions for obtaining 1H-tetrazole derivatives. The ease of operation, short time required, and high efficiency are other features of this method, which provides an efficient way to synthesize different heterocycles (Scheme 2).
 | ||
| Scheme 2 The synthesis of differently structured 5-substituted-1H-tetrazoles and 1-substituted-1H-tetrazoles in the presence of (Fe3O4@NFC@NSalophCu)CO2H in aqueous media and without solvent. | ||
Results and discussion
The (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst was prepared in multiple steps. In the first step, Fe3O4@NFC magnetic nanoparticles were synthesized via a sol–gel method. Then, the Fe3O4@NFC core–shell particles were reacted with (3-aminopropyl)triethoxysilane (APTES) as a spacer to obtain Fe3O4@NFC@APTES. After the reflux of an ethanolic solution of copper acetate with a Schiff base ligand, the salen complex was prepared. In this procedure, 5-chloromethyl salicylaldehyde was used to form 3,5-bis(((E)-5-(chloromethyl)-2-hydroxybenzylidene)amino)benzoic acid as the novel ligand, and this was then reacted with copper acetate to obtain complex V (Scheme 3). Finally, the salen complex [(5-Cl-Saloph)Cu(II)]CO2H was added to Fe3O4@NFC@APTES to generate the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst (Scheme 3). After the successful synthesis of the salen catalyst (Scheme 3), the magnetic nanocatalyst was characterized using various standard physicochemical techniques, such as Fourier-transform infrared spectroscopy (FT-IR), powder X-ray diffraction analysis (PXRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), thermogravimetric analysis (TGA), inductively coupled plasma optical emission spectrometry (ICP-OES), dynamic light scattering (DLS), vibrating-sample magnetometer (VSM), and energy-dispersive X-ray (EDS) analysis. FT-IR spectroscopy analysis, for investigating the steps of catalyst synthesis and to confirm the formation of the expected functional groups, has been done. The spectra of 5-chloromethyl salicylaldehyde, the salen Schiff base, [(5-Cl-Saloph)Cu(II)]CO2H, Fe3O4, Fe3O4@NFC, and the (Fe3O4@NFC@NSalophCu)CO2H nanocomposite are presented in Fig. 1 and 2. In Fig. 1a, the characteristic absorption bands observed at 731, 1107, 1450–1580, 1652, 2809, 2980, 3100, and 3250 cm−1 are related to C–Cl, C–O, CC aromatic, CO, C–H aldehyde, C–H aliphatic, C–H aromatic, and O–H bonds in 2-hydroxy-5-chloromethyl benzaldehyde. Additionally, in Fig. 1b, the signals located at around 1650 (CN), 1710 (CO; carboxylic acid), and 2500–3410 cm−1 (O–H; carboxylic acid) can be attributed to the structure of the Schiff base (5-Cl-Saloph)CO2H. What was expected to be observed is that the formation of the metal complex would cause the strong adsorption of the CN and O–H groups at 1650 and 3250 cm−1 seen in the spectrum in Fig. 1b to be moved to lower wavenumbers.
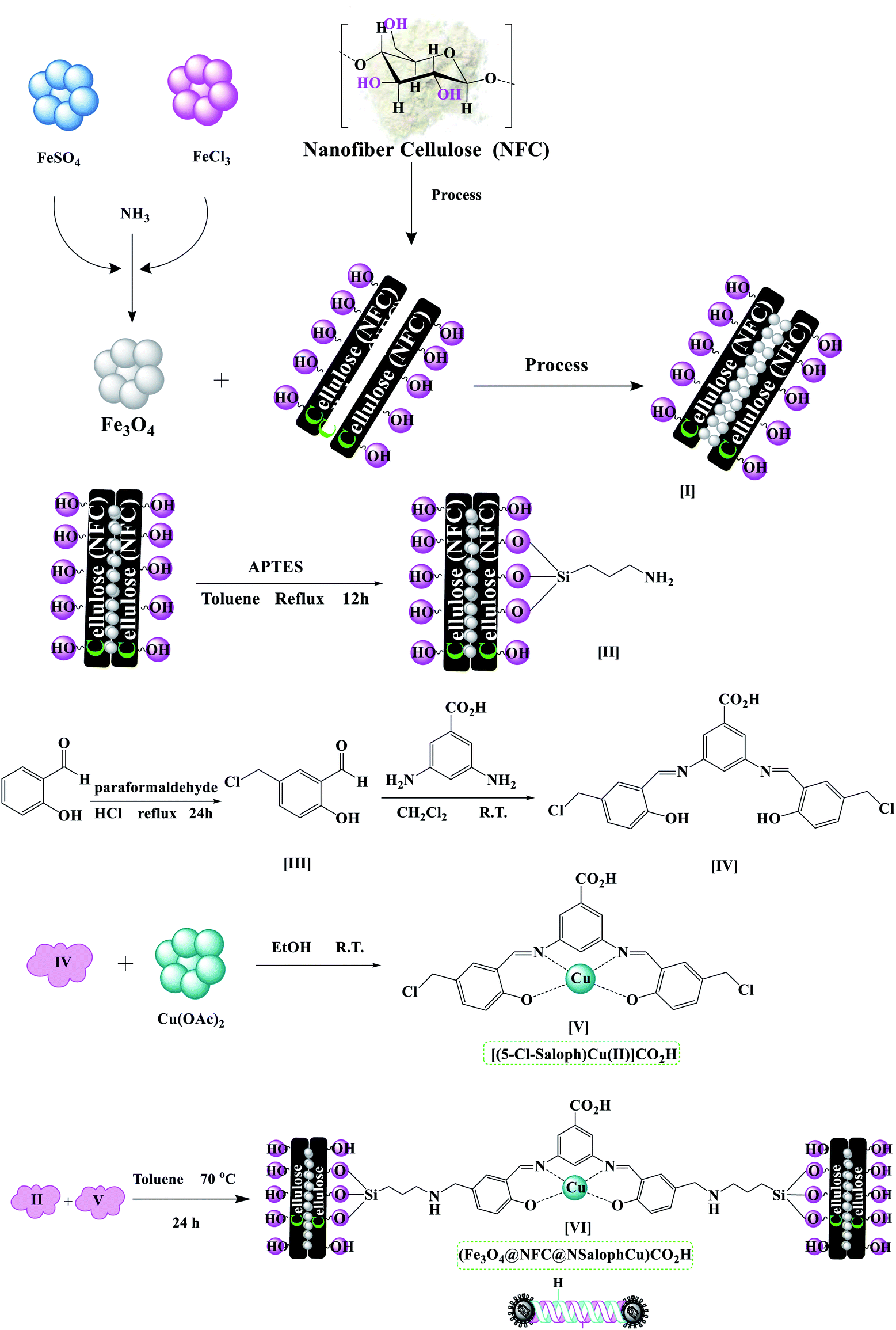 | ||
| Scheme 3 The preparation of the catalyst (Fe3O4@NFC@NSalophCu)CO2H. | ||
 | ||
| Fig. 1 FT-IR spectra of the synthesized nanocomposites: (a) 5-chloromethyl salicylaldehyde, (b) (5-Cl-Saloph)CO2H, and (c) (5-Cl-Saloph)Cu(II) CO2H. | ||
 | ||
| Fig. 2 FT-IR spectra of the synthesized nanocomposites: (d) Fe3O4, (e) Fe3O4@NFC, (f) Fe3O4@NFC@APTES, and (g) (Fe3O4@NFC@NSalophCu)CO2H. | ||
As observed in Fig. 1c, the positions of these adsorption peaks (CN and O–H) are reduced by 17 cm−1. This indicates the participation of azomethine nitrogen in bonding with metal ions. It also confirms the coordination of C-based double bonds with the metal through nitrogen. Also, the two absorption bands at 448 and 641 cm−1 could be assigned to Cu–O and Cu–N stretching, respectively (Fig. 2g), because the connection of each atom to the metal transmits electric charge to the metal and the empty orbitals of the metal-containing layer. This weakens the other bonds involving the corresponding atom. The FT-IR spectrum of Fe3O4 nanoparticles with two characteristic peaks at 3402 cm−1 and 578 cm−1 related to O–H and Fe–O stretching bands, respectively, confirms the formation of Fe3O4 nanoparticles (Fig. 2d). As mentioned earlier, NFC is a natural product, therefore, in the IR spectrum of Fe3O4@NFC (Fig. 2e), the stretching vibrations of C–O and C–H bonds, hydroxyl functional groups, and the Fe–O bonds of Fe3O4 should be observed.
Thus, in the Fe3O4@NFC spectrum (Fig. 2e), there are the absorption bands at 1100 and 2990 cm−1 (the stretching vibrations of C–O and C–H from cellulose), and 574 and 3200 cm−1 (the stretching vibrations of Fe–O and O–H in Fe3O4). As envisaged (Fig. 2f), several new bands become apparent at 1100, 1200, and 3200 cm−1 that correspond to the stretching vibrations of the C–N and Si–O functional groups and NH2 bonds, respectively, demonstrating the surface chemical modification of Fe3O4@NFC with (3-aminopropyl)triethoxysilane (APTES).
The crystalline structures of the Fe3O4 particles, Fe3O4@NFC, and (Fe3O4@NFC@NSalophCu)CO2H were determined via powder X-ray diffraction (PXRD) studies. As shown in Fig. 3, it can be seen that the characteristic broad diffraction peaks shown in the wide-angle PXRD patterns can be indexed to the cubic spinel magnetite Fe3O4 crystal structure, which shows diffraction peaks indicating a crystallized structure at 2θ values of 30.3°, 35.8°, 43.6°, 54.8°, 57.3°, and 63.2°, which can be assigned to the (220), (311), (400), (422), (511), and (440) planes respectively.42,43 Also, the XRD pattern of CuO (Fig. 3) shows peaks at 2θ values of 32.07°, 35.47°, 35.82°, 48.72°, 52.97°, 58.02°, 62.47°, 67.87°, and 69.32°, corresponding to the (110), (111), (022), (202), (202), (113), (022), (220), and (222) crystallographic phases.44,45 The low-angle PXRD pattern of (Fe3O4@NFC@NSalophCu)CO2H was also investigated, as depicted in Fig. 3. The PXRD pattern of (Fe3O4@NFC@NSalophCu)CO2H prepared via the Stöber process shows pronounced diffuse peaks from 20–25° that appear because of amorphous cellulose.46 The PXRD pattern of (Fe3O4@NFC@NSalophCu)CO2H, with immobilized Fe3O4 and Cu(II), shows characteristic peaks whose relative intensities match well with the reported PXRD pattern of Fe3O4 magnetite. In addition, these results indicate that the crystal structure of the (Fe3O4@NFC@NSalophCu)CO2H nanoparticles is not changed upon modification with [(5-Cl-Saloph)Cu(II)]CO2H, which means that the copper sites are in their Cu(II) state. With these explanations, these results imply that the spinel structure of Fe3O4 and the existence of Cu(II) have been retained during the process of catalyst preparation.
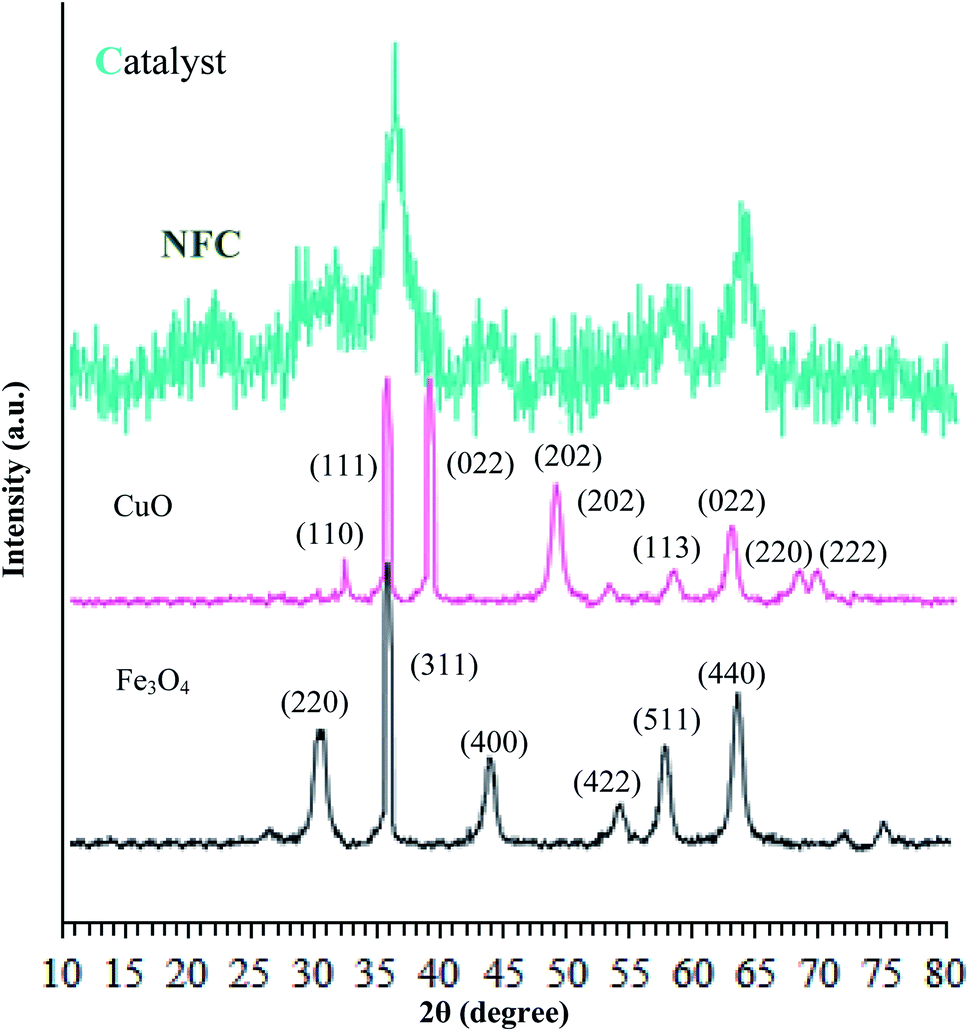 | ||
| Fig. 3 Low angle X-ray diffraction patterns of Fe3O4, Cu(II), and the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst. | ||
SEM and DLS analysis were employed to obtain visual images of the supported catalyst and to find out the shape, morphology, and particle distribution; also, EDX analysis was conducted to qualitatively determine the elements in the prepared samples. The SEM images, as shown in Fig. 4, reveal that Fe3O4 (magnetic), Fe3O4@NFC (core–shell), and (Fe3O4@NFC@NSalophCu)CO2H (the salen complex nanocatalyst) have almost uniform and spherical morphologies, ranging in size from 4 to 31 nm, indicating good agreement with the calculated results from the Debye–Scherrer equation. Some aggregation occurs due to the magnetic properties of the nanoparticles. From the EDX analysis of Fe3O4 (Fig. 4a; magnetic particles), Fe3O4@NFC (Fig. 4b; core–shell particles), and (Fe3O4@NFC@NSalophCu)CO2H (Fig. 4c; the salen complex nanocatalyst), the results displayed the presence of Cu, Fe, Si, O, N, and C elements, which could be acceptable evidence of the modification of the Fe3O4 surface by the nanocatalyst.
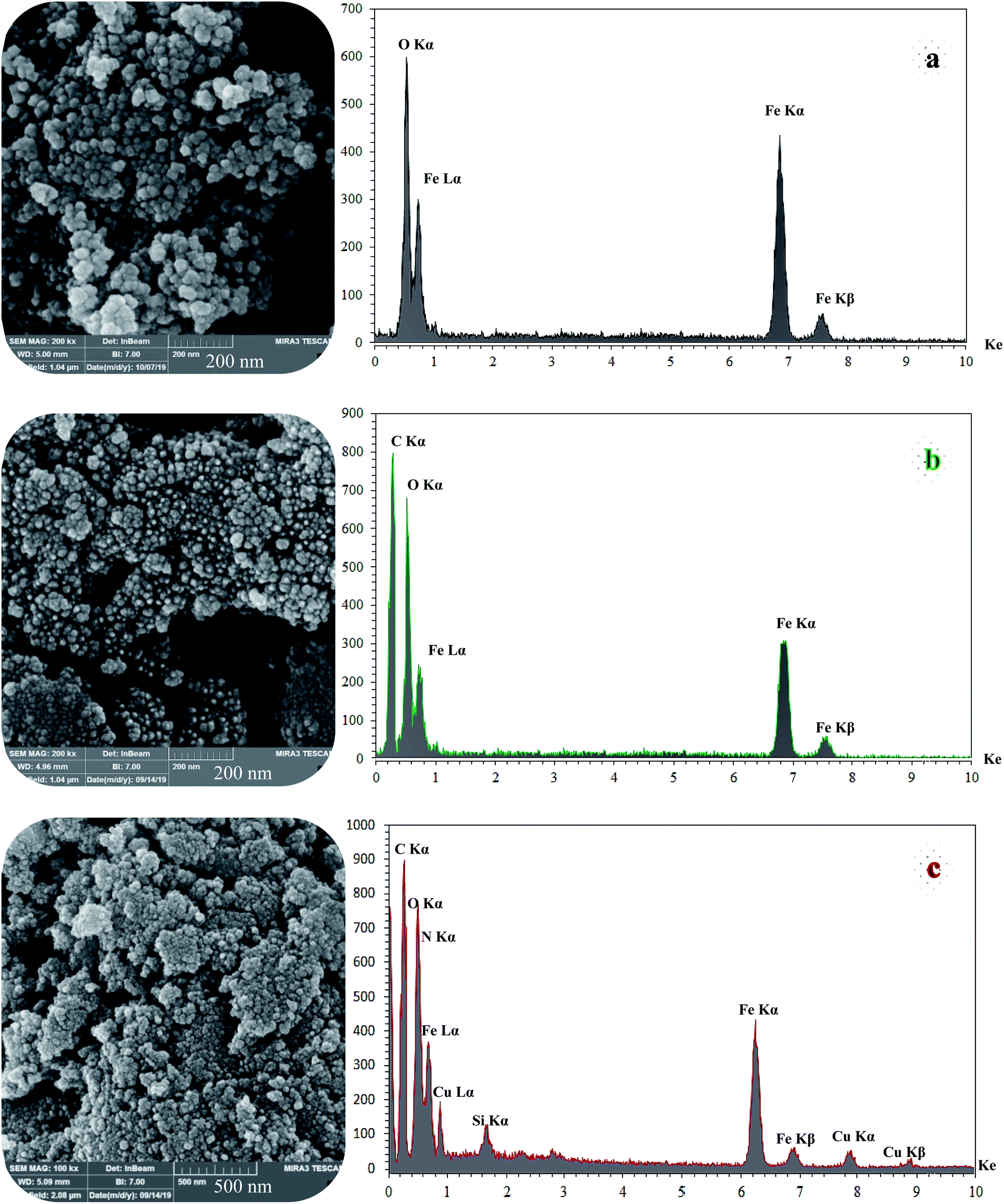 | ||
| Fig. 4 FE-SEM (left) and EDX (right) images of the different materials: (a) Fe3O4, (b) Fe3O4@NFC, and (c) (Fe3O4@NFC@NSalophCu)CO2H. | ||
Therefore, it can be inferred that the expected elements were loaded onto the magnetic surface (Fe3O4) in (Fe3O4@NFC@NSalophCu)CO2H (Fig. 4c). According to the results from ICP and EDX analysis, the amount of copper in the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst was calculated to be 1.47 mmol g−1. Also, the analyses gave heavy-metal percentages of 45.57 wt%, 2.35 wt%, and 6.95 wt% for Fe, Si, and Cu, respectively. In this study, to examine the size distributions of these nanoparticles, particle-size histograms were prepared via DLS analysis (Fig. 5a–c).
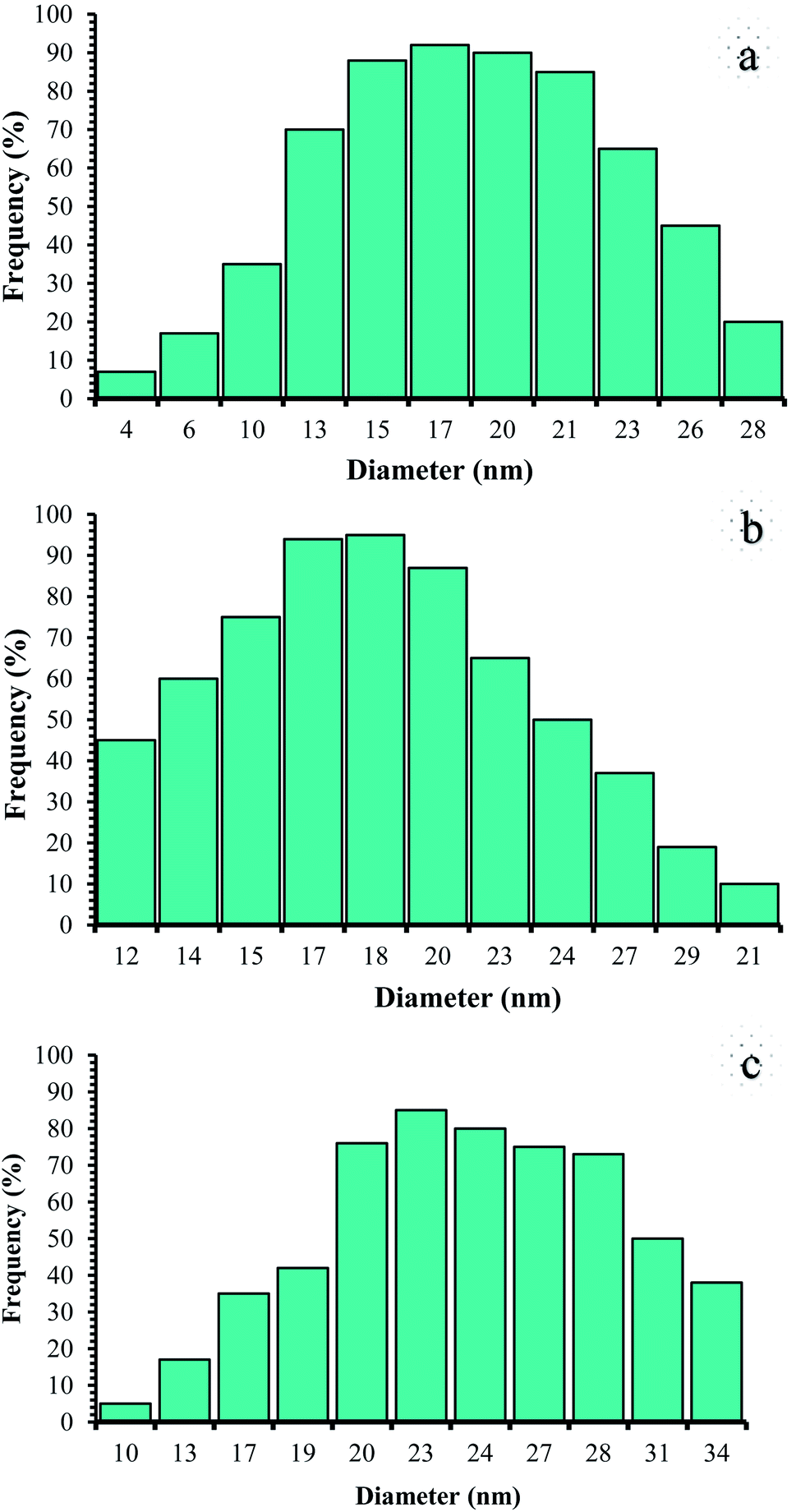 | ||
| Fig. 5 DLS analysis of (a) pure Fe3O4, (b) Fe3O4@NFC, and (c) (Fe3O4@NFC@NSalophCu)CO2H. | ||
The average diameters of the particles are evaluated to be about 17 nm for Fe3O4 (Fig. 5a), 18 nm for Fe3O4@NFC (Fig. 5b), and 23 nm for (Fe3O4@NFC@NSalophCu)CO2H (Fig. 5c).
The thermal behaviors of the synthesized samples in the final catalyst preparation pathway were studied using TGA analysis, and the obtained results are illustrated in Fig. 6. The total weight loss of uncoated Fe3O4 is 11% over the whole temperature range, which can be assigned to the removal of OH groups and water molecules from the surfaces of the Fe3O4 nanoparticles (Fig. 6a).47,48 From analysis, it is evident that the nanofiber cellulose structure degrades in three stages. In the first stage, at temperatures below 200 °C, degradation occurs due to the loss of water. The second stage involves significant weight loss that occurs between approximately 200 and 400 °C. This can generally be attributed to the destruction of the structure of natural cellulose under oxidation conditions. Decomposition at temperatures above 400 °C occurs due to the oxidation of rotten cellulose products (Fig. 6b).49,50 Fig. 6c also shows the thermal decomposition diagram of Fe3O4@NFC. At temperatures below 140 °C, gradual weight loss is shown, which can be attributed to the removal of water adsorbed on the nanocomposite surface. The second stage involves a sharp drop in weight from 150–450 °C, which is observed due to the decomposition of the cellulose structure; this form has good resistance up to about 650 °C. However, at temperatures above 650 °C, slight weight loss occurs, which may be due to the rupturing of the bonds between the nanofiber cells and the Fe3O4 nanoparticles, based on the thermal and mechanical properties of Fe3O4@NFC.51,52 In the thermogram of (Fe3O4@NFC@NSalophCu)CO2H (Fig. 6d), weight loss of less than 11% occurs in the temperature range of 25 to 280 °C, which is related to the volatilization of absorbed water on the surface of (Fe3O4@NFC@NSalophCu)CO2H. The next stage of weight loss in the temperature range of 325 to 475 °C is associated with the thermal decomposition of cellulose groups, an organic section. The weight loss of about 13% with a gentle slope at temperatures of 475–800 °C was assigned to the decomposition of immobilized organic moieties on the surfaces of the Fe3O4@NFC core–shell nanoparticles.
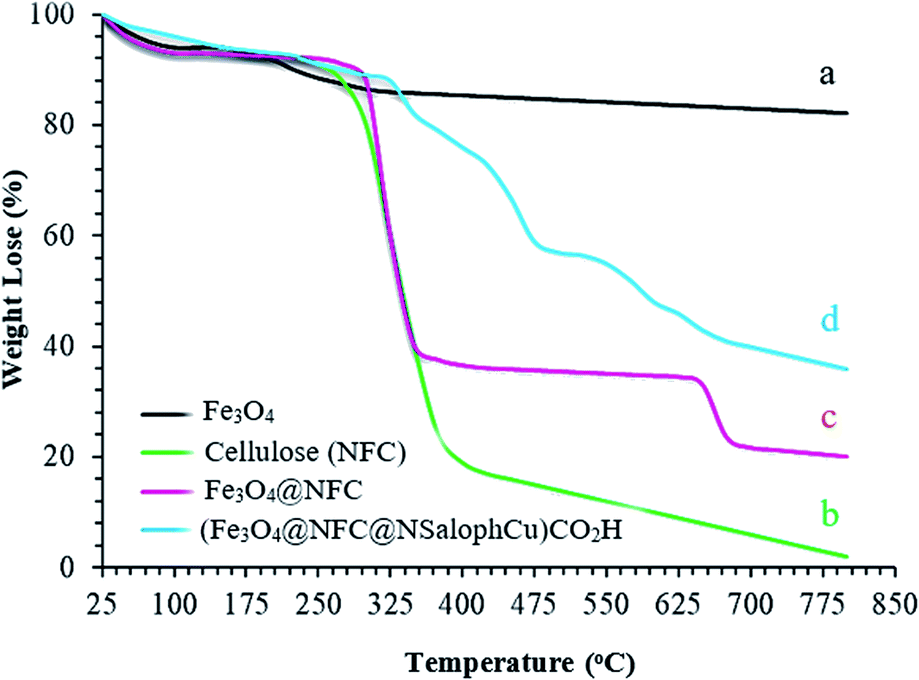 | ||
| Fig. 6 The TGA curves of the (a) Fe3O4 support, (b) nanofiber cellulose (NFC), (c) Fe3O4@NFC, and (d) the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst. | ||
Magnetic studies of all the prepared materials were carried out using a vibrating sample magnetometer (VSM) at room temperature and in an applied magnetic field, sweeping from −20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to +20000 Oe. The magnetization of the prepared samples based on the applied magnetic field can be observed in Fig. 7. It is reported that the saturation magnetization (Ms) value of bare Fe3O4 nanoparticles is 63.50 emu per g. The saturation magnetization (Ms) values of Fe3O4@NFC, Fe3O4@NFC@APTES, and (Fe3O4@NFC@NSalophCu)CO2H are 51.93, 46.84, and 41.97 emu per g, respectively. Therefore, the Ms value of Fe3O4 after coating with cellulose, APTES, and the [(5-Cl-Saloph)Cu(II)]CO2H salen complex decreases significantly from about 63.5 emu per g to 41.97 emu per g. The cellulose shell and silica that surround the Fe3O4 nanoparticles reduce the interactions between these particles. However, despite the reduction of the saturation magnetization, the catalyst can still be rapidly and easily separated from solution media via applying an external magnetic field.
000 to +20000 Oe. The magnetization of the prepared samples based on the applied magnetic field can be observed in Fig. 7. It is reported that the saturation magnetization (Ms) value of bare Fe3O4 nanoparticles is 63.50 emu per g. The saturation magnetization (Ms) values of Fe3O4@NFC, Fe3O4@NFC@APTES, and (Fe3O4@NFC@NSalophCu)CO2H are 51.93, 46.84, and 41.97 emu per g, respectively. Therefore, the Ms value of Fe3O4 after coating with cellulose, APTES, and the [(5-Cl-Saloph)Cu(II)]CO2H salen complex decreases significantly from about 63.5 emu per g to 41.97 emu per g. The cellulose shell and silica that surround the Fe3O4 nanoparticles reduce the interactions between these particles. However, despite the reduction of the saturation magnetization, the catalyst can still be rapidly and easily separated from solution media via applying an external magnetic field.
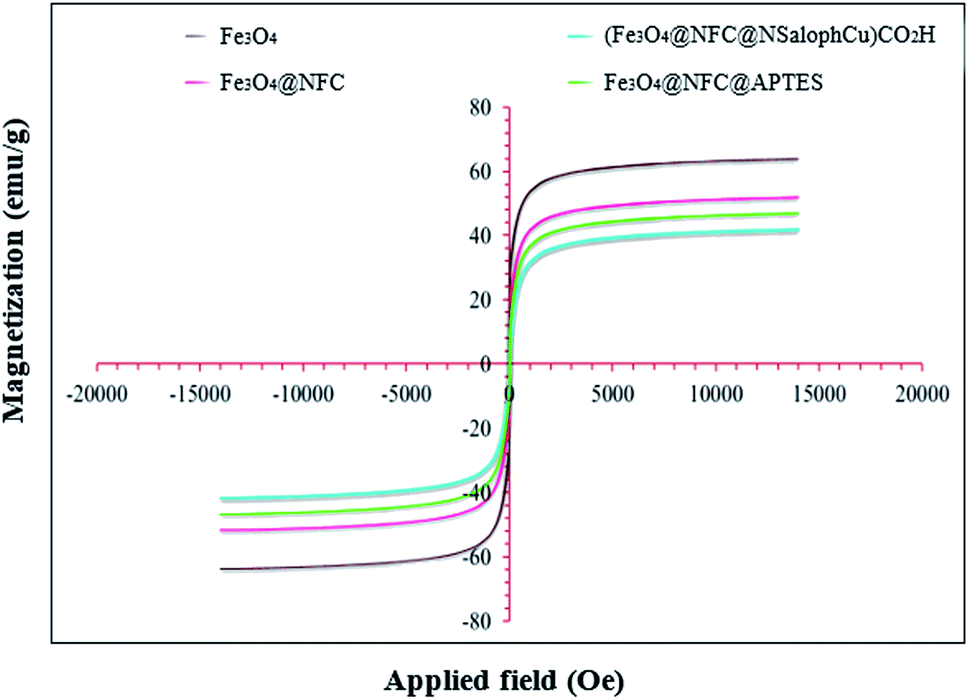 | ||
| Fig. 7 The magnetization curves at 300 K for Fe3O4, Fe3O4@NFC, Fe3O4@NFC-APTES, and the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst. | ||
The morphology and structure of the (Fe3O4@NFC@NSalophCu)CO2H salen complex were also observed using TEM (Fig. 8). TEM imaging reveals that the catalyst is made up of well-shaped spherical particles. The diameter of the NPs in the particle-size-distribution histogram is found to be about 17 nm (Fig. 8). In addition, spherically structured NPs with a diameter of about 17 nm are evident in the image in Fig. 8.
 | ||
| Fig. 8 A TEM image (top) and particle size distribution histogram (bottom) of the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst. | ||
The activity of the new (Fe3O4@NFC@NSalophCu)CO2H magnetic nanocatalyst for the synthesis of imidazole derivatives and tetrazoles was investigated. The reaction between benzaldehyde, hydroxylamine hydrochloride, and sodium azide in the presence of the mentioned catalyst was used as a model. The effects of various parameters on the catalytic efficiency of (Fe3O4@NFC@NSalophCu)CO2H were examined (Table 1). As shown in Table 1, the nature of the solvent was observed to profoundly affect both the catalyst activity and the product yield. CH3CN, DMF, and EtOH gave moderate yields, whereas solvent-free conditions were not suitable for this reaction (Table 1, entry 5). Further screening of the solvents revealed that when the reaction was carried out in H2O, the yield of the desired product increased significantly (Table 1, entry 1). Therefore, water, as the cheapest, safest, and most environmentally green solvent, is the best choice for this reaction. In the next step, the influence of the catalyst amount was investigated. The sample reaction was performed with different levels of catalyst at 80 °C. It was detected that 0.4 mol% catalyst was effectual at providing a high yield. Increasing the catalyst loading did not improve the production efficiency or lower the reaction time, while a lower catalyst loading decreased the reaction yield, even after a longer reaction time. Also, when the sample reaction was performed without catalyst or in the presence of Fe3O4 NPs, Fe3O4@NFC, or Cu(OAc)2 at 80 °C, the product was obtained but with very low efficiency.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Temp. (°C) | Time (min) | Yielda (%) |
| a Reaction conditions: aldehyde (1.0 mmol), hydroxylamine (1.2 mmol), and sodium azide (1.5 mmol). The catalyst was prepared using 0.4 mol% catalyst, as explained in the experimental section.b Reaction was performed in the presence of Fe3O4 as the catalyst.c Reaction was performed in the presence of Fe3O4@NFC as the catalyst.d Reaction was performed in the presence of Cu(OAc)2 as the catalyst.e Reaction was performed in the presence of [(5-Cl-Saloph)Cu(II)]CO2H as the catalyst. | |||||
| 1 | Water | 0.4 | 80 | 15 | 98 |
| 2 | EtOH | 0.4 | Reflux | 60 | 40 |
| 3 | CH3CN | 0.4 | Reflux | 30 | 55 |
| 4 | DMF | 0.4 | 80 | 30 | 80 |
| 5 | Neat | 0.4 | 80 | 120 | 20 |
| 6 | Water | No catalyst | 80 | 240 | 0 |
| 7 | Water | 0.2 | 80 | 45 | 70 |
| 8 | Water | 0.6 | 80 | 15 | 98 |
| 9 | Water | —b | 80 | 180 | Trace |
| 10 | Water | —c | 80 | 180 | 35 |
| 11 | Water | —d | 80 | 180 | 55 |
| 12 | Water | —e | 80 | 60 | 75 |
| 13 | Water | 0.4 | R.T. | 90 | 10 |
| 14 | Water | 0.4 | 40 | 60 | 60 |
| 15 | Water | 0.4 | 50 | 45 | 82 |
| 16 | Water | 0.4 | 60 | 15 | 97 |
| 17 | Water | 0.4 | 70 | 15 | 98 |
| 18 | Water | 0.4 | 90 | 15 | 95 |
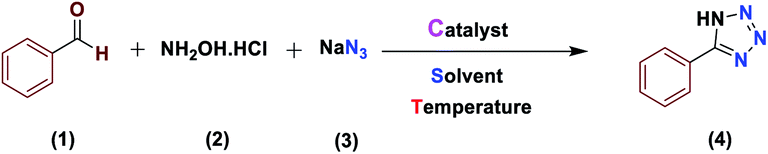
The use of the [(5-Cl-Saloph)Cu(II)]CO2H salen complex showed a higher product yield than the other cases (Table 1, entries 9–12). Subsequently, the effects of temperature on the reaction were explored, and a series of experiments was carried out at different temperatures (Table 1, entries 13–18). At room temperature, poor results are observed (Table 1, entry 13). The best relative performance was obtained at 60 °C (Table 1, entry 16).
After several experimental optimization studies, the optimal reaction conditions for the construction of tetrazole derivatives are given as follows: 0.4 mol% catalyst in H2O at 60 °C (Table 1, entry 16). After optimizing the reaction conditions, we further explored the scope and generality of the developed protocol for synthesizing 5-substituted-1H-tetrazoles. Various aldehydes with both electron-donating and electron-withdrawing substituents were employed, producing the corresponding tetrazoles in high to excellent yields (Table 2).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Aldehyde | Time (min) | Tetrazole | Yieldb (%) | Mp (°C) | TON | TOF | Ref. (lit) |
| a Reaction conditions: aldehyde (1.0 mmol), hydroxylamine (1.2 mmol), sodium azide (1.5 mmol), catalyst (0.4 mol%), and H2O (5 mL); 60 °C.b Isolated yield. | ||||||||
| 1a |  |
15 |  |
97 | 217–218 | 242.5 | 970 | 60 |
| 2b |  |
15 |  |
97 | 258–259 | 242.5 | 970 | 60 |
| 3c | 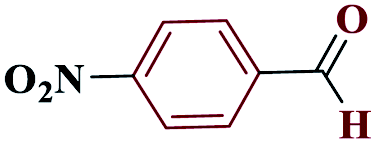 |
15 | 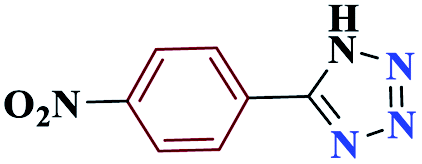 |
97 | 220–221 | 242.5 | 970 | 60 |
| 4d | 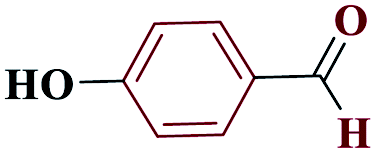 |
20 | 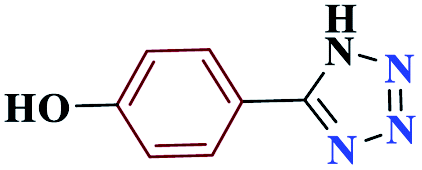 |
90 | 233–234 | 225 | 682 | 60 |
| 5e | 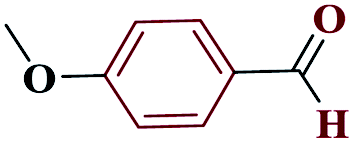 |
10 | 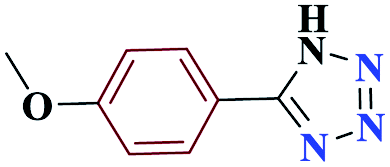 |
98 | 232–233 | 245 | 1476 | 60 |
| 6f | 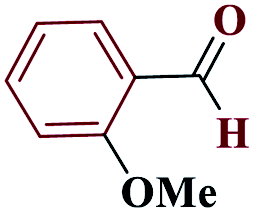 |
15 | 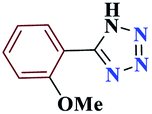 |
98 | 154–156 | 245 | 980 | 60 |
| 7g | 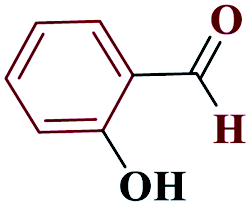 |
30 |  |
85 | 220–221 | 212.5 | 425 | 60 |
| 8h | 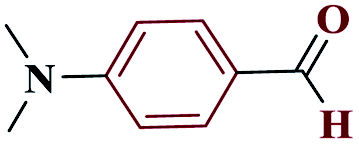 |
10 | 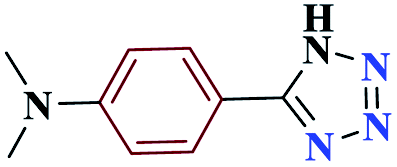 |
98 | 133–134 | 245 | 1476 | 60 |
| 9i |  |
15 | 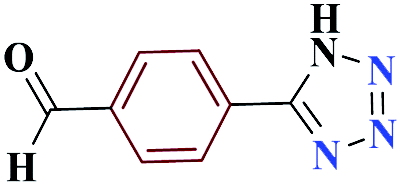 |
92 | 187–189 | 230 | 920 | 53 |
| 10j | 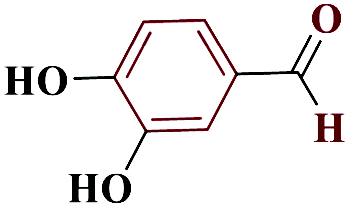 |
10 | 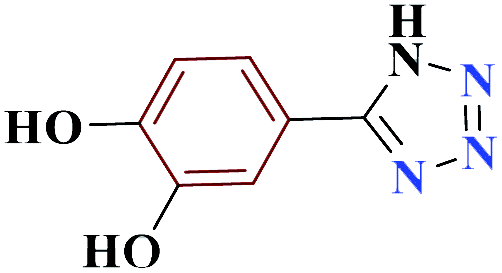 |
75 | 214–215 | 187.5 | 1130 | 60 |
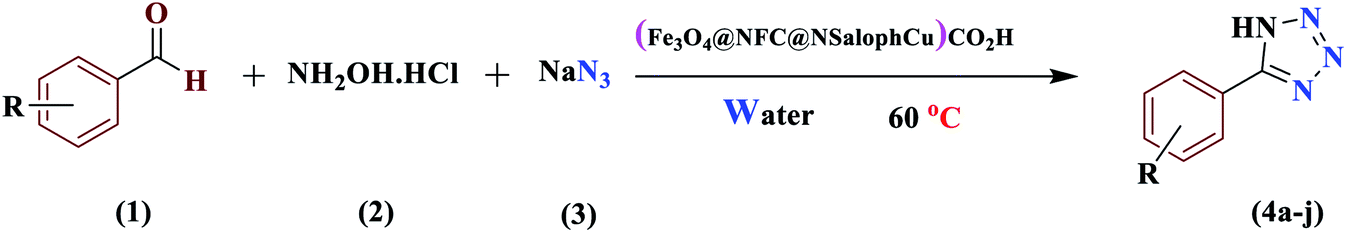
Generally, all the substrates resulted in products with high yields (Table 2, entries 1a–10j), but aldehydes with electron-donating substituents, due to the less electrophilic properties of the carbonyl group, required longer reaction times (Table 2), while electron-withdrawing groups had little effect on the reaction progress and showed the best performance. Moreover, steric effects were seen in this procedure. For example, 2-hydroxy and 2-methoxy benzaldehyde were transformed to the demanded products after a long time and with low yields in comparison with 4-hydroxy and 4-methoxy benzaldehyde (Table 2, entries 4d, 7g, 5e and 6f). Encouraged by the results from the 5-substituted-1H-tetrazole reactions, multi-component reactions leading to tetrazole compounds were investigated via reactions obtaining 1-substituted-1H-tetrazoles from amines. To determine the optimum conditions for MCRs between aromatic amines, triethyl orthoformate, and sodium azide at defined molar ratios, various solvents, temperatures, and catalytic quantities were tested. Practical information about the optimization process is provided in Table 3. At first, the reaction was carried out in various solvents. The data showed that the reaction efficiency after 100 min was negligible in all solvents in this experiment, and we continued the reaction for 180 minutes to observe the changes in the reaction efficiency. The details of these experiments are presented in Table 3 (entries 1–4). The catalyst has the effect of assisting and boosting this reaction.
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Temp. (°C) | Time (min) | Yielda (%) |
| a Reaction conditions: amine (1.0 mmol), triethyl orthoformate (1.2 mmol), and sodium azide (1.2 mmol). 0.3 mol% catalyst was primarily used, as explained in the experimental section.b Reaction was performed in the presence of Fe3O4 as the catalyst.c Reaction was performed in the presence of Fe3O4@NFC as the catalyst.d Reaction was performed in the presence of Cu(OAc)2 as the catalyst.e Reaction was performed in the presence of [(5-Cl-Saloph)Cu(II)]CO2H as the catalyst. | |||||
| 1 | Water | 0.3 | Reflux | 180 | 90 |
| 2 | EtOH | 0.3 | Reflux | 180 | 82 |
| 3 | CH3CN | 0.3 | Reflux | 180 | 50 |
| 4 | DMF | 0.3 | 100 | 180 | 75 |
| 5 | Neat | 0.3 | 100 | 20 | 98 |
| 6 | Neat | — | 100 | 300 | 0 |
| 7 | Neat | 0.1 | 100 | 45 | 75 |
| 8 | Neat | 0.5 | 100 | 15 | 95 |
| 9 | Neat | —b | 100 | 180 | Trace |
| 10 | Neat | —c | 100 | 180 | 30 |
| 11 | Neat | —d | 100 | 180 | 50 |
| 12 | Neat | —e | 100 | 60 | 77 |
| 13 | Neat | 0.3 | R.T. | 120 | 40 |
| 14 | Neat | 0.3 | 50 | 75 | 70 |
| 15 | Neat | 0.3 | 60 | 45 | 85 |
| 16 | Neat | 0.3 | 70 | 30 | 92 |
| 17 | Neat | 0.3 | 80 | 20 | 98 |
| 18 | Neat | 0.3 | 90 | 20 | 98 |
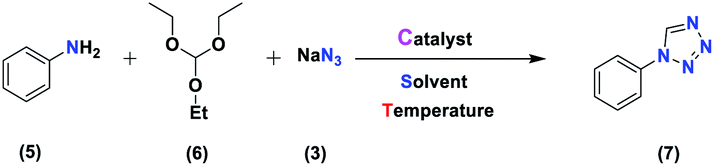
The reaction was then carried out under solvent-free conditions in the presence of different quantities of catalyst. The results showed that 0.3 mol% of catalyst was adequate to obtain a response; lower values resulted in reduced reaction efficiency and increasing the catalyst content to 0.5 mol% did not increase the reaction efficiency: instead it decreased slightly. In addition, in the presence of Fe3O4 NPs, Fe3O4@NFC, Cu (OAc)2, and [(5-Cl-Saloph)Cu(II)]CO2H at 80 °C, the demanded product was synthesized in low and good yields (Table 3, entries 9–12). To acquire the optimum temperature, the model reaction was done at different temperatures (Table 3, entries 13–18). A temperature of 80 °C led to the best results (Table 3, entry 17); the model reaction gave the best results when it was performed in a solvent-free fashion and at 80 °C in the presence of 6.5 mg (0.3 mol%) of catalyst (Table 3, entry 17). With the optimized reaction conditions in hand, we then studied the general use of our new catalytic system in the 1-substituted-1H-tetrazole reactions of some arylamines with triethyl orthoformate and sodium azide (Table 4). Under the optimized reaction conditions, different arylamines (electron-withdrawing and electron-donating groups) could be successfully converted to the corresponding tetrazole products. Based on previous reports53,54 and our observations, a suggested mechanism for the synthesis of 5-substituted-1H-tetrazole derivatives catalyzed by the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst is represented in Scheme 4. Initially, the coordination of the oxygen atom of the aldehyde with a catalyst acidic hydrogen increases the electrophilicity of the aldehyde [I]. Then, nucleophilic attack involving the nitrogen atom of hydroxylamine and the activated aldehyde causes the expulsion of water and the formation of an oxime intermediary [II]. After that, the oxime undergoes rearrangement followed by water removal to provide the corresponding nitrile intermediate [III]. Subsequently, the coordination of the nitrogen atom of the nitrile with the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst activates it toward azide ion attack at the electron-deficient carbon atom [IV]. Interplay between the Cu(II) catalyst and the nitrogen atom of the nitrile increases the electrophilic character of the nitrile.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Aldehyde | Time (min) | Tetrazole | Yieldb (%) | Mp (°C) | TON | TOF | Ref. (lit) |
| a Reaction conditions: aniline (1.0 mmol), triethyl orthoformate (1.2 mmol), sodium azide (1.2 mmol), catalyst (0.3 mol%), and solvent-free conditions; 80 °C.b Isolated yield. | ||||||||
| 1a |  |
20 | 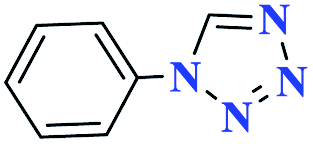 |
98 | 63 | 326.6 | 990 | 61 |
| 2b | 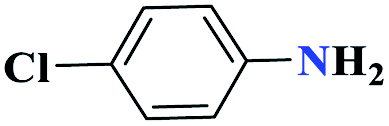 |
30 | 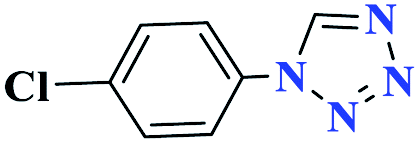 |
90 | 155 | 300 | 600 | 61 |
| 3c | 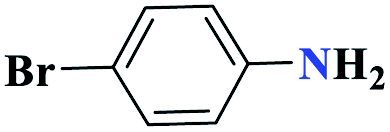 |
30 | 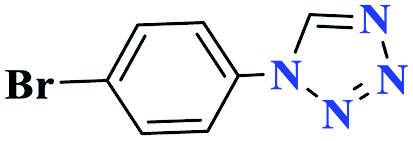 |
95 | 176–177 | 316.6 | 633 | 61 |
| 4d | 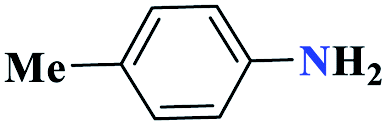 |
15 | 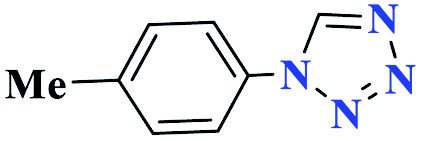 |
90 | 92–93 | 300 | 682 | 61 |
| 5e |  |
15 | 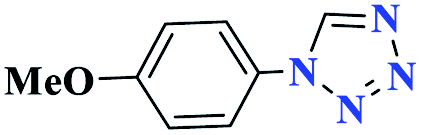 |
97 | 114–115 | 323.3 | 1293 | 61 |
| 6f | 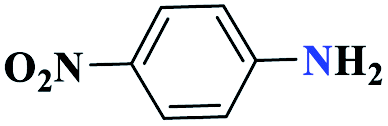 |
45 | 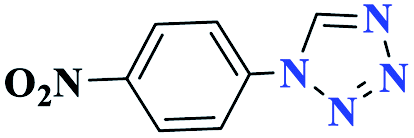 |
80 | 200 | 266.6 | 355.4 | 61 |
| 7g | 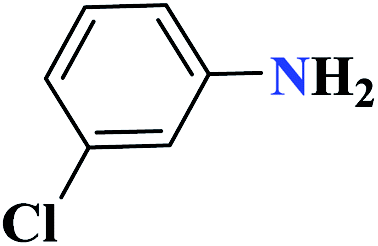 |
50 | 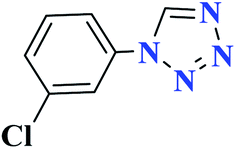 |
90 | 139–140 | 300 | 360 | 62 |
| 8h |  |
60 |  |
90 | 78–79 | 300 | 300 | 1 |
| 9i |  |
30 |  |
97 | 55 | 323.3 | 646.5 | 63 |
| 10j |  |
30 |  |
97 | 67–68 | 323.3 | 646.5 | 64 |

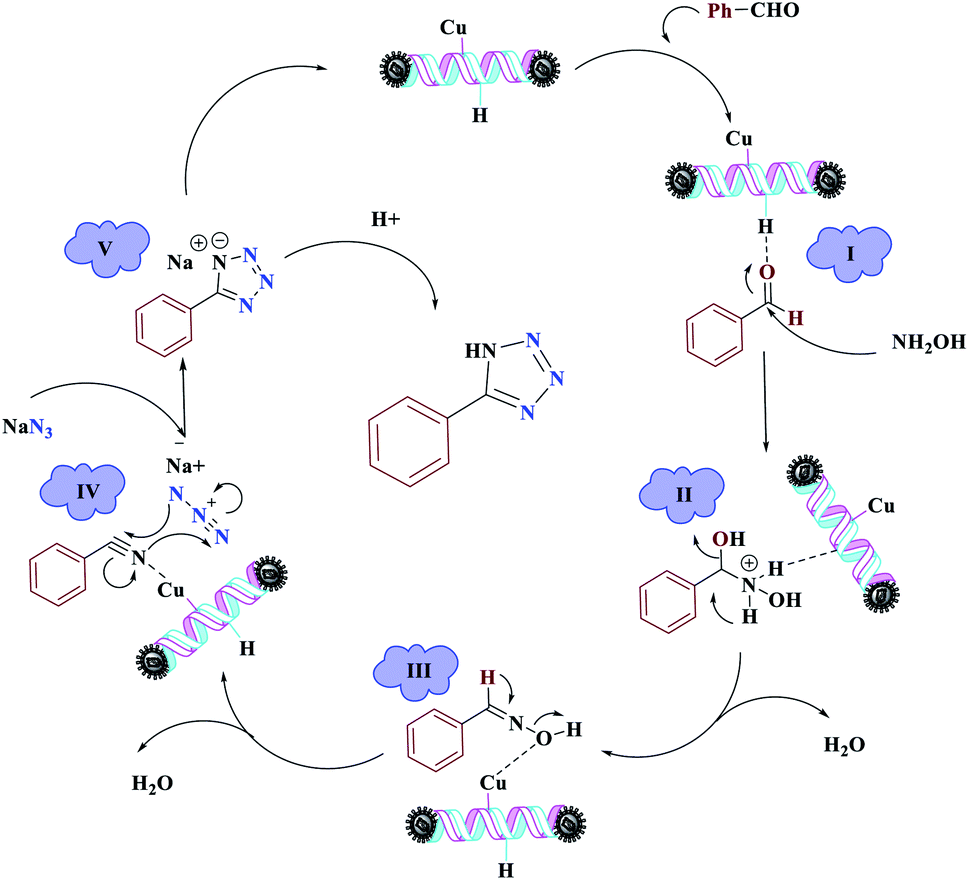 | ||
| Scheme 4 The suggested mechanism for the synthesis of 5-substituted-1H-tetrazoles in the presence of (Fe3O4@NFC@NSalophCu)CO2H. | ||
Subsequently, [3 + 2]-cycloaddition involving the activated nitrile and NaN3 results in the respective intermediate [V]. Finally, protonolysis of the intermediate [V] under the acidic conditions present in the reaction mixture produces the more stable product, a 5-substituted-1H-tetrazole, as a white solid. Due to Lewis acidity, first, the azide group coordinated with the copper nanocatalyst.
For the synthesis of 1-substituted-1H-tetrazoles, it is believed that the ethoxy group of TEOF is activated by (Fe3O4@NFC@NSalophCu)CO2H to initiate nucleophilic attack involving an amino-group nitrogen atom (Scheme 5). This facilitates the sequential removal of two ethanol molecules. The (Fe3O4@NFC@NSalophCu)CO2H-supported removal of ethanol in the presence of the nanocatalyst leads to the synthesis of the tetrazole. In a continuation of this research, the reusability and recyclability of the catalyst were investigated.
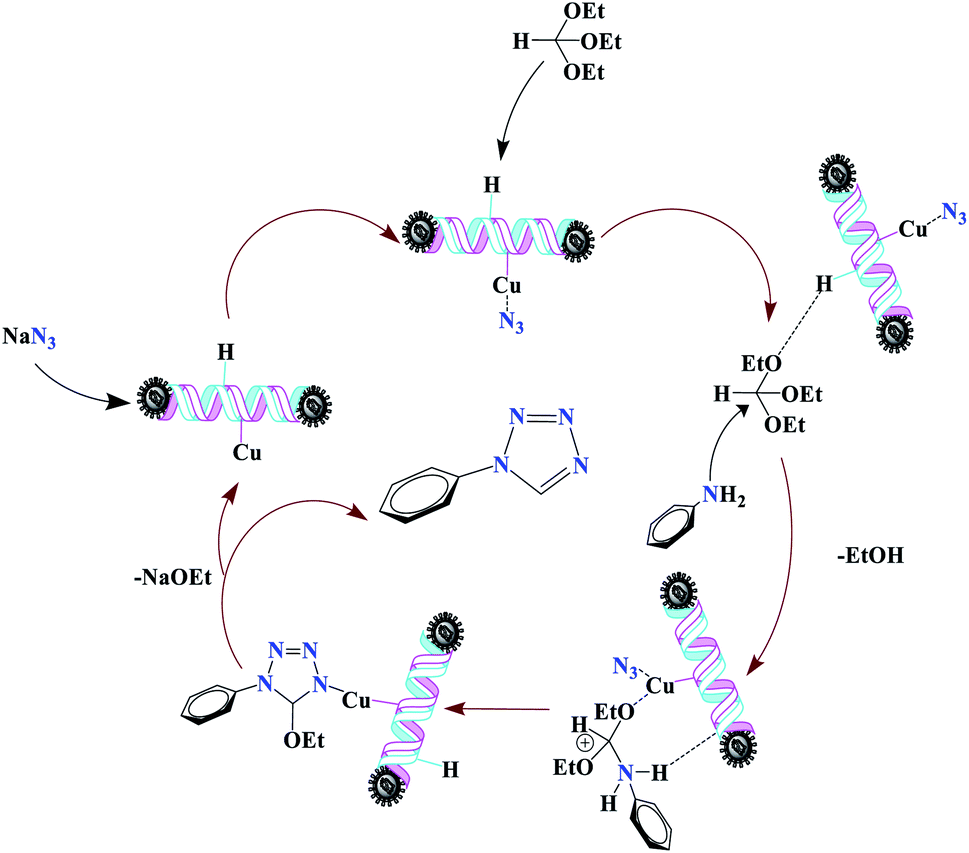 | ||
| Scheme 5 The suggested mechanism for the synthesis of 1-substituted-1H-tetrazoles in the presence of (Fe3O4@NFC@NSalophCu)CO2H. | ||
Catalyst recovery is considered necessary from various perspectives, such as environmental and commercial concerns.55,56 Therefore, the reusability of the (Fe3O4@NFC@NSalophCu)CO2H nanocatalyst was studied under the optimized conditions using the tetrazole model reaction. To investigate the stability of the nanocatalyst, after the first cycle, (Fe3O4@NFC@NSalophCu)CO2H was separated from the reaction mixture using a permanent magnet, washed with hot ethanol, and dried at 60 °C under vacuum. Afterward, the recycled catalyst was added to another vessel containing the starting materials and the next reaction cycle was carried out under the same conditions as discussed before. As exhibited in Fig. 9, the catalyst could be recovered and reused for at least seven consecutive runs. The yields of the 5-Ph-1H-tetrazole and 1-Ph-1H-tetrazole reactions reached 90% and 92% on the 7th run, meaning that only 7% and 6% drops in efficiency were observed compared to the corresponding fresh catalyst (97% for 5-Ph-1H-tetrazole and 98% for 1-Ph-1H-tetrazole). Also, the amounts of Cu that leached into solution after the 5-Ph-1H-tetrazole and 1-Ph-1H-tetrazole reactions in each cycle were measured via ICP.
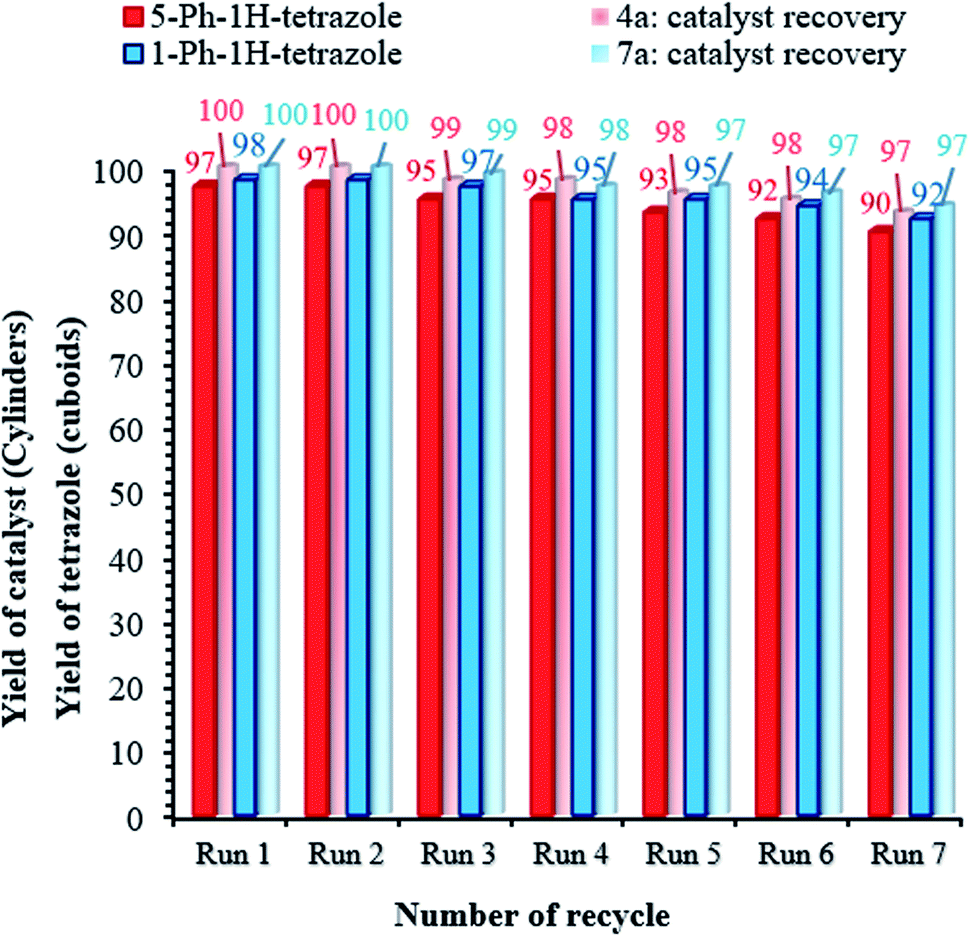 | ||
| Fig. 9 Catalyst recovery studies involving the use of (Fe3O4@NFC@NSalophCu)CO2H for the preparation of tetrazoles. | ||
The catalyst showed leaching in each cycle, with 3% metal leaching detected for the 4a and 7a reactions after the 7th run, reflecting the stability under the reaction conditions (Fig. 9, cylinders). In addition, the recovered catalyst from the 5-substituted-1H-tetrazole reaction was characterized via FT-IR, XRD, FE-SEM, and VSM analyses (Fig. 10a–d). FTIR and XRD spectra confirm that the structure of the recovered catalyst remained intact during recycling (Fig. 10a and c), which reflects its stability and durability.
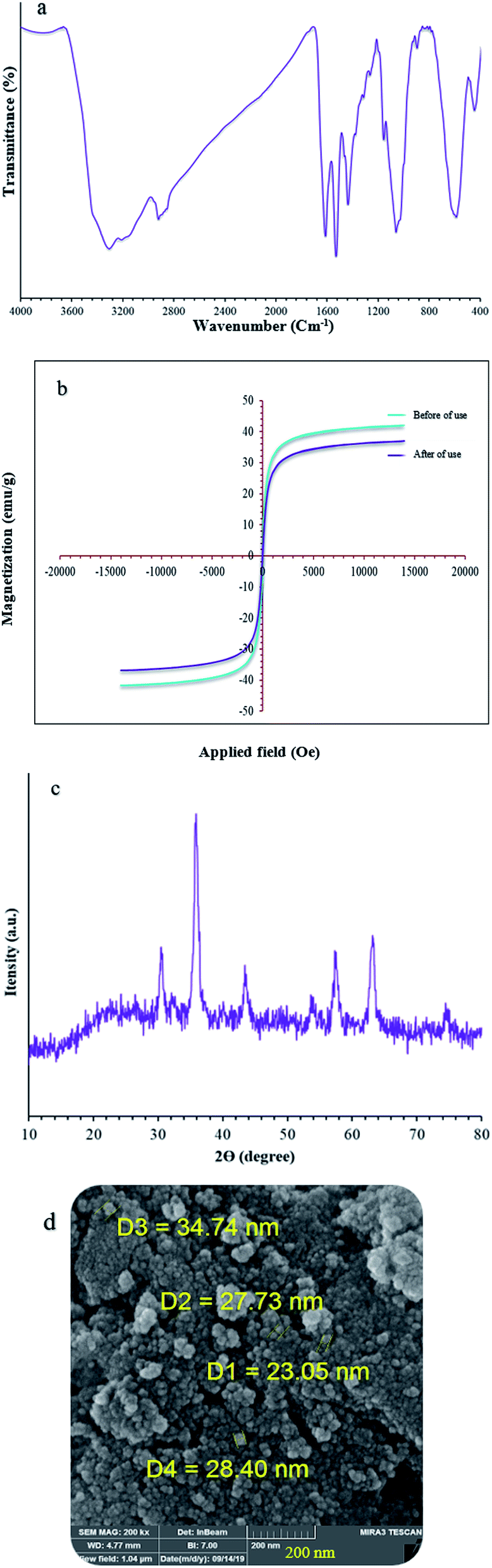 | ||
| Fig. 10 (a) FT-IR, (b) VSM, (c) XRD, and (d) Fe-SEM analysis of (Fe3O4@NFC@NSalophCu)CO2H after seven cycles. | ||
In addition, ICP analysis of the reusable catalyst demonstrated insignificant changes in the weight percentages of the elements compared with the corresponding fresh values, with values as follows: Fe, 45.54 wt%, Si, 2.33 wt%, and Cu, 6.8 wt%. In another study, to show the heterogeneous nature of the (Fe3O4@NFC@NSalophCu)CO2H magnetic nanocatalyst, hot filtration testing was applied during the preparation of a tetrazole under the optimum conditions. The reaction to obtain 5-substituted-1H-tetrazole was started in the presence of the catalyst.
After the reaction was half-completed, the reaction was stopped, and the catalyst was removed using an external magnet. The remaining mixture was maintained under optimized reaction conditions in the absence of catalyst. After 1 h, only small amounts of the product were formed (Fig. 11).
 | ||
| Fig. 11 Leaching experiments involving the use of (Fe3O4@NFC@NSalophCu)CO2H to obtain a 5-substituted-1H-tetrazole. | ||
This confirms the nature of the heterogeneous catalyst. To prove the efficiency of the catalyst for the production of 5-phenyl-1H-tetrazoles and 1-phenyl-1H-tetrazoles, we compared the results with other catalysts in the literature (Tables 5 and 6). The new proposed catalyst has high yields and short reaction times and, unlike some catalysts, does not require toxic and expensive solvents. In addition, the synthesized magnetic catalyst has properties such as recyclability and the ability to be separated using an external magnet. In recent years, various methods have been proposed for the synthesis of tetrazoles (Scheme 6, eqn (1)–(10)). From the viewpoint of atom economy (AE) and atom efficiency (AEf), the present methodology (Scheme 6, eqn (5) and (10)) was compared with those reported in the literature (Scheme 6, Tables 7 and 8).53 Each of the analyzed reactions has disadvantages, including high amounts of catalyst use, low efficiency, prolonged reaction times (24 h), harsh reaction conditions, and the use of high-boiling-point organic solvents (DMF and DMSO), leading to complicated isolation and recovery methods. Therefore, it is complicated to work with these materials. The advantages of the reactions demonstrated here are that none of the above disadvantages are observed (Scheme 7). Also, the use of (Fe3O4@NFC@ NSalophCu)CO2H as a natural, benign, cheap, and non-corrosive natural heterogeneous catalyst represents an interesting synthetic method with relatively high nuclear economy. Ethanol is the only by-product of this reaction. This solvent can be classified as a green solvent.
| Reaction | Catalyst | Reaction conditions | Time (h) | Yielda (%) | Ref. |
|---|---|---|---|---|---|
| a Isolated yield.b Reaction conditions: benzaldehyde (1.0 mmol), hydroxylamine (1.2 mmol), and sodium azide (1.5 mmol). | |||||
| 5-Substituted-1H-tetrazoleb | CuNO3·3H2O | DMSO/120 °C | 24 | 78 | 65 |
| Nano-Cu2O-MFR | DMF/100 °C | 8 | 92 | 66 | |
| Cu–MCM-41 | DMF/140 °C | 12 | 90 | 67 | |
| Copper(II) Schiff base | DMF/110 °C | 7 | 91 | 53 | |
| Bi(OTf)3 | DMF/120 °C | 24 | 87 | 68 | |
| Cu(OAC)2 | DMF/120 °C | 12 | 96 | 69 | |
| (NH4)4Ce(SO4)4–2H2O | DMF/reflux | 5 | 72 | 70 | |
| Humic acid | H2O/100 °C | 4 | 92 | 71 | |
| Fe3O4–CNT–TEA–Cu(II) | DMF/70 °C | 1.5 | 96 | 54 | |
| (Fe3O4@NFC@NSalophCu)CO2H | H2O/60 °C | 0.25 | 97 | This work | |
| Reaction | Catalyst | Reaction conditions | Time (h) | Yielda (%) | Ref. |
|---|---|---|---|---|---|
| a Isolated yield.b Reaction conditions: aniline (1.0 mmol), triethyl orthoformate (1.2 mmol), and sodium azide (1.2 mmol). | |||||
| 1-Substituted-1H-tetrazoleb | Natrolite zeolite | Neat/120 °C | 4 | 82 | 72 |
| In(OTf)3 | Neat/100 °C | 1.5 | 89 | 73 | |
| Silica sulfuric acid | Neat/120 °C | 5 | 95 | 74 | |
| Fe3O4@SiO2@salen@Cu | Neat/100 °C | 1 | 96 | 7 | |
| Fe3O4@quindiol@Cu | Neat/100 °C | 0.66 | 92 | 75 | |
| ZnS nanoparticles | DMF/R.T. | 0.5 | 92 | 76 | |
| Fe3O4@HT@AEPH2–CoII | H2O/90 °C | 1 | 95 | 77 | |
| ZnS nanoparticles | Neat/130 °C | 4.5 | 78 | 78 | |
| Cu NPs/bentonite | Neat/120 °C | 3 | 93 | 79 | |
| (Fe3O4@NFC@NSalophCu)CO2H | Neat/80 °C | 0.33 | 98 | This work | |
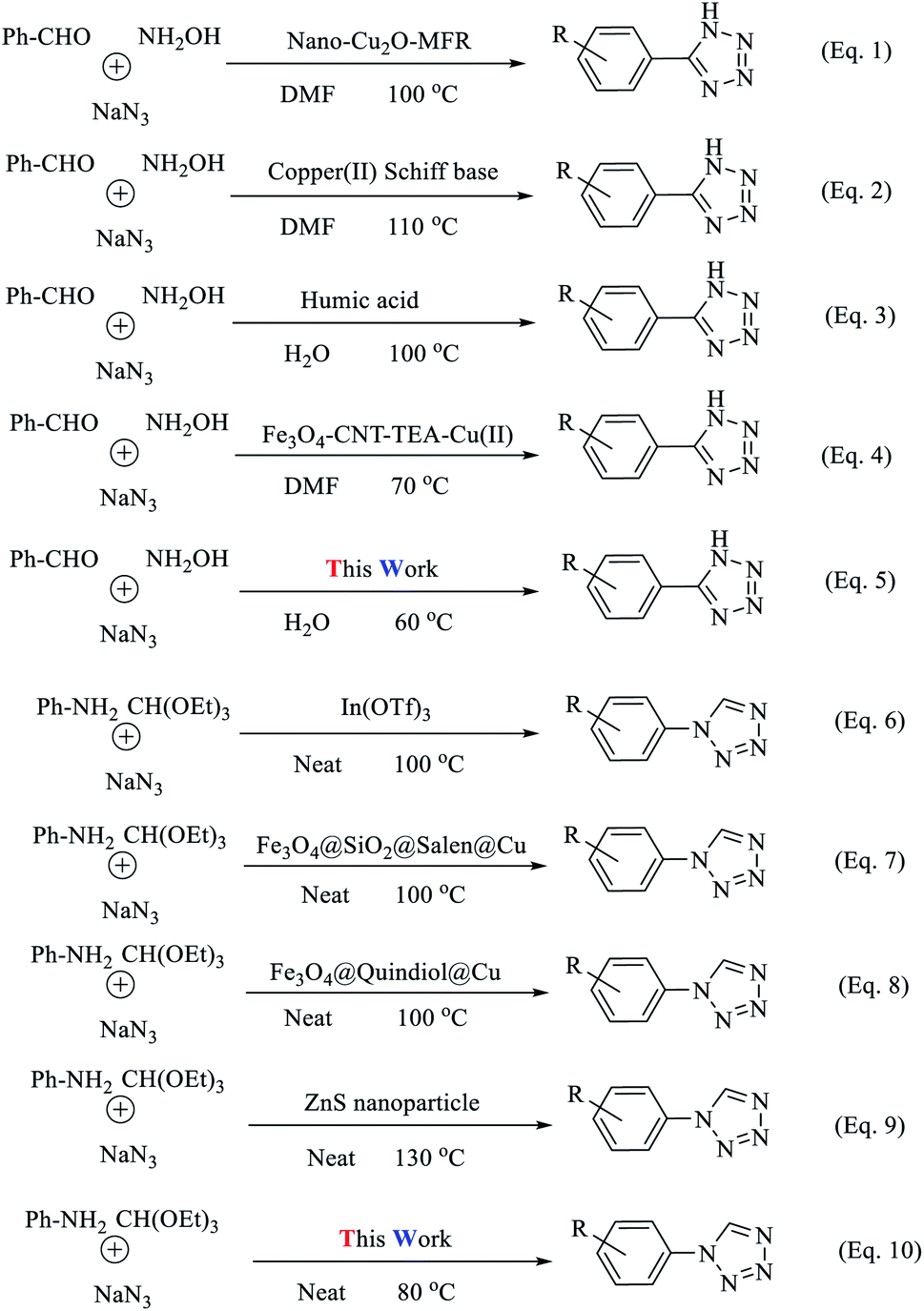 | ||
| Scheme 6 The synthesis of tetrazole derivatives with different catalysts. | ||
| Entry | Equation | Desired product | Time (h) | Yielda (%) | AE (%) | AEf |
|---|---|---|---|---|---|---|
| a Isolated yield. | ||||||
| 1 | 1 | 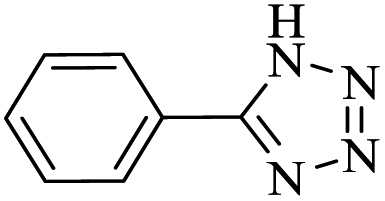 |
8 | 78 | 71.58 | 51.24 |
| 2 | 7 | 91 | 65.13 | |||
| 3 | 4 | 92 | 65.85 | |||
| 4 | 1 h 30 min | 96 | 68.71 | |||
| 5 | 15 min | 97 | 69.43 | |||
| 2 | 1 | 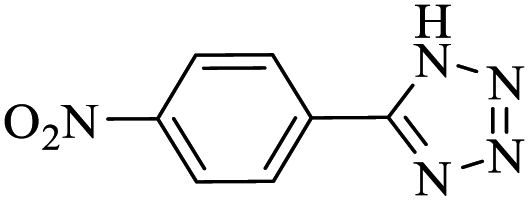 |
9 | 88 | 76.72 | 67.51 |
| 2 | 8 | 92 | 70.58 | |||
| 3 | 3 | 96 | 73.65 | |||
| 4 | 1 h 20 min | 96 | 73.65 | |||
| 5 | 15 min | 97 | 74.41 | |||
| 3 | 1 | 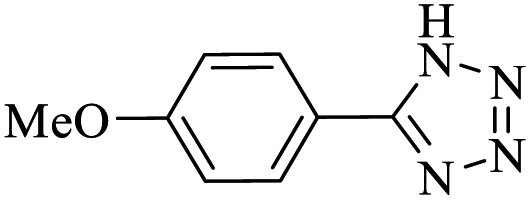 |
— | — | 75.23 | — |
| 2 | 8 | 90 | 67.07 | |||
| 3 | 5 | 90 | 67.07 | |||
| 4 | 2 h 30 min | 87 | 65.45 | |||
| 5 | 10 min | 98 | 73.72 | |||
| Entry | Equation | Desired product | Time (h) | Yielda (%) | AE (%) | AEf |
|---|---|---|---|---|---|---|
| a Isolated yield. | ||||||
| 1 | 6 |  |
5 | 67 | 47.70 | 31.95 |
| 7 | 1 | 92 | 43.88 | |||
| 8 | 40 min | 92 | 43.88 | |||
| 9 | 4 h 30 min | 78 | 37.20 | |||
| 10 | 20 min | 98 | 46.74 | |||
| 2 | 6 | 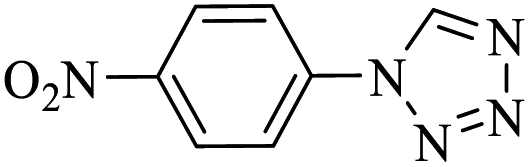 |
4 | 77 | 54.40 | 41.88 |
| 7 | 3 | 75 | 40.80 | |||
| 8 | 1 h 10 min | 92 | 50.04 | |||
| 9 | — | — | — | |||
| 10 | 45 min | 80 | 43.52 | |||
| 3 | 6 | 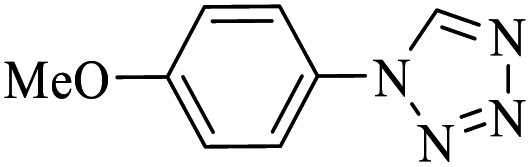 |
6 | 92 | 52.37 | 48.18 |
| 7 | 1 | 96 | 50.27 | |||
| 8 | 1 | 93 | 48.70 | |||
| 9 | — | — | — | |||
| 10 | 15 min | 97 | 50.79 | |||
 | ||
| Scheme 7 An overview of the (Fe3O4@NFC@NSalophCu)CO2H-catalyzed tetrazole reaction. | ||
Experimental
All chemical reagents and solvents were procured from Merck and Sigma-Aldrich and were used as received without further filtration. The purity of products and the progress of reactions were established based on TLC on silica gel Polygram STL G/UV 254 plates. The melting points of products were determined with electrothermal-type C14500 melting point apparatus. Fourier-transform infrared (FT-IR) spectra were recorded in pressed KBr pellet form using a JASCO FT/IR 4600 spectrometer in room temperature range from 400–4000 cm−1. NMR spectra were captured with a Bruker Avance DPX-250 spectrometer at 300 MHz, using DMSO-d6 solvent in the presence of tetramethylsilane as the internal standard. Thermogravimetric analysis (TGA) was carried out using a TGA Q600 TA thermogravimetric analyzer in the temperature range of 25–800 °C at a heating rate of 10 °C min−1 under an air atmosphere. High-resolution transmission electron microscopy (HRTEM) analysis was accomplished using a Tescan MIRA3 microscope. ICP experiments were carried out using a Varian Vista Pro CCD simultaneous ICP-OES instrument. Room-temperature magnetization isotherms were obtained using vibrating sample magnetometry (VSM; LakeShore Cryotronics 7407). Field-emission scanning electron microscopy (FE-SEM) images were obtained using Tescan MIRA3 apparatus. EDX spectroscopy was performed using a field-emission scanning electron microscope (JEOL7600F). The powder X-ray diffraction (XRD) studies were performed with Philips PW1730 apparatus with Cu Kα (λ = 0.154 nm) radiation.Preparation of Fe3O4@NFC@APTES core–shell NPs
Fe3O4@NFC was synthesized via a sol–gel method as reported earlier by us.57–59 The Fe3O4@NFC product (1 g) was sonicated in 15 mL of dry toluene for 30 min, and then (3-aminopropyl)triethoxysilane (APTES, 1.0 mmol) was added slowly to the mixture. The mixture was then stirred for 12 h at 80 °C. Fe3O4@NFC@APTES was separated using an external magnet, washed with toluene and Et2O, and dried at 75 °C in an oven under vacuum.Preparation of the 3,5-bis(((E)-5-(chloromethyl)-2-hydroxybenzylidene)amino)benzoic acid complex and its reaction with Cu(OAc)2: [(5-Cl-Saloph)Cu(II)]CO2H
5-chloromethyl salicylaldehyde (1) was synthesized via a method as reported earlier by us.55 Briefly, a mixture of salicylaldehyde (0.16 mol), paraformaldehyde (0.1 mol), and 100 mL of conc. HCl was refluxed under an air atmosphere at 70 °C for 24 h. A small amount of H2SO4 (2–3 drops) as a catalyst was added to the reaction mixture. After the reaction, the reaction mixture was cooled to room temperature and distilled water and CH2Cl2 (ratio 1:1) were added to the reaction mixture to separate the organic phase from the aqueous phase. Then the organic layer was separated and dried over MgSO4, and the solvent was removed under reduced pressure. The purple sediment was washed with sodium bicarbonate (50 mL) and then dried under an air atmosphere. The pure product, 5-chloromethyl salicylaldehyde (1), was obtained via recrystallization from EtOH (mp: 85–87 °C, 95% isolated yield). The product 1 (2.0 mmol) was dissolved in 15 mL of dichloromethane, and then 3,5-diaminobenzoic acid (1.0 mmol) was added slowly to the mixture. The mixture was then stirred for 6 h at room temperature. After the completion of the reaction, 3,5-bis(((E)-5-(chloromethyl)-2-hydroxybenzylidene)amino)benzoic acid ((5-Cl-Saloph)CO2H) was washed with dichloromethane and crystallized with ethanol. Then, for the formation of Cu(II) and the complex (5-Cl-Saloph)CO2H, the ligand (1.0 mmol) was dissolved in 15 mL of ethanol and then Cu(OAc)2·H2O (1.5 mmol) was added to the reaction mixture. The reaction mixture was stirred for 6 h under reflux conditions. The solution was allowed to cool to room temperature and then the resultant [(5-Cl-Saloph)Cu(II)]CO2H complex was filtered off and washed with cold ethanol.
Preparation of [(5-Cl-Saloph)Cu(II)]CO2H supported on Fe3O4@NFC@APTES [(Fe3O4@NFC@NSalophCu)CO2H]
The synthesized Fe3O4@NFC-APTES (1.5 g) was sonicated in dry toluene (15 mL) for 30 min. [(5-Cl-Saloph)Cu(II)]CO2H (4 mmol) was dissolved in dry toluene (15 mL) and added dropwise to the dispersed mixture, which was stirred at 70 °C for 24 h. The solid was separated using an external magnet, washed with distilled water and diethyl ether, and dried at 70 °C in an oven under vacuum for 12 h.Typical procedure for the synthesis of 5-substituted-1H-tetrazoles derivatives
An aldehyde (1.0 mmol), hydroxylamine (1.2 mmol), and sodium azide (1.5 mmol) were added to a 5 mL round-bottomed flask containing a magnetic stirrer, water (5 mL), and (Fe3O4@NFC@NSalophCu)CO2H (0.4 mol%; 8 mg). The reaction mixture was stirred at 60 °C, and the reaction progress was monitored via thin-layer chromatography. Upon the completion of the reaction, the catalyst was removed using an external magnet. The solvent was removed under reduced pressure and, for the removal of soluble impurities trapped in the solid, it was washed with water (5 mL). Then, 10 mL of hot ethanol (50 °C) was added and the mixture was stirred to afford the tetrazole in powder form. After completing the reaction, the precipitate formed was filtered and purified via recrystallization from ethanol to afford the desired product (Table 3).General procedure for the synthesis of 1-substituted-1H-tetrazoles from amines
An aniline (1.0 mmol), triethyl orthoformate (1.2 mmol), and sodium azide (1.2 mmol) were added to a test tube containing a magnetic stirrer and (Fe3O4@NFC@NSalophCu)CO2H (0.3 mol%; 6.5 mg). The reaction mixture was stirred at 80 °C without solvent, and the reaction progress was monitored via thin-layer chromatography. Upon the completion of the reaction, the mixture was cooled to ambient temperature and diluted with ethyl acetate (10 mL). The catalyst was removed using an external magnet, and then the resulting solution was washed with water, dried over anhydrous Mg2SO4, and evaporated. The residue was concentrated and recrystallized from EtOAc/n-hexane (ratio, 1:2). The products were characterized via a comparison of their melting points with the reported ones. The spectral data for selected compounds are given below.
Spectral data
Conclusions
In conclusion, a facile methodology was developed for the preparation of tetrazole derivatives using (Fe3O4@NFC@NSalophCu)CO2H nanoparticles as an efficient heterogeneous catalyst. The nanocatalyst presents outstanding activity under simple conditions. Good-to-high yields of products were obtained using a wide variety of substrates within short reaction times. The catalyst was characterized via FT-IR, EDX, FE-SEM, TEM, XRD, VSM, DLS, and TGA analyses. Hot filtration testing revealed the heterogeneous nature of the catalyst with low levels of metal leaching. The catalyst could be recycled for at least seven consecutive cycles with a negligible loss of efficiency. There are myriad advantages to using this method, such as the thermal stability, heterogeneous nature, short reaction times, clean and simple procedure, excellent yields, easy workup, eco-friendly nature, cost-effectiveness, and easy product separation and purification, making the nanocatalyst a suitable alternative for the preparation of tetrazole derivatives. Beneficially, this catalyst can easily be separated from the reaction environment using an external magnetic field and reused several times without significant loss of catalytic stability or activity.Author contributions
Pouya Ghamari Kargar: investigation, methodology, software, writing – original draft. Ghodsieh bagherzade: project administration, data curation, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the University of Birjand and financial support from the Research Council.References
- H. Sharghi, S. Ebrahimpourmoghaddam and M. M. Doroodmand, J. Organomet. Chem., 2013, 738, 41–48 CrossRef CAS.
- P. Ghamari Kargar, S. Aryanejad and G. Bagherzade, Appl. Organomet. Chem., 2020, e5965 CAS.
- P. Ghamari Kargar, M. Bakherad, A. Keivanloo and A. H. Amin, Iran. J. Catal., 2018, 8(3), 179–187 Search PubMed.
- A. K. Gupta, C. H. Song and C. H. Oh, Tetrahedron Lett., 2004, 45, 4113–4116 CrossRef CAS.
- F. Abrishami, M. Ebrahimikia and F. Rafiee, Appl. Organomet. Chem., 2015, 29, 730–735 CrossRef CAS.
- L. M. T. Frija, A. Ismael and M. L. S. Cristiano, Molecules, 2010, 15, 3757–3774 CrossRef CAS PubMed.
- F. Dehghani, A. R. Sardarian and M. Esmaeilpour, J. Organomet. Chem., 2013, 743, 87–96 CrossRef CAS.
- R. S. Upadhayaya, S. Jain, N. Sinha, N. Kishore, R. Chandra and S. K. Arora, Eur. J. Med. Chem., 2004, 39, 579–592 CrossRef CAS PubMed.
- K. Terashima, T. Tanimura, H. Shimamura, A. Kawase, K. Uenishi, Y. Tanaka, I. Kimura, T. Kamisaki, Y. Ishizuka and M. Sato, Chem. Pharm. Bull., 1995, 43, 1042–1044 CrossRef CAS PubMed.
- A. Rajasekaran and P. P. Thampi, Eur. J. Med. Chem., 2005, 40, 1359–1364 CrossRef CAS PubMed.
- A. Bielenica, D. Szulczyk, W. Olejarz, S. Madeddu, G. Giliberti, I. B. Materek, A. E. Koziol and M. Struga, Biomed. Pharmacother., 2017, 94, 804–812 CrossRef CAS PubMed.
- A. Nohara, H. Kuriki, T. Saijo, H. Sugihara, M. Kanno and Y. Sanno, J. Med. Chem., 1977, 20, 141–145 CrossRef CAS PubMed.
- P. J. Kothari, S. P. Singh, S. S. Parmar and V. I. Stenberg, J. Heterocycl. Chem., 1980, 17, 1393–1398 CrossRef CAS.
- A. S. Gundugola, K. L. Chandra, E. M. Perchellet, A. M. Waters, J.-P. H. Perchellet and S. Rayat, Bioorg. Med. Chem. Lett., 2010, 20, 3920–3924 CrossRef CAS PubMed.
- J. Li, S. Y. Chen, S. Tao, H. Wang, J. J. Li, S. Swartz, C. Musial, A. A. Hernandez, N. Flynn, B. J. Murphy, B. Beehler, K. E. Dickinson, L. Giupponi, G. Grover, R. Seethala, P. Sleph, D. Slusarchyk, M. Yan, W. G. Humphreys, H. Zhang, W. R. Ewing, J. A. Robl, D. Gordon and J. A. Tino, Bioorg. Med. Chem. Lett., 2008, 18, 1825–1829 CrossRef CAS PubMed.
- R. N. Butler, Adv. Heterocycl. Chem., 1977, 21, 323–435 CrossRef CAS.
- P. Camilleri, M. W. Kerr, T. W. Newton and J. R. Bowyer, J. Agric. Food Chem., 1989, 37, 196–200 CrossRef CAS.
- R. J. Nachman, G. M. Coast, K. Kaczmarek, H. J. Williams and J. Zabrocki, Acta Biochim. Pol., 2004, 51, 121–127 CrossRef CAS PubMed.
- C. N. S. S. P. Kumar, D. K. Parida, A. Santhoshi, A. K. Kota, B. Sridhar and V. J. Rao, MedChemComm, 2011, 2, 486 RSC.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945–7950 CrossRef CAS PubMed.
- D. R. Patil, Y. B. Wagh, P. G. Ingole, K. Singh and D. S. Dalal, New J. Chem., 2013, 37, 3261 RSC.
- J. Bonnamour and C. Bolm, Chem.–Eur. J., 2009, 15, 4543–4545 CrossRef CAS PubMed.
- G. Venkateshwarlu, A. Premalatha, K. C. Rajanna and P. K. Saiprakash, Synth. Commun., 2009, 39, 4479–4485 CrossRef CAS.
- P. Mani, A. K. Singh and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 1879–1882 CrossRef CAS.
- M. Parveen, F. Ahmad, A. Mohammed Malla and S. Azaz, New J. Chem., 2015, 39, 2028–2041 RSC.
- M. Abdollahi-Alibeik and A. Moaddeli, New J. Chem., 2015, 39, 2116–2122 RSC.
- W.-K. Su, Z. Hong, W.-G. Shan and X.-X. Zhang, Eur. J. Org. Chem., 2006, 2723–2726 CrossRef CAS.
- M. L. Kantam, K. B. Shiva Kumar and K. Phani Raja, J. Mol. Catal. A: Chem., 2006, 247, 186–188 CrossRef CAS.
- Y. Zhu, Y. Ren and C. Cai, Helv. Chim. Acta, 2009, 92, 171–175 CrossRef CAS.
- A. Ahmadi, T. Sedaghat, H. Motamedi and R. Azadi, Appl. Organomet. Chem., 2020, 34, e5572 CrossRef CAS.
- M. A. Ashraf, Z. Liu, C. Li and D. Zhang, Appl. Organomet. Chem., 2021, 35, e6133 CAS.
- L. Bosch and J. Vilarrasa, Angew. Chem., Int. Ed., 2007, 46, 3926–3930 CrossRef CAS PubMed.
- J. A. Glaser, Clean Technol. Environ. Policy, 2012, 14, 513–520 CrossRef CAS.
- H. Dong, X. Zhang, Y. Lu, Y. Yang, Y.-P. Zhang, H.-L. Tang, F.-M. Zhang, Z.-D. Yang, X. Sun and Y. Feng, Appl. Catal., B, 2020, 276, 119173 CrossRef CAS.
- T. Neuberger, B. Schöpf, H. Hofmann, M. Hofmann and B. von Rechenberg, J. Magn. Magn. Mater., 2005, 293, 483–496 CrossRef CAS.
- R. Mohammadi and M. Z. Kassaee, J. Mol. Catal. A: Chem., 2013, 380, 152–158 CrossRef CAS.
- J. Safari and Z. Zarnegar, J. Mol. Catal. A: Chem., 2013, 379, 269–276 CrossRef CAS.
- B. Karimi, F. Mansouri and H. Vali, Green Chem., 2014, 16, 2587 RSC.
- T. Li, M. Zhu, Z. Yang, J. Song, J. Dai, Y. Yao, W. Luo, G. Pastel, B. Yang and L. Hu, Adv. Energy Mater., 2016, 6, 1601122 CrossRef.
- U. Ray, S. Zhu, Z. Pang and T. Li, Adv. Mater., 2020, 2002504 CrossRef.
- Y. Li, H. Zhu, Y. Wang, U. Ray, S. Zhu, J. Dai, C. Chen, K. Fu, S.-H. Jang, D. Henderson, T. Li and L. Hu, Small Methods, 2017, 1, 1700222 CrossRef.
- S. Chidambaram, B. Pari, N. Kasi and S. Muthusamy, J. Alloys Compd., 2016, 665, 404–410 CrossRef CAS.
- H. Khashei Siuki, G. Bagherzade and P. Ghamari Kargar, ChemistrySelect, 2020, 5, 13537–13544 CrossRef CAS.
- H. Esmaeili-Shahri, H. Eshghi, J. Lari and S. A. Rounaghi, Appl. Organomet. Chem., 2018, 32, e3947 CrossRef.
- Z. A. Hajar Khanehzaei, M. B. Ahmad and K. Shameli, Int. J. Electrochem. Sci., 2014, 9, 8189–8198 Search PubMed.
- S. Azad and B. B. Fatameh Mirjalili, RSC Adv., 2016, 6, 96928–96934 RSC.
- N. Arsalani, H. Fattahi and M. Nazarpoor, eXPRESS Polym. Lett., 2010, 4, 329–338 CrossRef CAS.
- I. Karimzadeh, M. Aghazadeh, M. R. Ganjali, T. Doroudi and P. H. Kolivand, J. Magn. Magn. Mater., 2017, 433, 148–154 CrossRef CAS.
- I. Hasan, R. A. Khan, W. Alharbi, K. H. Alharbi and A. Alsalme, Nanomaterials, 2019, 9, 1687 CrossRef CAS PubMed.
- L. K. Kumawat, M. Kumar, P. Bhatt, A. Sharma, M. Asif and V. K. Gupta, Sens. Actuators, B, 2017, 240, 365–375 CrossRef CAS.
- M. Yadav, Compos. Commun., 2018, 10, 1–5 CrossRef.
- L. E. Low, B. T. Tey, B. H. Ong, E. S. Chan and S. Y. Tang, Carbohydr. Polym., 2017, 155, 391–399 CrossRef CAS PubMed.
- S. Layek, B. Agrahari, S. Dey, R. Ganguly and D. D. Pathak, J. Organomet. Chem., 2019, 896, 194–206 CrossRef CAS.
- P. Akbarzadeh, N. Koukabi and E. Kolvari, Mol. Diversity, 2020, 24, 319–333 CrossRef CAS PubMed.
- S. K. Nalilu, H. N. Pati, D. Varadaraji, S. S. Suban, V. R. Ramasamy, K. Kubendiran and J. S. K. G. Raguraman, Org. Commun., 2010, 3, 45–46 Search PubMed.
- P. Ghamari kargar, M. Noorian, E. Chamani, G. Bagherzade and Z. Kiani, RSC Adv., 2021, 11, 17413–17430 RSC.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 37086–37097 RSC.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2021, 11, 4339–4355 RSC.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 32927–32937 RSC.
- M. Kazemnejadi and A. R. Sardarian, RSC Adv., 2016, 6, 91999–92006 RSC.
- V. Khorramabadi, D. Habibi and S. Heydari, Green Chem. Lett. Rev., 2020, 13, 50–59 CrossRef CAS.
- F. Verma, A. Sahu, P. K. Singh, A. Rai, M. Singh and V. K. Rai, Green Chem., 2018, 20, 3783–3789 RSC.
- M. S. Ghasemzadeh and B. Akhlaghinia, ChemistrySelect, 2020, 5, 6440–6452 CrossRef CAS.
- V. P. Karavai, P. N. Gaponik and O. A. Ivashkevich, Magn. Reson. Chem., 1989, 27, 611–615 CrossRef CAS.
- S. Tao, in Transforming Teacher Quality in the Global South, Palgrave Macmillan UK, London, 2016, pp. 25–49 Search PubMed.
- S. Behrouz, J. Saudi Chem. Soc., 2017, 21, 220–228 CrossRef CAS.
- M. Abdollahi-Alibeik and A. Rezaeipoor-Anari, J. Magn. Magn. Mater., 2016, 398, 205–214 CrossRef CAS.
- M. Sridhar, K. Mallu, R. Jillella, K. Godala, C. Beeram and N. Chinthala, Synthesis, 2013, 45, 507–510 CrossRef CAS.
- M. Heravi, A. Fazeli, H. Oskooie, Y. Beheshtiha and H. Valizadeh, Synlett, 2012, 23, 2927–2930 CrossRef CAS.
- C. Ji and F. N. Egolfopoulos, Proc. Combust. Inst., 2011, 33, 955–961 CrossRef CAS.
- H. Wang, Y. Wang, Y. Han, W. Zhao and X. Wang, RSC Adv., 2020, 10, 784–789 RSC.
- D. Habibi, M. Nasrollahzadeh and T. A. Kamali, Green Chem., 2011, 13, 3499 RSC.
- T. Jin, S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 2004, 45, 9435–9437 CrossRef CAS.
- D. Habibi, H. Nabavi and M. Nasrollahzadeh, J. Chem., 2013, 2013, 1–4 Search PubMed.
- D. Habibi, N. Pakravan, A. Arabi and Z. Kaboudvand, Appl. Organomet. Chem., 2018, 32, e3988 CrossRef.
- H. Naeimi and F. Kiani, Ultrason. Sonochem., 2015, 27, 408–415 CrossRef CAS PubMed.
- M. Salimi, F. Esmaeli-nasrabadi and R. Sandaroos, Inorg. Chem. Commun., 2020, 122, 108287 CrossRef CAS.
- H. Naeimi, F. Kiani and M. Moradian, J. Nanopart. Res., 2014, 16, 2590 CrossRef.
- A. Rostami-Vartooni, M. Alizadeh and M. Bagherzadeh, Beilstein J. Nanotechnol., 2015, 6, 2300–2309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01913a |
| This journal is © The Royal Society of Chemistry 2021 |