
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of fluorinated graphene-modified epoxy coatings on the corrosion behaviour of 2024 aluminium alloy
Shixiong Zhao a,
Baojie Dou*acd,
Song Duana,
Xiuzhou Lin*ac,
Yingjun Zhangac,
Wilfred Emoriac,
Xiulei Gaob and
Zhiwen Fangb
a,
Baojie Dou*acd,
Song Duana,
Xiuzhou Lin*ac,
Yingjun Zhangac,
Wilfred Emoriac,
Xiulei Gaob and
Zhiwen Fangb
aCollege of Materials Science and Engineering, Sichuan University of Science and Engineering, Zigong 643000, China. E-mail: baojiedou@suse.edu.cn; linxiuzhou@suse.edu.cn
bZhongshan Photoelectric Materials Co., Ltd, Zibo 255138, China
cMaterial Corrosion and Protection Key Laboratory of Sichuan Province, Sichuan University of Science and Engineering, Zigong 643000, China
dChimie ParisTech, PSL Research University, CNRS, Institut de Recherche de Chimie Paris (IRCP), Paris, F-75005, France
First published on 13th May 2021
Abstract
This study provides an enhanced corrosion resistance of epoxy resin (EP) by embedding fluorinated graphene (FG) into the epoxy matrix. FG with different fluorine contents was obtained by reacting nitrogen trifluoride (NF3) gas with GO and then incorporated into the EP matrix to fabricate the different composites. Through a series of characterization methods, the chemical composition and microstructures of FG were systematically analyzed, and its corrosion resistance was also studied. Results revealed that F atoms were bonded to the GO surface to form C–F covalent bonds, and an FG lamellar thickness less than 2 nm. The contact angle of the coatings increased with the incorporation of FG, and the coating resistance of FG2/EP coating was 3 orders of magnitude more than that of the EP coating after immersion for 4080 h. Thus, the incorporation of FG into epoxy matrix significantly enhanced its hydrophobic properties and barrier performance, which was beneficial to improving the long-term corrosion resistance of the coating.
1. Introduction
Fluorinated graphene (FG) is a novel graphene derivative material. It is either partially fluorinated or completely fluorinated, and the fluorine atoms are supported on the surface of the graphene to form a C–F covalent bond with the structural transformation of C–C bonds from sp2 to sp3.1–3 FG has attracted growing attention due to its unique nanostructure and carbon fluorine bond. Some of its advantages are large surface area, wide band gap, high stability, low surface energy and hydrophobicity, in addition to its graphene-like two-dimensional lamellar structure.4–7 These advantages set FG apart for application as a new type of carbon material in thin high performance dielectric materials,8–11 thermoelectric devices,6,12 lubrication,13–15 nano-composite materials,16,17 and even stem cell substrates.18 Despite all the successes on its utilization in a wide range of industrial applications, scanty reports exist on its research in the field of anticorrosion, especially organic coatings. This is therefore the motivation of this study.Epoxy resins are one of the most widely applied materials in anticorrosion coatings due to some of the excellent properties they exhibit, such as chemical resistance, toughness, low shrinkage on curing, good mechanical properties, corrosion resistance, and their excellent adhesion to substrates.19–21 However, a pure epoxy resin usually contains holes and/or defects due to curing shrinkages. These holes become points of entry for corrosive species such as water, oxygen, and chloride ions, which ultimately disrupts the protection mechanism and reduces the corrosion resistance of the material.22 For this reason, pigments are often used in epoxy resin coatings to enhance the barrier performance and corrosion protective property.23–25 Traditional pigments and fillers include red lead,26 chromate,27 titanium dioxide,28–31 silicon dioxide,32,33 iron oxide34,35 and glass flakes.36 Nevertheless, traditional fillers are limited in their applications due to their toxicity, environmental hazards, and poor protection performance, etc.
Compared with traditional fillers, nanomaterials such as graphene and graphene oxide (GO), have received great attention from researchers in recent years because of their small size and large specific surface area, which play great roles in their barrier effects.37,38 However, micro-galvanic corrosion between graphene and metal matrixes is a common occurrence when graphene is used as filler in organic coatings.39 Also, there are limited research on GO nanosheets as filler materials because of its poor compatibility with organic coatings which usually results to severe agglomeration, eventually reducing the corrosion resistance of epoxy resins. In order to solve the problem of dispersion, Stankovich et al. prepared modified GO through isocyanate addition, and the obtained product was uniformly dispersed in polar solvents.40 Ramezanzadeh et al. modified graphene by using siloxane via sol–gel method, and the obtained composites improved the corrosion protection of epoxy coatings.41 Zheng et al. synthesized a novel GO-poly(urea-formaldehyde) (GUF) composites by anchoring a prepolymer of urea-formaldehyde (UF) resin onto GO through in situ polycondensation. The GUF composites exhibit superior compatibility in epoxy resin, which can significantly improve the long-term corrosion resistance of epoxy coatings.42 These studies prove that there are great potentials for improving the corrosion resistance of the epoxy resin coatings with the addition of the modified GO.
FG is obtained from graphite or GO modified by fluorination, which has excellent properties of lubrication, wear reduction and superhydrophobic. Yang et al. prepared FG coating with excellent superhydrophobic properties through pressing FG on the epoxy surfaces.43 Min et al. successfully fabricated polyimide/fluorinated graphene oxide (PI/FGO) nanocomposites by simple hydrothermal reaction. Wherein, the addition of FG significantly enhanced the lubrication performance of pure PI, and its friction coefficient and wear rate were reduced by 33.1% and 80.8%, respectively.44 These findings indicate that FG is an potential material in the coating field. However, the formation process of FG leads to a change in the hybrid orbital of graphene from sp2 to sp3, which might be causing FG with different microstructure under different fluorine contents. The intrinsic diversity in the surface morphology, composition and microstructure of FG with different F content may result in quite different compatibility and stability of the FG in the coating, and then lead to differences in the corrosion resistance of the FG-modified coatings. Herein, the difference between the composition and morphological structure of FG with different fluorine content were systematically studied in this paper. Simultaneously, the long-term corrosion resistance and protection mechanism of FG with different fluorine content modified epoxy coating were explored. The results of this research would provide a theoretical basis for FG modified coatings and new strategy to design novel long-term corrosion resistant coatings.
2. Materials and experimental methods
2.1 Materials
Natural graphite powder, dimethylbenzene, n-butyl alcohol, potassium permanganate, sulfuric acid (H2SO4, ≥95.0%) and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). The epoxy resin (E-44) and polyamide curing agent (PA-651) were purchased from Nantong Xingchen Synthetic Material Co., Ltd (Nantong, China). The 2024 aluminum alloy plate (4.36 Cu, 1.49 Mg, 0.46 Mn, 0.25 Fe, 0.14 Si and balance Al, in mass%) with a sample size of 50 mm × 40 mm × 5 mm, was used as the substrate. Before applying the coating on the substrate, the 2024 Al alloy was abraded with SiC paper to remove rust, and degreased with ethanol. All other materials were used without further purification.2.2 Synthesis of graphene oxide
Graphite flakes (GF) were oxidized to graphene oxide (GO) by the improved Hummer's method.45,46 1 g of GF was added to a mixed acid solution containing 90 ml H2SO4 and 10 ml H3PO4 in a 500 ml three-neck flask, and maintained below 5 °C while stirring. 6 g of KMnO4 was slowly added to the solution while maintaining the temperature for 1 h. The mixture was transferred to an oil bath to maintain a temperature of 50 ± 2 °C for 2 h. And 100 ml of deionized water was added slowly to the flask and the temperature was raised to 85 °C for 1 h. Add 120 ml deionized water and 15 ml H2O2 (30%) to end the experiment, and the solution was golden yellow. The suspension obtained above was washed repeatedly with a mixture of HCl solution and deionized water, and centrifuged at a speed of 5000 rpm for 10 min. Stop washing when pH reached 7, and the solution was placed in glass beakers and vacuum dried for 12 h at 85 °C to obtain GO.2.3 Synthesis of fluorinated graphene
The preparation process of fluorinated graphene (FG) is shown in Fig. 1. Firstly, 5 g of GO powder was placed in a reaction furnace under a flowing nitrogen environment. The reaction temperature was raised to 120 °C and maintained for 1 h. High purity nitrogen trifluoride (NF3) gas was passed in, and the temperature of the reaction furnace was adjusted to 400 °C, with reaction times of 5 h, 10 h and 15 h. Noteworthy, NF3 gas is decomposed into free fluorine atoms at high temperatures and these atoms react with GO to generate FG. Finally, the impure FG powder was repeatedly washed with 10% hydrofluoric acid solution and deionized water, and then centrifugally filtered to neutrality and freeze-dried in vacuum to obtained pure FG powder. The obtained FG prepared at different reaction times were named FG1, FG2, and FG3 for convenience.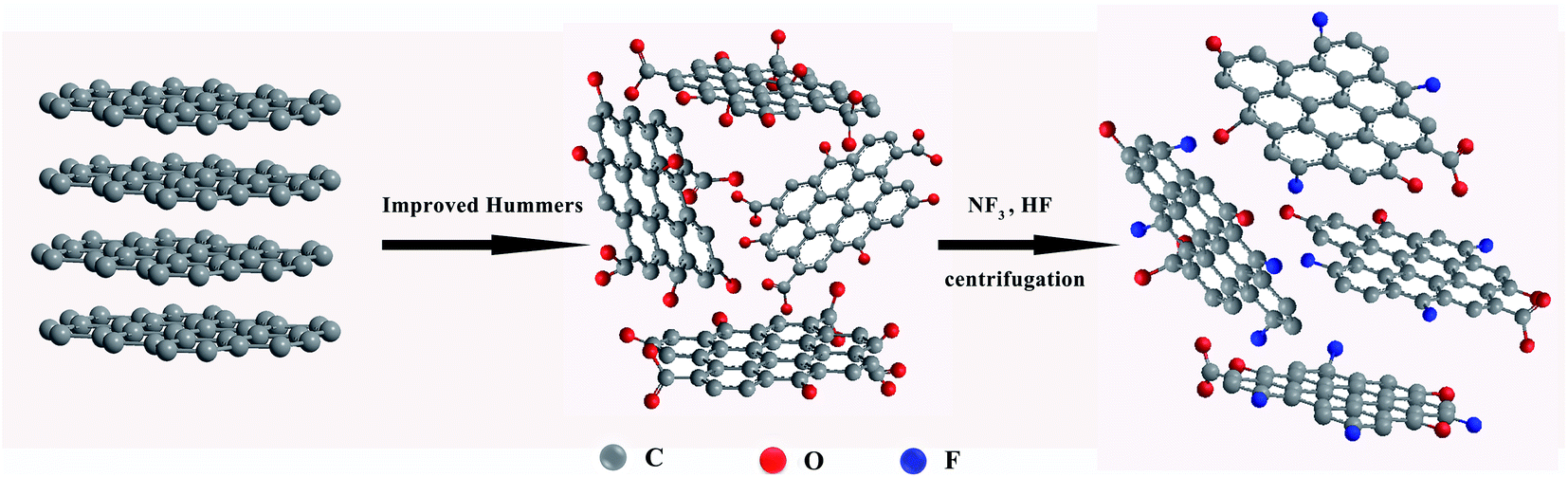 | ||
| Fig. 1 The preparation process of fluorinated graphene. | ||
2.4 Preparation of the coatings
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, with the solvent comprising of a mixture of dimethylbenzene and n-butyl alcohol. The mixture was stirred to dissolve the EP. Polyamide as curing agent (with EP to polyamide weight ratio of 2:1) was added to the resin mixture, and mechanically stirred for 10 min to obtain a uniform mix. Finally, the mixture was placed in a vacuum chamber at room temperature for 20 min to remove air bubbles, and then applied on the surface of pre-treated 2024 aluminum alloy panels by using a coater to control the thickness of the coatings. The specimens were subsequently cured in a drying oven at 40 °C for 24 h. The thickness of the dried coating was 100 ± 10 μm.
:3, with the solvent comprising of a mixture of dimethylbenzene and n-butyl alcohol. The mixture was stirred to dissolve the EP. Polyamide as curing agent (with EP to polyamide weight ratio of 2:1) was added to the resin mixture, and mechanically stirred for 10 min to obtain a uniform mix. Finally, the mixture was placed in a vacuum chamber at room temperature for 20 min to remove air bubbles, and then applied on the surface of pre-treated 2024 aluminum alloy panels by using a coater to control the thickness of the coatings. The specimens were subsequently cured in a drying oven at 40 °C for 24 h. The thickness of the dried coating was 100 ± 10 μm.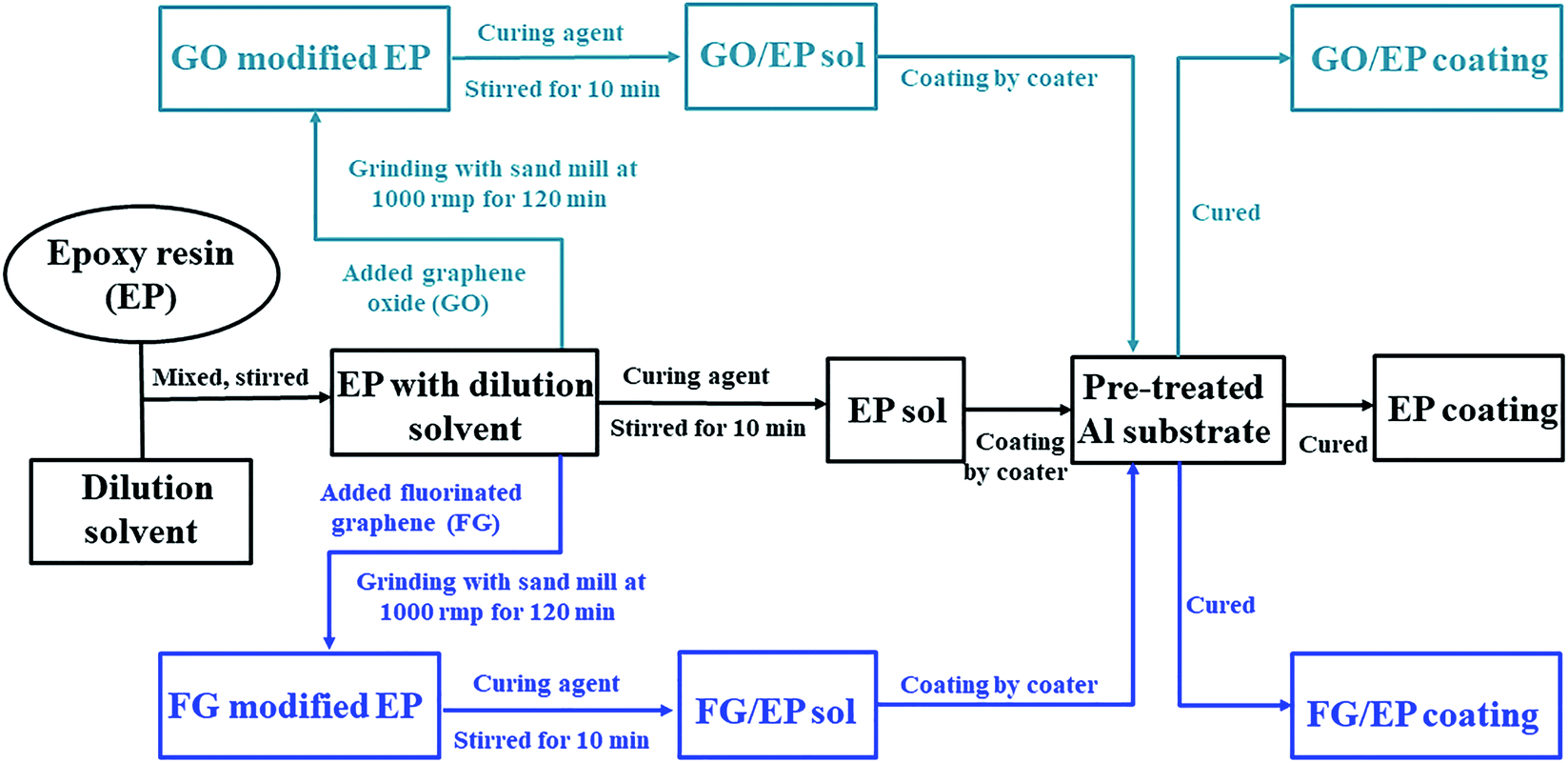 | ||
| Fig. 2 Schematic diagram of the EP, GO/EP and FG/EP coatings preparation process. | ||
2.5 Characterization and measurement
Scanning Vibrating Electrode Technique (SVET) was used to test the corrosion resistance of the coatings. The SVET measurement was carried out in 3.5 wt% NaCl solution by a VersaSCAN micro scanning electrochemical workstation (AMETEK, USA). The coating thickness was about 40 μm. The vertical current density was recorded on a lattice of 41 × 41 points over an area of 3 × 3 mm2 (step size 75 μm). The size of the probe tip (platinum–iridium) was 10 μm and the vibration amplitude was 30 μm with a frequency of 80 Hz. All the tests were repeated at least three times to ensure the repeatability.
3. Results and discussion
3.1 Characterization of graphite, GO and three FG
The synthesized samples were analyzed by XPS to evaluate their chemical states and elemental contents. Fig. 3(a) presents the survey scan spectra of all the samples, and the C 1s peak and O 1s peak of the GO sample is located at 284.8 eV and 533.05 eV, respectively. In comparing the scans for GO and graphite, a significant increase in oxygen atoms was observed, indicating that the graphite was successfully oxidized. After the reaction between GO and NF3, there were obvious additions of new characteristic peaks at 688.5 eV and 833.5, 861.5 eV for the FG samples. These correspond to F 1s and F Auger.47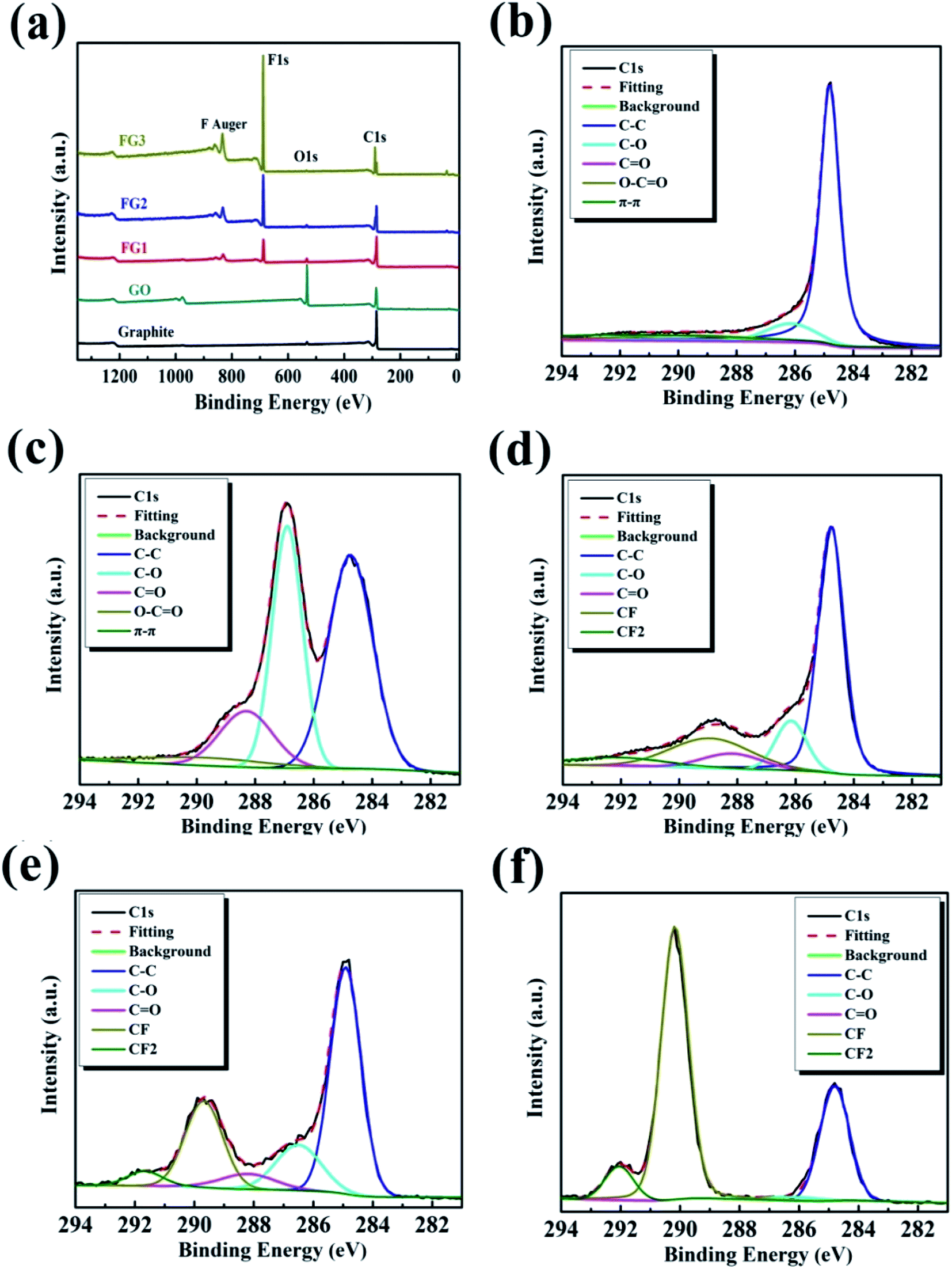 | ||
| Fig. 3 Full XPS spectra (a), high resolution C 1s spectra of graphite (b), GO (c), FG1 (d), FG2 (e), and FG3 (f). | ||
The high resolution XPS C 1s region spectra of all the samples are shown in Fig. 3(b–f) while their surface elemental analyses are presented in Table 1. As shown in Fig. 3(b), the deconvoluted C 1s peak of graphite showed peak binding energies of 284.83, 286.24, 288.28, 289.5, and 291.31 eV, corresponding to C–C, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, O–CO and π–π* bonds, respectively. However, after improved Hummer's method, the C 1s spectrum of GO was fitted into five peaks centered at about 284.74, 286.90, 288.30, 289.7, and 292.65 eV, corresponding to the C–C, C–O, CO, O–CO and π–π* bonds, respectively.42,46 By comparing Fig. 3(b) and (c), with the increase in oxygen content, the intensity of the C–C peak caused by the sp2 carbon bond in the graphite gradually decreased, while the intensity of the peaks for the oxygen-containing functional groups (such as hydroxyl, carboxyl and carbonyl) obviously increased.48 Table 1 reveals that the oxygen content increased by 28.95 at%. After fluorination reaction, Fig. 3(d) reveals the addition of two bonds at 288.93 and 292.10 eV, corresponding to C–F and –CF2 bonds.47,49 Similarly, Fig. 3(e) displays the deconvoluted C 1s peak of FG2 showing peak binding energies at 289.68 and 291.67 eV, representing C–F and –CF2 bonds. Furthermore, the deconvoluted C 1s peak of FG3 showed peak binding energies at 290.18 and 292.05 eV, representing C–F and –CF2 bonds, as shown in Fig. 3(f). Comparing the C 1s spectra of GO and the three FG samples, the oxygen-containing functional groups gradually depleted, leading to the formation of new bonds which were subsequently strengthened, indicating that the depletion was as a result of the reactions between NF3 and the oxygen-containing functional group on the surface of GO. At the end of the reaction, Table 1 shows that the content of F atom increased to 48.28 at%.
O, O–CO and π–π* bonds, respectively. However, after improved Hummer's method, the C 1s spectrum of GO was fitted into five peaks centered at about 284.74, 286.90, 288.30, 289.7, and 292.65 eV, corresponding to the C–C, C–O, CO, O–CO and π–π* bonds, respectively.42,46 By comparing Fig. 3(b) and (c), with the increase in oxygen content, the intensity of the C–C peak caused by the sp2 carbon bond in the graphite gradually decreased, while the intensity of the peaks for the oxygen-containing functional groups (such as hydroxyl, carboxyl and carbonyl) obviously increased.48 Table 1 reveals that the oxygen content increased by 28.95 at%. After fluorination reaction, Fig. 3(d) reveals the addition of two bonds at 288.93 and 292.10 eV, corresponding to C–F and –CF2 bonds.47,49 Similarly, Fig. 3(e) displays the deconvoluted C 1s peak of FG2 showing peak binding energies at 289.68 and 291.67 eV, representing C–F and –CF2 bonds. Furthermore, the deconvoluted C 1s peak of FG3 showed peak binding energies at 290.18 and 292.05 eV, representing C–F and –CF2 bonds, as shown in Fig. 3(f). Comparing the C 1s spectra of GO and the three FG samples, the oxygen-containing functional groups gradually depleted, leading to the formation of new bonds which were subsequently strengthened, indicating that the depletion was as a result of the reactions between NF3 and the oxygen-containing functional group on the surface of GO. At the end of the reaction, Table 1 shows that the content of F atom increased to 48.28 at%.
| Sample | Elemental content (at%) | O/C (%) | F/C (%) | ||
|---|---|---|---|---|---|
| C 1s | O 1s | F 1s | |||
| Graphite | 95.48 | 4.52 | — | 4.73 | 0 |
| GO | 71.05 | 28.95 | — | 40.75 | 0 |
| FG1 | 69.92 | 10.37 | 19.71 | 14.83 | 28.19 |
| FG2 | 67.49 | 3.49 | 29.01 | 5.17 | 42.98 |
| FG3 | 50.66 | 1.06 | 48.28 | 2.09 | 95.30 |
As shown in Table 1, the atomic ratio of fluorine to carbon (F/C) quantified by XPS were calculated to be 28.19%, 42.98% and 95.30% for FG1, FG2 and FG3, respectively. The ratio increased from 0% to 95.30% because NF3 was decomposed into free fluorine atoms and reacted with the oxygen-containing groups. The results show the successful preparation of FG with different fluorine contents, achieved by effective control of the reaction time of the reactants (GO and NF3). The increasing fluorine to carbon ratio with reaction time was also a notable observation.
In order to further analyze the formation of the different types of functional groups in FG. The samples were analyzed by FT-IR and the results are shown in Fig. 4. From the Fig. 4, the GO curve exhibited characteristic peaks at 3436, 1733, 1642 and 1051 cm−1, which are attributed to the stretching vibrations of O–H, CO in COOH, CC and C–O–C in Uron rings, respectively.42,46 Compared with graphite, the appearance of the CO group in GO proves the loading of oxygen-containing functional groups on the graphite surface by the improved Hummer's method. This provides more possibilities for the fluorination reaction between NF3 and graphite in contrast to the direct reaction. A new peak corresponding to the stretching vibration of C–F covalent bond at 1225 cm−1 and asymmetric stretching vibrations of CF2 groups at 1328 cm−1 appeared after the reaction of GO and NF3.50 The results demonstrate that F atoms were successfully grafted on the surface of GO by their reactions with the carboxyl groups. Meanwhile, the functional groups varied with the different reaction times of NF3, the peak intensity of the C–F covalent bond became stronger as the content of fluorine increased. The FT-IR results further indicate that FG was successfully produced by NF3 reaction, and these are consistent with the XPS results.
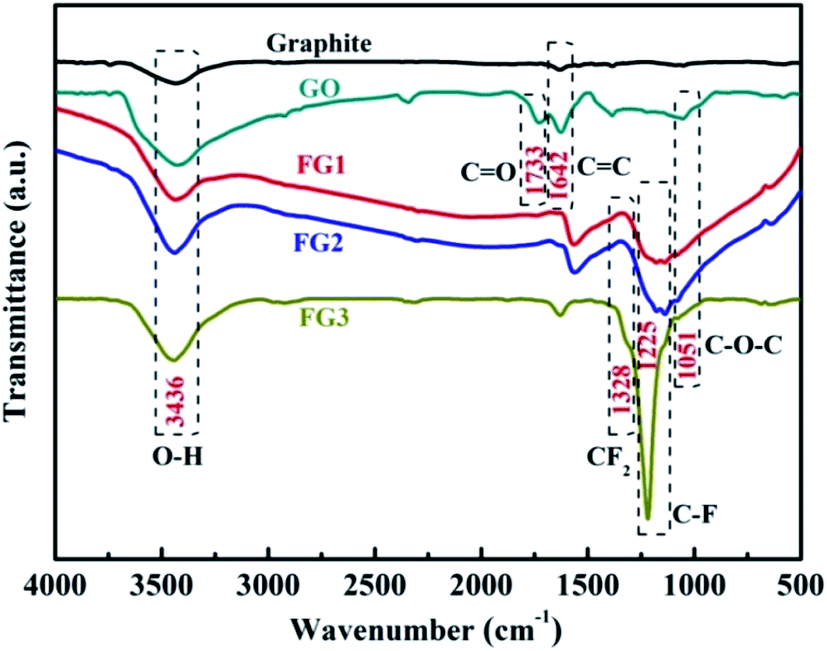 | ||
| Fig. 4 FT-IR spectra of graphite, GO, FG1, FG2, and FG3. | ||
Raman spectroscopy is commonly used to characterise variations in the degree of defects in graphene materials.14 As shown in Fig. 5, graphite, GO, FG1 and FG2 samples exhibit characteristic diffraction peaks at both the D band (1360 cm−1) and the G band (1580 cm−1). However, there is no obvious D and G peak in FG3. This may be due to the high fluorine content that caused the sp2 hybrid orbital change and a highly distorted graphite lattice.51 The G band is due to the sp2 of C vibrating in-plane and the D band represents the defective vibrational band of the graphene sample. The extent of the defect in the sample is characterized by the intensity ratio (ID:IG) of the D band to the G band.14,47 The ID:IG ratio of GO is 0.9 and higher than that of graphite at 0.277, due to the presence of oxygen atoms breaking the order of graphite. However, the ID:IG ratio of FG is higher than that of GO, and it increased with the increase of fluorine content. This result shows that the presence of fluorine atoms extremely affects the orderliness of graphite materials.
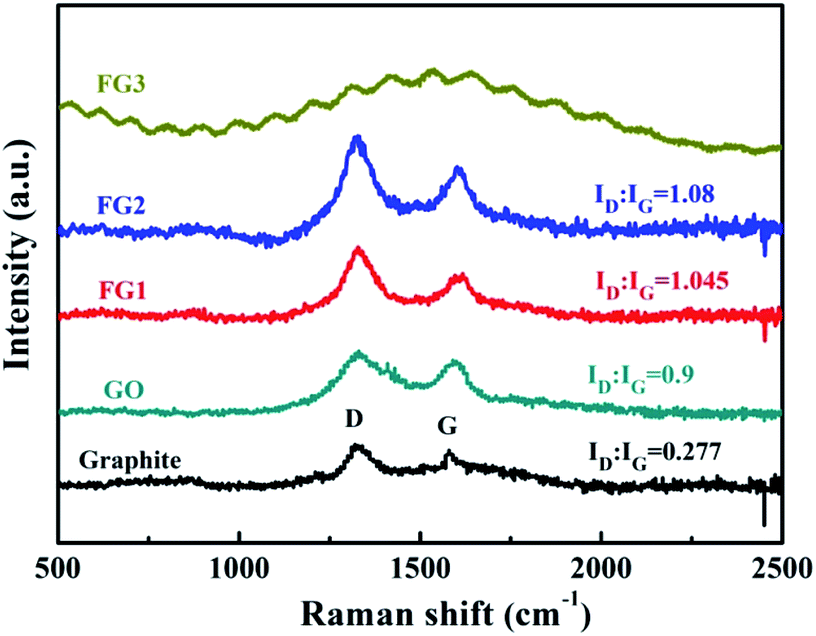 | ||
| Fig. 5 Raman spectra of graphite, GO, FG1, and FG2. | ||
The effect of the modifications by fluorination on the interlayer distance during the preparation of FG was studied by XRD analysis and the results are presented in Fig. 6. Graphite exhibited a typical (002) peak at 26.44° with a d-spacing of 0.339 nm. Through the improved Hummer's method, GO peak value shifted to 10.59° with a corresponding d-spacing of 0.835 nm. The results indicate that abundant oxygen atoms were inserted into the interlayer space, yielding a completely oxidized graphite.52
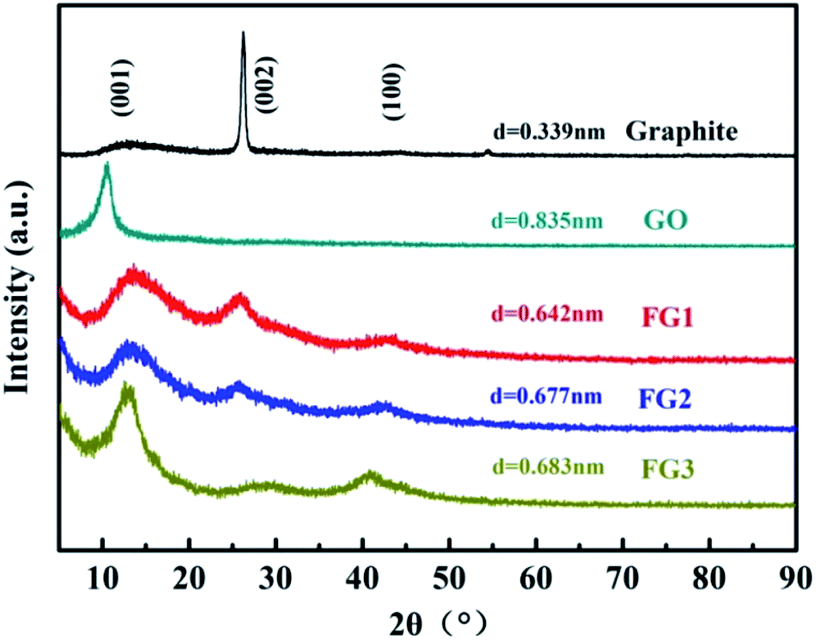 | ||
| Fig. 6 XRD patterns of graphite, GO, FG1, FG2, and FG3. | ||
Although having different fluorine contents, the same characteristic peaks of (001), (002) and (100) were observed for all the FG samples (Fig. 6). By the Bragg equation, the 2θ and d-spacing of FG1, FG2 and FG3 were 13.78°, 13.06° and 12.95°, and 0.642 nm, 0.677 nm and 0.683 nm, respectively. The novel peak could be indexed as the (001) reflection in a hexagonal system particular for compounds exhibiting very high F levels.53 Although, the decrease of the peak intensity at 10.59° indicates that the crystallites arranged with (001) orientation were disordered by fluorination under excess fluorine gas, this phenomenon is caused by chemical reactions between GO and NF3. Notably, the d-spacing calculated for GO and FG decreased from 0.835 nm to approximately 0.683 nm. The results further proves the successful modification of GO, and illustrates the strong interaction between GO and NF3. The very weak and broad peak around 26.44° could be assigned to the (002) reflection, confirming the existence of sp2 C in FG and the sample was poorly ordered. The peak around 41.95° could be assigned to the (100) reflection associated with the C–C in-plane length in the reticular system.54
The surface morphology of GO and FG nanosheets dispersed in ethanol were observed by SEM and AFM. Fig. 7 illustrates the SEM images showing the morphologies and microstructures of GO and the three FG samples. Fig. 7(a1) reveals that the expected smooth surface area of the GO sheet is large and complete, together with obvious wrinkles and overlaps. This observations are connected to the sp3 carbons and formation of oxygen-containing functional groups in the basal planes, as well as the various GO structural defects.55,56 Moreover, the thickness of GO was about 1.398 nm as shown in Fig. 7(a2 and a3).
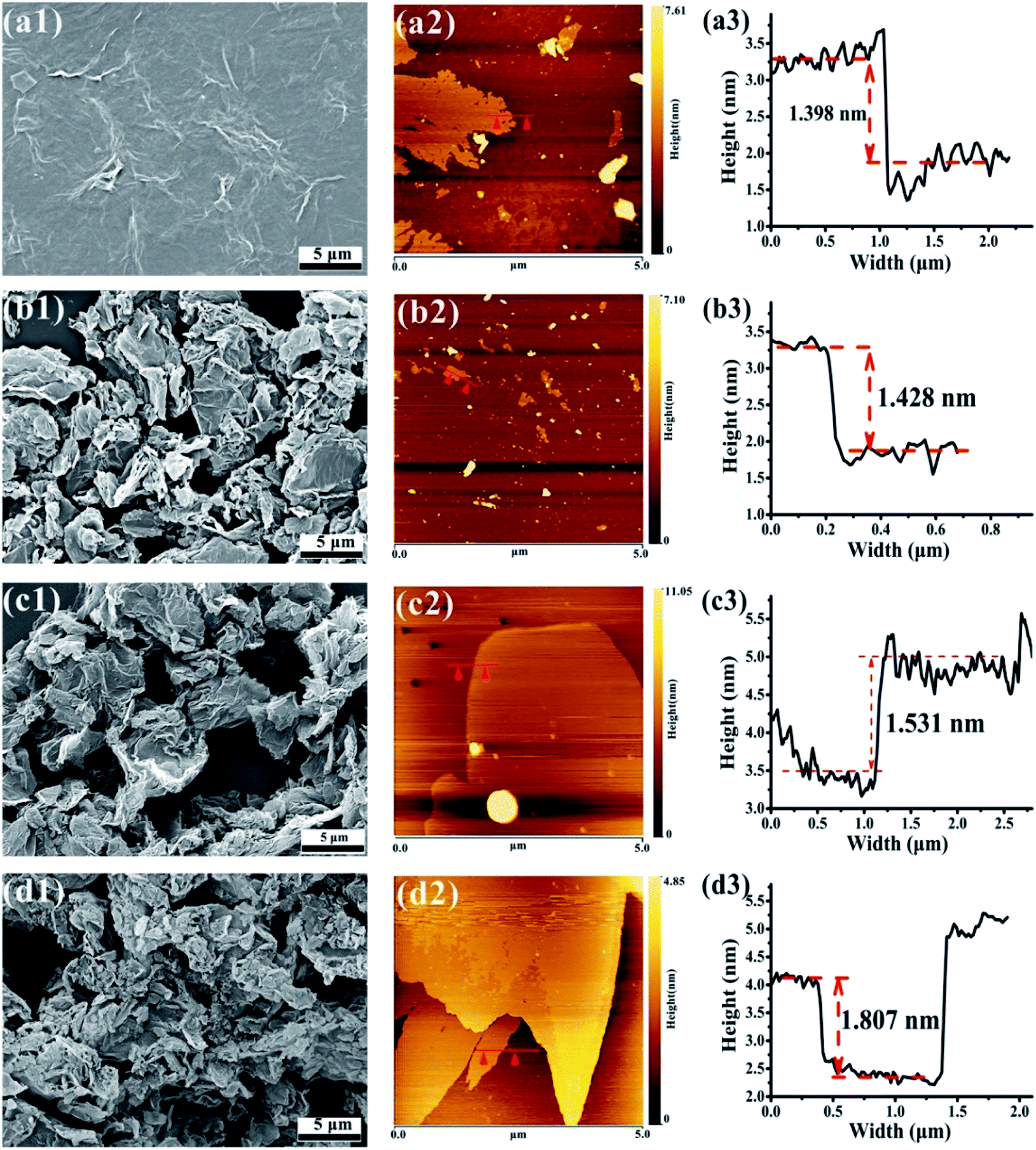 | ||
| Fig. 7 SEM images of GO (a1), FG1 (b1), FG2 (c1), and FG3 (d1). AFM images of GO (a2 and a3), FG1 (b2 and b3), FG2 (c2 and c3), and FG3 (d2 and d3). | ||
Fig. 7(b1, c1 and d1) shows that the three FG sheet materials were obviously loose and uniformly dispersed, and had different degrees of agglomeration and surface wrinkles compared with GO. Low surface energy of C–F bond existed on the surface or between the layers of FG, leading to obvious overlaps of the FG nanosheets. As the content of fluorine increased up to 48.28%, the agglomeration of FG resultantly increased. Meanwhile, Fig. 7(b3, c3 and d3) also shows that the thickness of the three FG nanosheets were 1.428 nm, 1.531 nm, and 1.807 nm, respectively. These observations are consistent with those from XPS, FT-IR, and XRD which authenticated the modification of GO and the formation of FG with different fluorine contents having two-dimensional structures like GO. However, due to the different fluorine contents in the FG samples, there are expected differences between the GO and GO-modified epoxy coatings. Therefore, the next focus will be on the effect of different fluorine contents in FG on the performance of epoxy resin coatings.
3.2 Characterization of FG/EP composites
Sedimentation test and SEM cross-sectional microstructure analysis were employed to determine the compatibility and stability of pigments in the epoxy resin solution. This was done to access the compatibility of the FG sheets in epoxy resin. The required sample volume in transparent glass vials was 10 ml. Fig. 8 shows the digital photos of FG powders dispersed in the epoxy resin solution taken through light transmission. After sand milling for 120 minutes, the GO and FG fillers were evenly dispersed in the epoxy resin as shown in Fig. 8. Consequently, Fig. 8(a) shows observed sedimentation up to 72 h. The bottom of the sedimentation flask changed from yellow to black in the GO/EP resin solution, indicating that sedimentation occurred. For observation up to 3000 h, FG1 and FG2 did not show any obvious settlement in epoxy resin as shown in Fig. 8(b and c). The results indicate that FG nanosheets can be uniformly dispersed and has good compatibility and stability in epoxy resin. However, Fig. 8(d) shows that FG3 was poorly dispersed in epoxy resin, with the observation of a slight yellow-brown precipitate at 1320 h. With the extension of the test time, the amount of precipitation significantly increased and the color became darker. On the whole, the compatibility and stability of FG in epoxy resin is better than that of GO. With increased degree of fluorination, FG with high fluorine content exhibited ease of agglomeration and sedimentation in the epoxy resin, thereby compromising the corrosion protection properties of the coating. | ||
| Fig. 8 Visual illustration of sheets dispersed in resin at different times (a) GO/EP, (b) FG1/EP, (c) FG2/EP, and (d) FG3/EP. | ||
Fig. 9 presents the cross-sectional microstructure characteristics of GO/EP and the three FG/EP coatings after brittle fracture. From Fig. 9(a), there were obvious agglomerations and a large number of cracks in the GO/EP coating, which indicates that GO was not well dispersed in the EP coating. However, the cross-sectional microstructure of the FG1/EP coating reveals a uniform and smooth plane, as shown in Fig. 9(b). No cracks were found on the coating surface and the surface between EP coating and pigment filler. This result signifies that FG1 was excellently dispersed in the epoxy coating. Similarly, no cracks were found in the cross-sectional structure of the FG2/EP coating as displayed in Fig. 9(c). Conversely, the cross-sectional microstructure of FG3/EP coating showed cracks and obvious agglomerations of fillers, as shown in Fig. 9(d). Combining the results of the sedimentation test and the SEM image of the coating cross-section, it is clear that the compatibilities of FG1 and FG2 nanosheets in epoxy resin are better than those of GO and FG3 nanosheets. These results further indicates that the better compatibilities of FG1 and FG2 may be beneficial to their contributions in the long-term corrosion resistance of the epoxy coatings, while the poor compatibilities of GO and FG3 may be disadvantageous.
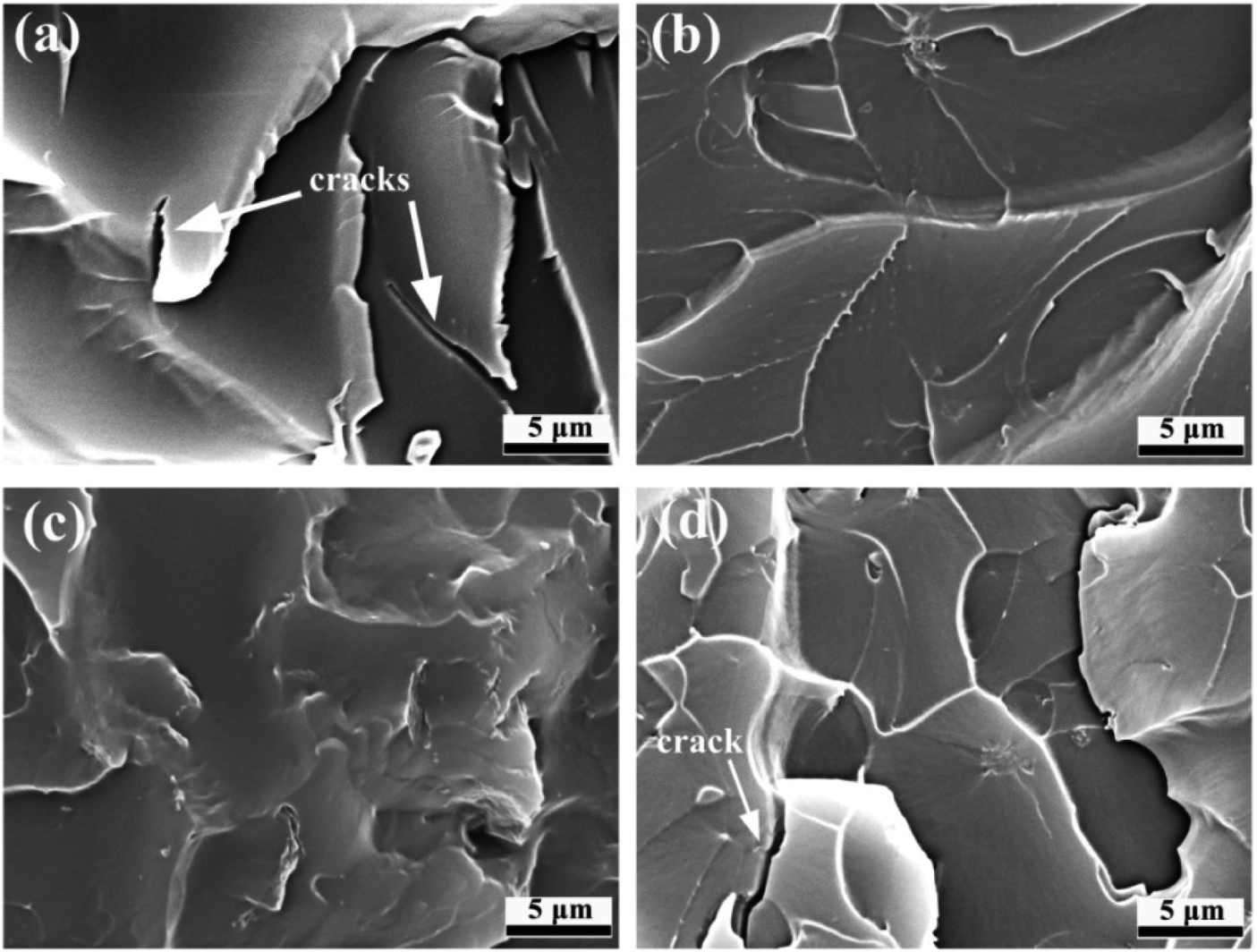 | ||
| Fig. 9 SEM images of the cross-sectional microstructures of (a) GO/EP, (b) FG1/EP, (c) FG2/EP, and (d) FG3/EP. | ||
The contact angle of a coating surface is an important index to evaluate the wettability of the coating.57 Fig. 10 displays the contact angle (θ), work of adhesion (WA), and surface free energy (γsv) results of EP, GO/EP, and the three FG coatings. The results for WA and γsv were calculated by Young's and Neumann equations.58
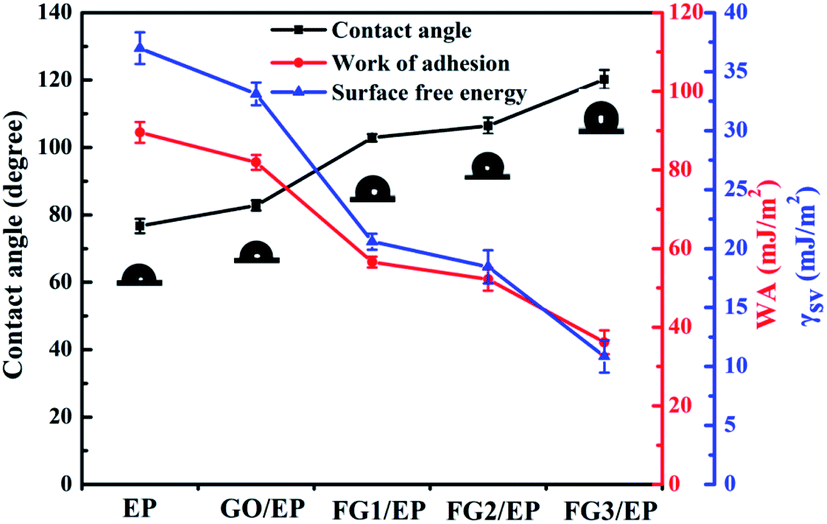 | ||
| Fig. 10 Plots of the contact angle (θ), work of adhesion (WA), and surface free energy (γsv) of EP, GO/EP, and three FG coatings. | ||
The contact angle of the epoxy coating was about 76.7°, while the contact angle of GO/EP and FG/EP coatings were larger than EP coating owing to the doping of GO and FG with different fluorine contents. It is worth noting that the contact angle of the FG/EP coatings (102.88°, 106.45° and 120.2° corresponding to FG1, FG2, and FG3, respectively) were significantly higher than that of the GO/EP coating (82.8°). These results indicate that FG can improve the hydrophobicity of EP coating as it also exhibited low values of WA and γsv. All of these can reduce the droplets left on the coating surface, reduce the possibility of wetting, enhance the barrier ability of the coating to liquids, and indirectly improve the corrosion resistance of the coating.
3.3 Corrosion protection properties of FG/EP coatings
The effect of the addition of GO and FG nanosheets on the barrier and corrosion protection properties of EP coating was discussed and compared using OCP and EIS. The samples were immersed in 3.5 wt% NaCl solution and tested at different immersion times. As shown in Fig. 11, the OCP values for the various coatings decreased with immersion time. At the initial stage of immersion, the OCP values dropped sharply to the lowest point. The shift of the OCP to negative directions indicates the increased risk of diffusion of water and corrosive species into the interface between the coating and metal. However, FG/EP coatings exhibited the more positive OCP values than EP and GO/EP coatings, indicating that FG nanosheets can provide excellent barrier performances in EP coating and minimize the risk of corrosion compared with the GO/EP coatings.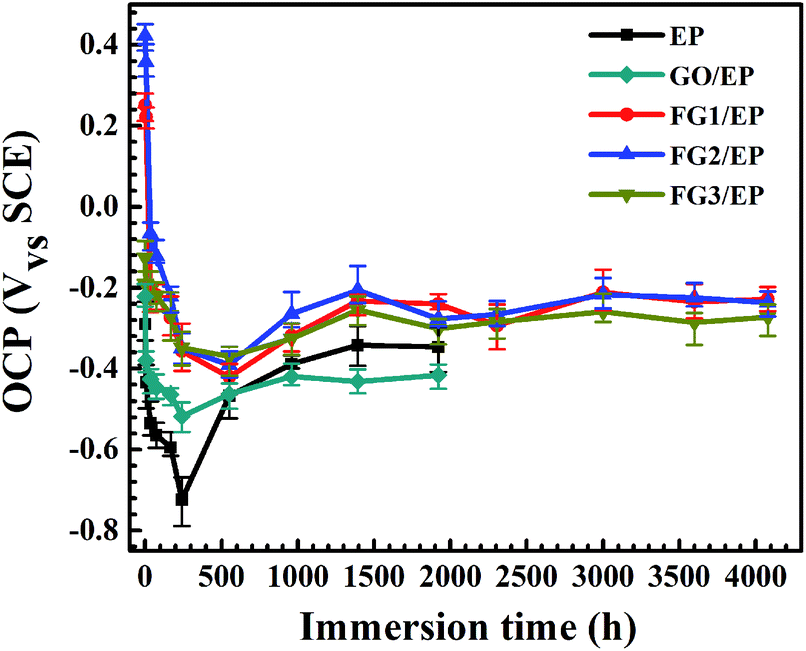 | ||
| Fig. 11 OCP values of EP, GO/EP and the three FG/EP coatings after different immersion durations in 3.5 wt% NaCl solution. | ||
According to previous research reports, the impedance modulus at low frequency (|Z|0.01Hz) is an important parameter to characterize the corrosion resistance of coating.59–61 Fig. 12 shows the |Z|0.01Hz of EP, GO/EP and FG/EP coatings immersed in 3.5 wt% NaCl solution at different durations. At the initial stage of immersion, the |Z|0.01Hz of all the samples decreased. The EP and GO/EP coatings had a |Z|0.01Hz value of 4.96 × 1010 Ω cm2 and 7.89 × 1010 Ω cm2, respectively. After 240 h immersion, the |Z|0.01Hz value of the EP coating increased significantly and fluctuated at about 7.09 × 107 Ω cm2 to 5.72 × 108 Ω cm2 at 1920 h immersion. Also, the |Z|0.01Hz value for the GO/EP coating increased from 1.29 × 109 Ω cm2 for 240 h immersion and stabilized at 4.69 × 109 Ω cm2 for 1920 h immersion. Therefore, the GO nanosheet has the potential to improve the corrosion resistance of the EP coating to a certain extent. However, the |Z|0.01Hz values of the three FG/EP coatings were 2 orders of magnitude higher than that of the EP coating. The |Z|0.01Hz value of FG2/EP coating stabilized at 8.57 × 1010 Ω cm2 to 7.27 × 1010 Ω cm2 after 4080 h immersion. The values for FG1/EP and FG3/EP coatings were similarly as stable as that of the FG2/EP coating. These results further confirm that the FG/EP coatings provide stronger corrosion protections than the EP and GO/EP coatings, thus impeding electrolyte diffusion towards the Al substrate.
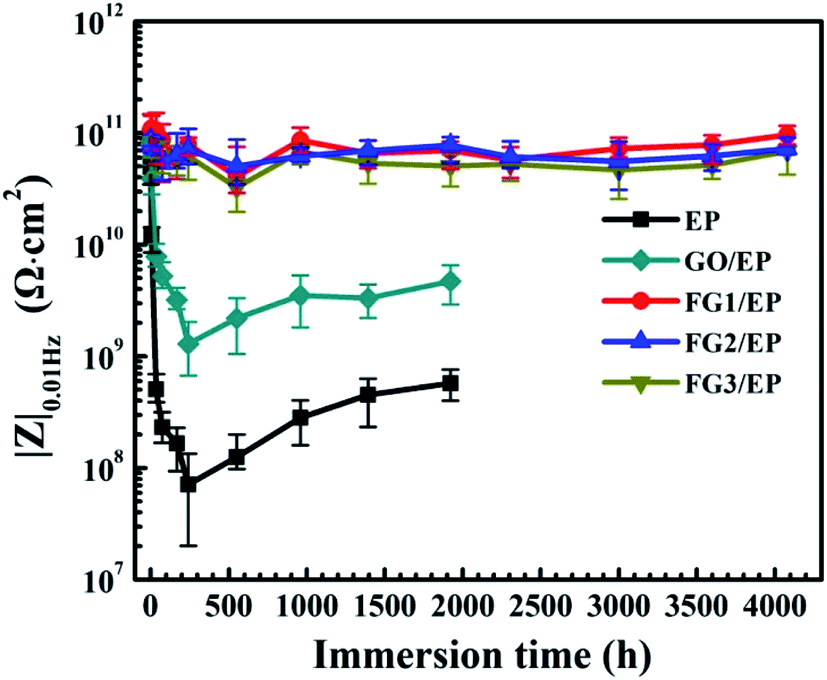 | ||
| Fig. 12 Evolutions of |Z|0.01Hz of the EP, GO/EP and three FG/EP coatings after different immersion duration in 3.5 wt% NaCl solution. | ||
Fig. 13 illustrates the Bode and Nyquist plots of EP, GO/EP and the FG/EP coatings immersed in 3.5 wt% NaCl solution for 0.5 h, 240 h, 960 h, 1920 h, and 4080 h. According to Fig. 13(a and b), the Bode and Nyquist plots of all the coatings exhibited one time constants for the early immersion period. The circuit diagram (model A) for the fitting of experimental data at this stage is shown in Fig. 14. The diagram suggests that at this stage the corrosive species penetrated into the coating but did not reach the coating/metal interface.23 Moreover, the low-frequency impedance modulus (|Z|0.01Hz) of all the coating samples exceeded 7.6 × 1010 Ω cm2, except for EP with 4.96 × 1010 Ω cm2. The exact values were 7.89 × 1010, 1.06 × 1011, 8.57 × 1010, and 7.62 × 1010 Ω cm2 for GO/EP, FG1/EP, FG2/EP, and FG3/EP coatings, respectively. The absolute impedance of the FG1/EP, FG2/EP, and FG3/EP coatings were independent of the frequency and involved a phase angle approaching 90° within a frequency range of 10–105 Hz. The three FG/EP coatings showed higher |Z|0.01Hz values than EP sample. Among of all the coating samples, only FG2/EP coating attained absolute impedance close to 80° at a frequency of 0.01 Hz (Fig. 13(a)). Nonetheless, they all displayed good barrier properties at the initial immersion period. After immersion for 240 h, the |Z|0.01Hz of the EP coating dropped to 7.09 × 107 Ω cm2, whereas its phase angle approached 80° within a frequency range of 102–105 Hz, which was 10° lower than that recorded at 0.01 Hz, as shown in Fig. 13(c). It can be observed from Fig. 13(d) that a new low frequency diffusion field appeared around the EP coating, which led to the inclusion of the Warburg impedance element (W) to the equivalent circuit (Fig. 14 model C). These results reveal that the electrolytes penetrated or diffused through the micro-pores and defects into the EP and GO/EP coatings, and the diffusion effects of the coating or coating/substrate interface dominated the corrosion attack at low frequency.62 The same observation was seen for the plot of the GO/EP coating. However, the |Z|0.01Hz values for FG1/EP, FG2/EP, and FG3/EP coatings decreased to 7.91 × 1010, 7.03 × 1010 and 6.15 × 1010 Ω cm2, respectively.
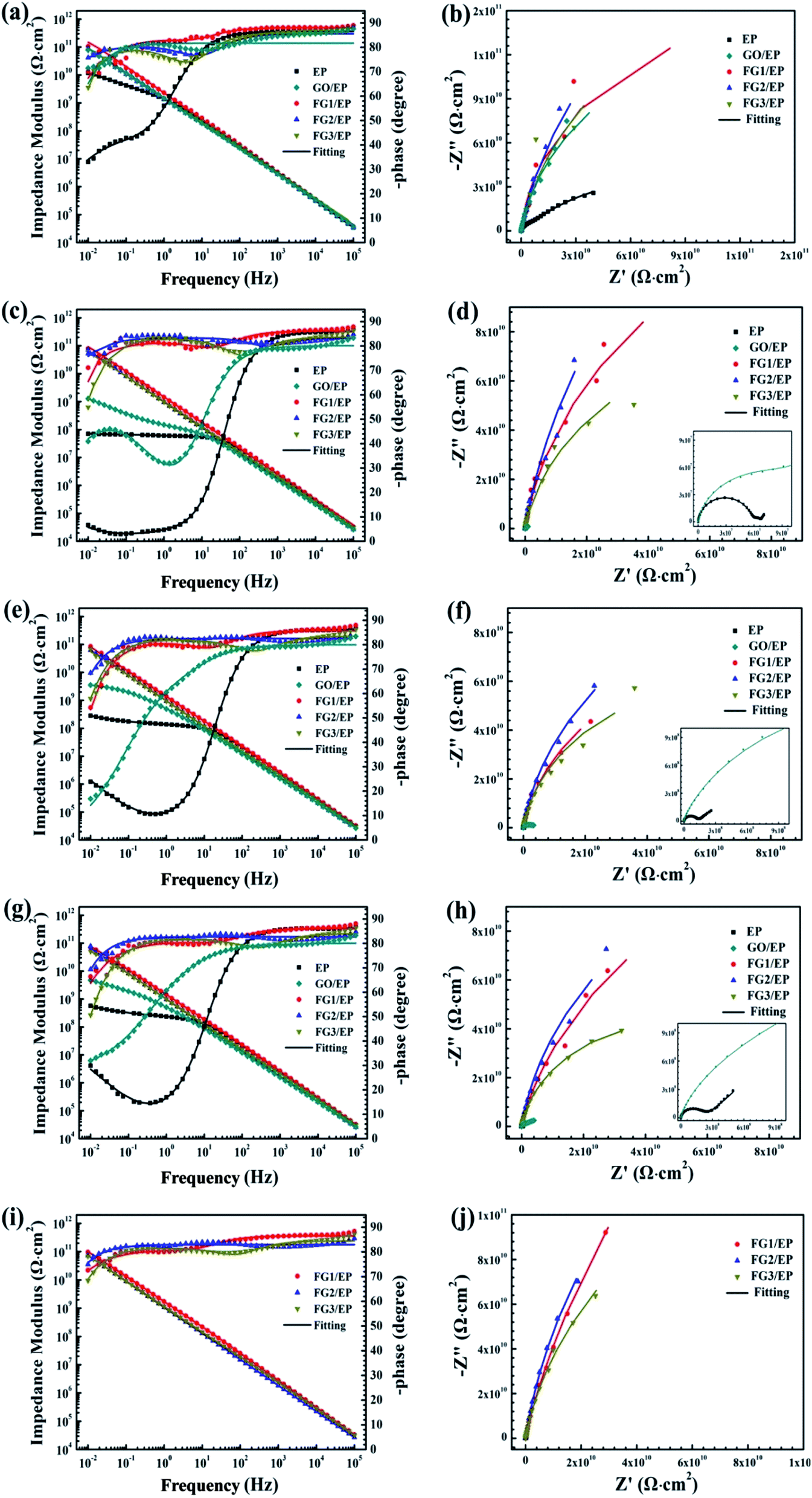 | ||
| Fig. 13 Impedance plots of EP and three FG/EP coatings immersed in 3.5 wt% NaCl solution for (a and b) 0.5 h, (c and d) 240 h, (e and f) 960 h, (g and h) 1920 h, and (i and j) 4080 h. | ||
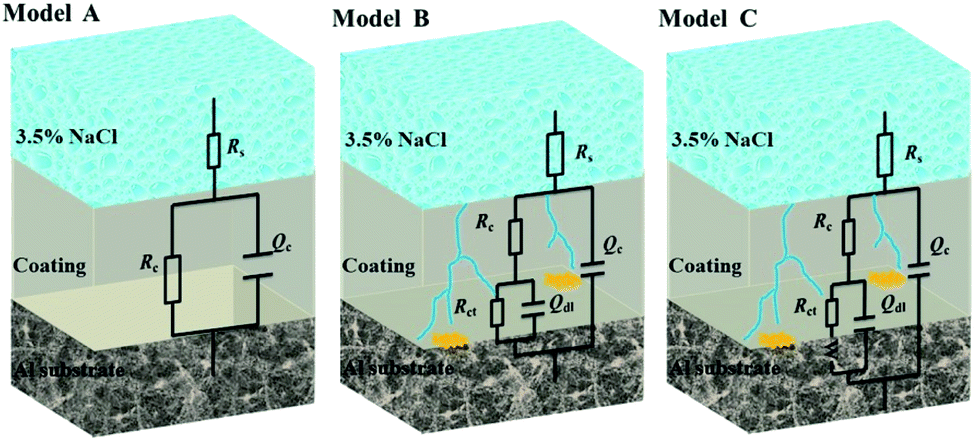 | ||
| Fig. 14 The equivalent electrical circuits used for the fitting of EIS data. | ||
As the immersion time increased to 960 h, the |Z|0.01Hz values and phase angle exhibited an upward trend (Fig. 13e and f). At this stage, corrosion products were formed due to the increased diffusion of the electrolytes within the coating and/or the coating/substrate interface, and the accumulation of the corrosion products blocked the holes and defects. For GO/EP coating, corrosion-induced acceleration of charge transfer resistance occurred at the interface between the metal substrate and the coating (Fig. 13e and f), especially with the second time constant at the low-frequency phase becoming obvious, so the model B equivalent circuit in Fig. 14 was still applied. After immersion for 1920 h (Fig. 13g and h), the |Z|0.01Hz values of the three FG/EP coatings were significantly higher than that of EP and GO/EP coatings. FG2/EP coating had the highest value (7.76 × 1010 Ω cm2) of all the coatings, which is more than one order and two orders of magnitude to that of the GO/EP and EP coatings, respectively. As the immersion time increased to 4080 h, Fig. 13(i and j) reveals that the results of the three FG/EP coatings remained relatively unchanged. In general, the FG nanosheets obviously enhanced the corrosion protection performance of the EP coating. Notably, the FG2/EP coating provided the best corrosion protection compared to the other FG/EP coatings.
The equivalent electrical circuits in Fig. 14 were used to fit the EIS data.62,63 From the Fig. 14, model A is composed of solution resistance (Rs), coating capacitance (Qc), and coating resistance (Rc). In addition to the aforementioned parameters, model B and C have the double layer capacitance (Qdl), the charge transfer resistance (Rct), and the Warburg impedance element (W). The Nyquist plots of all the samples at the initial immersion exhibited a purely capacitive loop (Fig. 13(b)). Thus, model A was used to fit the process. As the immersion time progressed, the corrosive species penetrated the EP, GO/EP, FG1/EP and FG3/EP coatings into the coating/substrate interface, and the processes were fitted with equivalent circuit model B or C, as were required.
In order to further understand the barrier effect of GO and FG nanosheets in epoxy resin, the fitting circuits parameters Rc, Rct, and Qc were separately discussed.64 Generally, Rc is used to represent the barrier and shielding effect of coatings on corrosive media. Rc decreasing with immersion time was due to the penetration of corrosive species through the coating micropores or defects. Fig. 15(a) displays the Rc values of EP, GO/EP, and the three FG/EP coating samples at different immersion times. The Rc value of the EP coating sharply decreased from 4.58 × 1010 Ω cm2 to 5.70 × 107 Ω cm2 within 240 h immersion. After 240 h immersion, the value increased slightly and finally stabilized at 1.66 × 108 Ω cm2 after 1920 h. Similarly, the Rc value of the GO/EP coating sharply decreased from 3.06 × 1011 Ω cm2 to 1.13 × 108 Ω cm2 within 240 h immersion. This may be due to the poor dispersion of GO in the epoxy matrix and the formation of cracks in the EP coating as shown in Fig. 9(a), and thus sharply decreasing the coating resistance. After 240 h immersion, the Rc value increased slightly, due to the formation of corrosion products and the slow diffusion effects, and finally stabilizing at 1.68 × 108 Ω cm2 after 1920 h. As the fluorine content increased, the Rc value of the FG/EP coating increased and then decreased. When the fluorine content in FG was 29.01%, FG2/EP coating had the largest Rc value, and the barrier effect of the coating was prominent within 4080 h immersion. The Rc values of FG1/EP coating decreased from 3.13 × 1011 Ω cm2 to 3.43 × 108 Ω cm2 while that of FG3/EP coating decreased from 4.02 × 1011 Ω cm2 to 3.54 × 107 Ω cm2 within 552 h immersion. After 552 h immersion, the Rc value fluctuated between 3.09 × 108 Ω cm2 and 2.60 × 107 Ω cm2. In addition, the Rc value of FG2/EP coating decreased from 5.8 × 1011 Ω cm2 to 2.76 × 1011 Ω cm2 after 4080 h immersion. The results imply that the FG2/EP coating exhibited an excellent physical barrier effect and restrained the penetration of corrosive species, which was as a result of its high compatibility in epoxy resin, as well as its hydrophobicity (Fig. 9(c) and 10).
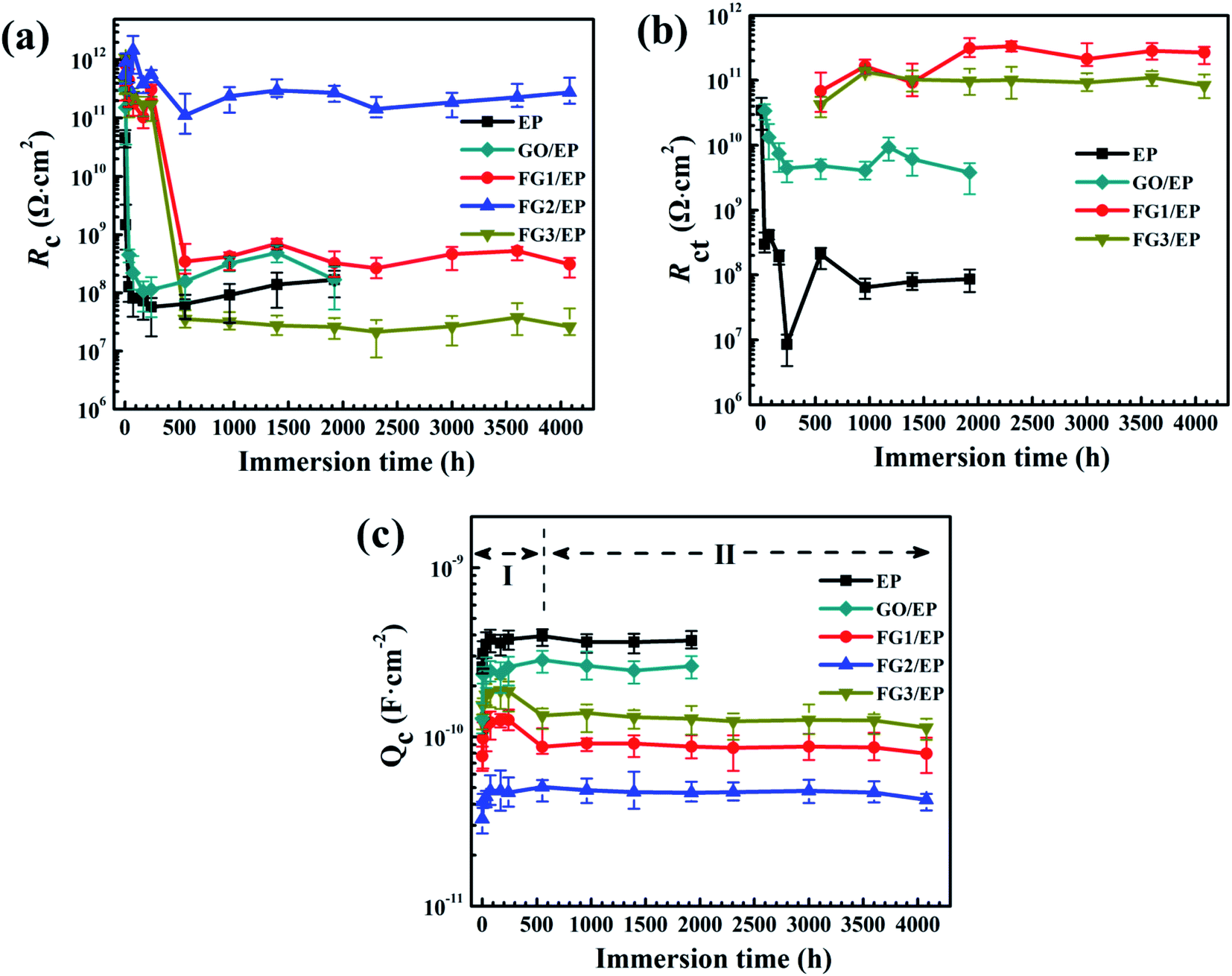 | ||
| Fig. 15 Plots of the coating resistance Rc (a), charge transfer resistances Rct (b), and coating capacitance Qc (c), as functions of the immersion time for EP, GO/EP and the three FG/EP coatings after different immersion durations in 3.5 wt% NaCl solution. | ||
Rct is usually measured at the interface between the coating and metal substrate, which is related to the degree of electron transfer across the metal surface, and it is inversely proportional to the corrosion rate of the metal.60,65 This means that the larger the Rct value, the more difficult it is for the metal to corrode, and vice versa. Fig. 15(b) shows the variation of Rct value with the immersion time for EP, GO/EP, and the three FG/EP coatings. The Rct value of the EP coating dramatically decreased from 3.44 × 1010 Ω cm2 to 8.47 × 106 Ω cm2 within 240 h immersion. This was the least Rct value recorded for this study. After 240 h immersion, the Rct value increased slightly and finally stabilized at 8.68 × 107 Ω cm2 for 1920 h immersion. Likewise, The Rct value of the GO/EP coating considerably decreased from 3.40 × 1010 Ω cm2 to 4.40 × 109 Ω cm2 within 240 h of immersion, and the value increased slightly with increase in immersion time before stabilizing at 3.80 × 109 Ω cm2 for the 1920 h immersion. However, the Rct values of FG1/EP and FG3/EP coatings did not emerge until 552 h immersion. The values of the coatings gradually increased from 6.83 × 1010 to 2.68 × 1011 Ω cm2 and 4.25 × 1010 to 8.42 × 1011 Ω cm2 within 4080 h immersion. This was due to the hydrophobic properties of the FG/EP coatings, which is better than those of the EP and GO/EP coatings as shown in Fig. 10. It is therefore became difficult for the electrolytes to penetrate the FG/EP coatings and diffuse to the coating/substrate interface within 552 h immersion, thereby accounting for the absence of Rct. As the immersion time progressed, the corrosion tendency of FG1/EP and FG3/EP coatings began to emerge, and the internal defects of the coating began to dominate the corrosion process. Notably, the Rct of the FG2/EP coating did not emerge within 4080 h immersion. The electrolytes could not permeate through the coating because of its excellent hydrophobic performance and the homogeneous nature of FG2 in EP matrix.
The Qc value is generally considered an important parameter in the determination of water absorption and water resistance of coatings.64 Water absorption of organic coatings reveals their protective properties as water on coating/metal interface may cause loss of adhesion and blistering. Fig. 15(c) shows the Qc values of EP, GO/EP, and the three FG/EP coatings at different immersion times. These Qc curves can be roughly divided into two stages, one is the water absorption and accumulation in the coatings caused by the penetration of the solution into the coatings at early immersion, while the second stage is the saturation state of the water absorption in the coatings.60 The Qc value of the EP coating increased from 2.59 × 10−10 to 3.78 × 10−10 F cm−2 within 240 h immersion. After 240 h immersion, the Qc value slightly decreased and stabilized at 3.70 × 10−10 F cm−2 for 1920 h immersion. Meanwhile, the Qc value of GO/EP coating increased from 1.28 × 10−10 to 2.59 × 10−10 F cm−2 within 240 h. After immersion for 240 h, the Qc value stabilized at 2.62 × 10−10 F cm−2 corresponding to the immersion at 1920 h. However, the Qc values of the three FG/EP coatings had much lower values than those of the EP and GO/EP coatings, indicating that FG/EP adsorbed less water or exhibited excellent water resistance. Notably, the Qc value of FG2/EP increased from 3.27 × 10−11 to 4.25 × 10−11 F cm−2 within 4080 h immersion. This is because the low surface energy of FG possibly endowed the FG/EP coating with hydrophobic characteristics, hence reducing the possibility of solution penetration, and inhibiting the transport of corrosive species into the EP coating, thus effectively preventing the metal from corrosion. The results further prove that the 29.01% FG nanosheets provided the best corrosion performance.
After the electrochemical impedance test, all the coating samples were photographed to estimate corrosion impacts on them, and the images are presented in Fig. 16. After 1920 h immersion, many corrosion products and blisters appeared on the EP coating (Fig. 16(a)). Simultaneously, some corrosion pittings were observed on the GO/EP coating (Fig. 16(b)). Comparatively, slight localized corrosion and blisters were observed on the FG1/EP and FG3/EP coatings after immersion for 4080 h as shown in Fig. 16(c and e). In contrast, Fig. 16(d) indicated that the FG2/EP coating surface showed no obvious corrosion effect. The results indicate that the FG nanosheets significantly improved the corrosion resistance of EP coating. Meanwhile, the FG2/EP coating with a fluorine content of 29.01% exhibited a long-term corrosion resistance effect due to its excellent hydrophobic characteristics and physical barrier performance in epoxy resin.
 | ||
| Fig. 16 Digital photographs taken at the end of the electrochemical impedance test showing corrosion effects on (a) EP, (b) GO/EP, (c) FG1/EP, (d) FG2/EP, and (e) FG3/EP coatings immersed in 3.5 wt% NaCl solution. | ||
SVET was used to measure the potential signal changes around the coating surface (3 × 3 mm2 areas), and the signals were converted to current density by applying Ohm's law.66 The current density was used to evaluate the local electrochemical corrosion degree of the metal substrate under the coating. Fig. 17 shows the current density distribution maps of coated Al alloys immersed in a 3.5 wt% NaCl solution. Usually, high anodic current densities are displayed in red, while low anodic current densities are displayed in blue.
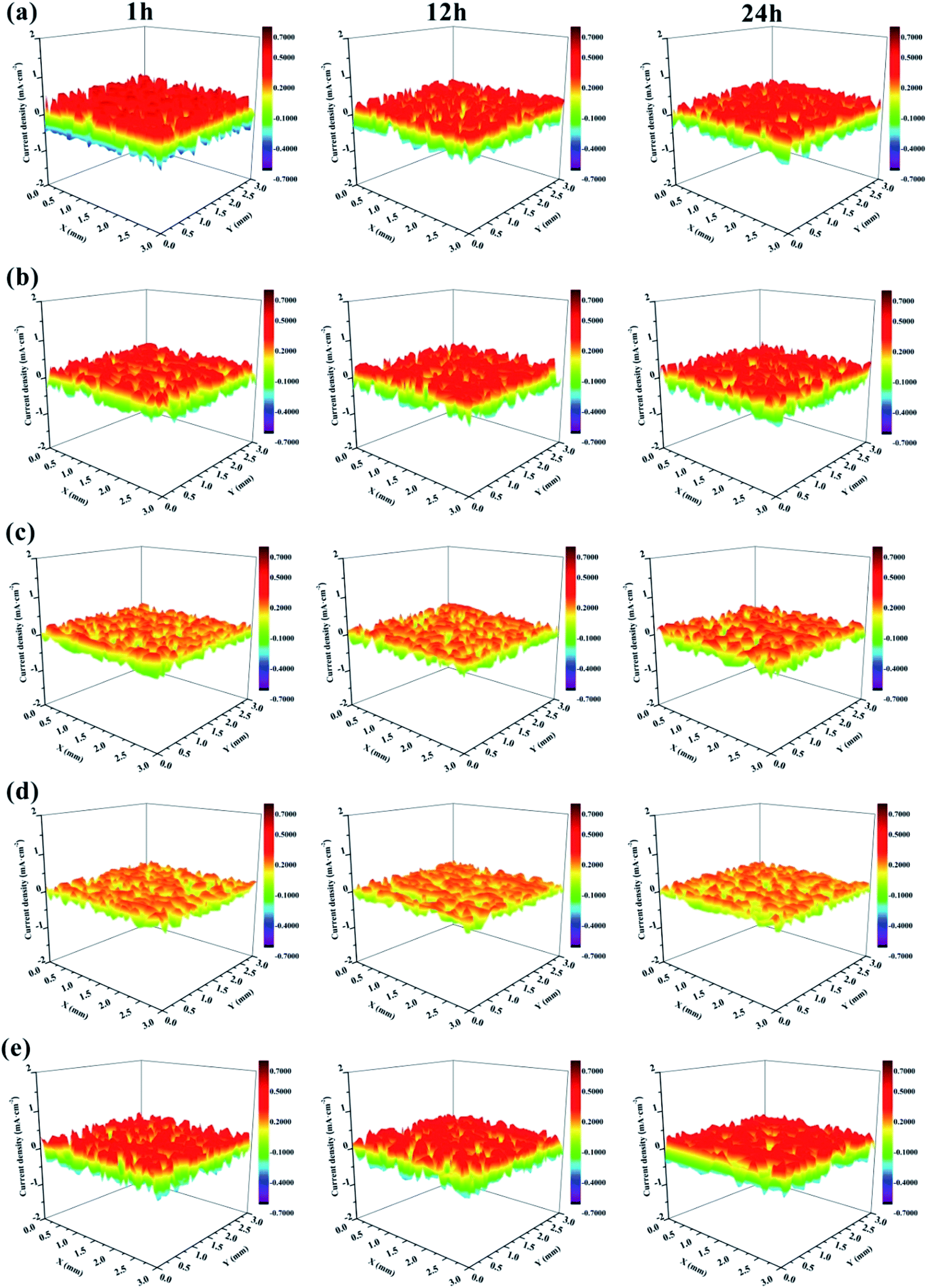 | ||
| Fig. 17 SVET maps of the current density for the defected surface of (a) EP, (b) GO/EP, (c) FG1/EP, (d) FG2/EP, and (e) FG3/EP coatings after different immersion durations in 3.5 wt% NaCl solution. | ||
It is obvious that the EP coating (Fig. 17(a)) exhibited the largest red portion. This translated to its very high anodic current density which was 0.575 mA cm−2 after 1 h immersion, and it decreased to 0.427 mA cm−2 after 12 h immersion. This may be explained by the penetration of corrosive species (in this case, chloride and oxygen) from the test solution (3.5 wt% NaCl solution) through the coating and reaching the metal surface, reacting with the surface to form corrosion products, which is expected to block the micropores and defects, and obstructing further transport of corrosive species to the metal surface. Ultimately, this would decrease the corrosion current density and reduce the rate of corrosion. For this reason, there was no obvious difference between the maps of EP coating immersed for 12 h to that immersed for 24 h. Fig. 17(b) shows that the corrosion current density of the GO/EP coating was lower than that of the EP coating. It reached 0.408 mA cm−2 after 1 h immersion, and slightly decreased to 0.399 mA cm−2 when the immersion time increased to 12 h. The results signify that the GO/EP coating was slightly corroded with few buildups of corrosion products on the surface as the GO nanosheets delayed the infiltration process of the corrosive species. Compared with the EP coating, the FG/EP coatings exhibited excellent corrosion resistances, with the FG2/EP coating having the highest value as shown in Fig. 17(c, d and e). At the initial immersion stage, the corrosion current densities of FG1/EP, FG2/EP and FG3/EP were 0.241, 0.223 and 0.427 mA cm−2, respectively. The values after 24 h immersion were 0.251, 0.223 and 0.408 mA cm−2, respectively. The experimental results showed that FG/EP exhibited better corrosion resistance than EP and GO/EP coatings. The results further illustrates that the barrier effect of FG was better than that of GO in the EP coating. However, excessive fluorination reduced the corrosion protection of FG/EP, which is attributed to the poor dispersion of FG3 leading to the formation of cracks in the EP coating. The SVET results generally reflect the same observations with the EIS measurements.
3.4 Protection mechanism of coatings
Based on the above results, the protection mechanism diagrams of the different coatings are proposed and presented in Fig. 18. The EP coating being hydrophilic in nature has a great ease to form micropores during its curing process. It is therefore more likely to allow the penetration of oxygen, water molecules, and ionic species through it and onto the coating/substrate interface, increasing the risk of metal corrosion and compromising the long-term protection of the metal. The GO/EP coating has a more improved corrosion protection compared to the EP coating. This is due to the observed “maze” effect of GO nanosheet structures in the epoxy matrix, slowing down the rapid penetration of oxygen, water molecules and ionic species to considerable extents. With extended immersion time, the GO/EP coating proved unreliable in the corrosion protection of the substrate. However, the FG/EP coatings exhibited the best prospects for long-term corrosion resistance compared with the EP and GO/EP coatings. Notably, the FG2/EP coating manifested the most excellent long-term corrosion resistance performance compared with the other FG/EP coatings. The enhancement of corrosion protection performance can be attributed to the following factors: (1) FG2 had a tremendous dispersivity and an exceptional compatibility in the epoxy matrix, which greatly reduced the micropores and/or defects in the EP coating, thus improving its integrity. (2) The low surface energy of FG improved the hydrophobicity of the FG/EP coating, which prevented the transportation of the corrosion medium through the coating and to the metal surface, thereby reducing the possibility of substrate corrosion. (3) The lamellar structure of FG can further impede the penetration of corrosive species through the coating. An obstruction to the penetration paths of corrosive solutions greatly extends the time for corrosion impact on the metal substrate, thereby improving their physical barrier effects. The dual influence of hydrophobicity and barrier effect accounts for the long-term protective effect of FG2/EP coating on metals.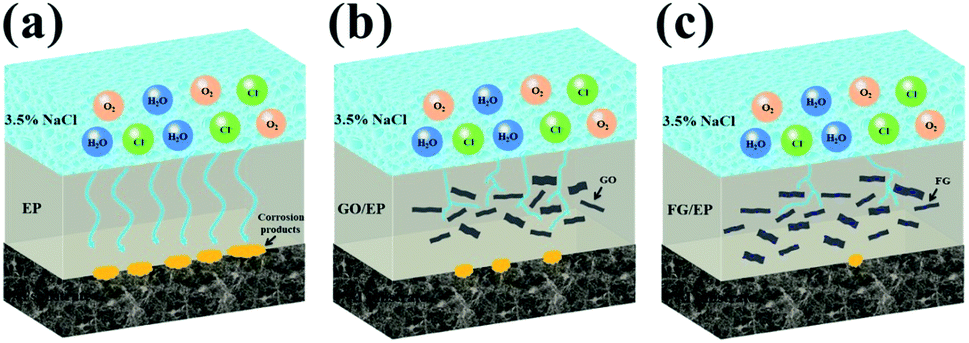 | ||
| Fig. 18 Corrosion protection mechanism of (a) EP, (b) GO/EP and (c) FG/EP coatings. | ||
4. Conclusions
In summary, FG with different fluorine contents were produced by time-controlled chemical reaction of GO with NF3 gas, and the products were incorporated into EP matrix to fabricate the composites. The chemical composition and microstructures of the FG samples were systematically analyzed, and their corrosion resistance studied. The results showed that FG was successfully fabricated, and its lamellar thickness was less than 2 nm. Meanwhile, the incorporation of FG into the epoxy matrix significantly enhanced its hydrophobicity and corrosion resistance performance. However, with increased fluorine content, the corrosion resistance of FG/EP coating increased and then decreased. This is attributed to the extremely low surface energy of FG with high fluorine contents, which caused ease of agglomeration and poor dispersivity in the epoxy resin, ultimately leading to cracks in the epoxy coating. These internal cracks in the coatings are believed to have accelerated the penetration of corrosive species, and encouraging the attack on metals. Furthermore, the barrier effects and anti-corrosion properties of FG2 (having a fluorine content of 29.01%) in EP coating were significantly superior to those of other FG/EP coatings. The Rc of the FG2/EP coating was 3 orders of magnitude more than that of the EP coating, and the SVET results also showed that its current density was less than 2 times that of the EP coating. Therefore, of all the studied composites, FG2 offered the optimal corrosion protection in EP coating.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 51901146), the Foundation of Sichuan Science and Technology Program (no. 2020JDRC0098 and 21ZDYF2921), the Key Projects of Science and Technology Plan of Zigong (no. 2019YYJC23) and the Research Foundation for the introduction of talent of Sichuan University of Science and Engineering (no. 2018RCL14). Dr Baojie Dou would also like to express his gratitude to China Scholarship Council (CSC) for financial support.References
- R. R. Nair, W. Ren, R. Jalil, I. Riaz, V. G. Kravets, L. Britnell, P. Blake, F. Schedin, A. S. Mayorov and S. Yuan, Small, 2015, 6, 2877–2884 CrossRef PubMed.
- R. Zboril, F. Karlicky, A. B. Bourlinos, T. A. Steriotis, A. K. Stubos, V. Georgakilas, K. Safarova, D. Jancik, C. Trapalis and M. Otyepka, Small, 2010, 6, 2885–2891 CrossRef CAS PubMed.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson and P. E. Sheehan, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- K. J. Jeon, Z. Lee, E. Pollak, L. Moreschini, A. Bostwick, C. M. Park, R. Mendelsberg, V. Radmilovic, R. Kostecki and T. J. Richardson, ACS Nano, 2011, 5, 1042–1046 CrossRef CAS PubMed.
- R. Tian, X. Jia, J. Yang, Y. Li and H. Song, Chem. Eng. J., 2020, 395, 125104 CrossRef CAS.
- S. K. Singh, S. G. Srinivasan, M. Neek-Amal, S. Costamagna, A. C. T. V. Duin and F. M. Peeters, Phys. Rev. B, 2013, 87, 104114 CrossRef.
- T. Bharathidasan, T. N. Narayanan, S. Sathyanaryanan and S. S. Sreejakumari, Carbon, 2015, 84, 207–213 CrossRef CAS.
- K. I. Ho, C. H. Huang, J. H. Liao, W. Zhang, L. J. Li, C. S. Lai and C. Y. Su, Sci. Rep., 2014, 4, 5893 CrossRef CAS PubMed.
- C. Sun, Y. Feng, Y. Li, C. Qin and W. Feng, Nanoscale, 2013, 6, 2634–2641 RSC.
- Y. Luan, J. Yin, K. Zhu, K. Cheng, J. Yan, K. Ye, G. Wang and D. Cao, Chem. Eng. J., 2020, 392, 123668 CrossRef CAS.
- W. Wang, Y. Li, Y. Feng, J. Han, F. Zhang, P. Long, C. Peng, C. Cao, Y. Cao, H. Yang and W. Feng, Chem. Eng. J., 2018, 349, 756–765 CrossRef CAS.
- W. Huang, Q. X. Pei, Z. Liu and Y. W. Zhang, Chem. Phys. Lett., 2012, 552, 97–101 CrossRef CAS.
- K. Fan, X. Chen, X. Wang, X. Liu, Y. Liu, W. Lai and X. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 28828–28838 CrossRef CAS PubMed.
- C. Min, Z. He, H. Song, H. Liang, D. Liu, C. Dong and W. Jia, Tribol. Int., 2019, 140, 105867 CrossRef CAS.
- L. Sun, Z. Yan, Y. Duan, J. Zhang and B. Liu, Dent. Mater., 2018, 34, e115–e127 CrossRef CAS PubMed.
- B. Wang, J. Wang and J. Zhu, ACS Nano, 2014, 8, 1862–1870 CrossRef CAS PubMed.
- X. Wang, Y. Dai, W. Wang, M. Ren, B. Li, C. Fan and X. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 16182–16188 CrossRef CAS PubMed.
- Y. Wang, W. C. Lee, K. K. Manga, P. K. Ang, J. Lu, Y. P. Liu, C. T. Lim and K. P. Loh, Adv. Mater., 2012, 24, 4285–4290 CrossRef CAS PubMed.
- H. Duan, Y. Chen, S. Ji, R. Hu and H. Ma, Chem. Eng. J., 2019, 375, 121916 CrossRef CAS.
- Y. Hao, F. Liu and E. H. Han, Prog. Org. Coat., 2013, 76, 571–580 CrossRef CAS.
- L. Xiao, Z. Liu, N. Li, S. Li, P. Fu, Y. Wang, J. Huang, J. Chen and X. Nie, New J. Chem., 2020, 44, 16856–16863 RSC.
- S. A. Haddadi, S. A. A. Ramazani, M. Mahdavian, P. Taheri and J. M. C. Mol, Ind. Eng. Chem. Res., 2019, 58, 3033–3046 CrossRef CAS.
- F. Meng, L. Liu, W. Tian, H. Wu, Y. Li, T. Zhang and F. Wang, Corros. Sci., 2015, 101, 139–154 CrossRef CAS.
- Y. Ma, H. Di, Z. Yu, L. Liang, L. Lv, Y. Pan, Y. Zhang and D. Yin, Appl. Surf. Sci., 2016, 360, 936–945 CrossRef CAS.
- J. R. Vilche, E. C. Bucharsky and C. A. Giúdice, Corros. Sci., 2002, 44, 1287–1309 CrossRef CAS.
- M. R. El-Sharif, Y. J. Su, C. U. Chisholm and A. Watson, Corros. Sci., 1993, 35, 1259–1265 CrossRef CAS.
- M. Costa and C. B. Klein, Crit. Rev. Toxicol., 2006, 36, 155–163 CrossRef CAS PubMed.
- B. D. Mert, Corros. Sci., 2016, 103, 88–94 CrossRef.
- H. P. Duong, C.-H. Hung, H. C. Dao, M. D. Le and C.-Y. Chen, New J. Chem., 2018, 42, 8745–8751 RSC.
- C. J. Weng, J. Y. Huang, K. Y. Huang, Y. S. Jhuo, M. H. Tsai and J. M. Yeh, Electrochim. Acta, 2010, 55, 8430–8438 CrossRef CAS.
- A. Ghosal and S. Ahmad, New J. Chem., 2017, 41, 4599–4610 RSC.
- D. In, N. Kayaman-Apohan and A. Güngr, Prog. Org. Coat., 2009, 65, 477–483 CrossRef.
- Y. Xia, Y. He, C. Chen, Y. Wu and J. Chen, Prog. Org. Coat., 2019, 132, 316–327 CrossRef CAS.
- X. Liu, Y. Shao, Y. Zhang, G. Meng, T. Zhang and F. Wang, Corros. Sci., 2015, 90, 451–462 CrossRef CAS.
- T. Liu, Y. Liu, Y. Ye, J. Li, F. Yang, H. Zhao and L. Wang, Prog. Org. Coat., 2019, 132, 455–467 CrossRef CAS.
- M. Ehsani, H. A. Khonakdar and A. Ghadami, Prog. Org. Coat., 2013, 76, 238–243 CrossRef CAS.
- G. Cui, Z. Bi, R. Zhang, J. Liu, X. Yu and Z. Li, Chem. Eng. J., 2019, 373, 104–121 CrossRef CAS.
- X. Luo, J. Zhong, Q. Zhou, S. Du, S. Yuan and Y. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 18400–18415 CrossRef CAS PubMed.
- S. Ren, M. Cui, W. Li, J. Pu, Q. Xue and L. Wang, J. Mater. Chem. A, 2018, 6, 24136–24148 RSC.
- S. Stankovich, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- B. Ramezanzadeh, S. Niroumandrad, A. Ahmadi, M. Mahdavian and M. H. M. Moghadam, Corros. Sci., 2016, 103, 283–304 CrossRef CAS.
- H. Zheng, M. Guo, Y. Shao, Y. Wang, B. Liu and G. Meng, Corros. Sci., 2018, 139, 1–12 CrossRef CAS.
- Z. Yang, L. Wang, W. Sun, S. Li, T. Zhu, W. Liu and G. Liu, Appl. Surf. Sci., 2017, 401, 146–155 CrossRef CAS.
- C. Min, Z. He, H. Liang, D. Liu, C. Dong, H. Song and Y. Huang, Polym. Compos., 2020, 41, 1624–1635 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- R. Al-Gaashani, A. Najjar, Y. Zakaria, S. Mansour and M. A. Atieh, Ceram. Int., 2019, 45, 14439–14448 CrossRef CAS.
- Z. Wang, J. Wang, Z. Li, P. Gong, X. Liu, L. Zhang, J. Ren, H. Wang and S. Yang, Carbon, 2012, 50, 5403–5410 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S. J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- P. Gong, Z. Wang, J. Wang, H. Wang, Z. Li, Z. Fan, Y. Xu, X. Han and S. Yang, J. Mater. Chem., 2012, 22, 16950–16956 RSC.
- K. Hou, P. Gong, J. Wang, Z. Yang, Z. Wang and S. Yang, RSC Adv., 2014, 4, 56543–56551 RSC.
- A. Mathkar, T. N. Narayanan, L. B. Alemany, P. Cox, P. Nguyen, G. Gao, P. Chang, R. Romero-Aburto, S. A. Mani and P. M. Ajayan, Part. Part. Syst. Charact., 2013, 30, 266–272 CrossRef CAS.
- O. C. Compton and S. B. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- K. Guérin, J. P. Pinheiro, M. Dubois, Z. Fawal and A. Hamwi, Chem. Mater., 2004, 16, 1786–1792 CrossRef.
- Z. Wang, J. Wang, Z. Li, P. Gong, J. Ren, H. Wang, X. Han and S. Yang, RSC Adv., 2012, 2, 11681–11686 RSC.
- H. P. Mungse and O. P. Khatri, J. Phys. Chem. C, 2014, 118, 14394–14402 CrossRef CAS.
- A. S. Dobrota, I. A. Pasti, S. V. Mentus and N. V. Skorodumova, Phys. Chem. Chem. Phys., 2016, 18, 6580–6586 RSC.
- F. Lei, B. Wu, H. Sun, F. Jiang, J. Yang and D. Sun, Ind. Eng. Chem. Res., 2018, 57, 16709–16717 CrossRef CAS.
- N. Parhizkar, T. Shahrabi and B. Ramezanzadeh, Corros. Sci., 2017, 123, 55–75 CrossRef CAS.
- Y. Chen, X. H. Wang, J. Li, J. L. Lu and F. S. Wang, Corros. Sci., 2007, 49, 3052–3063 CrossRef CAS.
- Y. Zhang, Y. Shao, T. Zhang, G. Meng and F. Wang, Corros. Sci., 2011, 53, 3747–3755 CrossRef CAS.
- J. H. Park, G. D. Lee and A. Nishikata, Corros. Sci., 2002, 44, 1087–1095 CrossRef CAS.
- M. Cui, S. Ren, H. Zhao, Q. Xue and L. Wang, Chem. Eng. J., 2018, 335, 255–266 CrossRef CAS.
- H. Zheng, Y. Shao, Y. Wang, G. Meng and B. Liu, Corros. Sci., 2017, 123, 267–277 CrossRef CAS.
- Y. Hao, F. Liu, E.-H. Han, S. Anjum and G. Xu, Corros. Sci., 2013, 69, 77–86 CrossRef CAS.
- B. Dou, Y. Wang, T. Zhang, G. Meng, Y. Shao, X. Lin and F. Wang, New J. Chem., 2018, 42, 9771–9782 RSC.
- D. Trinh, P. Dauphin Ducharme, U. Mengesha Tefashe, J. R. Kish and J. Mauzeroll, Anal. Chem., 2012, 84, 9899–9906 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |