
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRationalization of chirality transfer and fast conformational changes in a tris(2-pyridylmethyl)amine-based cage†
Gege Qiua,
Djamel Eddine Khatmiab,
Alexandre Martinez *a and
Paola Nava*a
*a and
Paola Nava*a
aAix Marseille Univ., CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: alexandre.martinez@centrale-marseille.fr; paola.nava@univ-amu.fr
bLaboratory of Computational Chemistry and Nanostructures, University of 08 May 45 Guelma, Algeria
First published on 13th April 2021
Abstract
The key features that govern the chirality transfer in a structurally contracted covalent cage, consisting of a northern chiral cyclotriveratrylene (CTV) connected to a southern tris(2-pyridyl-methyl)amine (TPA) unit by three methyl bridges, are described. The preferential orientation of the propeller arrangement of TPA is dictated by its compact structure, with an arm of the TPA unit pointing inside the cage, together with the relative positioning of the three pyridines regarding the chiral CTV cap. The diastereomers with P/P (or M/M) configurations for the CTV and TPA units adopt eclipsed structures and were found to be more stable by 40 kJ mol−1 than the P/M (or M/P) diastereomer which displays a staggered arrangement. The existence of isomerization pathways between isomers of the cage with low energy barriers (38 kJ mol−1) accounts for the 1H-NMR signal, which is consistent with an averaged C3 structure.
Introduction
Enantiopure artificial molecular containers arouse a growing interest as they can mimic the stereoselective processes found in nature, such as enantioselective enzymatic catalysis or chiral recognition.1 Among the covalent or self-assembled chiral cages, C3 symmetrical ones are of particular interest because they can lead to the formation of chiral propeller structures of triple-stranded helix, allowing the chirality to transfer from one side of the cage to its opposite. In particular, it was shown that the chirality of the cyclotriveratrylene unit (CTV), once included in a C3-symmetrical hemicryptophane cage, can propagate along the linkers to control the chirality of both the arms and the other unit located at the opposite side of the cage.2On the other hand, controlling the helicity of an achiral tris(2-pyridyl-methyl)amine (TPA) moiety is a challenge of particular interest, due to the versatility of this ligand for bio-inspired chemistry,3 catalysis,4 host–guest chemistry,5 and chiral sensing:6 by imposing the sense of its pyridine twist, it could be switched into a highly demanded chiral ligand7 for the development of complexes with potential applications as enantioselective sensors and catalysts. The remote control of the TPA helicity has been achieved through either a supramolecular functionalization through ionic contact,8 or covalent derivatization with chiral concave baskets.9
In this line, we have recently reported the covalent capping of the TPA ligand by a chiral cyclotriveratrylene unit as an efficient strategy to obtain predictable control of the clockwise/anticlockwise propeller arrangement of the TPA ligand.10 Indeed, the hemicryptophane cage 1, which consists in a TPA ligand covalently attached to a CTV unit through three methylene –CH2– bridges, displays a propeller arrangement of the southern pyridines dictated by the chirality of its northern CTV (Fig. 1a): the X-ray structure reveals that the P-CTV imposes a right-handed propeller orientation of the TPA moiety (the P/P-1 cage), whereas the M-CTV enantiomer induces a left-handed orientation of the TPA moiety (the M/M-1 cage).10,11
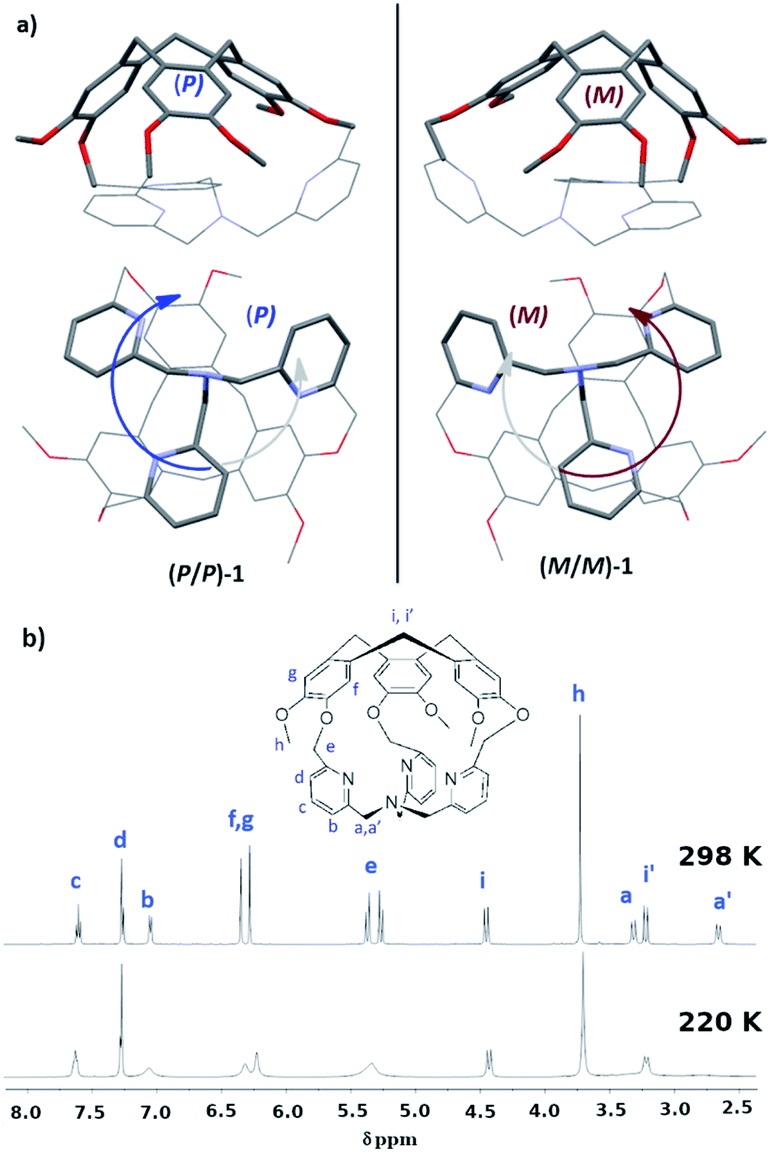 | ||
| Fig. 1 (a) Structure of cage 1 from XRD. The P/P enantiomer is shown on the left and the M/M on the right; (b) 1H-NMR at 298 and 220 K.10 | ||
The crystal structure of 1 is particularly compact: by observing the TPA part, Fig. 1a, we notice that one –CH2– unit connected to the nitrogen atom Nsp3 has its hydrogen atoms pointing inside the cage (one-arm-inside conformation). However, in solution the 1H-NMR of the cage displays an overall C3 symmetry (Fig. 1b). Furthermore, by decreasing the temperature up to 220 K, variable temperature 1H-NMR analysis do not allow for the observation of complete decoalescence of the signals that only appear broader in some cases, suggesting a particularly fast motion even at 220 K. A fast equilibrium between conformations presenting outward and inward orientations of the –CH2–N arm could account for this experimental result. Indeed, NOE correlations between the –CH2–N protons and the aromatic protons of the CTV unit were observed, suggesting that the self-inclusion process is certainly retained and that each arm goes in and out of the cavity at a rate that is fast on NMR time scale.10
As the static view of the X-ray diffraction analysis and the time-scale resolution of the NMR, result in a relatively simple view of the three-dimensional arrangement, we present here a detailed study of the chiral cage 1 based on experimental observations and computational techniques. Indeed, computational treatments represent a powerful tool that allows for exploring the conformational space that could hardly be evidenced experimentally.5,12
Our work proposes a new explanation for the transfer of chirality observed in 1. Furthermore, our study reveals the existence of isomerization pathways between three isomers of 1 that could not be observed by NMR analysis due to averaged signals. This work therefore elucidates the key parameters to achieve chirality transfer, and provide unprecedented insights into the dynamic behaviour of a structurally contracted hemicryptophane cage.
Results and discussion
Preliminary molecular dynamics calculations were performed to explore the potential energy surface of the neutral cage.13 Reported calculations were performed at the PBE0-D3/def2-TZVP level of theory (see the ESI for further details†).14–18Chirality transfer
The P/P isomer of 1, optimized from the XRD data, is shown in Fig. 2 (P/P-1). The one-arm-inside structure guarantees a staggered conformation of the three CH2 links connecting the nitrogen atom Nsp3 to the pyridine moieties (Fig. 2, bottom view). By considering the inside-arm, the pyridine ring is nearly orthogonal to the C(68)–Nsp3 bond and the CH2O link connecting the pyridine to the top CTV is oriented in such a way that the dihedral angle N(92)–C(67)–C(29)–O(2) is of 92.6° (computed structure), reducing the repulsion between the nitrogen and the oxygen lone pairs (Fig. 2, side view).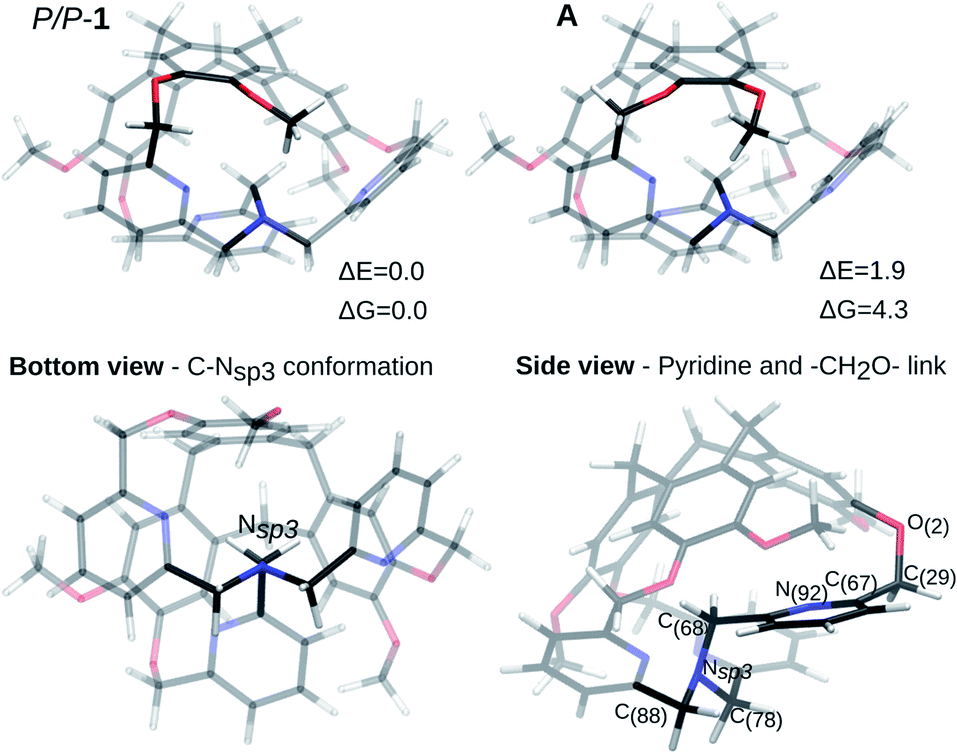 | ||
| Fig. 2 Optimized structures from XRD. Two possible conformations are shown. The bottom and side views refer to the most stable. ΔE and ΔG in kJ mol−1. | ||
In the XRD structure, some disorder is observed concerning the position of the –OMe groups of the CTV. The energy difference between two possible optimized structures (the P/P-1 and A isomers in Fig. 2) is negligible (ΔE = 1.9 kJ mol−1, ΔE = ZPE corrected energy difference, ZPE = zero-point energy, ΔG = 4.3 kJ mol−1) and the conversion barrier between them is also small (ΔE# = 25.9 kJ mol−1, ΔG# = 28.0 kJ mol−1). In the following, ZPE corrected electronic energy differences and Gibbs free energies differences are reported with respect to the most stable structure.
As only the P/P (or M/M) structure is experimentally observed and to better understand this remarkable chirality transfer, we optimized hypothetical conformations for the P/M cage (P→ CTV; M→ left-handed TPA). Results from the Density Functional Theory (DFT) calculations are reported in Fig. 3, where the structures are depicted from a top view, to underline the position of the aromatic rings of the CTV unit. The P/P cage is also reported for comparison.
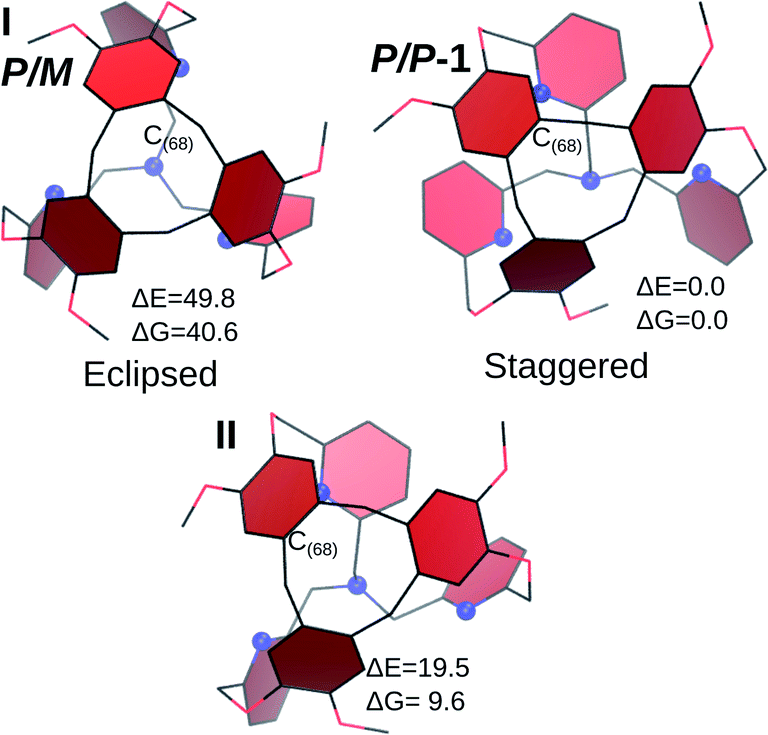 | ||
| Fig. 3 Optimized structures for hypothetical P/M isomers of 1. The P/P isomer is also reported. Blue spheres represent the nitrogen atoms. ΔE and ΔG in kJ mol−1. | ||
The first structure, I, is built by assuming a C3 arrangement of the cage (even if the optimization was conducted without imposing the symmetry constraint). This conformation is all-arm-outside, with an evident P/M pattern. The aromatic rings of the CTV are almost eclipsed with the pyridines of the bottom of the cage, whereas in the P/P cage they are clearly alternating, without superposition. This aspect contributes to the higher energy of I with respect to the P/P isomer (ΔE = 49.8 kJ mol−1, ΔG = 40.6 kJ mol−1).
From I, a rotation was applied to generate an isomer with the C(68)H2 inside the cage. The geometry optimization procedure leads to a tilt of the pyridine ring connected to this CH2 link, resulting into structure II. In this case, it is no more possible to associate a P or M descriptor for the bottom unit, as the pyridine of the inside-arm shows the same orientation than in the PP cage, while the two other pyridines are still oriented similarly than in I. This structure is slightly less stable than the crystal structure (ΔE = 19.5 kJ mol−1, ΔG = 9.6 kJ mol−1) and it readily converts to a P/P cage (ΔE# = 19.6 kJ mol−1, ΔG# = 17.4 kJ mol−1, see the ESI for further details†).
Finally, these results suggest that the preferential formation of P/P (or M/M) diastereomers compared to the P/M (or M/P) structures arises from the less stable eclipsed structure observed in the latter case. A key parameter that induces the chirality transfer in 1 is the preferential staggered positioning of the TPA pyridines compared to the CTV aromatic rings. As a result, the chirality transfer might be dependent on the distance between the CTV and the TPA unit. This finding is in good agreement with previous observations on a bigger TPA-based hemicryptophane owning phenyl linkages, where no chirality transfer was observed between the CTV and the southern TPA unit while the phenyl linkers were helically arranged (see Fig. S3 in the ESI†).19
Dynamic aspects
As presented in Fig. 1, the XRD analysis shows a non-symmetrical structure for cage 1, while the 1H-NMR suggests a C3 symmetry, possibly due to a signal averaging caused by fast conformational changes. We present here some computed pathways that could account for this averaging effect.Cage 1 is formed by three arms connecting the southern tertiary amine to the northern methylene –CH2– units of the CTV. These arms are not equivalent: in Fig. 4 three isoenergetic isomers are depicted, where the inside arm contains respectively C(68) (A, red arm inside, same structure as in Fig. 2), C(88) (B, green arm inside), and C(78) (C, cyan arm inside). In Fig. 4, we present as well three computed pathways for the conversion of A into B and/or C and their Gibbs free energy reaction profiles. The conversions of A to B and to C are achieved by following respectively the AB and AC pathways. An alternative mechanism is proposed that implies an intermediate where the three bottom arms are equivalent (all-arms-outside configuration) and it can therefore evolve to either A, or B, or C (path AD). These three pathways share the first step, located at 63.8 kJ mol−1, which is a rotation of the –C(68)H2– moiety leading to intermediate 1.
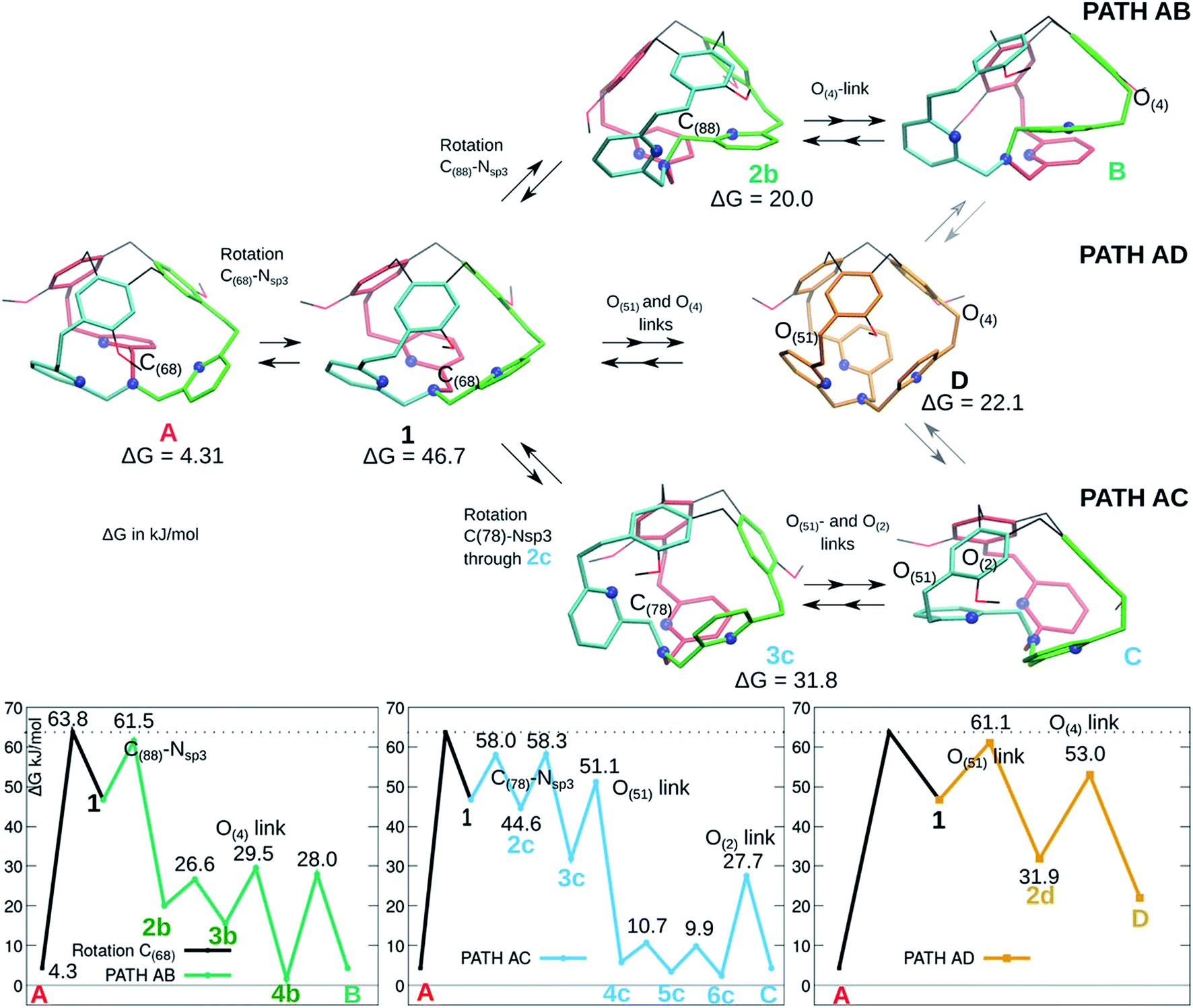 | ||
| Fig. 4 Isomerization pathways between isoenergetic structures of 1: A (red arm inside), B (green arm inside), C (cyan arm inside). Selected intermediates are depicted (see the ESI for more details†). The atoms involved in each step (excluded the –OMe rotations) are indicated. The blue spheres represent the N atoms. The ΔG values relative to the most stable P/P isomer are indicated for the displayed intermediates. | ||
-The AB pathway implies a step to rotate the arm containing C(88) into the cage, resulting in the intermediate 2b, where all the pyridines have already approximately the final conformation. The transition state corresponding to this transformation is located at 61.5 kJ mol−1. The following steps are rearrangements to reorient the OMe groups and the oxygen link of the new inside-arm.
-The AC pathway implies two successive steps to rotate the arm C(78) into the cage. A first transition state, located at 58 kJ mol−1, implies a rearrangement of the pyridine connected to C(78) and leads to 2c, which readily evolves to 3c (see Fig. S4 in the ESI†). In this intermediate 3c, the pyridine ring connected to C(78) is in an unfavourable position, with the nitrogen atom pointing inside the cage. In the next step a rotation occurs that adjusts simultaneously the positions of the pyridine rings and the oxygen link of the new inside arm (O(51)). From there, the following steps are rearrangements to reorient the OMe groups and the oxygen link O(2). With the exception of the first step, the transition states are all lower than 60 kJ mol−1 in ΔG.
-In the AD path, two rotations implying the oxygen links O(51) (transition state located at 61.1 kJ mol−1) and O(4) leads to intermediate D, which presents a ‘C3 symmetry’ and it can therefore evolve to each of the three isoenergetic A, B and C structures.
All over, the highest transition states are located at 60–64 kJ mol−1 above the computed Gibbs free energy of the crystal structure. The computed pathways do not differ significantly from an energetic point. The AC path appears slightly preferred; however, differences are tiny. We notice here that the D intermediate is more stable than 1, even if the two structures share a similar conformation of the bottom part of the cage (all-arm-outside conformation). The optimized structures of 1 and D are compared in Fig. 5, to illustrate general structural parameters that contribute to stabilize conformations. Differently than in A, the –CH2– links connected to the bottom Nsp3 have almost eclipsed conformations in 1 and D. The differences between 1 and D are related to the positions of the –CH2O– links connecting the pyridines to the top CTV (O(4), O(51) and O(2)). In D, all these links are optimally oriented, as the relative positions of the oxygen atoms with respect to the nitrogen atoms allow for reducing the repulsion due to their lone pairs. This is not the case for O(4) and O(51) in 1; moreover, even if the O(2) atom is properly oriented with respect to the nitrogen atom, one hydrogen atom is eclipsed with the pyridine ring.
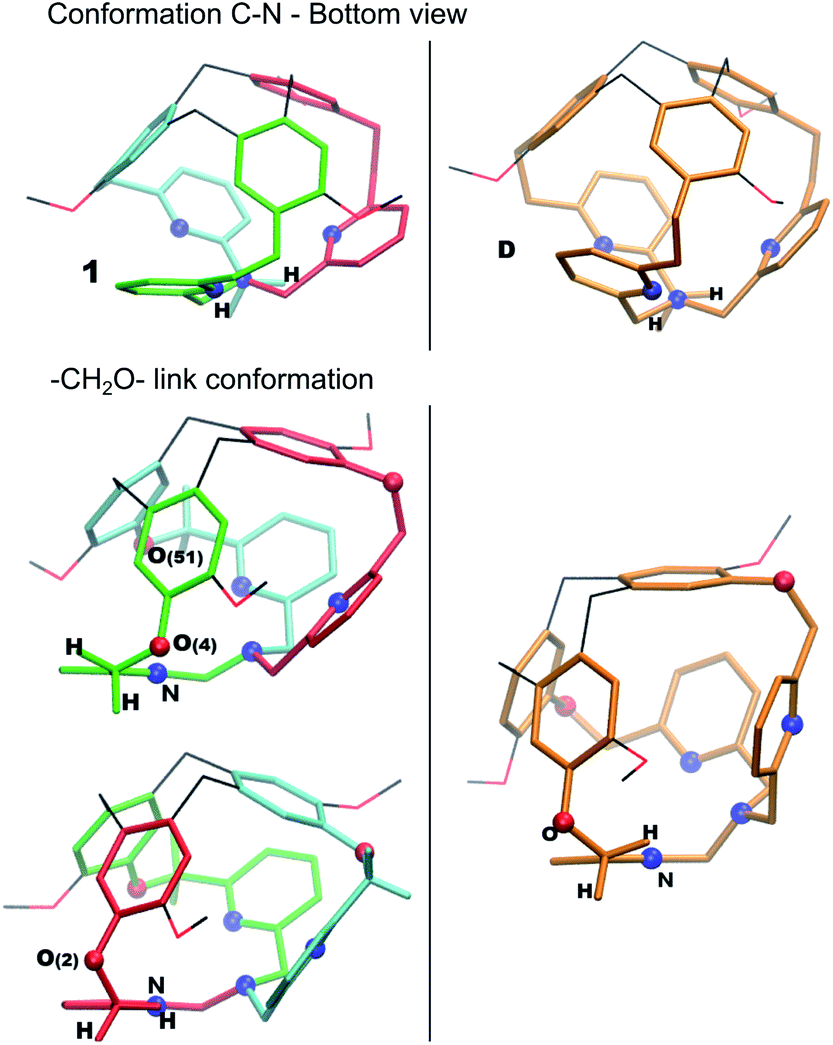 | ||
| Fig. 5 Optimized structures of 1 and D. | ||
The fact that the 1H-NMR signals suggest an overall C3 symmetry even at 220 K, motivated us to look for a further mechanism with lower barriers. A fourth pathway AE is presented in Fig. 6. It differs from the previous ones, as the initial step is a low-barrier conformational change that leads to a structure where the inner arm is conserved, as well as the right-handed TPA orientation.
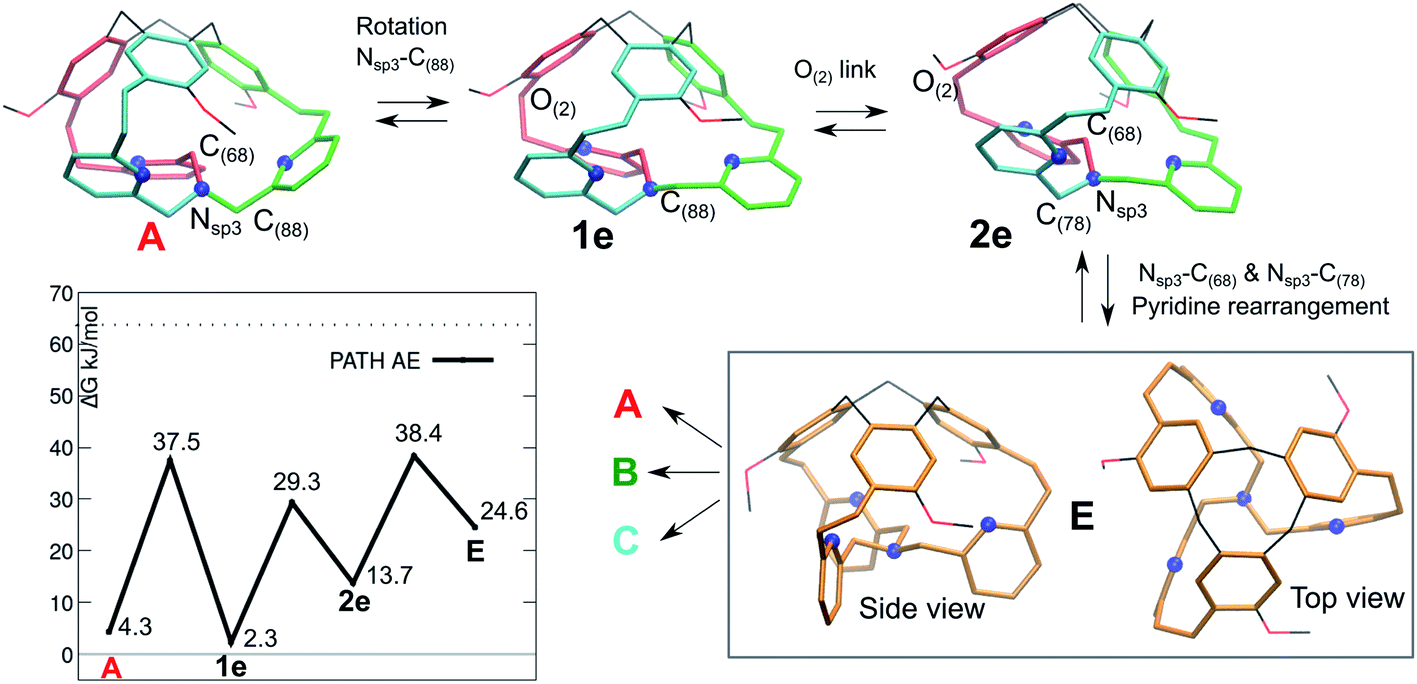 | ||
| Fig. 6 Isomerization pathway AE between isoenergetic structures of cage 1: A (red arm inside), B and C. The atoms involved in each step are indicated. The blue spheres represent the N atoms. | ||
The resulting intermediate 1e, very close in energy to A, is of relevance: the conformation change impacts the orientation of the C(88)H2 unit connected to the bottom Nsp3 (see Fig. 6), and, notably, it implies a configuration inversion of this bottom Nsp3. In the crystal structure the nitrogen lone pair is pointing inside the cage, while in 1e it is pointing outside the cage, as shown by the molecular orbitals depicted in Fig. 7.
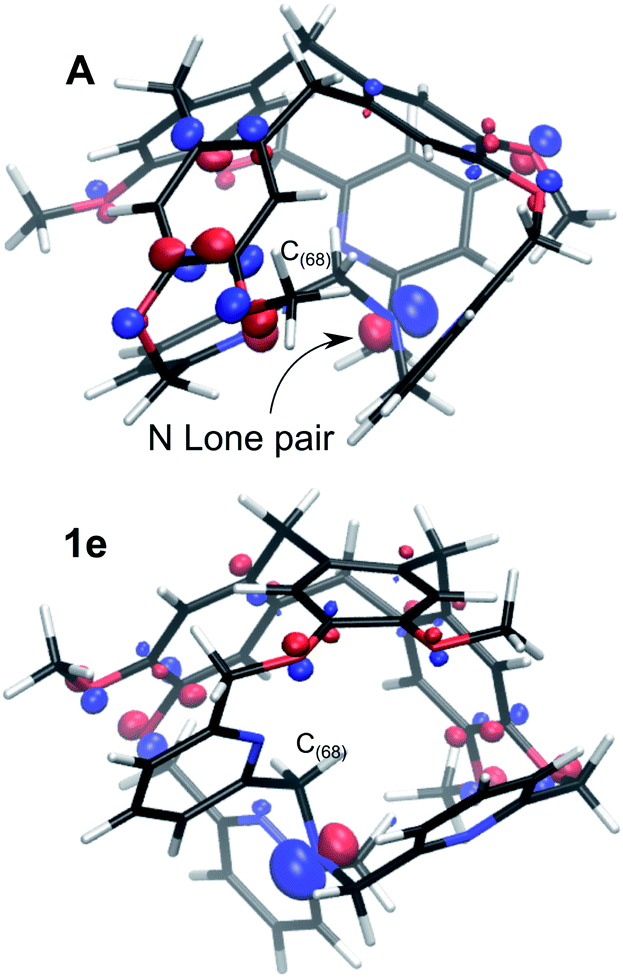 | ||
| Fig. 7 Molecular orbital representations of the lone pair of the bottom Nsp3, for the P/P isomers in their conformations A and 1e (isovalue 0.07 a.u.). | ||
After a minor conformation adjustment that implies the oxygen link –CH2O(2)–, a combined rotation of the inner arm –C(68)H2– and of the –C(78)H2– moieties leads to a rearrangement of the pyridines orientation. The resulting intermediate E has a ‘C3 symmetry’ and therefore can evolve to either A, B or C. This intermediate E differs from the ‘C3 symmetrical’ D intermediate, as it presents a compact structure, with the lone-pair of the bottom NNsp3 pointing inside the cage, as described earlier for the tribenzylamine hemicryptophane reported by N. S. Kahn et al.12
In E, the nitrogen atoms of the pyridines are directing their lone pairs toward the inside of the cage, as well. Interestingly, we cannot associate a P or M descriptor to the orientation of the TPA unit (see bottom view). However, the intermediate E, which is less stable than the crystal structure, readily transforms into A, B or C.
Finally, the computed isomerization barriers are compatible with fast conversion on NMR time scale among the A, B or C isomers, even at 220 K. This is consistent with an averaged 1H-NMR signal. Among the computed pathways, the AE mechanism is the preferred, with the highest transition state is located at only 38.4 kJ mol−1 (Gibbs free energy difference with respect to P/P-1).
Conclusions
This work describes a computational analysis that rationalizes the peculiar properties of the TPA-based hemicryptophane cage 1 in terms of chirality transfer and fast conformational changes. The remote control of the TPA helicity, dictated by the chirality of the CTV cap of 1, has been investigated by combining experimental characterization (XRD), with a computational treatment. Calculations indicate that the one-arm-inside structures (P/P-1, A, B and C) are the most stable among the computed ones as they possess optimal conformations, minimizing eclipsed bonds and showing a favourable orientation of the heteroatom lone pairs (nitrogen and oxygen atoms). Furthermore, several possible conformations of 1 have been optimized. Although some structures where the P or M descriptors were not applicable to the TPA unit (such as II) could exist, the P/M (or M/P) isomer of 1 was found to be much less stable that the P/P (or M/M) counterpart. These results are in good agreement with the experimental data as only M/M-1 and P/P-1 could be experimentally observed (XRD). Interestingly, a careful analysis of the structural parameters of these computed structures reveals that the more stable conformation M/M (or P/P) arises from a preferential staggered orientation of the pyridines regarding the CTV moiety. Therefore, the staggered or eclipsed conformations between the TPA pyridines and the CTV aromatics is key in order to achieve a robust chirality transfer.Finally, the missing link between the single picture given by the solid-state XRD analysis and the average configuration experimentally observed by the 1H-NMR studies, has been elucidated by evidencing favourable isomerization pathways between isoenergetic isomers of 1. Indeed, some pathways have been proposed for the conversion of one P/P structure into another conformation (A, B or C), suggesting that equilibria are possible among them. As a consequence, the 1H-NMR signal is averaged on all these possible structures.
Overall, our computational modelling highlights (i) the key structural feature that are required to achieve chirality transfer between one chiral CTV and another C3 symmetrical unit in a contracted cage, and (ii) the occurrence of fast conformational changes. Based on these results this computational approach will be further applied to the rational design of new chiral cages displaying chirality transfer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the French National Research Agency (ANR, France, OH Risque grant ANR-14-OHRI-0015-03).Notes and references
- (a) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (b) T. Brotin, L. Guy, A. Martinez and J.-P. Dutasta, Top. Curr. Chem., 2013, 341, 177–230 CrossRef CAS PubMed; (c) L. J. Chen, H. B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC; (d) H. Jędrzejewska and A. Szumna, Chem. Rev., 2017, 117, 4863–4899 CrossRef; (e) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed; (f) Y. Li, L. Bouteiller and M. Raynal, ChemCatChem, 2019, 11, 5212–5226 CrossRef CAS; (g) S. Roland, J. M. Suarez and M. Sollogoub, Chem.–Eur. J., 2018, 24, 12464–12473 CrossRef CAS PubMed.
- (a) G. Qiu, P. Nava, C. Colomban and A. Martinez, Front. Chem., 2020, 8, 994 Search PubMed; (b) H. Sato, J. A. Bender, S. T. Roberts and M. J. Krische, J. Am. Chem. Soc., 2018, 140, 2455–2459 CrossRef CAS PubMed; (c) A. U. Malik, F. Gan, C. Shen, N. Yu, R. Wang, J. Crassous, M. Shu and H. Qiu, J. Am. Chem. Soc., 2018, 140, 2769–2772 CrossRef CAS PubMed; (d) S. Séjourné, A. Labrunie, C. Dalinot, A. Benchohra, V. Carré, F. Aubriet, M. Allain, M. Sallé and S. Goeb, Inorganics, 2020, 8, 1 CrossRef; (e) A. Schaly, Y. Rousselin, J.-C. Chambron, E. Aubert and E. Espinosa, Eur. J. Inorg. Chem., 2016, 832–843 CrossRef CAS; (f) L. Hu, S. Polen, A. M. Hardin, Y. Pratumyot, C. M. Hadad and J. D. Badjić, Eur. J. Org. Chem., 2015, 6832–6840 CrossRef CAS; (g) L. You, G. Pescitelli, E. V. Anslyn and L. Di Bari, J. Am. Chem. Soc., 2012, 134, 7117–7125 CrossRef CAS PubMed.
- (a) M. Hee Lim, J. U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que Jr, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665–3670 CrossRef PubMed; (b) R. L. Peterson, R. A. Himes, H. Kotani, T. Suenobu, L. Tian, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2011, 133, 1702–1705 CrossRef CAS PubMed; (c) N. Le Poul, B. Colasson, G. Thiabaud, D. Jeanne Dit Fouque, C. Iacobucci, A. Memboeuf, B. Douziech, J. Řezáč, T. Prangé, A. de la Lande, O. Reinaud and Y. Le Mest, Chem. Sci., 2018, 9, 8282–8290 RSC.
- I. Garcia-Bosch and M. A. Siegler, Angew. Chem., Int. Ed., 2016, 55, 12873–12876 CrossRef CAS PubMed.
- L. Zhiquan, H. Xie, S. E. Border, J. Gallucci, R. Z. Pavlović and J. D. Badjić, J. Am. Chem. Soc., 2018, 140, 11091–11100 CrossRef CAS PubMed.
- (a) L. You, J. S. Berman and E. V. Anslyn, Nat. Chem., 2011, 3, 943 CrossRef CAS PubMed; (b) C. Bravin, G. Mason, G. Licini and C. Zonta, J. Am. Chem. Soc., 2019, 141, 11963–11969 CrossRef CAS PubMed.
- J. W. Canary, C. S. Allen, J. M. Castagnetto and Y. Wang, J. Am. Chem. Soc., 1995, 177, 8484–8485 CrossRef.
- L. Zhiquan, S. Polen, C. M. Hadad, T. V. RajanBabu and J. D. Badjić, J. Am. Chem. Soc., 2016, 138, 8253–8258 CrossRef CAS PubMed.
- R. Z. Pavlovic, L. Zhiquan, M. Güney, R. F. Lalisse, R. G. Hopf, J. Gallucci, C. Moore, H. Xie, C. M. Hadad and J. D. Badjić, Chem.–Eur. J., 2019, 25, 13124–13130 CrossRef CAS PubMed.
- G. Qiu, C. Colomban, N. Vanthuyne, M. Giorgi and A. Martinez, Chem. Commun., 2019, 55, 14158–14161 RSC.
- D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900–4942 CrossRef CAS PubMed.
- (a) N. S. Khan, J. M. Perez-Aguilar, T. Kaufmann, P. A. Hill, O. Taratula, O.-S. Lee, P. J. Carroll, J. G. Saven and I. J. Dmochowski, J. Org. Chem., 2011, 76, 1418–1424 CrossRef CAS PubMed; (b) H. Zimmermann, P. Tolstoy, H.-H. Limbach, R. Poupko and Z. Luz, J. Phys. Chem. B, 2004, 108, 18772–18778 CrossRef CAS; (c) C. García-Simón, C. Colomban, Y. A. Çetin, A. Gimeno, M. Pujals, E. Ubasart, C. Fuertes-Espinosa, K. Asad, N. Chronakis, M. Costas, J. Jiménez-Barbero, F. Feixas and X. Ribas, J. Am. Chem. Soc., 2020, 142, 16051–16063 CrossRef PubMed; (d) M. Q. E. Mubarak and S. P. de Visser, Inorg. Chem., 2019, 58, 15741–15750 CrossRef CAS PubMed; (e) Ł. Szyszka, P. Cmoch, A. Butkiewicz, M. A. Potopnyk and S. Jarosz, Org. Lett., 2019, 21, 6523–6528 CrossRef PubMed; (f) Ł. Szyszka, P. Cmoch, M. Górecki, M. Ceborska, M. A. Potopnyk, and S. Jarosz, Eur. J. Org. Chem., 2021, 6, 897–906 Search PubMed.
- (a) J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS; (b) N. L. Allinger, J. Am. Chem. Soc., 1977, 99, 8127–8134 CrossRef CAS.
- (a) P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, 123, 714–733 CrossRef CAS; (b) J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS; (c) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244 CrossRef PubMed; (d) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed; (e) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- TURBOMOLE V7.2, 2017, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com Search PubMed.
- S. A. Ikbal, C. Colomban, D. Zhang, M. Delecluse, T. Brotin, V. Dufaud, J. P. Dutasta, A. B. Sorokin and A. Martinez, Inorg. Chem., 2019, 58, 7220–7228 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, results of the molecular dynamics runs, further explanatory schemes and xyz coordinates of DFT optimized geometries. See DOI: 10.1039/d1ra01761f |
| This journal is © The Royal Society of Chemistry 2021 |