
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-energy optical switching of SO2 linkage isomerisation in single crystals of a ruthenium-based coordination complex†
Jacqueline M. Cole *abcd,
David J. Gosztolad and
Sven O. Sylvestera
*abcd,
David J. Gosztolad and
Sven O. Sylvestera
aCavendish Laboratory, Department of Physics, University of Cambridge, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK. E-mail: jmc61@cam.ac.uk
bISIS Neutron and Muon Source, STFC Rutherford Appleton Laboratory, Harwell Science and Innovation Campus, Didcot, OX11 0QX, UK
cDepartment of Chemical Engineering and Biotechnology, University of Cambridge, West Cambridge Site, Philippa Fawcett Drive, Cambridge, CB3 0AS, UK
dArgonne National Laboratory, 9700 South Cass Avenue, Lemont, Illinois 60439, USA
First published on 8th April 2021
Abstract
Single crystals that behave as optical switches are desirable for a wide range of applications, from optical sensors to read–write memory media. A series of ruthenium-based complexes that exhibit optical switching in their single-crystal form via SO2 linkage photoisomerisation are of prospective interest for these technologies. This study explores the optical switching behaviour in one such complex, trans-[Ru(SO2)(NH3)4(H2O)]tosylate2 (1), in terms of its dark and photoinduced crystal structure, as well as its light and thermal decay characteristics, which are deduced by photocrystallography, single-crystal optical absorption spectroscopy and microscopy. Photocrystallography results reveal that a photoisomerisation level of 21.5(5)% is achievable in 1. Biphasic photochromic crystals of 1 were generated by applying green and then red light to switch on and off the η2-(OS)O photoisomer in different regions of a crystal. Heat is a known alternative to its thermal decay, whereby a method is demonstrated that employs optical absorption spectra to determine its activation energy of 30 kJ mol−1. This low-energy barrier to optical switching agrees well with computational studies on 1, as well as being comparable to activation energies in ruthenium-based nitrosyl linkage photoisomers that also display solid-state optical switching.
Introduction
Materials that exhibit optical switching are of great interest given their wide-ranging applications as light-driven molecular rotors,1 photocatalysts,2 protein nanovalves,3 and read–write optical memory media,4 to name just a few. Optical switching in a crystalline material is particularly helpful for solid-state device technologies, as a crystal is a pure form of solid-state media; as such, it can be incorporated into a device with modest fabrication or processing effort. Single-crystal optical switches are particularly appealing where their functional origins lie in light-induced structural changes. This is because such changes carry long-range effects throughout the material, given the periodic nature of a crystal-lattice environment. This may lead to correlated photoswitching effects, whose ‘out-of-equilibrium’ manifestations have been coined as “the real terra nova of solid-state chemistry”.5Single-crystal optical switching has been observed in a wide range of coordination complexes via the process of linkage photoisomerisation.6,7 The light-sensitive ligand switches from its dark-state configuration to a photoisomer upon light activation, to afford a binary encoding in the crystal. The coordination complex, trans-[Ru(SO2)(NH3)4(H2O)]tosylate2, (1) is one in a family of ruthenium sulfur-dioxide based (hereafter denoted [RuSO2]) materials that exhibit SO2 linkage photoisomerisation.8–20 Photocrystallography21–25 studies have shown that 11.1(1)% of an η2-(OS)O photoisomer can be generated within the crystal structure of 1 at 90 K,9 while 36% of the less thermally stable η1-OSO photoisomer can form at 13 K, albeit split over two disordered orientations with 24% and 12% photoconversion.10 Scheme 1 illustrates these photoisomerisation processes. Note that the dark-state η1-SO2 isomer remains the predominant species in 1 once photoisomerised, owing to photoconversion limits that are imposed by crystal-lattice strain.26
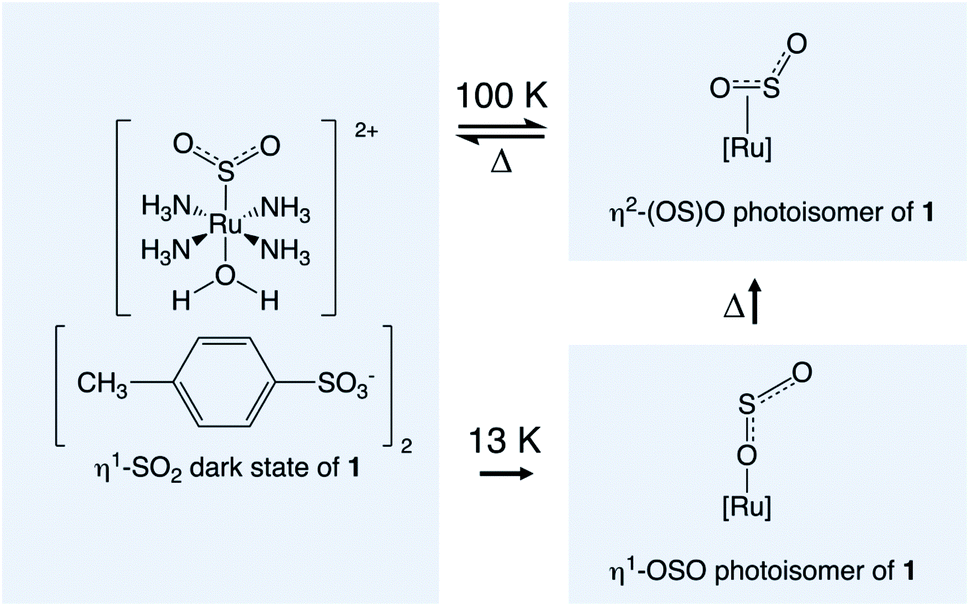 | ||
| Scheme 1 Compound 1 in its dark-state configuration and its SO2 photoisomeric configurations. | ||
While these distinct isomeric structures of 1 have been confirmed crystallographically, their single-crystal optical switching behaviour remains unknown. In particular, their single-crystal optical absorption properties have not been explored, nor have the kinetics associated with the photoisomerisation process of 1. The single-crystal photochromism characteristics of 1 have also not been ascertained, despite their potential application in optical memory devices.4 Its thermally-induced reverse isomerisation process has been tracked by multi-temperature infrared (IR) spectroscopy, yielding an activation energy, Ea = 58.4 kJ mol−1.9 However, these results are on the powdered form of 1; those of the single-crystal phase are expected to differ since the light-absorption and scattering characteristics of a material depend inherently on its form.
This paper thus seeks to gain insights into the light-induced optical and thermal switching properties of 1. Before embarking on a study of these optical switching properties, though, we undertake a new photocrystallographic determination of 1 because we need to compare the optical switching behaviour of 1 with more optimal light-induced crystal structure information. The previous photocrystallographic reports on 1 did not employ optimum light-induced experimental conditions. This is because the light-induced crystal structure of 1 was originally determined during the early phase of photocrystallographic developments, whose technical aspects were then still very much exploratory; the method has now been refined considerably. Indeed, the originally reported 11.1(1)% photoconversion fraction of 1 to its η2-(OS)O isomer at N2-based cryogenic temperatures is anomalously low when compared with light-induced crystal structures of more recently determined [RuSO2] complexes.8–20 We will show how almost double the photoconversion fraction of η2-(OS)O isomer can be achieved in 1 through this work. We then present a fundamental demonstration of the optical read and write capabilities in a single-crystal of 1 via its display of photochromism. The thermal stability and decay rates of 1 are then explored using single-crystal optical absorption spectroscopy as a metric to afford the activation energy, Ea. The use of this optical property metric, rather than a purely structural metric which has been the focus of previous evaluations of Ea,9 allows the optical properties of 1 to be discerned as a function of temperature in concert with the Ea determination. We will show that 1 exhibits a low-energy barrier to optical switching, which has implications for its prospective application to optical read–write memory-based technologies.
Experimental methods
Materials
The coordination complex, trans-[Ru(SO2)(NH3)4(H2O)]tosylate2, (1) was synthesised from the precursor, trans-[Ru(SO2)(NH3)4Cl]Cl, which was synthesised according to a literature procedure.26 4.4 mg of this precursor was dissolved in 300 μL of distilled water with 3.2 mg of p-tosylic acid (>98% purity, Sigma Aldrich) to form an aqueous solution of 1. Red plate-like crystals precipitated from solution; they were isolated through vacuum filtration and washed with methanol.Characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 576 or 25589 reflections for the dark- or light-induced data collections, respectively. Data for the dark-state crystal structure were first obtained. The crystal was then maintained at 100 K on the diffractometer and held static while 505 nm light was shone onto its thinnest face for 2 h, using a Thorlabs M505L3 light emitting diode (LED) whose head power output was 1000 mA constant current and 3.3 V forward voltage. The crystal was then illuminated for an additional 15 min at three rotated ϕ orientations, 90°, 180° and 270° from its thinnest face, i.e. the crystal was photo-stimulated by 2 h 45 min in total for the photo-crystallography experiment. This light was switched off before acquiring data for the light-induced crystal structure. See the ESI† for further experimental details, as well as structure solution and refinement information. A detailed overarching description of the specialised photocrystallography aspects of these experiments is given elsewhere.21–25
576 or 25589 reflections for the dark- or light-induced data collections, respectively. Data for the dark-state crystal structure were first obtained. The crystal was then maintained at 100 K on the diffractometer and held static while 505 nm light was shone onto its thinnest face for 2 h, using a Thorlabs M505L3 light emitting diode (LED) whose head power output was 1000 mA constant current and 3.3 V forward voltage. The crystal was then illuminated for an additional 15 min at three rotated ϕ orientations, 90°, 180° and 270° from its thinnest face, i.e. the crystal was photo-stimulated by 2 h 45 min in total for the photo-crystallography experiment. This light was switched off before acquiring data for the light-induced crystal structure. See the ESI† for further experimental details, as well as structure solution and refinement information. A detailed overarching description of the specialised photocrystallography aspects of these experiments is given elsewhere.21–25To induce photoisomerisation, the crystal was illuminated with 505 nm light from a ThorLabs M505F1 fiber optically coupled light-emitting diode (LED). The light from the LED was collimated and then coupled into the microscope through a side port, focusing it onto the back aperture of the objective, thus filling the field of view and evenly illuminating the entire crystal. The excitation power measured at the objective was typically 588 μW giving an estimated 27 μW mm−2 illuminating the field of view.
To remove photoisomerisation by illumination, 633 nm red light from a HeNe laser was substituted for the 505 nm photoisomerisation light and followed the same beam path. The excitation power measured at the objective was typically 482 μW giving an estimated 22 μW mm−2 illuminating the field of view.
Optical absorption spectra were recorded by imaging the crystal on the entrance slit (75 μm) of the spectrometer and dispersing the light, using a 150 line per mm grating, onto the detector. The image was positioned such that 10 rows of the detector were illuminated with light that passed through the crystal (IT); whereas 10 rows of the detector directly above the crystal recorded light that passed through only the sapphire substrate (I0). Absorption spectra were then calculated as log10(IT/I0).
A more detailed description of this custom instrument set up and its operational pipeline, as provisioned to support photocrystallography, is given elsewhere.27
Results and discussion
Light-induced structural changes in 1
The dark and photoinduced structures of 1 at 100 K, determined by photocrystallography, are given in Fig. 1. Fig. 1(a) shows that the dark-state crystal structure of 1 mirrors that of its previous structure determinations.9,10 However, unlike previous work, we realised a much higher η2-(OS)O photo-isomer photoconversion fraction of 21.5(5)% in 1, as shown in Fig. 1(b). This photoconversion fraction is almost double that of the original photocrystallographic determination of 1 (cf. 11.1(1)% η2-(OS)O).9 The η2-(OS)O configuration revealed by each study appears to be comparable, albeit that its disorder was fully modelled in this study. Thereby, anisotropic displacement parameters (ADPs) were refined on all SO2 atoms of the η2-(OS)O photoisomer, through which the sulfur was found to be split in a 60:40 ratio across two atomic positions, S1A and S1B in Fig. 1. Both of its adjoining oxygen atoms display oblate ADPs that essentially project along a vector which joins S1A and S1B, i.e. the η2-(OS)O photoisomer is either poorly coordinated to the Ru ion via dynamic disorder or it exists as several static η2-(OS)O photoisomers that are distinct but have very similar configurations. ADPs on S and one of the O atoms were modelled in the original photocrystallographic study on 1 and showed similar native characteristics: the maximum principal component of each of their ADPs was U22, i.e. their ADPs projected in the same direction, whose magnitudes were 0.043 Å2 and 0.063 Å2, respectively. However, the η2-(OS)O photoisomer of 1 in that previous study9 was not fully modelled, presumably because the much lower photoconversion fraction achieved therein would have restricted its explicit refinement.
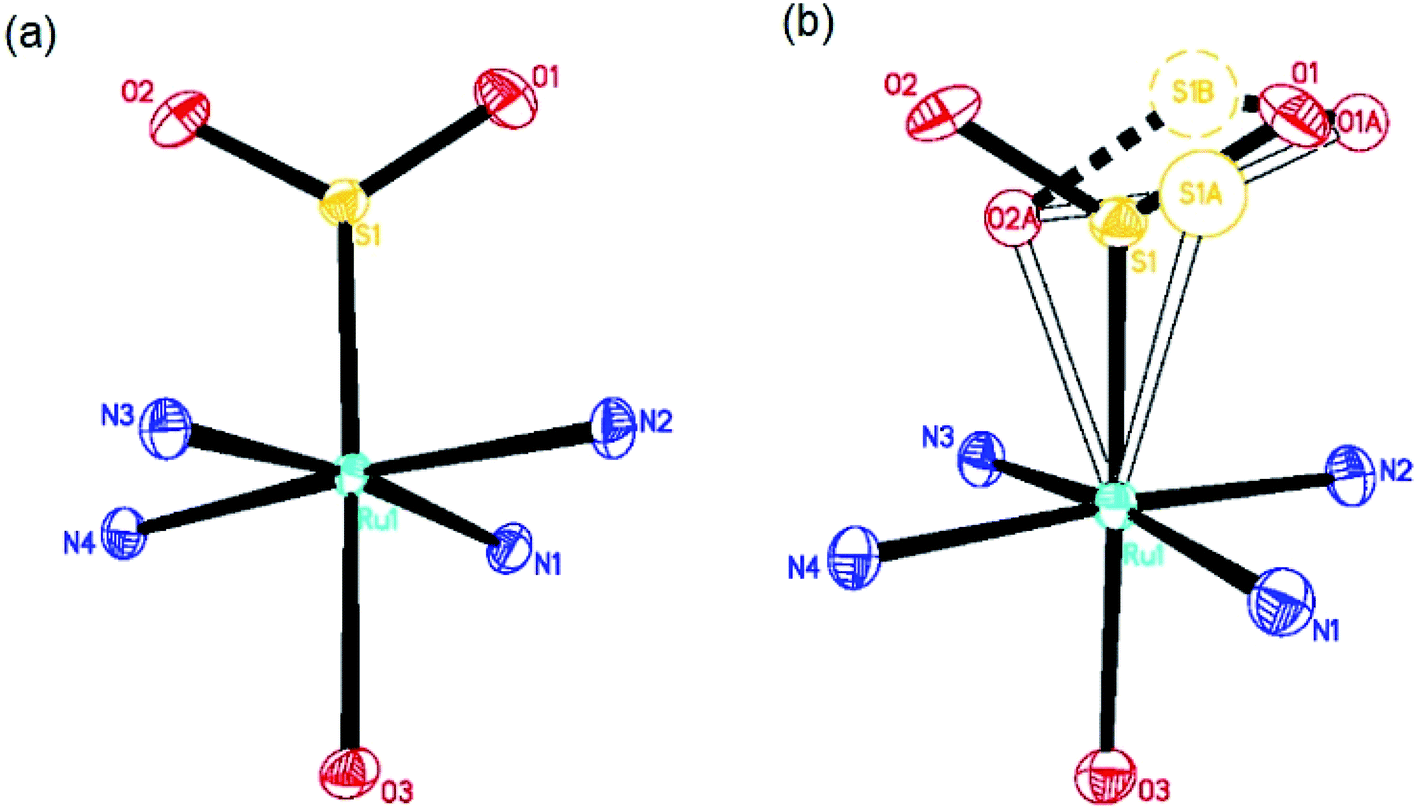 | ||
| Fig. 1 (a) Dark-state crystal structure of 1; (b) light-induced crystal structure of 1. Hollow atoms and bonds denote the light-induced photoisomers. The dashed S1B atom and associated lines represent a disordered component of the η2-(OS)O configuration. Ammine hydrogen atoms and the tosylate counterions are omitted for the purposes of clarity. | ||
No evidence was found for the existence of any η1-OSO photoisomer in this or the previous photocrystallography study9 at 100 K and 90 K, respectively. However, this configuration has been observed in 1 via a different photocrystallography study on 1 that was conducted previously at a much lower temperature (13 K);10 moreover, η1-OSO configurations have been found at 100 K in other members of this [RuSO2] family of complexes.12–20 Given this history of η1-OSO photoisomers in similar complexes, and that the subtle and complex nature of light-induced SO2 disorder within these [RuSO2] compounds could potentially mask other minor photoisomeric components, single-crystal Raman spectroscopy of 1 was employed. This served as an independent check that the current photocrystallography study has not missed any evidence for the existence of a η1-OSO photoisomer at 100 K. The spectroscopy corroborated our photocrystallography results, as judged by the absence of a Raman shift at 595 cm−1 at 90 K in 1 (see ESI†), a peak that has been attributed to this η1-OSO photoisomeric species in the closely related coordination complex, trans-[Ru(SO2)(NH3)4Cl]Cl, at 90 K.27
The single-crystal Raman spectroscopy results also complemented the photocrystallography results in that they evidenced a range of vibrational modes that are due to local photostructural changes in 1. Resonance Raman effects are prevalent in ruthenium ions28 and the high intensities seen in this experiment are consistent with the ruthenium cation in 1 exhibiting such phenomena. Accordingly, Raman shifts in 1 correspond entirely to the ruthenium-based cation of 1, while any contributions from tosylate ions are too weak to observe. A summary of the peak assignments to the Raman spectral shifts for the light-induced structure of 1 is given in Table 1. These assignments were made from the multi-temperature Raman spectra that are provided in the ESI.†
| Raman shift/cm−1 (dark) | Raman shift/cm−1 (light) | Classification of vibrational mode | Refs to related classifications |
|---|---|---|---|
| 353 (w) | 354 (w) | Ru–S stretching mode | 28 and 29 |
| — | 405 (s) | Ru–(OS)O stretching mode | — |
| 462–485 (w) | 475–490 (w) | Ru–N ammine bending mode | 30 |
| 564 (w) | 555 (s) | Deformation mode of SO2 | 30 |
| 802 (m) | 806 (m) | Ammine rocking mode | 26 |
| — | 889 (s) | η2-(OS)O symmetric stretching mode (part of doublet) | — |
| — | 958 (m) | η2-(OS)O symmetric stretching mode (part of doublet) | 10 and 31 |
| 1121 (s) | 1116/1123 (m) | η1-SO2 (dark-state) symmetric stretching mode | 30 |
| 1212 (w) | 1212 (m) | η1-SO2 (dark-state) asymmetric stretching mode | 30 |
| — | 1301 | A mode involving ammine ligands (tentative assignment) | 27 |
| — | 1450 | A mode involving ammine ligands (tentative assignment) | 27 |
| 1600 (w) | 1600 (w) | H2O bending mode | 32 and 33 |
| — | 1800 | A mode involving ammine ligands (tentative assignment) | 27 |
| — | 1843 | A mode involving ammine ligands (tentative assignment) | 27 |
| 2900–3100 (s) | 2900–3100 (m) | H2O stretching modes | 32 and 33 |
| 3200–3350 (m) | 3200–3350 (w) | Ru–N ammine stretching modes | 26 |
Light- and thermally-induced optical switching properties of 1
The much higher photoconversion fraction of 1 to the η2-(OS)O photoisomer observed herein at 100 K, compared to the original photocrystallography study,9 is presumably a result of the different light sources and light application methods used in the different diffraction experiments, as well as the benefit of auxiliary information in this study that was available from single-crystal optical absorption spectroscopy and Raman spectroscopy and microscopy. In particular, a series of multi-temperature single-crystal optical absorption spectra were acquired from measurements that explored the effect of different light sources and the duration of their light exposure on 1, as well as assessed its thermal stability. These series of experiments largely followed the optical spectroscopy methodologies that have been developed by Cole et al. to support photocrystallography.27 The reader is referred to Experimental methods for specific technical detail about the measurements conducted herein.The duration of light exposure on a crystal of 1 at 100 K from a 505 nm LED source was tracked by single-crystal optical absorption spectroscopy and microscopy (Fig. 2). The crystal was deemed to be saturated with light after 180 min of photostimulation, judging from its unchanging optical absorption spectrum profile between successive spectral captures by this point. A final spectrum was acquired after a subsequent time lapse of 30 min in order to check that the photoisomerised species that had formed in 1 at 100 K was metastable. The resulting spectrum was indistinguishable from the spectrum that was taken immediately after 180 min of light exposure, thereby assuring metastability of the η2-(OS)O photoisomer in 1 at 100 K.
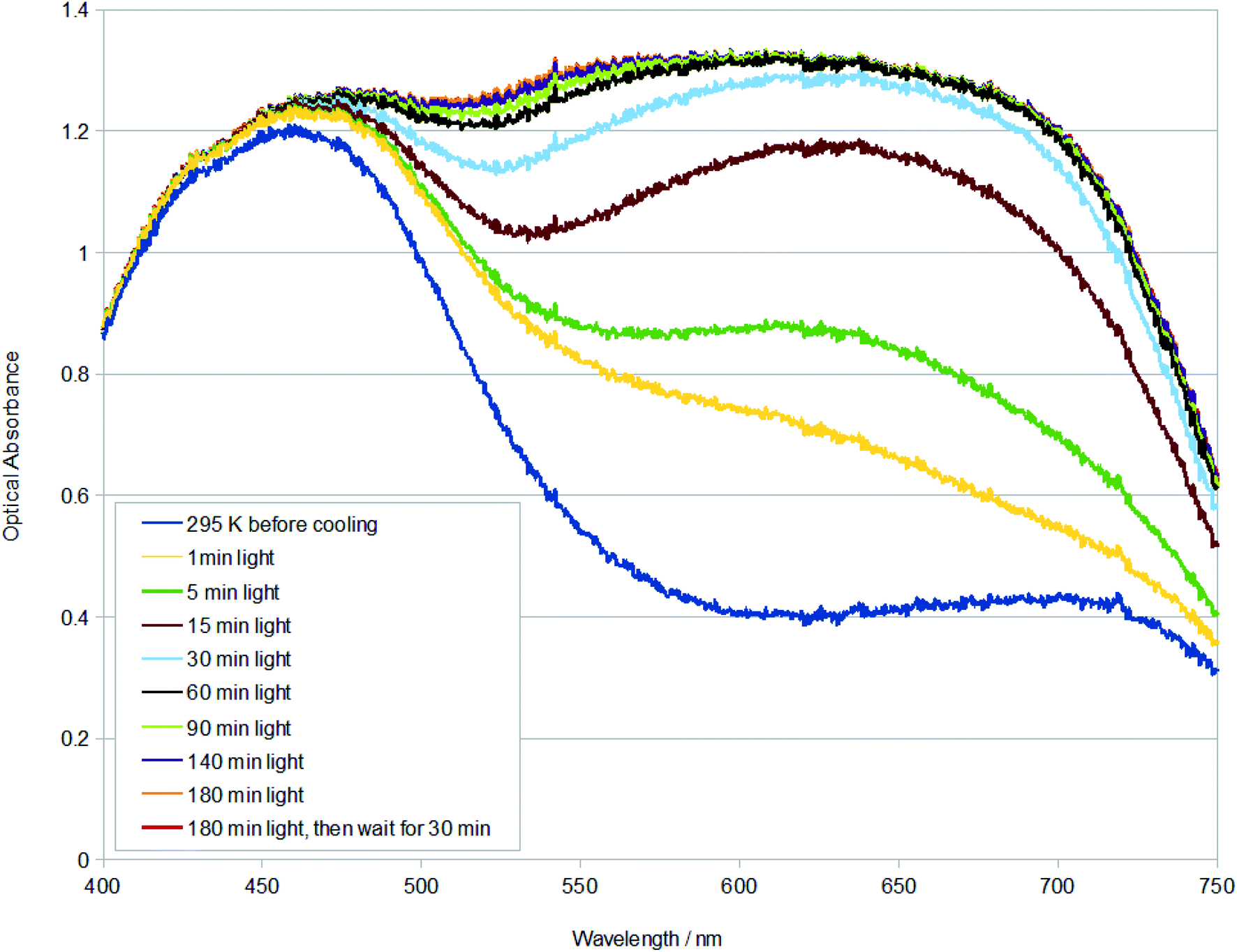 | ||
| Fig. 2 Single-crystal optical absorption spectra of 1 after t minutes of light exposure at 100 K. A reference dark-state spectrum of 1 at 295 K is provided. | ||
The changing spectral profiles of 1 as a function of 505 nm LED light exposure are broadly similar to those observed for trans-[Ru(SO2)(NH3)4Cl]Cl.27 Thereby, the optical absorption initially rises substantially across the green-red region of the visible spectrum, levelling off soon after its optical absorbance exceeds that of the dominant dark-state absorption peak. Accordingly, the fully light-saturated optical absorption spectrum of 1 is essentially a ‘black body absorber’ within the visible range down to c. 450 nm, where it starts to downturn. The rate by which 1 is saturated with light was calculated from the single-crystal optical absorption spectra shown in Fig. 2, via a four-step process: (i) the active area under each spectrum in Fig. 2 (400–715 nm) was numerically integrated using the composite Simpson's rule, which is well suited to this task given that there are a large number of finely-spaced data points; (ii) the area of the dark-state spectrum was subsequently subtracted from that of each light-induced spectrum, yielding the metric, area (light–dark); (iii) the order of the photoisomerisation process was then determined by exploring the relationship between temperature and the light-driven change in area. This revealed that the photoisomerisation is first order, judging from the linear fit to a plot of T vs. ln(area (light–dark)). The first-order classification of this photoisomerisation process is consistent with previous findings from IR spectroscopy measurements on 1,9 as well as IR spectroscopy and photocrystallography experiments on other [RuSO2] complexes.9,13–17 (iv) The negative gradient of this plot was thus determined, yielding a rate constant, k = 8.280 × 10−5 s−1 at 100 K, for the η1-SO2 (dark) to η2-(OS)O (light) photoisomerisation process in 1, from a linear fit whose coefficient of determination, R2 = 0.999.
This photoisomerisation process in 1 is one-and-a-half orders-of-magnitude slower than that observed in the related complex, trans-[Ru(SO2)(NH3)4Cl]Cl,27 where k = 3.074 × 10−3 s−1 at 100 K (R2 = 0.995). This stands to reason given that H2O will be a stronger electron-donating ligand than Cl in this series of complexes, while 1 also has a larger counterion which will further stabilize the H2O ligand on 1. Accordingly, 1 will be much more resistant to SO2 photoisomerisation than trans-[Ru(SO2)(NH3)4Cl]Cl, and so its rate constant is much slower. The starkly contrasting large and small trans influences observed in the light-induced crystal structure of trans-[Ru(SO2)(NH3)4Cl]Cl9,27 and 1,9 respectively, corroborate these rate-constant differences. Despite this corroboration, it is worth noting that the photoisomerisation rate constant for trans-[Ru(SO2)(NH3)4Cl]Cl at 100 K is complicated by its additional presence of some η1-OSO photoisomer.27
In fact, 1 is even closer chemically to trans-[Ru(SO2)(NH3)4(H2O)]chlorobenzenesulfonate2, differing from 1 by just one substituent: Cl versus CH3 in its anion composition; yet, unlike 1, it does not form η2-(OS)O photoisomer upon light exposure; instead, it undergoes a η1-SO2 to η2-OSO photoconversion,14 whose k = 7.90 × 10−3 s−1 at 100 K is even faster than that of trans-[Ru(SO2)(NH3)4Cl]Cl. This suggests that the rate constant in this family of [RuSO2] complexes could be determined primarily by η1-OSO photoisomer formation where it occurs. Such a contrast in the type of photoisomer formation observed in 1, compared with trans-[Ru(SO2)(NH3)4(H2O)]chlorobenzenesulfonate2, despite such a small chemical change is presumably structural in origin. Hirshfeld surfaces for crystal structures of these two complexes (see ESI†) suggest that the methyl group of one tosylate anion in the crystallographic asymmetric unit of 1 has close interactions with the SO2 ligand of its cation, while no close interactions are observed between any chloro group and the SO2 ligand in trans-[Ru(SO2)(NH3)4(H2O)]chlorobenzenesulfonate2. The reaction cavity of the η1-SO2 ligand in 1 will thus be more restricted,11 which seemingly limits its ability to photoisomerise to just the η2-(OS)O ligand when exposed to light at 100 K. Nonetheless, 1 can photoconvert to the η1-OSO configuration at a much lower temperature of 13 K;10 thus, this structural variation in 1 has a subtle but important bearing on its photoisomerisation characteristics.
Photochromic effects in a crystal of 1 were shown to be associated with its photoisomerization process, as evidenced via optical microscopy that was performed in concert with the single-crystal optical absorption spectroscopy.27 The microscopy photographs in Fig. 3 (top) show that the crystal of 1 transitions from a red to essentially black colour, which is consistent with the spectral changes observed in the optical absorption spectra of 1 (Fig. 2). Fig. 3 (top) necessarily displays a restricted number of photographs. However, the full set of photographs which accompany every optical absorption spectrum were manufactured into a video that tracks these photochromic effects on 1 as a function of light-exposure duration and its subsequent thermal decay (see ESI†). Heat was applied since this is known to reverse the photoisomerisation process in this family of complexes. Thereby, once the crystal had been fully saturated with light, it was warmed to 160 K and thence monitored via optical microscopy (Fig. 3 (middle)). These results show that the crystal returns to its dark-state configuration upon heating and does so in a non-destructive fashion. The fully reversible nature of this photoisomerisation is an attractive feature of these [RuSO2] compounds towards an ultimate read–write memory device application.25
 | ||
| Fig. 3 Single-crystal optical microscopy images of the crystal of 1 (top) upon the application of t minutes of 505 nm light at 100 K; which was then (middle) warmed to 160 K and monitored as a function of time elapsed at 160 K; (bottom) upon the application of 30 min of 505 nm light while at 100 K, to which t min of 633 nm light was then applied. | ||
Nevertheless, the use of heat to reverse photoisomerisation will afford a slow optical switching process, while a read–write memory device operation would need to be fast, as well as precisely controlled. The alternative use of red light to deactivate the light-induced species in 1 was postulated, given that photochemical observations on trans-[Ru(SO2)(NH3)4Cl]Cl suggest that such deactivation may be possible in that similar complex.31 Accordingly, 633 nm light from a HeNe laser was applied to the crystal of 1 once it had been photoexcited by 505 nm light for 30 min at 100 K, to establish its metastable photoisomeric state at close to light saturation levels, judging from Fig. 2. Single-crystal optical absorption spectra were acquired between progressive applications of red light of a certain time duration, until the cumulative duration of red-light exposure reached 60 min. The results displayed in Fig. 3 (bottom) reveal that red light successfully deactivates the photoisomerisation completely, judging from the indistinguishable optical absorption spectra of the dark-state species at 100 K, and the 30 min green-irradiated species after 60 min red-light application at 100 K. Fig. 3 (bottom) also shows that the crystal has nearly fully returned to its dark state after only 10 minutes of red-light application.
Microscopy images of the crystal were acquired in concert with these optical absorption spectra. A video that tracks the changes observed in the crystal across all acquired images is given in the ESI.† Fig. 3 (bottom) provides a selection of these images, which reveals that the crystal exhibits a black-to-orange photochromic change quickly but unevenly in terms of the rate per location on the crystal. The effect is observed as a wave of change that begins at the bottom-right corner of the crystal and progresses up and left until the entire crystal becomes orange, as shown in Fig. 3 (bottom). A green tinge is associated with this wavefront while the overall black colour lightens into a deep-green hue en route to its final orange appearance. The image acquired after 10 min red-light exposure carries a distinct biphasic crystal colouring; this is very interesting because it demonstrates the viability of a macroscopic binary encoding of dark and light-induced SO2 structural isomers in crystals of 1. Naturally this biphasic formation would need to be controlled in an eventual application. Yet, this fundamental work can already offer some suggestions to this end. For example, it would seem that the red light fully bathed the crystal in this experiment, but its highest intensity was centered towards the bottom-right end of the crystal, i.e. the area from which the photochromic change stemmed. The precise location and intensity of this red-light application onto the crystal would therefore seem to affect the nature of this biphasic appearance of a crystal of 1. This location specificity also indicates that red light could even be used to pattern crystals of 1 using suitable optical stencils. While such device-engineering considerations are beyond the scope of this work, these foundation-level findings can hopefully motivate this effort.
From a metrological perspective, it is important to note that the optical absorption spectra in this experiment were acquired from a detector array whose window observed the top-right part of the crystal; thus, the aforementioned spectral indication that 10 min of red-light application had almost fully deactivated the photoisomerisation. While this observation remains true, the potential uneven nature of photochromic changes, and factors that cause such unevenness, also need to be considered. This highlights the importance of a concerted optical absorption spectroscopy and microscopy instrument setup that was custom-built for this type of research;27 without such a setup, we would have been unable to tally the uneven photochromic changes observed in the crystal with the spectral findings. Furthermore, while the photoisomerisation decay from red-light application is uneven in this experiment, and we have seen that this may pose a practical benefit, its light arrangement could presumably be altered to create a uniform rate of change if required.
The alternative thermal decay route to enact photoisomerisation decay does not have the same ready opportunity to pattern a crystal in the way that this is an option for red-light application. This is illustrated in Fig. 3 (middle), whereby the thermal decay process affords a uniform photochromic change throughout the entire crystal. This uniformity is inevitable given that the crystal has to be raised above its metastable temperature in order to thermally decay; no part of the crystal can thus remain in its photoisomerised state any longer.
The ease by which 1 can be switched was also assessed given its prospective ultimate application. Thereby, the activation energy, Ea, of the η2-(OS)O to η1-SO2 isomerisation process in 1 was determined. A bespoke methodology was developed to meet this task. In the first instance, seven series of measurements were conducted at a range of decay temperatures that span 145–163 K in 3 K increments. For each series, 60 min of 505 nm light was initially applied to the crystal of 1 in order to afford the photoisomerised species at 100 K; the temperature was then raised to one that was beyond the temperature at which 1 becomes metastable, thereby invoking thermal decay. Single-crystal optical absorption spectra were then acquired as a function of elapsed time from the moment that the crystal reached the decay temperature in question. These data enabled seven rate constants to be determined from the aforementioned four-step data-analysis process that was devised in order to determine a rate constant, k, from such spectra, to assess the rate of photoisomerisation as a function of light-exposure duration (vide supra). These seven rate constants were then employed in a plot of 1/T versus lnk, given that the photoisomerisation of 1 is known to undergo a first-order process.9
Ea is determined from the gradient of a linear fit to this plot, following the Arrhenius equation:
| lnk = −(Ea/RT) + lnA |
All seven series of single-crystal optical absorption spectra acquired at each decay temperature are given in the ESI,† together with their cognate microscopy images which are presented in the form of seven videos that track each thermal-decay process. The rate constants that are derived from these spectra are provided in Table 2, which are all used in the Arrhenius plot of 1/T versus lnk that is displayed in Fig. 4, from which an Ea = 30(1) kJ mol−1 was afforded.
| Temperature/K (error ± 2.5 K) | Rate constant, k/s−1 | Coefficient of determination, R2 |
|---|---|---|
| 145 | 8.897 × 10−4 | 0.997 |
| 148 | 1.130 × 10−3 | 0.998 |
| 151 | 3.367 × 10−3 | 1.000 |
| 154 | 4.520 × 10−3 | 0.999 |
| 157 | 3.981 × 10−3 | 0.998 |
| 160 | 1.258 × 10−2 | 0.997 |
| 163 | 1.271 × 10−2 | 0.996 |
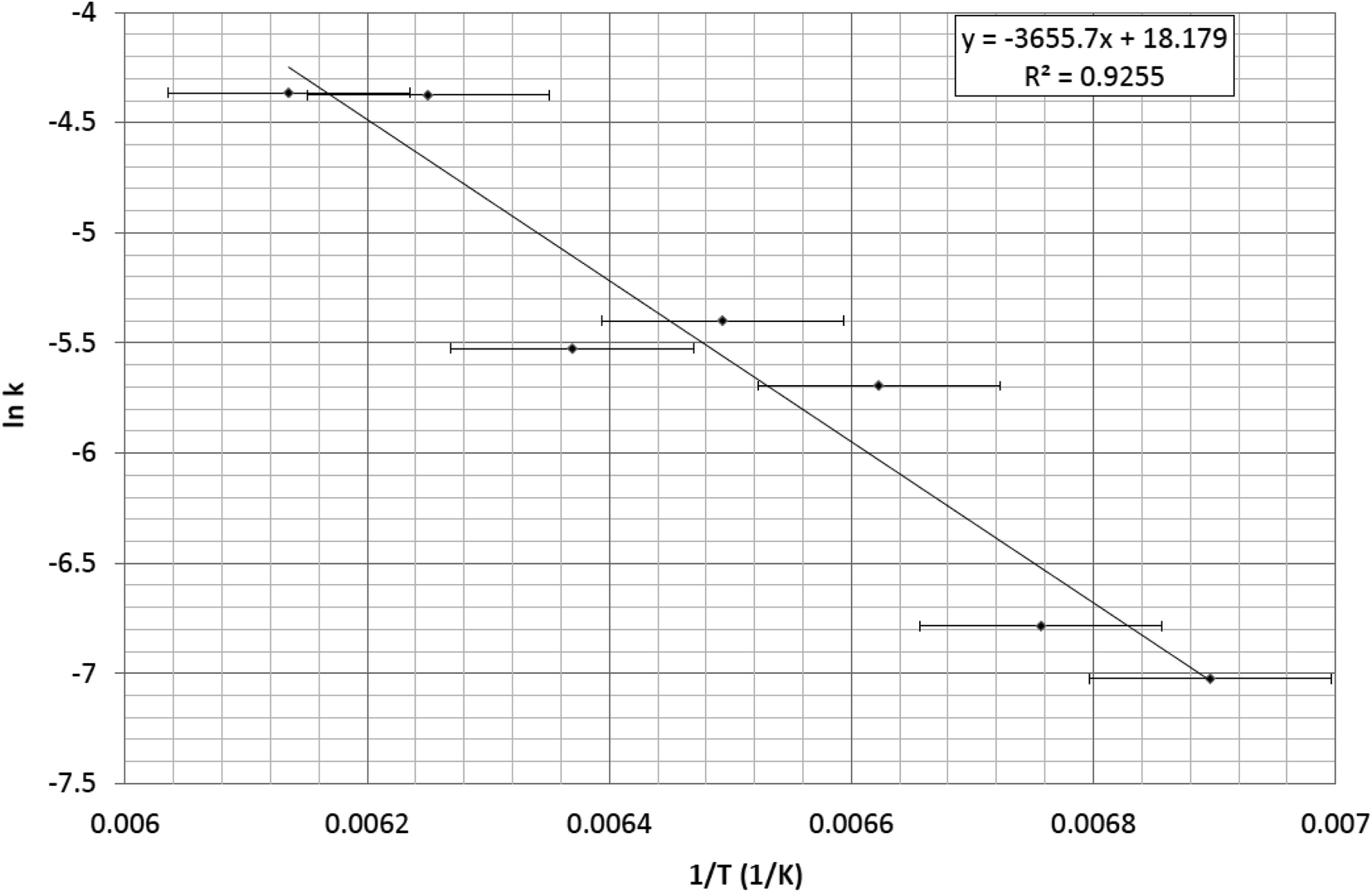 | ||
| Fig. 4 Arrhenius plot of 1/T versus lnk whose gradient (Ea/R) yields an activation energy, Ea = 30(1) kJ mol−1. | ||
The error for the Ea stems primarily from the temperature value which is very accurate in its reading; yet, this is compromised by the position of the thermistor in the cryostat which measures the temperature of the coldfinger of the cryostat in which the single-crystal sample is held; this is close to the crystal but there will nonetheless be a modest thermal gradient. Empirical temperature calibration tests were performed on the instrument set up to correct for this error, using the known thermal transitions of other crystals within the [RuSO2] family of complexes as a reference. This reduced the temperature error to ±2.5 K, which is reasonable for many experiments. Nonetheless, this error is significant when working with such small values of 1/T in the current study; although, the linear fit in Fig. 4 shows that the data analysis accounts well for this error. This is the primary error that propagates to the Ea value.
In principle, there might be an observable error associated with the reference dark-state spectrum that is subtracted from the light-induced spectra, given that the last data point in each series of spectra is used as the dark-state reference. This data point is associated with full recovery of the dark state of 1; yet, the dark state will range from 78.5–100% depending upon the level of photoconversion in 1 at a given point. i.e. this method assumes a 100% fraction of dark-state in the area at all times. However, the optical absorption spectra of the dark state will diminish only slightly, as judged by Fig. 2, which shows that light-stimulated changes dominate the spectral contributions, even after just a few minutes of light exposure. A possible error pertaining to this concern was nonetheless checked by integrating the area of the dark and light-induced spectra using three different integration windows of (i) 400–715 nm, (ii) 475–715 nm, and (iii) 600–715 nm. These ranges were chosen for this test since they correspond to the (i) full active spectral area that can be considered to be reliable with the band-pass filters housed in the instrument set up; (ii) the active spectral area from which has been subtracted the region where only the dark-state signal contributes; (iii) the active spectral area from which has been subtracted the region that contains the peak profile, such that an essentially flat and featureless dark-state signal remains. Ea for 1 was calculated using data from each window, affording values of (i) 30.4(1.0) kJ mol−1; (ii) 30.0(1.0) kJ mol−1; (iii) 30.8(9) kJ mol−1. This confirmed that the photoisomerisation effects dominate the single-crystal optical absorption spectra in 1 such that any offset in dark state owing to the assumption of the 100% dark-state fraction has a negligible effect as they are less than the aforementioned primary source of error.
The Ea value of 30(1) kJ mol−1 (0.311 eV) for 1 obtained from these results compares well with results of density functional theory (DFT) from the ‘basis set 2’ model realised by Kovalevsky et al.,9 which predicted that the η2-(OS)O bound photoisomer of 1 was 0.27 eV more stable than its η1-SO2 dark-state configuration. Our Ea value for 1 is also comparable to, but generally less than, the Ea values of other Ru-based complexes that exhibit linkage photoisomerisation on another type of (NO) ligand.34 Those Ea values were determined from experiments on polycrystalline samples, from a series of rate constants that were obtained using light-induced changes in specific peak intensities of vibrational spectra as a function of elapsed time, once the sample had been raised to one of a series of temperatures which are above the point where the NO photoisomer in each complex is metastable. Kovalevsky et al.9 also reported an Ea value of 58.4 kJ mol−1 for the thermal decay of the η2-(OS)O photoisomer in 1, using infra-red spectroscopy. Thereby, peak-intensity changes of the dark-state η1-SO2 symmetric stretching mode of 1 were tracked with elapsed time at 7–10 temperature points which are above that where it is metastable. Their Ea value from infra-red spectroscopy is nearly double that of their DFT value,9 so the results herein stand to offer some assurance to their DFT studies.
Conclusions
This study has showcased fundamental structural and optical property findings of 1 which indicate that it could have prospects for read–write memory applications. The dark-state and light-induced crystal structures of 1 have been redetermined via this work in order to show that it can hold a higher photoconversion level of the η2-(OS)O photoisomer (21.5(5)%) than was previously realised.9 The kinetic characteristics of the light-exposure process for 1 have been presented, using single-crystal optical absorption spectra to determine a rate constant, k = 8.280 × 10−5 s−1 for η1-SO2 to η2-(OS)O photoisomerisation with green (505 nm) light. This is one-and-a-half orders-of-magnitude slower than the same rate constant for the closely related complex, trans-[Ru(SO2)(NH3)4Cl]Cl (k = 3.074 × 10−3 s−1 at 100 K), which also forms an η2-(OS)O photoisomer. Our study found that both red (633 nm) light and heating of 1 will fully restore the complex to its dark state. Optical microscopy supported the absorption spectroscopy experiments, displaying photochromic changes in the crystal of 1 in response to various light and thermal stimuli. We also showed that 1 can be optically patterned in a fashion that is switched on and off by green and red light, respectively. Only LEDs were required for this photostimulation of 1, which offers a technical simplicity to any ultimate applications of this optical switching behaviour. The deactivation of photoisomerisation via red light is also more rapid and controllable than the alternative heating process. 1 was found to exhibit a low-energy barrier to deactivation (Ea = 30 kJ mol−1), as was determined by numerical integration of a series of optical absorption spectra that were acquired at seven different decay temperatures. This Ea value is on the low end of those stated in the aforementioned report on ruthenium-based NO photoisomerisation.34 Given that metal-nitrosyl linkage photoisomers have already been prospected for their read–write memory applications,35–37 it would thus seem that [RuSO2] complexes could also be viable for such applications: they can be readily photoexcited at similar energies to metal-nitrosyl complexes.Author contributions
J. M. C. performed all of the photo-crystallography, optical microscopy and absorption spectroscopy and Raman spectroscopy work, with experimental assistance from D. J. G. in setting up the Raman spectroscopy, optical microscopy and absorption spectroscopy apparatus. J. M. C. carried out the data analysis. J. M. C. was the PhD supervisor of S. O. S. who synthesized the material. J. M. C. drafted the manuscript. All authors provided input and agreed on the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. M. C. is grateful for the BASF/Royal Academy of Engineering Research Chair in Data-Driven Molecular Engineering of Functional Materials, which is partly supported by the STFC via the ISIS Neutron and Muon Source. J.M.C. also thanks the 1851 Royal Commission of the Great Exhibition for the 2014 Fellowship in Design, hosted by Argonne National Laboratory where work done was supported by the U.S. Department of Energy (DOE) Office of Science, Office of Basic Energy Sciences, and used research resources of the Center for Nanoscale Materials, Office of Science User Facilities operated for the DOE Office of Science by Argonne National Laboratory, supported by the U.S. DOE, all under contract no. DE-AC02-06CH11357. S. O. S. acknowledges the Cambridge Commonwealth Trust for a PhD Scholarship.References
- N. Koumura, R. Zijlstra, R. van Delden, N. Harada and B. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- A. Koçer, M. Walko, W. Meijberg and B. L. Feringa, Science, 2005, 309, 755–758 CrossRef PubMed.
- D. D. Dashitsyrenova, A. A. Adonin, I. D. Gorokh, O. A. Kraevaya, A. V. Pavlova, P. A. Abramov, L. A. Frolova, M. N. Sokolov, V. P. Fedin and P. A. Troshin, Chem. Commun., 2020, 56, 9162–9165 RSC.
- A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19–30 RSC.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- L. E. Hatcher, J. M. Skelton, M. R. Warren and P. R. Raithby, Acc. Chem. Res., 2019, 52, 1079–1088 CrossRef CAS PubMed.
- A. Yu. Kovalevsky, K. A. Bagley, J. M. Cole and P. Coppens, Inorg. Chem., 2003, 42, 140–147 CrossRef CAS PubMed.
- A. Yu. Kovalevsky, K. A. Bagley and P. Coppens, J. Am. Chem. Soc., 2002, 124, 9241–9248 CrossRef CAS PubMed.
- K. F. Bowes, J. M. Cole, S. L. G. Husheer, P. R. Raithby, T. Savarese, H. A. Sparkes and J. E. Warren, Chem. Commun., 2006, 2448–2450 RSC.
- A. E. Phillips, J. M. Cole and T. d'Almeida, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 155118 CrossRef.
- A. E. Phillips, J. M. Cole and T. d'Almeida, Inorg. Chem., 2012, 51, 1204–1206 CrossRef CAS PubMed.
- S. O. Sylvester, J. M. Cole and P. G. Waddell, J. Am. Chem. Soc., 2012, 134, 11860–11863 CrossRef CAS PubMed.
- J. M. Cole, D. J. Gosztola, J. J. Velazquez-Garcia, S.-Y. G. Wang and Y.-S. Chen, Chem. Commun., 2021, 57, 1320–1323 RSC.
- S. O. Sylvester and J. M. Cole, Adv. Mater., 2013, 25, 3324–3328 CrossRef CAS PubMed.
- S. O. Sylvester and J. M. Cole, J. Phys. Chem. Lett., 2013, 4, 3221–3226 CrossRef CAS.
- S. O. Sylvester, J. M. Cole, P. G. Waddell, H. Nowell and C. Wilson, J. Phys. Chem. C, 2014, 118, 16003–16010 CrossRef CAS.
- A. E. Phillips, J. M. Cole, K. S. Low and G. Cibin, J. Phys.: Condens. Matter, 2013, 25, 085505 CrossRef PubMed.
- J. M. Cole, J. J. Velazquez-Garcia, D. J. Gosztola, S.-Y. G. Wang and Y.-S. Chen, Chem. Mater., 2019, 31, 4927–4935 CrossRef CAS.
- K. T. Mukaddem, J. M. Cole, K. A. Beyer and S. O. Sylvester, J. Phys. Chem. C, 2020, 124, 10094–10104 CrossRef CAS.
- P. Coppens, D. V. Fomitchev, M. D. Carducci and K. Culp, J. Chem. Soc., Dalton Trans., 1998, 6, 865–872 RSC.
- J. M. Cole, Chem. Soc. Rev., 2004, 33, 501–513 RSC.
- J. M. Cole, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 259–271 CrossRef CAS PubMed.
- J. M. Cole, Analyst, 2011, 136, 448–455 RSC.
- J. M. Cole, Z. Kristallogr., 2008, 223, 363–369 CAS.
- L. H. Vogt, J. L. Katz and S. E. Wiberley, Inorg. Chem., 1965, 4, 1157–1163 CrossRef CAS.
- J. M. Cole, D. J. Gosztola, J. J. Velazquez-Garcia and Y.-S. Chen, J. Phys. Chem. C, 2020, 124, 28230–28243 CrossRef CAS.
- J. K. Hurst, J. Zhou and Y. Lei, Inorg. Chem., 1992, 31, 1010–1017 CrossRef CAS.
- M. Lauter, D. K. Breitinger, R. Breiter, J. Mink and E. Bencze, J. Mol. Struct., 2001, 563–564, 383–388 CrossRef CAS.
- D. K. Breitinger and R. Breiter, J. Mol. Struct., 1995, 349, 45–48 CrossRef CAS.
- D. A. Johnson and V. C. Dew, Inorg. Chem., 1979, 18, 3273–3274 CrossRef CAS.
- B. Kolesov, Am. Mineral., 2006, 91, 1355–1362 CrossRef CAS.
- D.-Y. Wu, S. Duan, X.-M. Liu, Y.-C. Xu, Y.-X. Jiang, B. Ren, X. Xu, S. H. Lin and Z.-Q. Tian, J. Phys. Chem. A, 2008, 112, 1313–1321 CrossRef CAS PubMed.
- K. Ookubo, Y. Morioka, H. Tomizawa and E. Miki, J. Mol. Struct., 1996, 379, 241–247 CrossRef CAS.
- D. Schaniel, M. Imlau, M. Weismoeller, T. Woike, K. Kraemer and H. U. Guedel, Adv. Mater., 2007, 19, 723–726 CrossRef CAS.
- M. Imlau, Th. Woike, R. Schieder and R. A. Rupp, Europhys. Lett., 2001, 53, 471–477 CrossRef CAS.
- Th. Woike, S. Hausshul, B. Sugg, R. A. Rupp, J. Beckers, M. Imlau and R. Schieder, Appl. Phys. B, 1996, 63, 243–248 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2040379 and 2040380. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01696b |
| This journal is © The Royal Society of Chemistry 2021 |