DOI:
10.1039/D1RA01649K
(Paper)
RSC Adv., 2021,
11, 18246-18251
A computational study for the reaction mechanism of metal-free cyanomethylation of aryl alkynoates with acetonitrile†
Received
2nd March 2021
, Accepted 12th May 2021
First published on 20th May 2021
Abstract
A computational study of metal-free cyanomethylation and cyclization of aryl alkynoates with acetonitrile is carried out employing density functional theory and high-level coupled-cluster methods, such as coupled-cluster singles and doubles with perturbative triples [CCSD(T)]. Our results indicate that the reaction of aryl alkynoates with acetonitrile in the presence of tert-butyl peroxybenzoate (TBPB) under metal-free conditions tends to proceed through cyanomethylation, spirocyclization and ester migration of the kinetically favoured coumarin derivatives. 1,2-Ester migration in the spiro-radical intermediate 10 does not proceed via the formation of the carboxyl radical 11 suggested by Sun and co-workers. Our results also demonstrate that the t-butoxy radical is substantially responsible the formation of the cyanomethyl radical by the abstraction of a hydrogen atom from acetonitrile.
Introduction
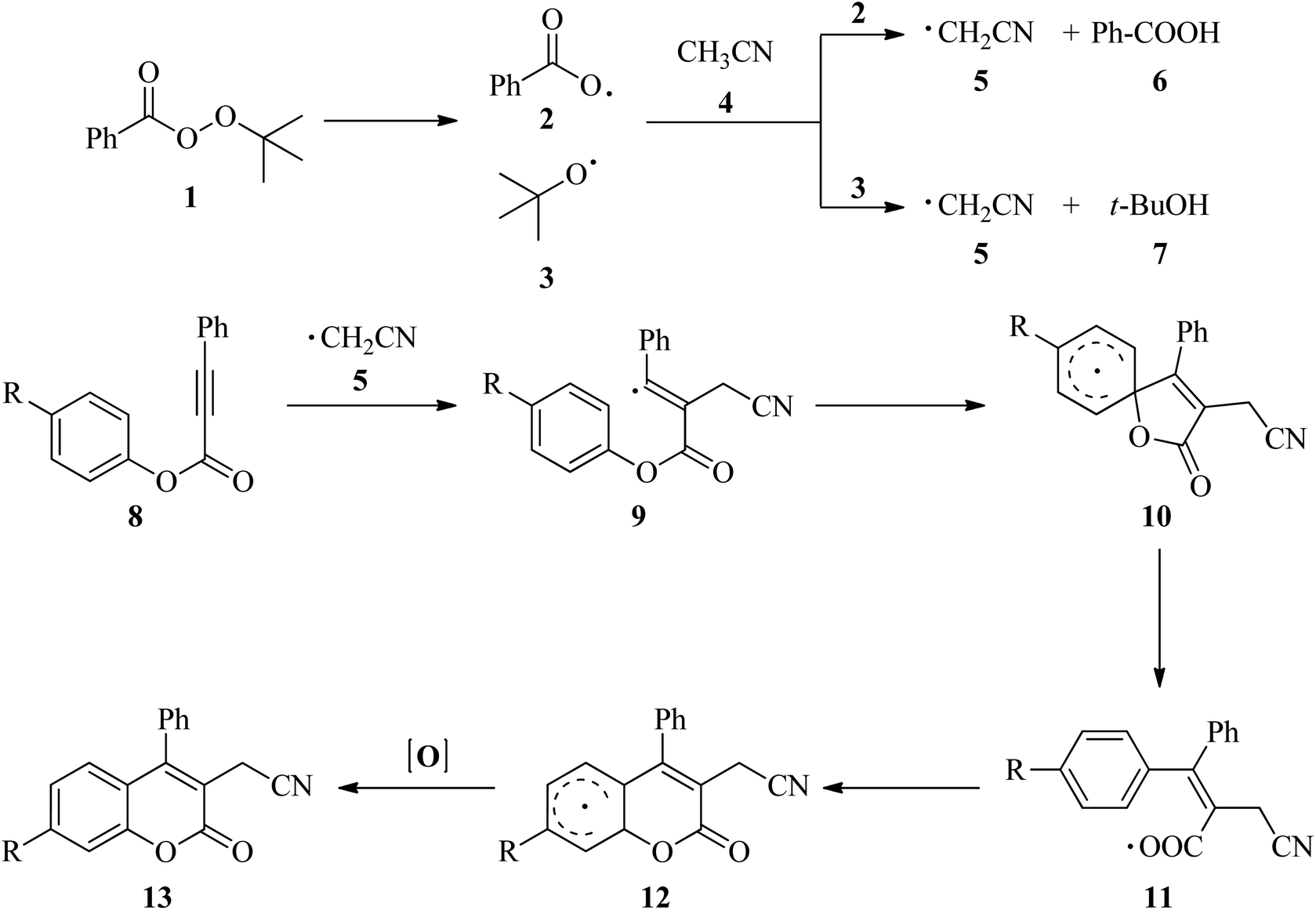
Cyanomethylation is a very useful reaction to synthesize cyano-containing compounds in medicinal and synthetic organic chemistry due to easy conversion of the cyano group into primary amines, ketones, carboxylic acids, esters, amides and even tetrazoles.1–3 In addition, cyanomethyl moieties obtained via cyanomethylation are found as versatile motifs in some important bioactive natural products and drugs.4,5 A variety of protocols have been reported, including transition-metal-catalyzed methods6–16 and metal-free catalyzed methods17–22 for the synthesis of cyanomethyl-containing compounds. Compared to the toxic metal used, metal-free catalyzed methods used acetonitrile9–12,17,18 and its analogs such as bromoacetonitrile,13–16 cyanoacetic acid19,20 and ethyl cyanoacetate5 have drawn considerable attention due to its inexpensive and environmental-friendly properties. In recent years, metal-free cyanomethylation using of acetonitrile as a cyanomethyl source in the synthesis of the cyano-containing compounds was reported.17,18,21 In 2016, Sun and co-workers18 demonstrated a convenient and efficient method for the synthesis of 3-cyanomethylated coumarins, a class of very important heterocyclic compounds in pharmaceuticals and dyes, via cyanomethylation and cyclization of aryl alkynoates using acetonitrile as the cyanomethyl source in the presence of tert-butyl peroxybenzoate (TBPB) under metal-free conditions. A control experiment carried out in the presence of 2,6-di-tert-butyl-4-methylphenol (BHT) or 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) showed that the thermal cyanomethylation and cyclization of aryl alkynoates proceeds via a radicalic process as described in Scheme 1.
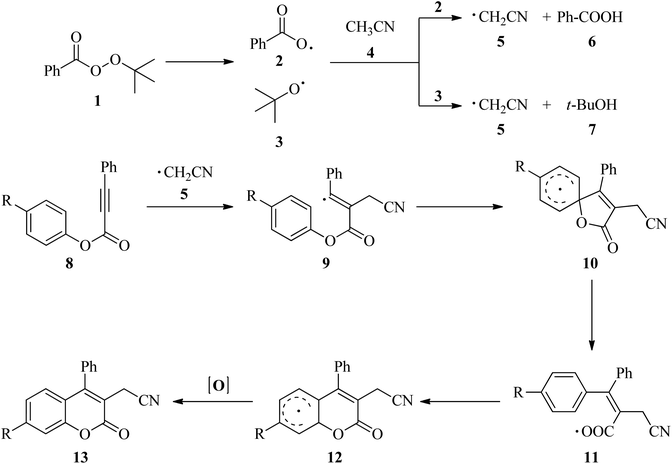 |
| | Scheme 1 Proposed mechanism by Sun and co-workers.18 | |
According to the mechanism proposed by Sun and co-workers,18 at first, TBPB (1) decomposed into the benzoyloxy (2) and tert-butoxy radical (3) at high temperature. One of these radicals abstracted a hydrogen atom from acetonitrile (4) to generate the cyanomethyl radical (5). An electrophilic attack of cyanomethyl radical (5) to alkynoate (8) yields the alkenyl radical 9. Then, an intramolecular spirocyclization of the alkenyl radical 9 gives the spiro-radical intermediate 10. Next, the ester migration took place via a carboxyl radical 11 to yield the radical 12. Finally, the radical 12 was oxidized to a carbocation or a radical followed by deprotonation or abstraction of a hydrogen radical to yield the cyano-containing coumarin derivatives 13. Additionally, in these reactions, benzoic acid (6) and tert-butanol (7) are also formed as by-product.
Result and discussion
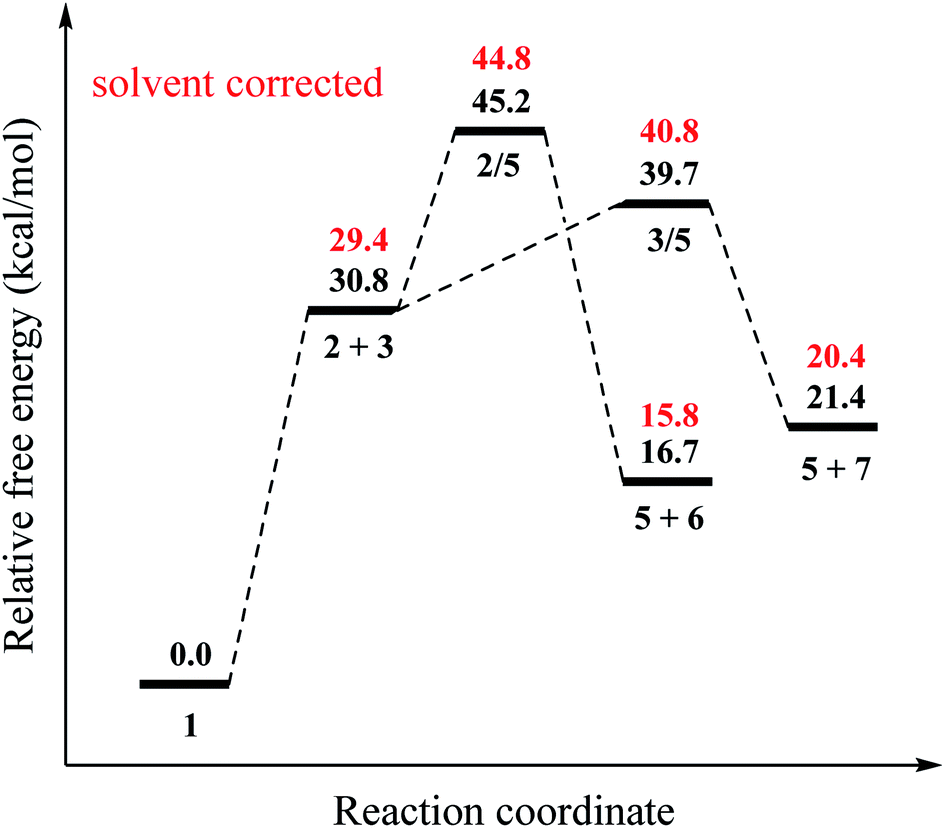
Our results for the mechanism proposed by Sun and co-workers are reported in Fig. 1 and 2. For the initiation of the radicalic process, the 1 → 2 + 3 conversion, the computed reaction free energy is 30.8 kcal mol−1 (Fig. 1). Zhou23 was reported that the bond dissociation energy (BDE) (Gibbs free energy at 383.15 K) of TBPB is 11.2 kcal mol−1 at M06-L-D3/6-31+G(d,p) level and no any transition state (TS) for this step. We did not also find any TS for this step. The decomposing of TBPB (1) into benzoyloxy (2) and t-butoxy radical (3) was carried out at high temperature, which provide enough energy for this dissociation process. To generating the cyanomethyl radical (5), a hydrogen atom from acetonitrile (4) is abstracted by one of these radicals. For the abstraction of a hydrogen atom from acetonitrile (4) by benzoyloxy radical (2), 2 → 5, the reaction free energy and barrier are −14.1 and 14.4 kcal mol−1, respectively. For the other path for the formation of cyanomethyl radical (5), 3 → 5, the reaction free energy and barrier are −9.4 and 8.9 kcal mol−1, respectively. The barrier of the abstraction of a hydrogen atom from acetonitrile (4) by t-butoxy radical (3) is 5.5 kcal mol−1 lower than that of benzoyloxy radical (2) (Fig. 1). These results show that t-butoxy radical (3) is substantially responsible the abstraction of hydrogen atom.
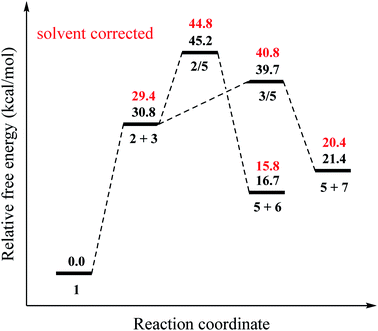 |
| | Fig. 1 Relative free energy profile (at 403.15 K) for the formation of cyanomethyl radical (5) shown in Scheme 1 at the DLPNO-CCSD(T)/cc-PVTZ//B3LYP-6-311G(d,p) level. | |
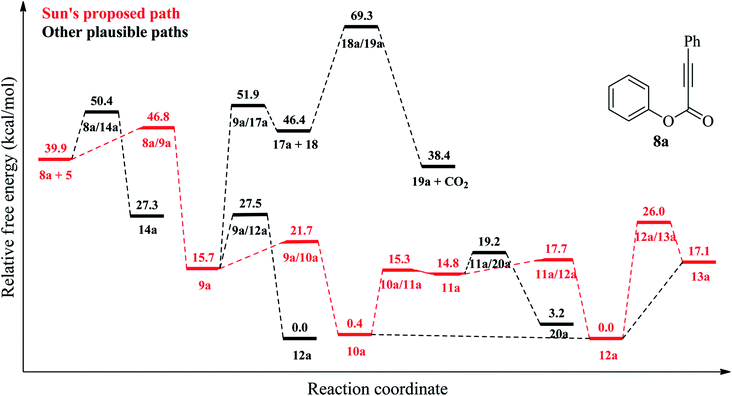 |
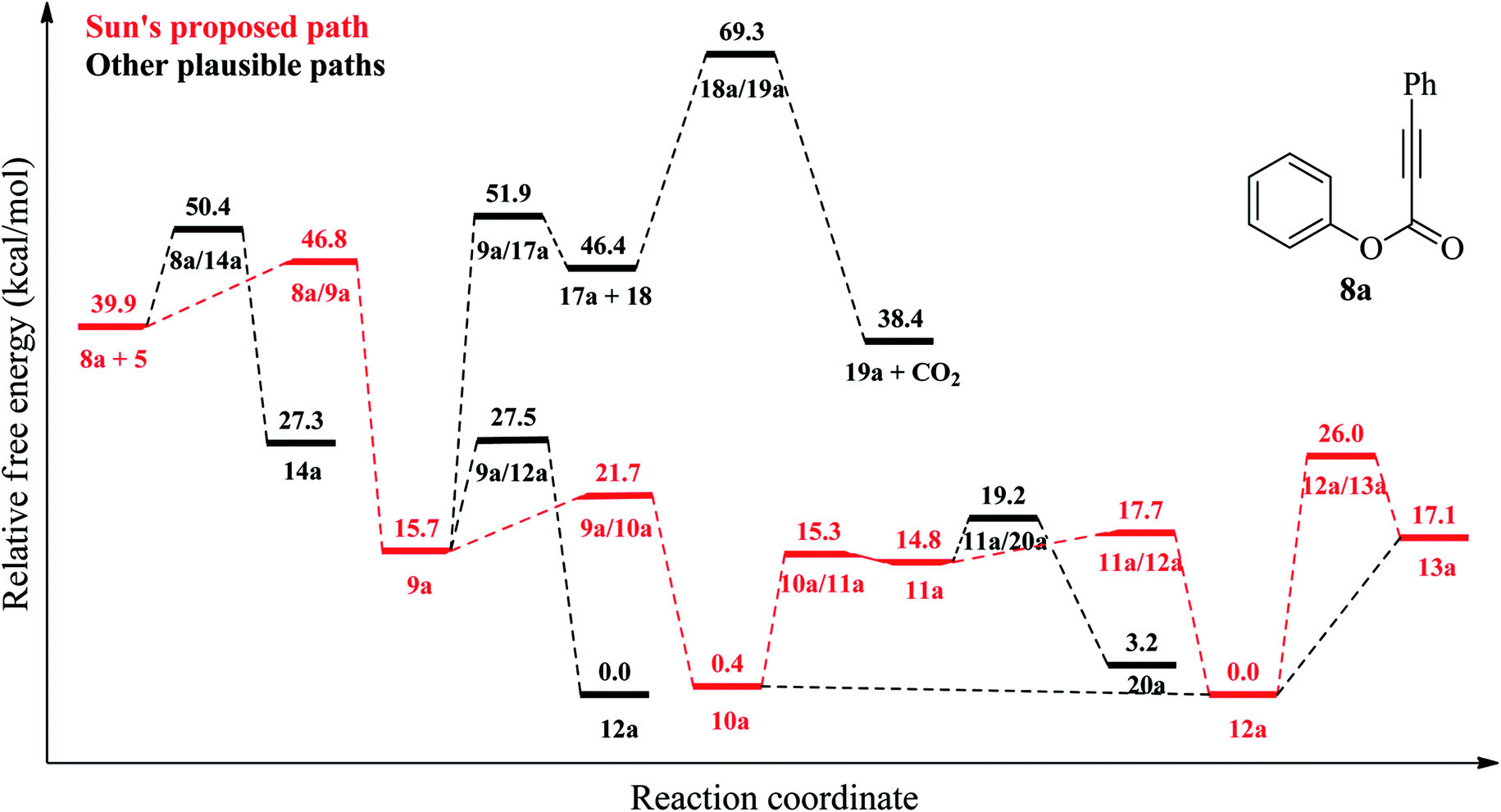
| | Fig. 2 Relative free energy profile (at 403.15 K) for reaction mechanism of 8a shown in Schemes 1 and 2 at the DLPNO-CCSD(T)/cc-PVTZ//B3LYP-6-311G(d,p) level. | |
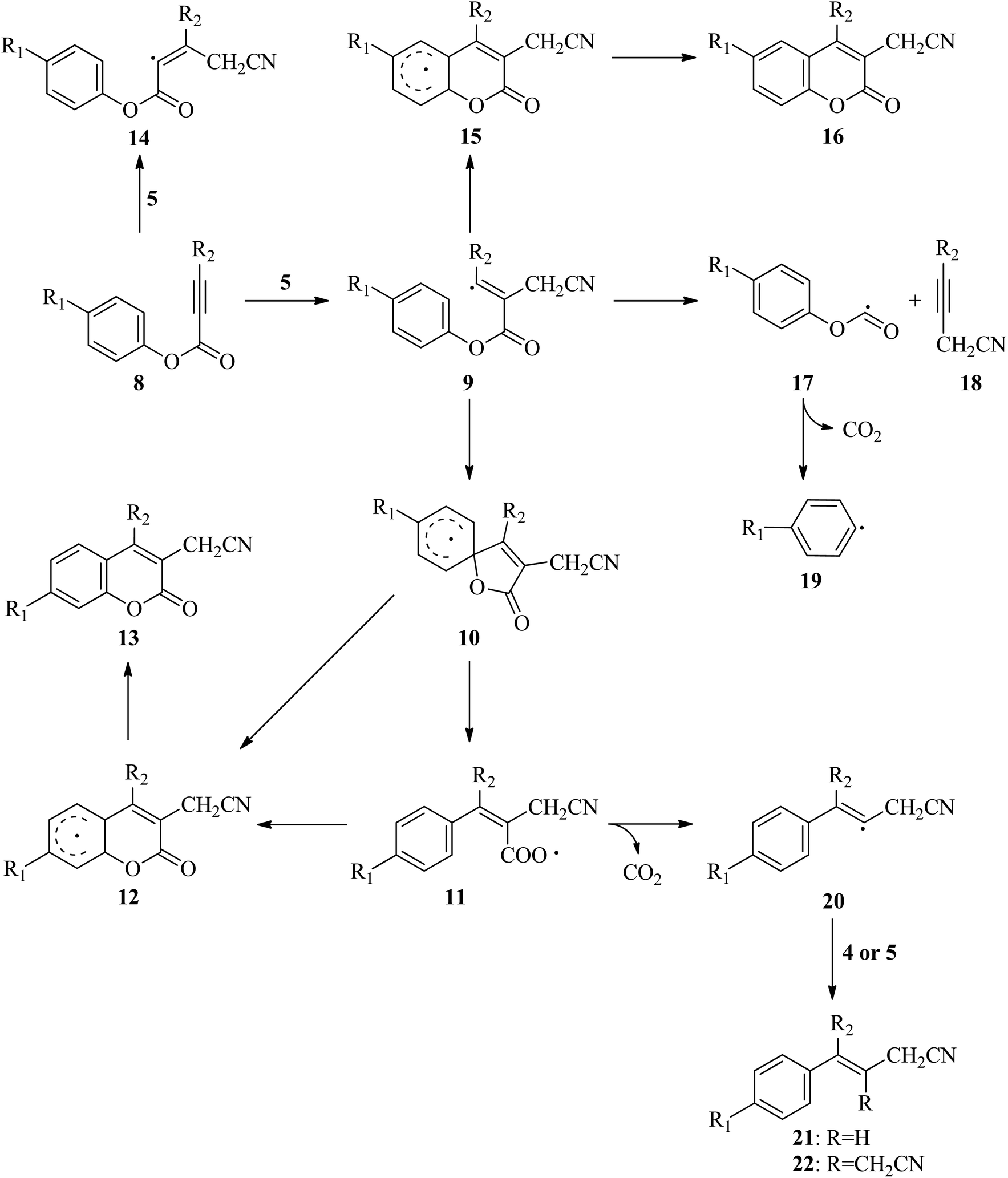
In the literature, there are many studies for the reaction of aryl alkynotes with a variety of radicals. As a result of these studies, coumarin derivatives (13 (ref. 18 and 24–35) or 16 (ref. 36–45)) or styrene derivatives (21 (ref. 46–48) or 22 (ref. 49)) were obtained under different reaction conditions. All plausible mechanism for aryl alkanoates 8 with cyanomethyl radical (5) is proceed as described in Scheme 2. There are two possible electrophilic attack of cyanomethyl radical (5) to the C–C triple bond of aryl alkynoate derivatives 8. In the first one, the cyanomethyl radical (5) is reacted with alkynes, 8, to form the intermediate 9. The alternative electrophilic attack of cyanomethyl radical (5) to alkynes, 8, is formed the intermediates 14. For the electrophilic attack of cyanomethyl radical (5) to the C–C triple bond of aryl alkynoate derivative 8a, 8a → 9a, the reaction free energy and barrier are −24.2 and 6.9 kcal mol−1, respectively. For the alternative electrophilic attack of cyanomethyl radical (5) to the alkyne 8a, 8a → 14a, the reaction free energy and barrier are −12.6 and 10.5 kcal mol−1, respectively. Both the intermediate 9a is more stable than the intermediate 14a (the relative energy of 9a is lower 11.6 kcal mol−1 than that of 14a) and the barrier for the formation of 9a is lower 3.6 kcal mol−1 than the barrier for the formation of 14a. This result clarifies why the electrophilic attack of cyanomethyl radical (5) to alkynes, 8, is formed the formation of the intermediates 9. The intramolecular spirocyclization of the alkenyl radical 9 gives the spiro-radical intermediate 10, while the intramolecular cyclization of the alkenyl radical 9 gives the intermediate 15. For the intramolecular spirocyclization of the alkenyl radical 9, 9a → 10a, the reaction free energy and barrier are −15.3 and 6.0 kcal mol−1, respectively. For the intramolecular cyclization of the alkenyl radical 9, 9a → 12a, the reaction free energy and barrier are −15.7 and 11.8 kcal mol−1, respectively. Also, decarboxylation of the intermediate 17 forming with release acetylene derivative 18 generates phenyl radical 19. For the formation of the intermediate 17, 9a → 17a, the reaction free energy and barrier are 30.7 and 36.2 kcal mol−1, respectively. For the decarboxylation of the intermediate 17, 17a → 19a, the reaction free energy and barrier are −8.0 and 22.9 kcal mol−1, respectively. While the aryl migration via cleavage of C–O bond yields the carboxyl radical 11, the ester migration process without the formation of the carboxyl radical 11 affords the intermediate 12. For 1,2-aryl migration, 10a → 11a, the reaction free energy and barrier are 14.4 and 14.9 kcal mol−1, respectively. For the 10a → 12a conversion, the reaction energy is −0.4 kcal mol−1 and 1,2-ester migration proceeds barrierless. The cyclization of carboxyl radical 11 yields the intermediate 12 oxidized by abstraction of a hydrogen radical to yield the desire product, coumarin derivatives 13. For the cyclization of carboxyl radical 11, 11a → 12a, the reaction free energy and barrier are −14.8 and 2.9 kcal mol−1, respectively. For the oxidation by generating a hydrogen radical of the intermediate 12, the reaction free energy and barrier are 17.1 and 26.0 kcal mol−1, respectively. However, the oxidation of the intermediate 12 with single electron oxidation followed by deprotonation, or abstraction of the hydrogen atom by benzoyloxy radical (2) or t-butoxy radical (3) proceeds barrierless. The decarboxylation with release of carbon dioxide of the carboxyl radical 11 yields alkenyl radical 20, which is ready to afford the formation of styrene derivatives 21 and 22. For the decarboxylation of the carboxyl radical 11, 11a → 20a, the reaction free energy and barrier are −11.6 and 4.4 kcal mol−1, respectively.
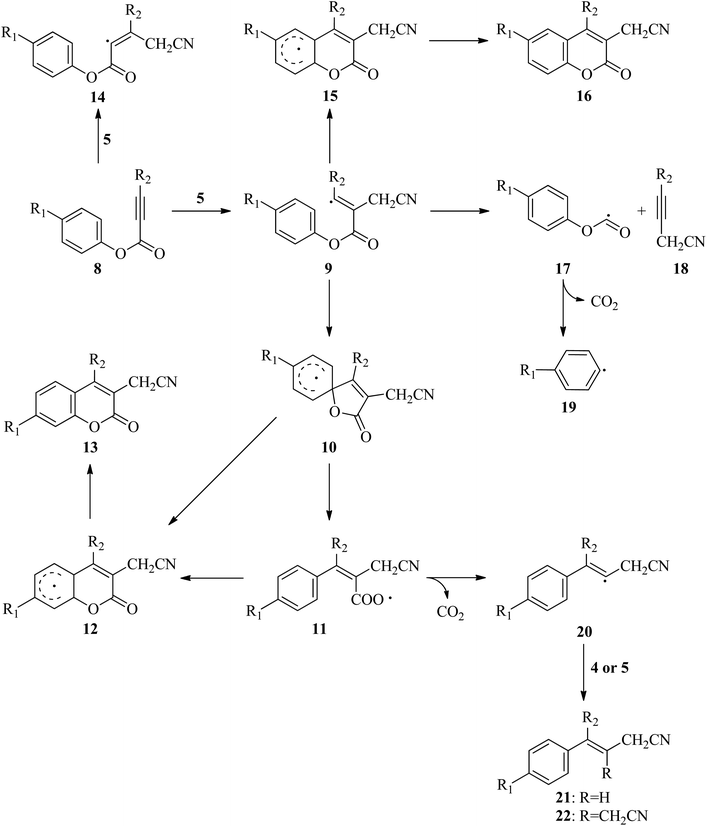 |
| | Scheme 2 Plausible mechanism for aryl alkanoates with cyanomethyl radical. | |
Conclusion
In this study, all plausible mechanism for metal-free cyanomethylation of aryl alkynoates with acetonitrile has been investigated with high-level coupled-cluster methods, such as DLPNO-CCSD(T), along with the cc-pVTZ basis set. According to our computations, the decomposition of TBPB, which is the initial step of the radicalic reaction, include the highest reaction energy step and show that the rate-determining step is the formation of the benzoyloxy (2) and tert-butoxy radical (3) for the reaction scheme. 1,2-Ester migration in the spiro-radical intermediate 10 does not proceed via the formation of the carboxyl radical 11 suggested by Sun and co-workers because the relative energy of carboxyl radical 11 is nearly same that of TS 10/11 and activation free energy for the 11a → 20a conversion is very low for the proposed reaction condition. Also, solvent corrected free energies demonstrated that carboxyl radical 11 is not formed in the reaction conditions (see ESI†). Additionally, no observed styrene derivatives support that the intermediates 11 are not formed during the reaction. Our results demonstrate that the reaction of aryl alkynoates using acetonitrile as the cyanomethyl source in the presence of TBPB tends to yield the kinetically favoured product. Our results also indicate that t-butoxy radical (3) is substantially responsible in the formation of cyanomethyl radical (5). The computations are compatible with experimental results. Moreover, our computations provide useful insight into the reaction mechanism of aryl alkynotes with a variety of radicals.
Computational methods
Geometry optimizations and harmonic vibrational frequency computations for the structures considered were carried out with the density functional theory (DFT), the B3LYP functional.50–53 For this purpose, a Pople-type polarized triple-ζ split-valence basis sets, 6-311G(d,p), was used.54–56 Single point energies were computed at the optimized geometries with the coupled-cluster singles and doubles with perturbative triples method employing the domain based local pair natural orbitals approach [DLPNO-CCSD(T)].57,58 In DLPNO-CCSD(T) computations Dunning's correlation consistent triple-ζ split valence basis set, cc-pVTZ, is employed.59,60 DFT computations were carried out by using the Gaussian 09 program,61 while the ORCA package62,63 is employed for DLPNO-CCSD(T) computations. The solvation effects were considered employing the polarizable continuum model (PCM) in acetonitrile.64 For the transition state (TS) between species A and B, the A/B notation is used throughout the article.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by Hakkari University, Scientific Research Project Coordination Unit (BAP; Grant no. FM21AYP1).
References
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, New York, NY, USA, 2001 Search PubMed.
- R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 1989 Search PubMed.
- V. Y. Kukushkin and A. J. L. Pombeiro, Inorg. Chem. Acta, 2005, 258, 1–21 CrossRef.
- R. Lopez and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170–13184 CrossRef CAS PubMed.
- K. Imamura, N. Tomita, Y. Kawakita, Y. Ito, K. Ono, N. Nii, T. Miyazaki, K. Yonemori, M. Tawada, H. Sumi, Y. Satoh, Y. Yamamoto, I. Miyahisa, M. Sasaki, Y. Satomi, M. Hirayama, R. Nishigaki and H. Maezaki, Bioorg. Med. Chem., 2017, 25, 3768–3779 CrossRef CAS PubMed.
- J. Velcicky, A. Soicke, R. Steiner and H. G. Schmalz, J. Am. Chem. Soc., 2011, 133, 6948–6951 CrossRef CAS PubMed.
- J. R. Donald and S. L. Berrell, Chem. Sci., 2019, 10, 5832–5836 RSC.
- M. J. Tom and P. A. Evans, J. Am. Chem. Soc., 2020, 142, 11957–11961 CrossRef CAS PubMed.
- W. Zhang, S. Yang and Z. Shen, Adv. Synth. Catal., 2016, 358, 2392–2397 CrossRef CAS.
- Y. Liu, K. Yang and H. Ge, Chem. Sci., 2016, 7, 2804–2808 RSC.
- E. Wada, T. Takeuchi, Y. Fujimura, A. Tyagi, T. Kato and H. Yoshida, Catal. Sci. Technol., 2017, 7, 2457–2466 RSC.
- S. Zhang, Z. Shen and H. Jian, J. Org. Chem., 2020, 85, 6143–6150 CrossRef CAS PubMed.
- E. R. Welin, A. A. Warkentin, J. C. Conrad and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 9668–9672 CrossRef CAS PubMed.
- Q. Chang, Z. Liu, P. Liu, L. Yu and P. Sun, J. Org. Chem., 2017, 82, 5391–5397 CrossRef CAS PubMed.
- C. J. O'Brien, D. G. Droege, A. Y. Jiu, S. S. Gandhi, N. A. Paras, S. H. Olson and J. Conrad, J. Org. Chem., 2018, 83, 8926–8935 CrossRef PubMed.
- W. Zhang, C. Yang, Y. L. Pan, X. Li and J. P. Cheng, Org. Biomol. Chem., 2018, 16, 5788–5792 RSC.
- Z. Ni, X. Huang, J. Wang and Y. Pan, RSC Adv., 2016, 6, 522–526 RSC.
- Y. Yu, S. Zhuang, P. Liu and P. Sun, J. Org. Chem., 2016, 81, 11489–11495 CrossRef CAS PubMed.
- K. Balaraman, M. Moskowitz and C. Wolf, Adv. Synth. Catal., 2018, 360, 4705–4709 CrossRef CAS PubMed.
- V. M. Tkachuk, S. V. Melnykov, A. V. Vorobei, V. A. Sukach and M. V. Vovk, Chem. Heterocycl. Compd., 2019, 55, 66–71 CrossRef CAS.
- G. Hong, P. D. Nahide and M. C. Kozlowski, Org. Lett., 2020, 22, 1563–1568 CrossRef CAS PubMed.
- Z. Li, C. Rao, L. Chen, C. Fu, T. Zhu, X. Chen and C. Liu, Dyes Pigm., 2020, 173, 107967 CrossRef CAS.
- D. G. Zhou, Mol. Catal., 2020, 498, 111246 CrossRef CAS.
- Y. Liu, Q. L. Wang, C. S. Zhou, B. Q. Xiong, P. L. Zhang, S. J. Kang, C. A. Yang and K. W. Tang, Tetrahedron Lett., 2018, 59, 2038–2041 CrossRef CAS.
- M. J. Bu, G. P. Lu and C. Cai, Catal. Commun., 2018, 114, 70–74 CrossRef CAS.
- X. Wu, M. Jia, M. Huang, J. K. Kim, Z. Zhao, J. Liu, J. Xi, Y. Li and Y. Wu, Org. Biomol. Chem., 2020, 18, 3346–3353 RSC.
- Z. Wang, L. Wang, Z. Wang and P. Li, Asian J. Org. Chem., 2019, 8, 1448–1457 CrossRef CAS.
- D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925–11929 CrossRef CAS PubMed.
- L. Liu, Q. Ding, Q. Zong and G. Qui, Org. Chem. Front., 2015, 2, 670–673 RSC.
- S. Ni, J. Cao, H. Mei, J. Han, S. Li and Y. Pan, Green Chem., 2016, 18, 3935–3939 RSC.
- S. Feng, J. Li, Z. Liu, H. Sun, H. Shi, X. Wang, X. Xie and X. She, Org. Biomol. Chem., 2017, 15, 8820–8826 RSC.
- M. Zhu, W. Fu, Z. Wang, C. Xu and B. Ji, Org. Biomol. Chem., 2017, 15, 9057–9060 RSC.
- Q. Wang, C. Yang and C. Jiang, Org. Biomol. Chem., 2018, 16, 8196–8204 RSC.
- W. Fu, Y. Sun and X. Li, Synth. Commun., 2020, 3, 388–398 CrossRef.
- K. S. Kanyiva, D. Hamada, S. Makino, H. Takano and T. Shibata, Eur. J. Org. Chem., 2018, 5905–5909 CrossRef CAS.
- X. Zhang, Y. Li, X. Hao, K. Jin, R. Zhang and C. Duan, Tetrahedron, 2018, 74, 7358–7363 CrossRef CAS.
- W. Fu, M. Zhu, G. Zou, C. Xu, Z. Wang and B. Ji, J. Org. Chem., 2015, 80, 4766–4770 CrossRef CAS PubMed.
- W. C. Gao, T. Liu, B. Zhang, X. Li, W. L. Wei, Q. Liu, J. Tian and H. H. Chang, J. Org. Chem., 2016, 81, 11297–11304 CrossRef CAS PubMed.
- S. Feng, X. Xie, W. Zhang, L. Liu, Z. Zhong, D. Xu and X. She, Org. Lett., 2016, 18, 3846–3849 CrossRef CAS PubMed.
- D. Liu, J. Q. Chen, X. Z. Wang and P. F. Xu, Adv. Synth. Catal., 2017, 359, 2773–2777 CrossRef CAS.
- W. Yang, S. Yang, P. Li and L. Wang, Chem. Commun., 2015, 51, 7520–7523 RSC.
- Y. F. Zeng, D. H. Tan, Y. Chen, W. X. Lv, X. G. Liu, Q. Li and H. Wang, Org. Chem. Front., 2015, 2, 1511–1515 RSC.
- C. Pan, R. Chen, W. Shao and J. T. Yu, Org. Biomol.
Chem., 2016, 14, 9033–9039 RSC.
- X. Mi, C. Wang, M. Huang, Y. Wu and Y. Wu, J. Org. Chem., 2015, 80, 148–155 CrossRef CAS PubMed.
- K. Yan, D. Yang, W. Wei, F. Wang, Y. Shuai, Q. Li and H. Wang, J. Org. Chem., 2015, 80, 1550–1556 CrossRef CAS PubMed.
- C. Pan, Y. Chen, S. Song, L. Li and J. T. Yu, J. Org. Chem., 2016, 81, 12065–12069 CrossRef CAS PubMed.
- S. Ni, Y. Zhang, C. Xie, H. Mei, J. Han and Y. Pan, Org. Lett., 2015, 17, 5524–5527 CrossRef CAS PubMed.
- D. L. Kong, L. Chen, H. R. Wu, Y. X. Li, D. Wang and L. Liu, Org. Biomol. Chem., 2016, 14, 2210–2217 RSC.
- S. Ni, W. Sha, L. Zhang, C. Xie, H. Mei, J. Han and Y. Pan, Org. Lett., 2016, 18, 712–715 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chem. Acc., 1973, 28, 213–222 Search PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- Y. Guo, C. Riplinger, U. Becker, D. G. Liakos, Y. Minenkov, L. Cavallo and F. Neese, J. Chem. Phys., 2018, 148, 011101 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1995, 103, 4572–4585 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries of the transition states with selected interatomic distances, Cartesian coordinates for computed structures. See DOI: 10.1039/d1ra01649k |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab