
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure-induced stability and polymeric nitrogen in alkaline earth metal N-rich nitrides (XN6, X = Ca, Sr and Ba): a first-principles study†
Zhipeng Liu,
Shuli Wei *,
Yanhui Guo,
Haiyang Sun,
Hao Sun,
Qiang Chang and
Yuping Sun*
*,
Yanhui Guo,
Haiyang Sun,
Hao Sun,
Qiang Chang and
Yuping Sun*
School of Physics and Optoelectronic Engineering, Shandong University of Technology, 250049 Zibo, China. E-mail: weishuli@sdut.edu.cn; sunyuping@sdut.edu.cn
First published on 11th May 2021
Abstract
Multi-nitrogen or polynitrogen compounds can be used as potential high energy-density materials, so they have attracted great attention. Nitrogen can exist in alkaline earth metal nitrogen-rich (N-rich) compounds in the form of single or double bonds. In recent years, to explore N-rich compounds which are stable and easy to synthesize has become a new research direction. The N-rich compounds XN6 (X = Ca, Sr and Ba) have been reported under normal pressure. In order to find other stable crystal structures, we have performed XN6 (X = Ca, Sr and Ba) exploration under high pressure. We found that SrN6 has a new P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) phase at a pressure of 22 GPa and an infinite nitrogen chain structure, and BaN6 has a new C2/m phase at 110 GPa, with an N6 ring network structure. Further, we observed that the infinite nitrogen chain and the N6 ring network structure contain typical covalent bonds formed by the hybridization of the sp2 and sp3 orbitals of N, respectively. It is found that both SrN6 and BaN6 are semiconductor materials and the N-2p orbital plays an important role in the stability of the crystal structure for P-SrN6 and C2/m-BaN6. Because of the polymerization of nitrogen in the two compounds and their stabilities under high pressure, they can be used as potential high energy-density materials. The research in this paper further promotes the understanding of alkaline earth metal N-rich compounds and provides new information and methods for the synthesis of alkaline earth metal N-rich compounds (XN6, X = Ca, Sr and Ba).
phase at a pressure of 22 GPa and an infinite nitrogen chain structure, and BaN6 has a new C2/m phase at 110 GPa, with an N6 ring network structure. Further, we observed that the infinite nitrogen chain and the N6 ring network structure contain typical covalent bonds formed by the hybridization of the sp2 and sp3 orbitals of N, respectively. It is found that both SrN6 and BaN6 are semiconductor materials and the N-2p orbital plays an important role in the stability of the crystal structure for P-SrN6 and C2/m-BaN6. Because of the polymerization of nitrogen in the two compounds and their stabilities under high pressure, they can be used as potential high energy-density materials. The research in this paper further promotes the understanding of alkaline earth metal N-rich compounds and provides new information and methods for the synthesis of alkaline earth metal N-rich compounds (XN6, X = Ca, Sr and Ba).
1 Introduction
The nitrogen molecule has a strong triple bond and high bond energy, and it is difficult to react under environmental conditions.1 Polynitrogen compounds and polymer nitrogen are favored because of their excellent energy density, energy storage characteristics,2 unique chemical character, and thermodynamic stability.3 As is known, there are significant energy differences between the nitrogen–nitrogen single bond (160 kJ mol−1), nitrogen–nitrogen double bond (418 kJ mol−1), and nitrogen–nitrogen triple bond (954 kJ mol−1). When multi-nitrogen compounds containing single bonds and double bonds are converted into nitrogen molecules, the bonding method between nitrogen and nitrogen changes, which will release a lot of energy.4High energy-density materials (HEDMs) are generally based on the energy density of HMX (5.7 kJ g−1).5 High energy-density materials usually refer to high-energy explosives.3 Generally, an ideal HEDM should have the following characteristics: (i) high density, (ii) positive heat of formation, (iii) high detonation performance, (iv) low sensitivity towards external stimuli, (v) good thermal stability. In previous studies, for instance, scientists successfully synthesized cubic gauche nitrogen (cg-N)6 containing only single bonds under the condition of (110 GPa, 2000 K),7,8 it can be used as a potential high energy-density material. Therefore, N-rich compounds containing a large amount of nitrogen and nitrogen single bond and double bonds may become potential high energy-density materials. Above the stability field of cg-N, a layered polymer (LP)9 with a similar Pba2 phase10 between 120–180 GPa and a hexagonal layered polynitrogen phase10 with a pressure around 250 GPa were also synthesized. In order to stabilize the polynitrogen compounds11 as well as reduce the pressure and conditions for their synthesis, recently, an effective way is to add a small amount of metal atoms to pure nitrogen system through high pressure technology to stabilize the single and double bonds structure to form N-rich compounds.12 Using this research method, scientists have obtained a large number of N-rich metal compounds, for example, alkali metal and alkaline earth metal N-rich compounds.13,14 Metal atoms will cause the redistribution of electrons, leading to changes in chemical bonds, which can reduce the pressure of synthesis and increase energy storage.14
The high pressure compression to form novel compounds has become an valuable method,15,16 and the advantage is that it is easy to control and can help obtain more structures of N-rich nitrides to form stable polynitrogen. High pressure can change the bonding mode, leading to different bond lengths and hybridization modes, forming a rich multi-center covalent polymerization landscape.4 The change in the crystal structure caused by pressure has been proved by predicting the structural diversity of metal nitrides: N3,15 N4,17 N5,18 cyclo-N6,17 N8,19 N10,20 infinite nitrogen chain21 and nitrogen network structure,22 which provide an effective way to obtain N-rich compounds. In recent years, alkaline earth metal N-rich compounds have been theoretically predicted with important research significance as high energy-density materials under high pressure. Compared with alkali metals (Li,23 K,24 Na,25 Rb26 and Cs27), alkaline earth metals (Mg,17 Be,28 Ca,19 Sr29 and Ba30) and transition metals (Sc,31 Fe,32 Zn,33 Ag34 and Ir35) has more valence electrons than alkali metals, which are easier to improve the diversity of polymerized nitrogen forms. We have summarized the pressure ranges that exist the stable structures of alkali metals, alkaline earth metals and transition metals, in the Table S1 in ESI.† We also explained the reason for the transition pressure to decrease for the formation of polymeric nitrogen when we move from ionic azide to covalent azide, in the ESI.†
In this paper, we have predicted the stable phase structures of XN6 (Sr, Ba) alkaline earth metal N-rich compounds, analyzed the phase stability, and further studied their potential applications as high energy-density materials.3 The theoretical prediction found that the new phase of P-CaN6 is unstable under high pressure, while the new phase of P-SrN6 is metastable under pressure, and C2/m-BaN6 has been reported to be a metastable phase.30 In this paper, the P-SrN6 at 22 GPa and C2/m-BaN6 at 110 GPa were predicted to be stable under the corresponding high pressure conditions. After theoretical prediction and calculation, we found that the infinite nitrogen chain structure in the P-SrN6 structure is formed by polymerization when the Fddd-SrN6 structure is under pressure of 22 GPa. And the six-membered annular network structure in the C2/m-BaN6 structure is formed by polymerization when the Fmmm-BaN6 structure is under pressure of 110 GPa. We calculated the electronic band structure and projected density of states for SrN6 and BaN6, and they are all semiconductor materials. By Zintl–Klemm theory36 and calculating the electronic localization function,37 we know that the bonding manner of nitrogen atoms for P-SrN6 in high pressure is nitrogen–nitrogen single bond and double bonds and C2/m-BaN6 in high pressure the bonding manner of nitrogen atoms is nitrogen–nitrogen single bond. Due to their content of a large number of nitrogen–nitrogen single bond and double bonds, they can be used as a potential high energy-density material. The research in this paper provides new perspective for the exploration of XN6 system in the future.
2 Computation details
In order to find the stable structures of XN6 (X = Ca, Sr and Ba) alkaline earth metal N-rich compounds in the pressure range of 0–200 GPa, we used the CALYPSO38 structure prediction method based on swarm intelligence.39,40 We selected a point every 10 GPa in the pressure range of 0–200 GPa, and performed high pressure theoretical predictions and optimization on XN6 (X = Ca, Sr, Ba) alkaline earth metal N-rich compounds. The CALYPSO structure prediction method has been successfully applied to various systems from elementary solids to binary and ternary compounds.41 The optimization of the structure and the calculation of the electronic structure is performed within the framework of density functional theory (DFT),42 implemented by the VASP (Vienna Ab initio Calculation Simulation Package) code,43 and the (GGA) generalized gradient approximation44 PBE functional is used for calculation. The projector-augmented wave (PAW)45 method and the Sr, Ba and N potentials were adopted from the VASP potential library, and 5s2, 6s2 and 2s2 2p3 were treated as valence electrons for Sr, Ba and N atoms to calculate the electron–ion interaction. A plane-wave basis set cutoff of 800 eV, a Monkhorst–Pack k mesh spacing of 2π × 0.03 Å−1 in the Brillouin zone46 was selected to ensure that all enthalpy calculations converged to less than 1 meV per atom. The relative thermodynamic stability of different XN6 (X = Ca, Sr and Ba) alkaline earth metal N-rich compounds is calculated as follows:| ΔH (XN6) = [H (XN6) − H (X) − 6H (N2)/2]/(1 + 6) |
3 Results and discussions
In recent studies, we have found that metal nitrides have attracted more and more attention.50,51 Because metal nitrides are more stable than pure nitrogen compounds in terms of kinetics, and have the characteristics of low synthesis pressure and superior physical and chemical properties, they can be widely used in the research of high energy-density materials.33.1 CaN6 with P![[1 with combining macron]](https://www.rsc.org/images/entities/h3_char_0031_0304.gif) structure
structure
Since the Ca element is adjacent to the K element and can provide one more electron than the K element, this is more conducive to the diversity of nitrogen forms under high pressure. In previous studies, KN3 has been successfully predicted theoretically and can be used as a potential high energy-density material.52 This section mainly studies the structural properties and stability of CaN6, we found that a new transition phase of CaN6 occurred, and the Fddd structure under normal pressure was changed to the P structure at 32 GPa up to 100 GPa, as shown in Fig. 1(a). The infinite nitrogen chain structure in the P-CaN6 structure is composed of multiple nitrogen atoms, and its structural feature is an infinite armchair-shaped nitrogen chain,53 as shown in Fig. 1(d). It is polymerized by Fddd-CaN6 structure under 32 GPa, and Fddd-CaN6 structure was shown in Fig. 1(c).
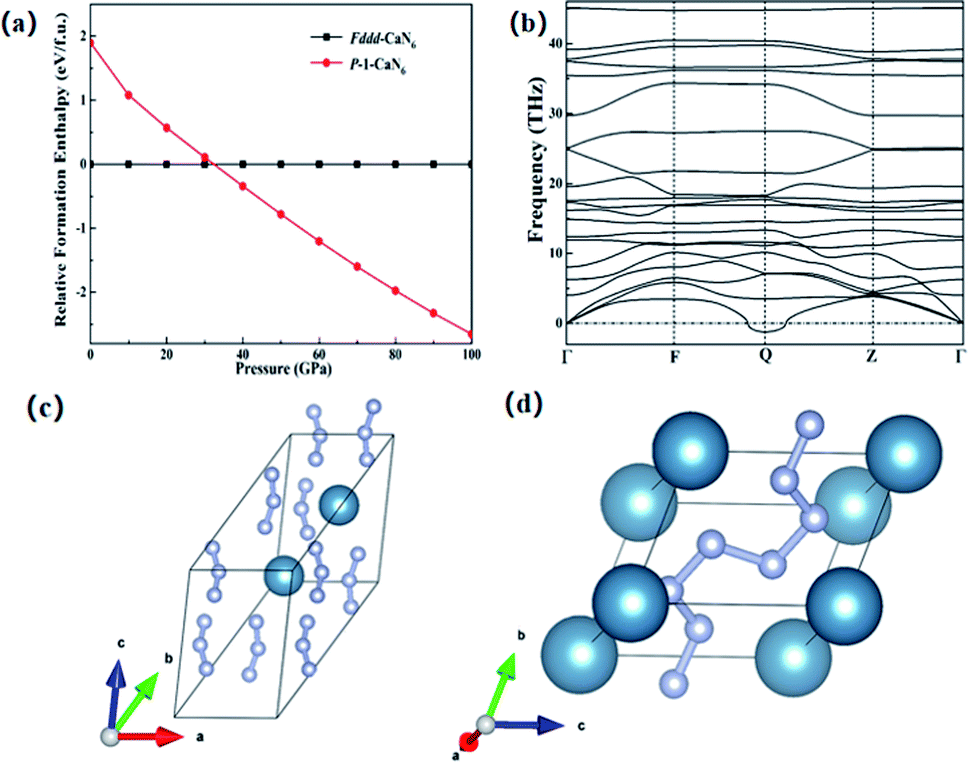 | ||
| Fig. 1 (a) Relative formation enthalpy of P-CaN6 structure and Fddd-CaN6 structure under different pressures. (b) The phonon spectrum of P-CaN6 structure. (c) Schematic diagram of the CaN6 crystal structure under normal pressure. (d) Schematic diagram of the P-CaN6 crystal structure under high pressure, the large blue balls represent Ca atoms, and the small balls represent nitrogen atoms. | ||
Referring to previous theoretical studies, we generally consider that the length of a single (N–N) bond is 1.45 Å, a double bond (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) equals 1.25 Å,18 and the length of a triple bond (N
N) equals 1.25 Å,18 and the length of a triple bond (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) is 1.10 Å at ambient conditions.33 The bond length between nitrogen and nitrogen in P-CaN6 is between 1.23 Å and 1.35 Å, surviving in the alternating range of single and double bonds. In P-CaN6 structure, the smallest unit formed by six nitrogen atoms can be equivalent to N62− anion, this is caused by the calcium atom transfers two valence electrons into the N6 structure, showing in the form of Ca2+(N6)2−. In N62− unit, the nitrogen atom satisfies the Zintl–Klemm theory,36 the covalent bonds are formed between the adjacent nitrogen atom, and form nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds coexistence status. In P-CaN6, the weight ratio of nitrogen is about 67%, due to the presence of single bond and double bonds of nitrogen and nitrogen, when it generates N2, it will release a lot of energy, which can theoretically be used as a potential high energy-density material. We further analyzed the dynamic stability of the P-CaN6 structure, and obtained the phonon dispersion curves after calculation, as shown in Fig. 1(b). It can be seen that imaginary frequencies appear in the entire Brillouin zone, indicating the P-CaN6 structure are not kinetically stable, so the P-CaN6 does not stable structure. However, our analysis of N62− anion has provided valuable information for the study of other metal polynitrides.
N) is 1.10 Å at ambient conditions.33 The bond length between nitrogen and nitrogen in P-CaN6 is between 1.23 Å and 1.35 Å, surviving in the alternating range of single and double bonds. In P-CaN6 structure, the smallest unit formed by six nitrogen atoms can be equivalent to N62− anion, this is caused by the calcium atom transfers two valence electrons into the N6 structure, showing in the form of Ca2+(N6)2−. In N62− unit, the nitrogen atom satisfies the Zintl–Klemm theory,36 the covalent bonds are formed between the adjacent nitrogen atom, and form nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds coexistence status. In P-CaN6, the weight ratio of nitrogen is about 67%, due to the presence of single bond and double bonds of nitrogen and nitrogen, when it generates N2, it will release a lot of energy, which can theoretically be used as a potential high energy-density material. We further analyzed the dynamic stability of the P-CaN6 structure, and obtained the phonon dispersion curves after calculation, as shown in Fig. 1(b). It can be seen that imaginary frequencies appear in the entire Brillouin zone, indicating the P-CaN6 structure are not kinetically stable, so the P-CaN6 does not stable structure. However, our analysis of N62− anion has provided valuable information for the study of other metal polynitrides.
3.2 SrN6 with P structure
This section mainly studies the structural properties and stability of SrN6, and other forms of strontium nitrogen compounds SrNn (n = 1–6) have been reported.51 In previous theoretical prediction, many alkali metal nitrides have been studied and proved to be good energy storage materials, such as RbN3.26 Because Sr element is adjacent to Rb element, Rb and its compounds have excellent physical and chemical properties such as easy ionization, radiation resistance, energy storage, etc., and are used in various fields.54 Because RbN3 contains nitrogen–nitrogen double bonds and exists stably, it can be used as a potential high energy-density material, that makes strontium nitrogen compound important research significance.55 After theoretical prediction and calculation, we found that the Fddd-SrN6 structure under ambient pressure can be transformed into a P-SrN6 structure at 22 GPa, and a new polymerization phase of N occurred, as shown in Fig. 2(a). The Fddd-SrN6 structure contains an independent unit consisting of every three nitrogen atoms, as shown in Fig. 2(b), and the infinite nitrogen chain structure in the P-SrN6 structure is formed by polymerization when the Fddd-SrN6 structure is under pressure of 22 GPa, as shown in Fig. 2(c). The structural parameter information of P-SrN6 is shown in Table 1.
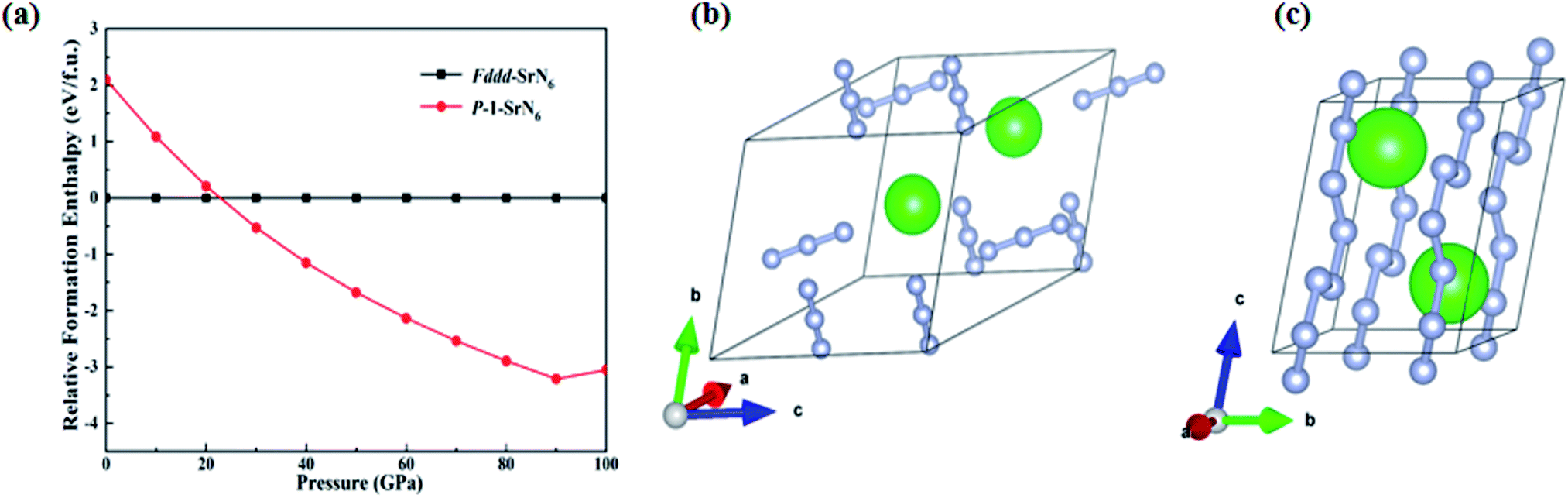 | ||
| Fig. 2 (a) Relative formation enthalpy of the high-pressure phase P-SrN6 structure relative to the normal pressure phase Fddd-SrN6 structure under different pressures. (b) Schematic diagram of Fddd-SrN6 structure. (c) Schematic diagram of P-SrN6 structure. The large green balls are Sr atoms and the small balls are nitrogen atoms. | ||
| Phase | P (GPa) | Z | Space group | Lattice parameters (Å, °) | N–N distance/average (Å) | Atomic coordinates (fractional) |
|---|---|---|---|---|---|---|
| SrN6 | 22 | 2 | P |
a = 5.28 | 1.32 | Sr (0.608, 0.294, 0.780) |
| b = 5.29 | N (0.179, 0.210, 0.690) | |||||
| c = 5.57 | (0.735, 0.741, 0.687) | |||||
| α = 91.85 | (0.130, 0.197, 0.904) | |||||
| β = 112.27 | (0.944, 0.247, 0.479) | |||||
| γ = 62.78 | (0.684, 0.743, 0.900) | |||||
| (0.008, 0.748, 0.736) | ||||||
| BaN6 | 110 | 2 | C2/m | a = 4.43 | 1.38 | Ba (0.500, −0.500, 0.500) |
| b = 4.78 | N (1.196, −0.500, 0.774) | |||||
| c = 6.25 | (1.818, −0.763, 0.917) | |||||
| α = 90.00 | ||||||
| β = 121.04 | ||||||
| γ = 90.00 |
We further studied the dynamic stability of P-SrN6 structure, and we obtained the phonon dispersion curves of P-SrN6 structure. It can be seen from the Fig. 3(a) that no virtual frequency appears in the entire Brillouin zone, indicating that P-SrN6 structure is dynamically stable. The infinite nitrogen chain structure is joined together by covalent bonds, and the valence electrons of Sr atoms are transferred to the N atoms, which plays a very important role in its stability. We calculated the electronic band structure and projected density of states for SrN6, and found that SrN6 is a semiconductor material, as shown in Fig. 3(b) and (c). In the projected density of states diagram, we can observe that the 2p state of N occupies a large proportion of the entire energy range, indicating that the valence electron state of N-2p also plays an important role in the stability of the P-SrN6 crystal structure, as shown in Fig. 3(c). As we all know, the polymerized form of nitrogen plays an important role in high energy-density material. In the infinite nitrogen chain of the P-SrN6 structure, every six nitrogen atoms form a unit and combine with one Sr atom. Since the Sr atom loses two electrons to form a Sr2+ cation, the nitrogen atoms in each unit get electrons combine with Sr atom. In a unit formed by every six nitrogen atoms, the nitrogen atom satisfies the Zintl–Klemm theory,36 which makes the nitrogen atom produce bond with the surrounding atoms, showing in the form of Sr2+(N6)2−, so the nitrogen and the nitrogen form nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds coexistence status. Due to the existence of nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds in the P-SrN6 structure and the nitrogen content reaches 49%, so it is easy to form polymeric nitrogen and conducive to obtaining high energy-density material.
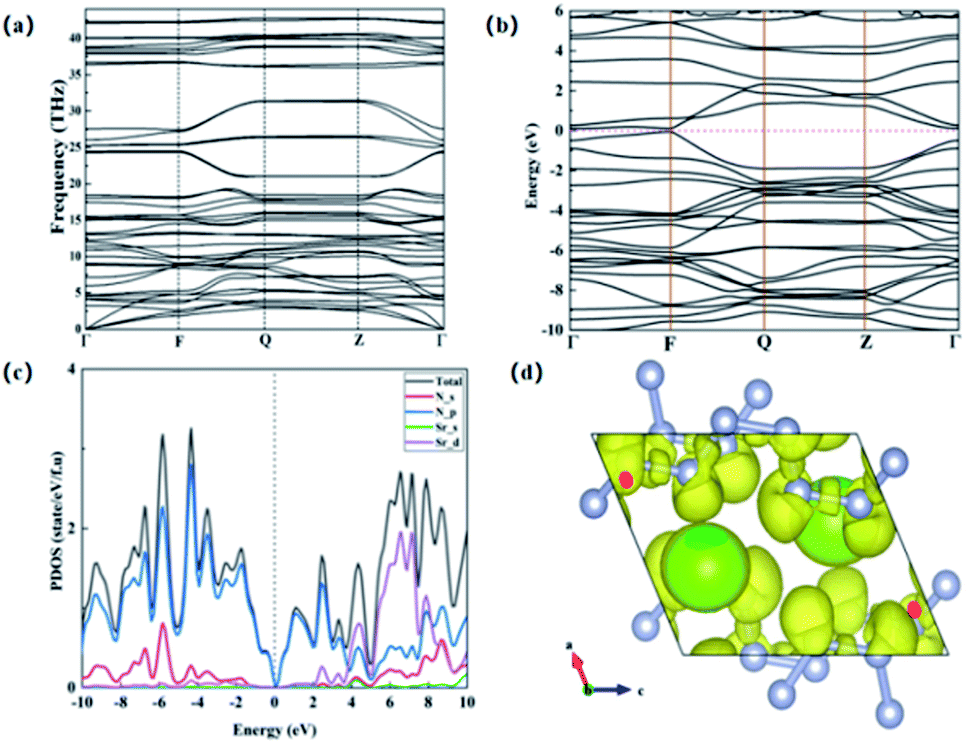 | ||
| Fig. 3 (a) The phonon spectrum of P-SrN6 structure. (b) Calculated electronic band structure diagram. (c) The calculated projected density of states (PDOS) at 22 GPa. (d) SrN6 electronic local function graph. | ||
In order to confirm this conclusion and gain a deeper understanding of the microscopic bonding mechanism of SrN6, we calculated the electronic localization function diagram and found that the nitrogen atoms have strong electronic localization and form typical covalent bonds, as shown in Fig. 3(d). The electronic local function is a three-dimensional spatial function used to characterize the distribution of electronic positioning, and its value is 0 to 1. When ELF = 1, it means the electrons are completely localized, and when ELF = 0, it means the electrons are completely delocalized, and when ELF = 0.5, it means that electrons are free. The ELF can explain the bonding and electronic distribution between the atoms, and can further explain microscopic bonding mechanisms. In Fig. 3(d), the nitrogen atoms form a chained structure, and they participate in bond formation in the form of sp2 hybridization. The two sp2 hybrid orbitals of all nitrogen atoms form covalent bond with one sp2 hybrid orbital of two adjacent nitrogen atoms. The remaining one sp2 hybrid orbitals are filled with isolated electrons. In the [–N62−–], due to the sp2 hybridization of the nitrogen atom, there are nitrogen–nitrogen single bonds and nitrogen–nitrogen double bonds between nitrogen and nitrogen.33 After calculation, in the infinite nitrogen chain, the distance between nitrogen and nitrogen is between 1.28 Å and 1.38 Å, there is surviving in the alternating range of single and double bonds. It is further proved that the structure of P-SrN6 contains nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds. In the previously reported article, the N-rich compounds with the infinite nitrogen chain structures have high energy density, in the ESI.† In the above analysis, we can judge that the content of nitrogen–nitrogen single bond and double bonds in P-SrN6 is relatively high, which a large amount of energy will be released when N2 is generated, and the synthesis pressure is low, so P-SrN6 can theoretically be used as a potential high energy-density material.
3.3 BaN6 with C2/m structure
Recently, the latest research on alkaline earth metal nitrides found that Ba and N can form a variety of compounds under high pressure.30 In this section, we mainly studied the crystal structure properties of the N-rich alkaline earth metal compound BaN6. In previous theoretical predictions, the alkaline earth metal N-rich compounds formed by alkaline earth metals Be,28 Mg,17 Ca19 and non-metallic N elements can be used as potential high energy-density materials. The chemical properties of Ba element are very active and can react with most non-metals, such as O2, N2, H2, etc. Ba element has strong reducibility and can form +2 cations, which is easy to combine with the N element under high pressure, so that the Ba element can better promote the diversity of poly-nitrogen forms.56 Therefore, exploring the alkaline earth metal N-rich compound BaN6 as a potential high energy-density material has important research significance.After theoretical calculations, the relative formation enthalpies of the five structures of BaN6 are obtained under different pressures, as shown in Fig. 4(a). After theoretical prediction, it is found that there is a P(I)–BaN6 structure with the lowest enthalpy value in the range of 21–110 GPa. This indicates that the Fmmm-BaN6 structure can be transformed to P(I)–BaN6 structure at P = 21 GPa. Our prediction of the P(I)–BaN6 structure is consistent with the previous predictions of Huang et al.,30 which also shows that our high-pressure theoretical predictions and calculation results are accurate. Further by theoretically prediction to high-pressure 200 GPa, it is found that a new monoclinic transition phase appears under the pressure of 110 GPa, the C2/m-BaN6 structure, as shown in Fig. 4(c). This indicates that the Fmmm-BaN6 structure can be transformed to C2/m-BaN6 structure at P = 110 GPa. The nitrogen atoms in the Fmmm-BaN6 structure exists as an independent unit for every two nitrogen atoms, as shown in Fig. 4(b). In the P(I)–BaN6 structure, under the pressure of 21 GPa, the polymerization form of nitrogen is formed by the polymerization of nitrogen five-rings and one nitrogen atom, as shown in Fig. 4(d). Under the pressure of 110 GPa, the nitrogen atoms polymerize to form the N6 ring network structure22 in C2/m-BaN6, as shown in Fig. 4(c). It can be seen from the crystal structure diagram that with the increase of pressure, the degree of polymerization of nitrogen in the three structures gradually increases, indicating that pressure can cause the polymerization of nitrogen to form nitrogen polymers. Here, we mainly calculated and analyzed the performance of C2/m-BaN6 structure. The structure parameter information of C2/m-BaN6 is given in Table 1.
 | ||
| Fig. 4 (a) The relative enthalpy of formation of BaN6 with five structures under different pressures. (b) Fmmm-BaN6 crystal structure diagram. (c) C2/m-BaN6 crystal structure diagram. (d) P(I)–BaN6 crystal structure diagram. The large green balls are Ba atoms, and the small balls are nitrogen atom. | ||
We further studied the dynamic stability of C2/m-BaN6 structure, and obtained the phonon dispersion curves by theoretical calculations, and found that there was no imaginary frequency phenomenon in the entire Brillouin zone, indicating that C2/m-BaN6 structure it is dynamically stable, as shown in Fig. 5(a). Previous studies have shown that the monoclinic C2/m-BaN6 structure exists in the form of metastable state.30 In order to analyze the electronic properties of C2/m-BaN6, so we calculated the electronic band structure and projected density of states for BaN6 and we found that BaN6 is a semiconductor material, as shown in Fig. 5(b) and (c). Because Ba element has a higher ionization energy, it is easy to combine with N element, so that valence electrons can be easily transferred to the N6 ring network structure. In Fig. 5(c), it can be seen that the N-2p orbital occupies a large proportion of the entire energy map, which indicates that the N-2p orbital plays an important role in maintaining the entire crystal structure, thereby improving the stability of the overall structure and the energy storage content. We calculated the bond length between nitrogen and nitrogen in the C2/m-BaN6 structure and found that the bond length between nitrogen and nitrogen in the N6 ring network structure is between 1.37–1.40 Å. Because this values are close to the bond length of the nitrogen–nitrogen single bond,18 which indicates that the N6 ring network structure contains a large number of nitrogen and nitrogen single bonds. In the C2/m-BaN6 structure, the N atom in the N6 ring satisfies the Zintl–Klemm theory36 to achieve charge balance. Since the Ba atom loses two electrons becomes +2 valence cations and combined with the N6 ring networks, the N atoms in the N6 ring get electrons and combine with Ba showing in the form of (Bax2+–(N6)x2−). In addition, in a –(N6)x2−, due to the sp3 hybridization of the nitrogen atom, each N atom in the N6 ring forms two or three σ bonds to connect to the surrounding N atoms, so that nitrogen–nitrogen single bond is formed between N and N. Therefore, we can get that the C2/m-BaN6 structure contains a large number of nitrogen–nitrogen single bonds.
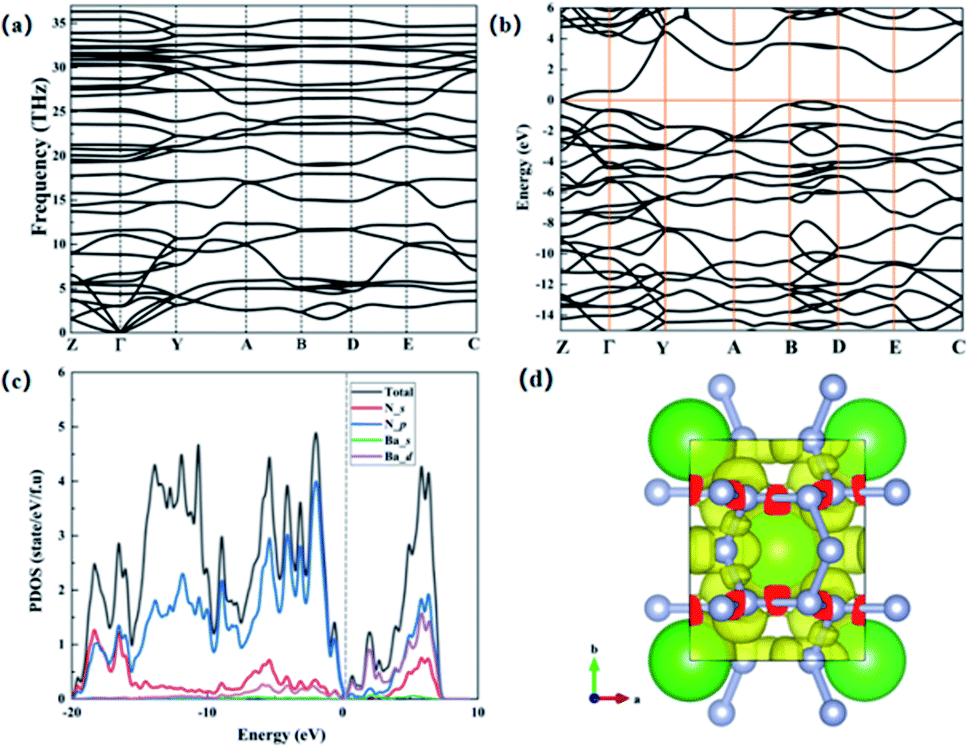 | ||
| Fig. 5 (a) The phonon spectrum of C2/m-BaN6 structure. (b) Calculated electronic band structure diagram. (c) The calculated projected density of states (PDOS) at 110 GPa. (d) BaN6 electronic local function graph. | ||
We have carried out theoretical calculations on the microscopic bonding mechanism of the C2/m-BaN6 structure, as shown in Fig. 5(d). After theoretical calculations, it is found that in the connected N6 ring network structures there are strong electronic localization and isolated electrons around N atoms. In Fig. 5(d), a nitrogen six-membered ring network structure is formed among nitrogen atoms, and they participate in bond formation in the form of sp3 hybridization, forming typical covalent bonds. It contains two types of nitrogen atoms, the three sp3 hybrid orbitals of N1 type atoms form three N–N single bonds with the one sp3 hybrid orbital of the adjacent three nitrogen atoms. The remaining one sp3 hybrid orbital is filled with isolated electrons. For every N2 type atom, the two sp3 hybrid orbitals form two σ bonds with one sp3 hybrid orbital of the surrounding two nitrogen atoms. Particularly, there are two remaining sp3 hybrid orbits are not involved in bonding, the remaining two sp3 hybrid orbitals are filled with isolated electrons. By analyzing the ELF of C2/m-BaN6 structure, we can conclude that the degree of N atoms polymerization in C2/m-BaN6 is relatively higher than other polynitrogen structures, so it has higher energy densities. Therefore, it also can be judged that nitrogen–nitrogen single bond is formed between nitrogen and nitrogen. In the previously reported article, the N-rich compounds with the N6 ring network structures have high energy density, in the ESI.† Since the N6 ring network structure contains a large number of nitrogen–nitrogen single bonds, and release a large amount of energy when N2 is generated. Therefore, the C2/m-BaN6 structure can theoretically be used as a potential high energy-density material.
4 Conclusions
This paper mainly studied the pressure-induced stability and polymeric nitrogen in alkaline earth metal N-rich nitrides (XN6, X = Ca, Sr and Ba), and the theoretical calculations were carried out by the first principles and CALYPSO theoretical prediction methods. After theoretical calculation, P-SrN6 at 22 GPa and C2/m-BaN6 at 110 GPa were predicted to be stable under the corresponding high pressure conditions. Further theoretical calculations found that the nitrogen in the P-SrN6 structure is polymerized in the form of infinite nitrogen chain, and the nitrogen in the monoclinic C2/m-BaN6 structure is polymerized in the form of an N6 ring network. Both P-SrN6 and C2/m-BaN6 structures are semiconductor materials. And in the P-SrN6 structure, the nitrogen atom is sp2 hybridized, lead to nitrogen and the nitrogen form nitrogen–nitrogen single bond and nitrogen–nitrogen double bonds coexistence status. In the C2/m-BaN6 structure, the nitrogen atom is sp3 hybridized, indicating that the nitrogen atom forms two or three σ bonds with the adjacent nitrogen atom. This electronic structure analysis can theoretically indicate that P-SrN6 and C2/m-BaN6 may be potential high energy-density materials. The research in this article is helpful to understand the potential properties of alkaline earth metal N-rich compounds under high pressure conditions, and provides a new theoretical basis for obtaining high energy-density materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2020QA059, ZR2019MA020), Shandong University of Technology Science and Technology PhD Funding.References
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC
.
- J. Uddin, V. Barone and G. E. Scuseria, Mol. Phys., 2006, 104, 745–749 CrossRef CAS
- T. M. Klapötke, in High Energy Density Materials, ed. T. M. Klapötke, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 85–121, DOI:10.1007/430_2007_057
- L. Zhang, Y. Wang, J. Lv and Y. Ma, Nat. Rev. Mater., 2017, 2, 17005 CrossRef CAS
- H. Lin, Q. Zhu, C. Huang, D.-D. Yang, N. Lou, S.-G. Zhu and H.-Z. Li, Struct. Chem., 2019, 30, 2401–2408 CrossRef CAS
- M. I. Eremets, A. G. Gavriliuk, I. A. Trojan, D. A. Dzivenko and R. Boehler, Nat. Mater., 2004, 3, 558–563 CrossRef CAS PubMed
- D. Plašienka and R. Martoňák, J. Chem. Phys., 2015, 142, 094505 CrossRef PubMed
- M. I. Eremets, R. J. Hemley, H.-k. Mao and E. Gregoryanz, Nature, 2001, 411, 170–174 CrossRef CAS PubMed
- D. Tomasino, M. Kim, J. Smith and C.-S. Yoo, Phys. Rev. Lett., 2014, 113, 205502 CrossRef PubMed
- D. Laniel, G. Geneste, G. Weck, M. Mezouar and P. Loubeyre, Phys. Rev. Lett., 2019, 122, 066001 CrossRef CAS PubMed
- F. Zahariev, S. V. Dudiy, J. Hooper, F. Zhang and T. K. Woo, Phys. Rev. Lett., 2006, 97, 155503 CrossRef CAS PubMed
- M. Bykov, S. Chariton, E. Bykova, S. Khandarkhaeva, T. Fedotenko, A. V. Ponomareva, J. Tidholm, F. Tasnádi, I. A. Abrikosov, P. Sedmak, V. Prakapenka, M. Hanfland, H.-P. Liermann, M. Mahmood, A. F. Goncharov, N. Dubrovinskaia and L. Dubrovinsky, Angew. Chem., 2020, 59, 10666 CrossRef CAS
- R. J. Bruls, H. T. Hintzen and R. Metselaar, J. Mater. Sci., 1999, 34, 4519–4531 CrossRef CAS
- X. Zhang, J. Yang, M. Lu and X. Gong, RSC Adv., 2015, 5, 21823–21830 RSC
- J. Jiang, P. Zhu, D. Li, Y. Chen, M. Li, X. Wang, B. Liu, Q. Cui and H. Zhu, J. Phys. Chem. B, 2016, 120, 12015–12022 CrossRef CAS PubMed
- C. Ji, F. Zhang, D. Hou, H. Zhu, J. Wu, M.-C. Chyu, V. I. Levitas and Y. Ma, J. Phys. Chem. Solids, 2011, 72, 736–739 CrossRef CAS
- S. Yu, B. Huang, Q. Zeng, A. R. Oganov, L. Zhang and G. Frapper, J. Phys. Chem. C, 2017, 121, 11037–11046 CrossRef CAS
- F. Peng, Y. Yao, H. Liu and Y. Ma, J. Phys. Chem. Lett., 2015, 6, 2363–2366 CrossRef CAS PubMed
- S. Zhu, F. Peng, H. Liu, A. Majumdar, T. Gao and Y. Yao, Inorg. Chem., 2016, 55, 7550–7555 CrossRef CAS PubMed
- K. Xia, X. Zheng, J. Yuan, C. Liu, H. Gao, Q. Wu and J. Sun, J. Phys. Chem. C, 2019, 123, 10205–10211 CrossRef CAS
- J. Zhang, A. R. Oganov, X. Li and H. Niu, Phys. Rev. B, 2017, 95, 020103 CrossRef
- X. Wang, Y. Wang, M. Miao, X. Zhong, J. Lv, T. Cui, J. Li, L. Chen, C. J. Pickard and Y. Ma, Phys. Rev. Lett., 2012, 109, 175502 CrossRef PubMed
- K. Ramesh Babu, C. Bheema Lingam, S. P. Tewari and G. Vaitheeswaran, J. Phys. Chem. A, 2011, 115, 4521–4529 CrossRef CAS PubMed
- K. Ramesh Babu and G. Vaitheeswaran, Chem. Phys. Lett., 2012, 533, 35–39 CrossRef CAS
- M. Zhang, K. Yin, X. Zhang, H. Wang, Q. Li and Z. Wu, Solid State Commun., 2013, 161, 13–18 CrossRef CAS
- X. Wang, J. Li, N. Xu, H. Zhu, Z. Hu and L. Chen, Sci. Rep., 2015, 5, 16677 CrossRef PubMed
- X. Wang, J. Li, H. Zhu, L. Chen and H. Lin, J. Chem. Phys., 2014, 141, 044717 CrossRef PubMed
- S. Wei, D. Li, Z. Liu, W. Wang, F. Tian, K. Bao, D. Duan, B. Liu and T. Cui, J. Phys. Chem. C, 2017, 121, 9766–9772 CrossRef CAS
- S. Wei, L. Lian, Y. Liu, D. Li, Z. Liu and T. Cui, Phys. Chem. Chem. Phys., 2020, 22, 5242–5248 RSC
- B. Huang and G. Frapper, Chem. Mater., 2018, 30, 7623–7636 CrossRef CAS
- J. Lin, D. Peng, Q. Wang, J. Li, H. Zhu and X. Wang, Phys. Chem. Chem. Phys., 2021, 23, 6863–6870 RSC
- L. Wu, R. Tian, B. Wan, H. Liu, N. Gong, P. Chen, T. Shen, Y. Yao, H. Gou and F. Gao, Chem. Mater., 2018, 30, 8476–8485 CrossRef CAS
- Z. Liu, D. Li, F. Tian, D. Duan, H. Li and T. Cui, Inorg. Chem., 2020, 59, 8002–8012 CrossRef CAS PubMed
- N. Yedukondalu, G. Vaitheeswaran, P. Modak and A. K. Verma, Solid State Commun., 2019, 297, 39–44 CrossRef CAS
- X. Du, Y. Yao, J. Wang, Q. Yang and G. Yang, J. Chem. Phys., 2021, 154, 054706 CrossRef CAS PubMed
- F. Wang and G. J. Miller, Inorg. Chem., 2011, 50, 7625–7636 CrossRef CAS PubMed
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS
- Q. Tong, J. Lv, P. Gao and Y. Wang, Chin. Phys. B, 2019, 28, 106105 CrossRef CAS
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Phys. Rev. Lett., 2011, 106, 015503 CrossRef PubMed
- B. Delley, J. Phys.: Condens. Matter, 2010, 22, 384208 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- X. Blanc, É. Cancès and M.-S. Dupuy, C. R. Math., 2017, 355, 665–670 CrossRef
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef
- S. D. Dabhi and P. K. Jha, Polymer, 2015, 81, 45–49 CrossRef CAS
- K. Xia, J. Yuan, X. Zheng, C. Liu, H. Gao, Q. Wu and J. Sun, J. Phys. Chem. Lett., 2019, 10, 6166–6173 CrossRef CAS PubMed
- B. Adivaiah, E. Narsimha Rao and G. Vaitheeswaran, J. Phys.: Condens. Matter, 2019, 31, 475402 CrossRef CAS PubMed
- G. Vaitheeswaran and K. R. Babu, J. Chem. Sci., 2012, 124, 1391–1398 CrossRef CAS
- D. Laniel, B. Winkler, E. Koemets, T. Fedotenko, M. Bykov, E. Bykova, L. Dubrovinsky and N. Dubrovinskaia, Nat. Commun., 2019, 10, 4515 CrossRef PubMed
- W. C. Butterman and R. G. Reese Jr, Mineral Commodity Profiles -- Rubidium, Report 2003-45, 2003 Search PubMed
- H. Zhu, X. Han, P. Zhu, X. Wu, Y. Chen, M. Li, X. Li and Q. Cui, J. Phys. Chem. C, 2016, 120, 12423–12428 CrossRef CAS
- A. A. L. Michalchuk, S. Rudić, C. R. Pulham and C. A. Morrison, Phys. Chem. Chem. Phys., 2018, 20, 29061–29069 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01631h |
| This journal is © The Royal Society of Chemistry 2021 |