
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of triethoxysilylated cyclen derivatives, grafting on magnetic mesoporous silica nanoparticles and application to metal ion adsorption†
Hao Li‡
ab,
Mathilde Ménard‡ a,
Ani Vardanyanc,
Clarence Charnaya,
Laurence Raehma,
Erwan Olivieroa,
Gulaim A. Seisenbaeva*c,
Roser Pleixats*b and
Jean-Olivier Durand*a
a,
Ani Vardanyanc,
Clarence Charnaya,
Laurence Raehma,
Erwan Olivieroa,
Gulaim A. Seisenbaeva*c,
Roser Pleixats*b and
Jean-Olivier Durand*a
aICGM, Univ. Montpellier, CNRS, ENSCM, Case 1701, Place Eugène Bataillon, CEDEX 05, 34095 Montpellier, France. E-mail: jean-olivier.durand@umontpellier.fr
bDepartment of Chemistry and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universitat Autònoma de Barcelona, 08193-Cerdanyola del Vallès, Barcelona, Spain. E-mail: Roser.Pleixats@uab.cat
cDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, 750 07 Uppsala, Sweden. E-mail: Gulaim.Seisenbaeva@slu.se
First published on 12th March 2021
Abstract
The synthesis through click chemistry of triethoxysilylated cyclen derivative-based ligands is described. Different methods were used such as the copper catalyzed Huisgen's reaction, or thiol–ene reaction for the functionalization of the cyclen scaffold with azidopropyltriethoxysilane or mercaptopropyltriethoxysilane, respectively. These ligands were then grafted on magnetic mesoporous silica nanoparticles (MMSN) for extraction and separation of Ni(II) and Co(II) metal ions from model solutions. The bare and ligand-modified MMSN materials revealed high adsorption capacity (1.0–2.13 mmol g−1) and quick adsorption kinetics, achieving over 80% of the total capacity in 1–2 hours.
Introduction
The anchoring of cyclen derivatives on silica-based materials has attracted attention over the last two decades. The main application concerns magnetic resonance imaging1–9 or radiolabelling10,11 with metal-DOTA-functionalized silica-based nanoparticles for theranostic applications, but not only. Other applications relate to ESR studies with Cu(II),12 or Cu(II) catalysis,13 Pd(II) recovery,14 or photoresponsive materials with Eu(III) or Tb(III).15,16 Therefore applications of silica-based materials functionalized with cyclens are very diverse, promising, and present a high interest. Two ways are used for the functionalization of silica materials with cyclen derivatives. The first one consists of anchoring a reactive function such as a chloro or amino group on silica using silanes such as chloro or aminopropyltriethoxysilane and then to react this function with an activated acid of DOTA derivative for aminopropyltriethoxysilane or with an amino of cyclen derivative for chloropropyltriethoxysilane. The drawback of this strategy is that the coupling reaction is usually not quantitative and the remaining reactive function on the surface of the material is not completely functionalized. The second possibility consists in preparing triethoxysilylated cyclen derivatives and then coupling the derivatives on the surface of the materials. The preparation involves the coupling reaction of silanes (iodopropyltriethoxysilane, aminopropyltriethoxysilane) with cyclen or activated DOTA derivatives.Magnetic mesoporous silica nanoparticles (MMSN) have gained much attention the last decade for bio-application, sensing or catalysis and the field has been comprehensively reviewed.17–22 In the course of our studies on functionalized MMSN for metal complexation,23 we were interested in the preparation of triethoxysilylated cyclen-based ligands, through click Cu-catalyzed azide–alkyne Huisgen's reaction24 with azidopropyltriethoxysilane or click thiol–ene reaction25 with mercaptopropyltriethoxysilane, which have not been investigated for this purpose. The cyclen derivatives were then grafted on the surface of MMSN and the materials were used for the extraction and separation of Ni(II) and Co(II) ions.
Experimental
Materials and methods
Cyclen (CAS 294-90-6, Fluorochem), di-tert-butyl dicarbonate (CAS 24424-99-5, Sigma Aldrich), allyl bromide (CAS 106-95-6, Sigma Aldrich), propargyl bromide (CAS 106-96-7, Fluorochem), (3-mercaptopropyl)triethoxysilane (CAS 4420-74-0, Sigma Aldrich), (3-azidopropyl)triethoxysilane (CAS 83315-69-9, Abcr GmbH), iron oxide (CAS 20344-49-4, Sigma Aldrich), oleic acid (CAS 112-80-1, Sigma Aldrich), n-docosane (CAS 629-97-0, Acros), oleylamine (CAS 112-90-3, Acros), tetraethyl orthosilicate TEOS (CAS 78-10-4, Sigma Aldrich), cetyltrimethylammonium bromide CTAB (CAS 57-09-0, Sigma Aldrich), pentane (CAS 109-66-0, Fisher), chloroform (CAS 67-66-3, Sigma Aldrich), trifluoroacetic acid (CAS 76-05-1, Fluorochem), triethylamine (CAS 121-44-8, Merck), dichloromethane (CAS 76-09-2, Carlo Erba), ethyl acetate (CAS 141-78-6, VWR), ethanol and acetone (Honeywell).FTIR spectra were recorded in the 4000–400 cm−1 range using 32 scans at a nominal resolution of 4 cm−1 with a Perkin Elmer 100 FT spectrophotometer equipped with an ATR unit. TEM images were recorded with JEOL 1200 EXII microscope and an JEOL 2200 FS (JEOL Europe SAS, Croissy Sur Seine, France). For the purpose of TEM analysis, the sample particles were dispersed in ethanol and then dropped onto copper grids covered with porous carbon films. TGA analyses were performed with a thermal analyser STA 409 Lux® (Netzsch) in the range 25–800 °C by a heating speed of 5 °C min−1.
Preparation of MMSN
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (2:1, v/v) to the as-synthesized product; (2) by adding 2 mL of pentane and 30 mL of a mixture of ether:ethanol (2:1, v/v) to the collected black product; (3) by adding 30 mL of a mixture of ether:ethanol (1:1, v/v) to the collected black product. Finally, the MIONs were stored in 15 mL of chloroform and stabilized by adding 200 μL of oleylamine.
:ethanol (2:1, v/v) to the as-synthesized product; (2) by adding 2 mL of pentane and 30 mL of a mixture of ether:ethanol (2:1, v/v) to the collected black product; (3) by adding 30 mL of a mixture of ether:ethanol (1:1, v/v) to the collected black product. Finally, the MIONs were stored in 15 mL of chloroform and stabilized by adding 200 μL of oleylamine.Synthesis of ligand
Preparation of MMSN grafted with Ln-Boc/tBu
In a 50 mL round-bottom flask equipped with a Dean–Stark apparatus, ligand Ln-Boc/tBu (0.1 mmol), MMSN (100 mg), H2O (500 μL) and Et3N (25 μL) were sonicated for 2 h, and then the mixture was heated in DMF (10 mL) at 120 °C for 24 h. After this time, the suspension was centrifugated (20000 rpm at 25 °C for 15 min). The solid was washed successively with H2O (2 × 30 mL), ethanol (2 × 30 mL) and acetone (2 × 30 mL) (sonication for 10 min, 10 min for centrifugation), dried under vacuum, and finally crushed to give the grafted material MMSN-Ln-Boc/tBu as black solid.
Preparation of MMSN-Ln
The MMSN-Ln-Boc/tBu were dissolved in TFA/CH2Cl2 (5:1) solution, the mixture was stirred at room temperature for 60 minutes. Then the NPs were collected by centrifugation (20000 rpm at 25 °C for 10 min). The material MMSN-L1 was washed successively with EtOH (2 × 30 mL), saturated NaHCO3 solution (2 × 30 mL), distilled water (2 × 30 mL), acetone (2 × 30 mL) and EtOH (2 × 30 mL). The material MMSN-L2 was washed successively with EtOH (2 × 30 mL), acetone (2 × 30 mL) and EtOH (2 × 30 mL). Finally, both MMSN-Ln-were dried under vacuum for several hours.
For isotherm experiments stock solutions of Ni2+ and Co2+ (50 mM) were prepared using nitrate salts and the final concentrations were adjusted by dilution with Milli-Q water (varying between 0.5–25 mM). Samples (10 mg) of magnetic nanoparticles (MNPs) were mixed with 10 mL of metal cation solution in plastic tubes of 50 mL and put on a shaker for 24 hours. After each experiment, MNPs were centrifuged (7000g) for 10 min and an aliquot (1 mL) was separated to determine the metal concentration in the remaining solution. The samples were first diluted 5–10 times (depending on the initial metal concentration) and titrated afterwards with EDTA using xylenol orange as an indicator. For each sample, the titrations were repeated 3 times, and the average concentration was calculated.
The uptake of metal cation by the magnetic nanoparticles was calculated according to the equation:
| Up = (C0 − Ce) × V/m |
For kinetic tests, the magnetic nanoadsorbents (40 mg) were mixed with 40 mL of metal cation solutions (with 10 mM initial metal concentration) and the uptake was measured after set interval of times. For that purpose, an aliquot of 1 mL was separated, diluted 10 times and the remaining metal concentration was evaluated by titration with EDTA and xylenol orange.
For selectivity test, mixed metal solution was prepared with final concentration of 5 mM + 5 mM Ni and Co ions. The overall concentration was measured by titration with EDTA and xylenol orange as an indicator. For adsorption experiment, 20 mg magnetic nanoparticles were mixed with 20 mL metal solution in 50 mL falcon tubes, and the tubes were put on a shaker for 24 hours. After 24 hours, the nanoparticles were separated from metal solution by centrifugation (7000g) and the remaining metal content was measured by titration (Table 2). The magnetic nanoparticles were dried under nitrogen atmosphere and the adsorbed metal distribution was determined by EDS analysis.
For the study of stability and reuse, 30 mg of each nanoparticle samples were mixed in 30 mL metal solution with 10 mM metal ion concentration for 24 hours. Afterwards, the solutions with nanoparticles were centrifuged (7000 rpm) and the metal content was estimated by titrations with EDTA and xylenol orange as an indicator. The desorption was carried out by adding 30 mL nitric acid (1 mM) and put on a shaker for 24 hours. Nanoparticles were centrifuged for 15 min (7000 rpm) and the amount of desorbed metal ions was estimated by titrations with EDTA. Afterwards, the nanoparticle samples were washed with MilliQ water and second round of adsorption experiment was carried out, with the same metal ion concentration and volume. Adsorption and desorption experiments were repeated 3 times.
Results and discussion
Ligand L1-Boc was synthesized in quantitative yield by a copper-catalyzed alkyne–azide cycloaddition reaction (CuAAC) of the alkyne 128,29 with commercial (3-azidopropyl)triethoxysilane under anhydrous conditions,30 using CuBr(PPh3)3 as catalyst in dry THF at 60 °C (Scheme 1).
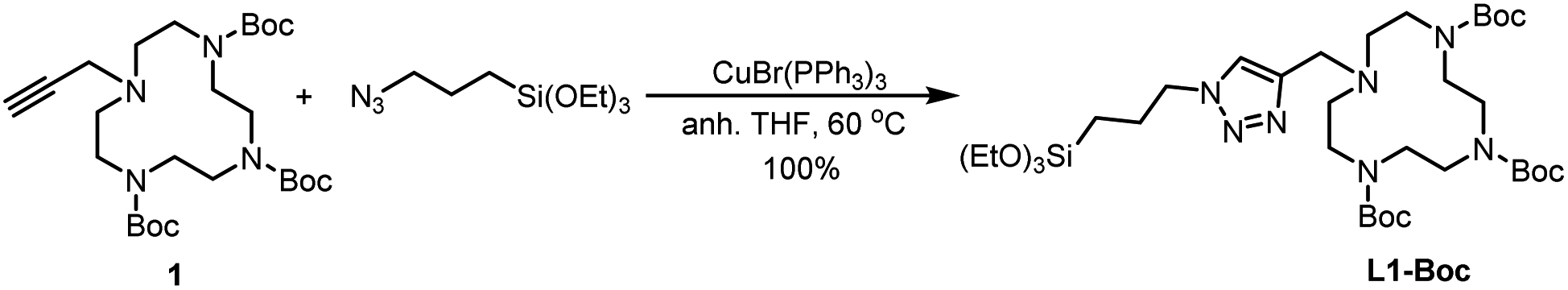 | ||
| Scheme 1 Synthesis of L1-Boc. | ||
Ligand L2-tBu was obtained through a thiol–alkene click reaction between the suitable alkene and 3-mercaptopropyltriethoxysilane. The synthesis of L2-tBu is summarized in Scheme 2. The commercial cyclen 2 was treated with three equivalents of tert-butyl 2-bromoacetate in the presence of sodium acetate to afford compound 3,31 which was reacted with allyl bromide in acetonitrile at room temperature in the presence of potassium carbonate to give 4 in an overall 60% yield from 2. The alkene 4 and (3-mercaptopropyl)triethoxysilane were irradiated at 365 nm using 2,2-dimethoxy-1,2-diphenylethanone (DMPA) as initiator, and the desired ligand L2-tBu was obtained in quantitative yield.
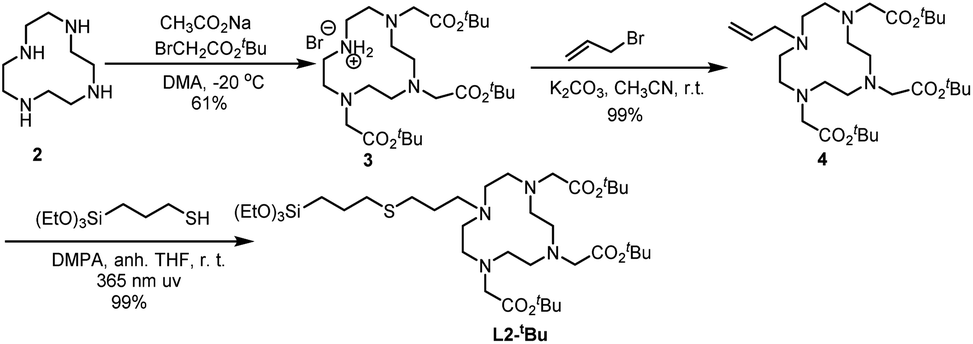 | ||
| Scheme 2 Synthesis of L2-tBu. | ||
Through these two synthesis routes, two new cyclen-derivatives that are, to the best of our knowledge, not described in the literature, were successfully synthesized. Nevertheless, these syntheses, although successful, have reaction times lasting several days. Thus, a modification of the synthesis procedure was explored to synthesize L1-Boc by click chemistry under microwaves, which allows to obtain the molecule in less than 1 h versus 48 hours through standard protocol. Briefly, precursor 1, the silylated azide and 6 mol% of catalyst were dissolved in dry THF and put in the microwaves at 100 °C, 200 Wmax, for 3 cycles of 10 min. The obtaining of the right molecule was assessed through HNMR analysis. Since the microwave approach was successful to synthesize L1-Boc the protocol was adapted for the synthesis of the derivative named L3-tBu, which could not be obtained through standard approaches. Thereby precursor 5 (see ESI† for the synthesis of 5),32 the silylated azide and 12 mol% of catalyst were dissolved in dry THF and stirred under microwaves at 100 °C (200 Wmax) for 4 cycles of 10 min (Scheme 3). This new cyclen derivative was then identified by 1H NMR, 13C NMR, 29Si NMR and HRMS. During the experiments, to set up the best protocols for microwave assisted synthesis of L1-Boc and L3-tBu two key parameters were identified: the catalyst amount and the number of 10 min cycles. Indeed, it was observed that with less catalyst the reaction did not occur fully and a mixture of starting and expected molecules was obtained. In addition, increasing the reaction time (6 cycles of 10 min for example) tended to damage the molecules: the 1H NMR signal at 2.63 ppm (4H) for L1-Boc and at 2.59 ppm (4H) for L3-tBu representative of the cyclen were missing.
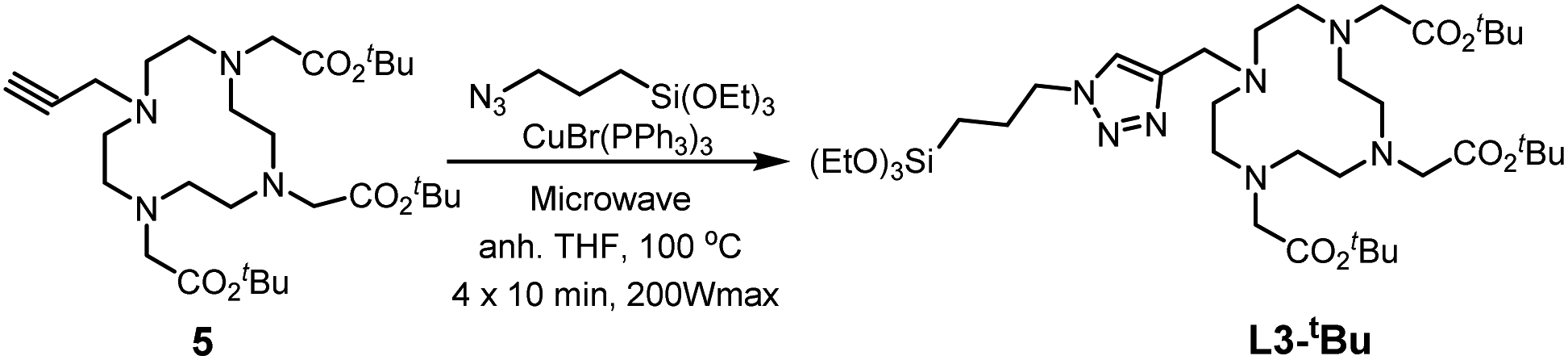 | ||
| Scheme 3 Microwave assisted synthesis of L3-tBu. | ||
Thus, after establishing the best protocol, microwaves allowed the efficient synthesis of L1-Boc and L3-tBu and represented a promising approach toward simple, cheaper and faster cyclen derivatives synthesis.
Core–shell nanoparticles were synthesised using ∼20 nm iron oxide cores obtained through thermal decomposition. A silica mesoporous shell was then grown around the core. These nanoparticles exhibited an overall diameter of 102 ± 4 nm by TEM images (Fig. 1a and b), and HRTEM showed the mesoporosity (Fig. 1c and d). The mesoporosity was further confirmed with N2 adsorption–desorption (BET) (Fig. 1e and f). MMSN showed type I–IV isotherm characteristic of micro–mesoporous materials. The specific surface area was 302 m2 g−1 and pore diameters (DFT method) showed two populations at 1.3 nm (microporosity) and a larger pic at 2.5 nm (mesoporosity). TEM was in agreement with the average hydrodynamic diameter (183 nm) of the nanoparticles determined by DLS (Fig. S4a†). These nanoparticles were also analyzed by zeta potential, TGA and IR (Fig. S4b–S6†).
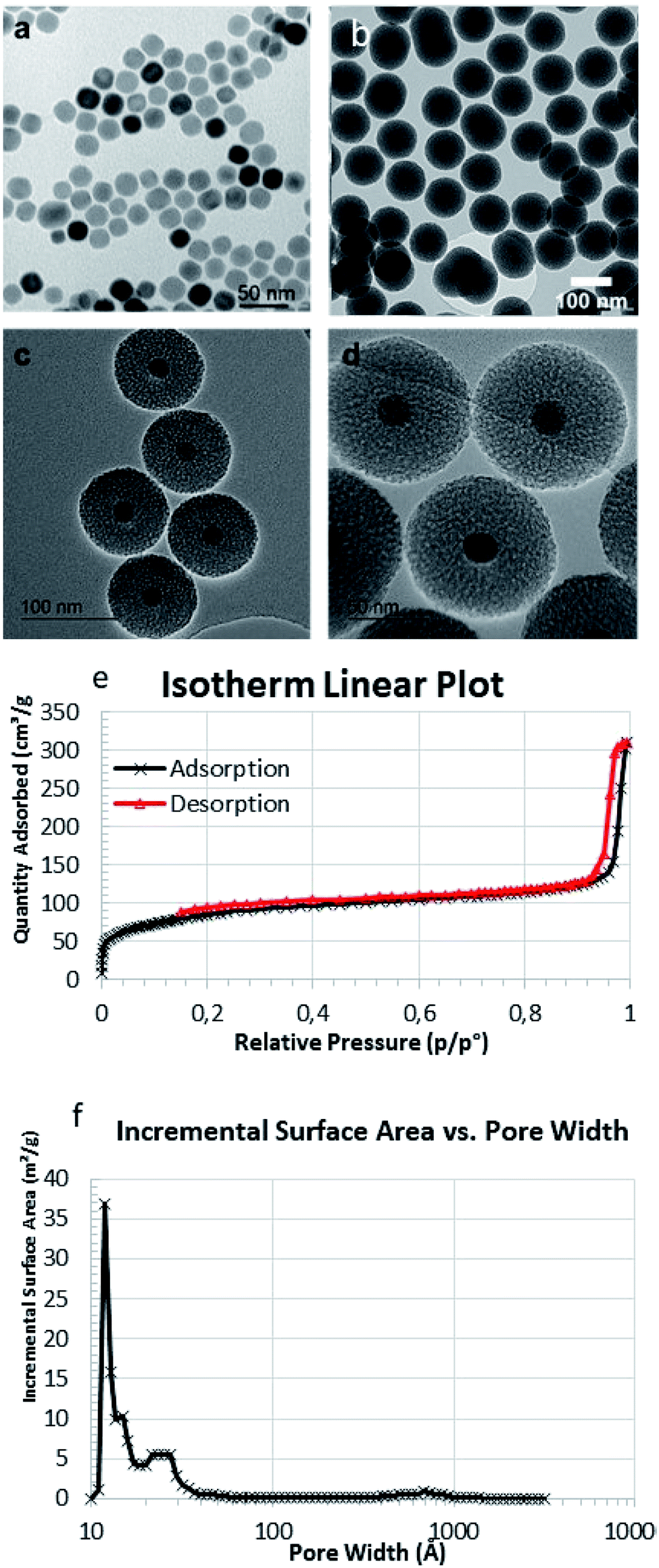 | ||
| Fig. 1 TEM image of (a) MIONs and (b) MMSN, (c and d) HRTEM images of MMSN showing the mesoporosity. (e and f) N2 adsorption–desorption analysis (BET) and pore diameters (DFT) of MMSN. | ||
Then ligands (L1-Boc and L2-tBu) were used to modify the surface of MMSN by post-grafting (Fig. 2). We initially investigated different grafting conditions with ligand L2-tBu, and the reaction was monitored by FTIR (γC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1657 cm−1) (see Table S1 and Fig. S7 in ESI†). In refluxing toluene no reaction occurred. The best conditions were established as follows: in DMF at 120 °C with 2 mmol L2-tBu per g MMSN, using water and Et3N as catalysts. The same procedure was used with L1-Boc. The BOC and t-Bu groups were then classically cleaved using TFA to lead to MMSN-L1 and MMSN-L2, respectively. To further assess and estimate the surface functionalisation the nanoparticles were characterised by DLS (Fig. S8†), zeta potential (Fig. S9†), FTIR (Fig. S10 and S11†), TGA (Fig. S12†), and elemental analysis. The data are summarised in Table 1.
O 1657 cm−1) (see Table S1 and Fig. S7 in ESI†). In refluxing toluene no reaction occurred. The best conditions were established as follows: in DMF at 120 °C with 2 mmol L2-tBu per g MMSN, using water and Et3N as catalysts. The same procedure was used with L1-Boc. The BOC and t-Bu groups were then classically cleaved using TFA to lead to MMSN-L1 and MMSN-L2, respectively. To further assess and estimate the surface functionalisation the nanoparticles were characterised by DLS (Fig. S8†), zeta potential (Fig. S9†), FTIR (Fig. S10 and S11†), TGA (Fig. S12†), and elemental analysis. The data are summarised in Table 1.
 | ||
| Fig. 2 Cyclen functionalized magnetic mesoporous core–shell silica nanoparticles (MMSN). | ||
| Sample | DLSa (nm) | pH | Zeta potential (mV) | EAb (mmol g−1) | TGAc (wt%) |
|---|---|---|---|---|---|
| a Hydrodynamic diameters from DLS.b EA: elemental analysis, experimental value of N content.c Determined by mass loss with TGA (a heating rate of 5 °C min−1 from 30 to 800 °C).d Not determined. | |||||
| MMSN | 183 | 7.24 | −32 | — | Reference |
| MMSN-L1-Boc | 216 | 6.85 | −6 | ndd | 5.31 wt% |
| MMSN-L1 | nd | 6.99 | 39 | 0.15 | 4.34 wt% |
| MMSN-L2-tBu | 237 | 6.75 | −14 | nd | 7.76 wt% |
| MMSN-L2 | nd | 6.65 | −36 | 0.18 | 7.19 wt% |
DLS showed a slight increase of the hydrodynamic diameters from MMSN to MMSN-L1-Boc and MSN-L2-tBu in agreement with a successful grafting of the ligand on the surface of MMSN (Fig. S8†). This also indicates that the NPs keep their colloidal stability in water even after their surface modification. The grafting was also confirmed by zeta potential measurements performed around neutral pH 7. From raw NPs to grafted NPs, the zeta-potential increased due to the presence of organic coating (Fig. S9†). After TFA treatment, zeta potential became highly positive with MMSN-L1 in agreement with the presence of ammoniums at neutral pH whereas a strong decrease was noticed with MMSN-L2 as carboxylate groups were deprotected. The high zeta-potential positive value for MMSN-L1 and negative one for MMSN-L2 also indicate a good colloidal stability of these nano-objects at a neutral pH. Finally, the grafting content was determined by TGA analysis (Fig. S11†) and elemental analysis (Table 1). TGA analysis showed a grafting of 4.34 wt% for MMSN-L1 which corresponds to a grafting of 0.12 mmol g−1 and 7.19 wt% for MMSN-L2 which corresponds to a grafting of 0.15 mmol g−1. These results were confirmed by elemental analysis on N which indicated a grafting of 0.15 and 0.18 mmol g−1 for MMSN-L1 and MMSN-L2, respectively.
The prepared magnetic core–shell silica nanoparticles were studied for the adsorption of Ni2+ and Co2+ from aqueous solutions at room temperature.
All the magnetic nanoparticles showed similar maximum adsorption capacity (varying between 1–2.13 mmol g−1), MMSN having the lowest capacity for both heavy metals (Table 2). For both heavy metals, adsorption isotherms were best fit with Langmuir curves model (Fig. 3), having higher correlation coefficients compared to Freundlich (Fig. S13†), indicating similar mechanisms with uniform binding sites in a monolayer adsorption.
| Entry | Sample | Ni | Co | Ni:Co = 1:1 |
|---|---|---|---|---|
| 1 | MMSN | 1.00 | 1.00 | 1.01 |
| 2 | MMSN-L1 | 2.13 | 2.00 | 0.997 |
| 3 | MMSN-L2 | 1.16 | 1.33 | 0.94 |
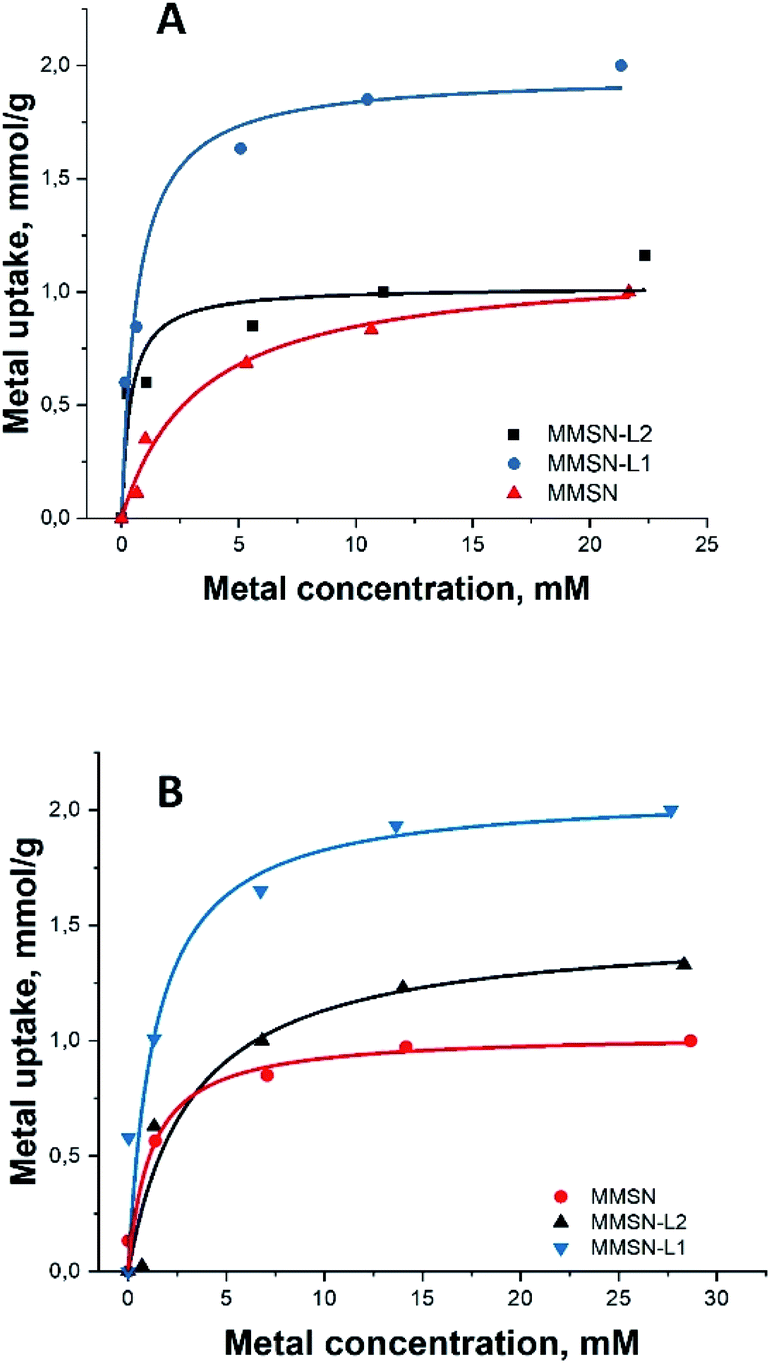 | ||
| Fig. 3 Metal adsorption isotherms: (A and B) Langmuir isotherms for Ni2+ and Co2+, respectively (for Freundlich isotherms for Ni2+ and Co2+, respectively, please, see ESI Fig. S13†). | ||
Note that the measurements for the isotherm and the kinetics were carried out under different conditions. Final concentrations in the measurements of isotherms were considerably higher (25 mM) along with lower amount of the applied adsorbent (10 mg) to achieve saturation (Fig. 3). In case of kinetic measurements the solution was much less concentrated (10 mM), while the amount of adsorbent was, in contrast, higher in order to distinguish the first steps in adsorption. The saturation of the adsorbent was not achieved in the kinetic measurements (Fig. 4).
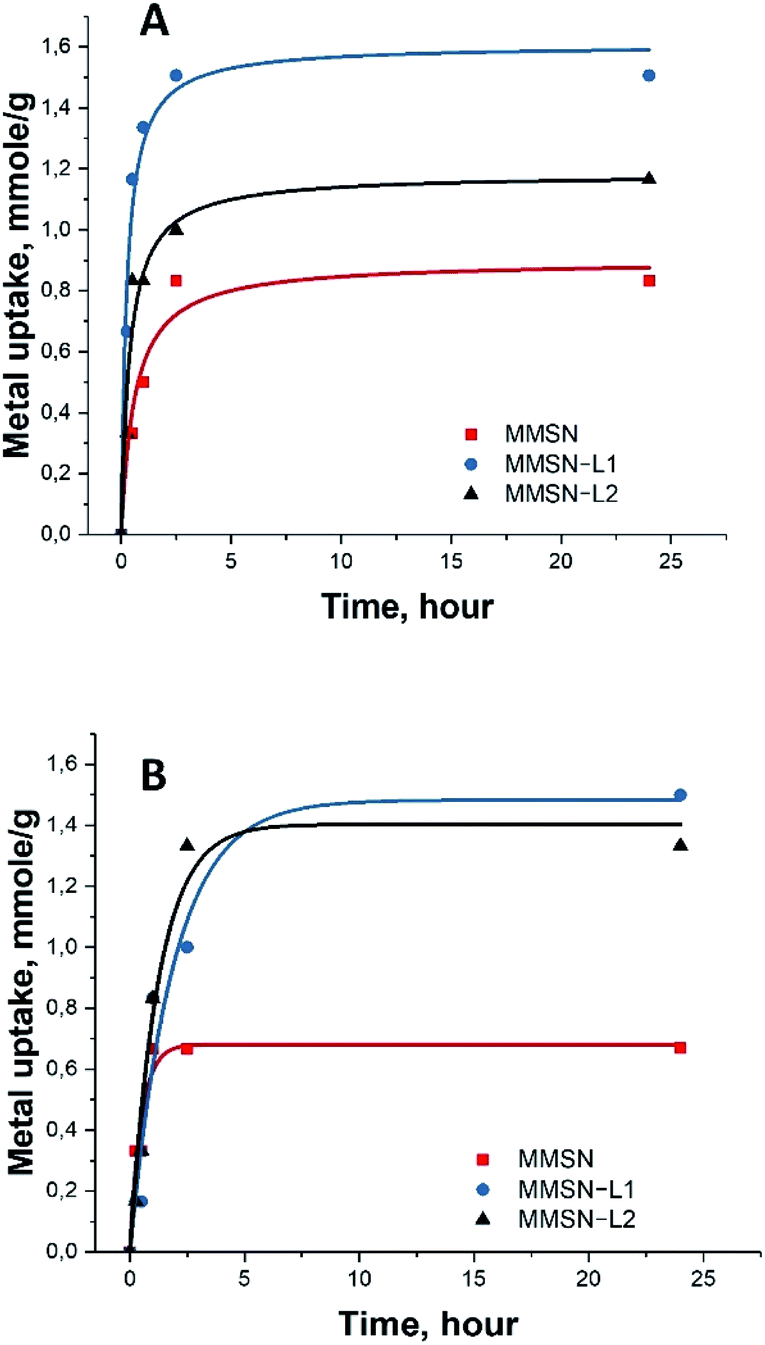 | ||
| Fig. 4 Adsorption kinetics of two heavy metals for (A) nickel(II), (B) cobalt(II). | ||
The kinetic test results showed fast uptake at the first 1–2 hours of metal interaction with magnetic nanoparticles, reaching over 80% of the total capacity. Slower uptake was found after these 2 hours and the equilibrium was reached after 5 hours (Fig. 4). This indicated quick and unhindered diffusion through the mesopores.
It is interesting to note that bare, not surface modified MMSN display high capacity in uptake of both Ni(II) and Co(II) cations, most probably due to their very high negative surface charge as manifested by strong zeta-potential (−32 mV). Particles modified by L2, acting presumably as a predominantly hard base in Pearson classification, display almost the same capacity as bare silica. This can be explained by some consumption and shielding of the silicate functions after grafting of the cyclen functions, which is compensated by the carboxylate ones in reactivity. On the contrary, the MMSN modified by L1, a soft base in Pearson classification reveal strongly enhanced adsorption characteristics, indicating that the ligand is acting for heavy metal cations as an azo-crown functionality forming stable surface complexes with soft Pearson acids such as Ni(II) and Co(II).
From the results of the selectivity tests with Ni:Co = 1:1 mixture it is apparent that magnetic nanoparticles have lower adsorption capacity when introduced to mixed metal solution. This could be explained by possible competition for the adsorption sites between the two metal ions, making it more difficult for the ligand to bind the metals. Ni2+ and Co2+ cations can possess different coordination geometry when binding to soft ligands in Pearson classification. Ni2+ cations are prone to Jahn–Teller distortion while Co2+ usually display undistorted octahedral coordination. On the contrary, MMSN nanoparticles without any ligands revealed similar results with previous experiments (with single metals only), showing that there is no competition between Ni and Co without any ligands attached to nanoparticles.
EDS analysis gave similar results in regards with selectivity of magnetic nanoparticles (see ESI, Tables S2–S5†). The numbers for L2 and MMSN for contents of both metals are rather similar and in general relatively low, being approximately twice higher for L1. This ratio is reflecting the data on total capacity of the materials (see Fig. 3). This indicates that the enhanced surface of silica itself makes also a strong contribution to adsorption along with complexation to ligands.
Recyclability of the materials was studied, testing the uptake and release capacity in 4 consecutive adsorption–desorption cycles. The results, summarised in Fig. S14,† show unaltered functional characteristics for ligand-free reference material MMSN and functionalized MMSN-L1. In the case of MMSN-L2 it was, however, possible to see some loss of capacity.
Conclusions
In summary, click chemistry such as thiol–ene or Huisgen's reaction was very efficient for the syntheses of cyclen-based ligands possessing triethoxysilyl group. Associated with microwaves, Huisgen's reaction showed successful preparation of challenging L3-tBu ligand. L1-Boc and L2-tBu ligands were grafted on magnetic mesoporous silica nanoparticles (MMSN) and protective groups were cleaved in order to extract and separate metal ions Ni(II) and Co(II) from model solutions. The kinetic of metal(II) uptake was very quick reaching 80% of the capacity after 1–2 hours. The adsorption capacity of MMSN was very high, up to 2.13 mmol g−1 of Ni(II) with ligand L1 grafted on MMSN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by ERA-MIN 2 MetRecycle project supported in Sweden by Research Council Vinnova grant no. 2018-00739, in France by ADEME, Ministerio de Ciencia, Innovación y Universidades (MICIU) of Spain (Project RTI2018-097853-B-I00 and RED2018-102387-T), DURSI-Generalitat de Catalunya (Project SGR2017-0465) and China Scholarship Council (CSC) (H. L., No. 201606890025).References
- V.-L. Tran, V. Thakare, F. Rossetti, A. Baudouin, G. Ramniceanu, B.-T. Doan, N. Mignet, C. Comby-Zerbino, R. Antoine, P. Dugourd, F. Boschetti, F. Denat, C. Louis, S. Roux, T. Doussineau, O. Tillement and F. Lux, J. Mater. Chem. B, 2018, 6, 4821–4834 RSC.
- V. Rao N, H. S. Han, H. Lee, V. Q. Nguyen, S. Jeon, D.-W. Jung, J. Lee, G.-R. Yi and J. H. Park, Nanoscale, 2018, 10, 9616–9627 RSC.
- S. Sinha, W. Y. Tong, N. H. Williamson, S. J. P. McInnes, S. Puttick, A. Cifuentes-Rius, R. Bhardwaj, S. E. Plush and N. H. Voelcker, ACS Appl. Mater. Interfaces, 2017, 9, 42601–42611 CrossRef CAS PubMed.
- V. S. Marangoni, O. Neumann, L. Henderson, C. C. Kaffes, H. Zhang, R. Zhang, S. Bishnoi, C. Ayala-Orozco, V. Zucolotto, J. A. Bankson, P. Nordlander and N. J. Halas, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6960–6965 CrossRef CAS PubMed.
- C. Guo, J. Hu, A. Bains, D. Pan, K. Luo, N. Li and Z. Gu, J. Mater. Chem. B, 2016, 4, 2322–2331 RSC.
- T. Nakamura, H. Matsushita, F. Sugihara, Y. Yoshioka, S. Mizukami and K. Kikuchi, Angew. Chem., Int. Ed., 2015, 54, 1007–1010 CrossRef CAS PubMed.
- F. Carniato, M. Muñoz-Úbeda, L. Tei and M. Botta, Dalton Trans., 2015, 44, 17927–17931 RSC.
- J. J. Davis, W.-Y. Huang and G.-L. Davies, J. Mater. Chem., 2012, 22, 22848–22850 RSC.
- H. Benachour, A. Seve, T. Bastogne, C. Frochot, R. Vanderesse, J. Jasniewski, I. Miladi, C. Billotey, O. Tillement, F. Lux and M. Barberi-Heyob, Theranostics, 2012, 2, 889–904 CrossRef CAS PubMed.
- X. Zhang, F. Chen, M. Z. Turker, K. Ma, P. Zanzonico, F. Gallazzi, M. A. Shah, A. R. Prater, U. Wiesner, M. S. Bradbury, M. R. McDevitt and T. P. Quinn, Biomaterials, 2020, 241, 119858 CrossRef CAS PubMed.
- S.-H. Cheng, D. Yu, H.-M. Tsai, R. A. Morshed, D. Kanojia, L.-W. Lo, L. Leoni, Y. Govind, L. Zhang, K. S. Aboody, M. S. Lesniak, C.-T. Chen and I. V. Balyasnikova, J. Nucl. Med., 2016, 57, 279–284 CrossRef CAS PubMed.
- C. Gros, F. Rabiet, F. Denat, S. Brandès, H. Chollet and R. Guilard, J. Chem. Soc., Dalton Trans., 1996, 1209–1214, 10.1039/DT9960001209.
- H. F. Bereczki, L. Daróczi, I. Fábián and I. Lázár, Microporous Mesoporous Mater., 2016, 234, 392–400 CrossRef CAS.
- F. Wu, G. Ye, R. Yi, T. Sun, C. Xu and J. Chen, Dalton Trans., 2016, 45, 9553–9564 RSC.
- B. Yan and Q.-P. Li, Microporous Mesoporous Mater., 2014, 196, 284–291 CrossRef CAS.
- E. Delgado-Pinar, M. T. Albelda, J. C. Frías, O. Barreiro, E. Tejera, V. Kubíček, L. J. Jiménez-Borreguero, F. Sánchez-Madrid, É. Tóth, J. Alarcón and E. García-España, Dalton Trans., 2011, 40, 6451–6457 RSC.
- S. Liu, B. Yu, S. Wang, Y. Shen and H. Cong, Adv. Colloid Interface Sci., 2020, 281, 102165 CrossRef CAS PubMed.
- N. Z. Knezevic, I. Gadjanski and J.-O. Durand, J. Mater. Chem. B, 2019, 7, 9–23 RSC.
- T. Zhao, N.-T. Nguyen, Y. Xie, X. Sun, Q. Li and X. Li, Front. Chem., 2017, 5, 118 CrossRef PubMed.
- D. Ling, N. Lee and T. Hyeon, Acc. Chem. Res., 2015, 48, 1276–1285 CrossRef CAS PubMed.
- N. Z. Knezevic, E. Ruiz-Hernandez, W. E. Hennink and M. Vallet-Regi, RSC Adv., 2013, 3, 9584–9593 RSC.
- J. Liu, S. Z. Qiao, Q. H. Hu and G. Q. Lu, Small, 2011, 7, 425–443 CrossRef CAS PubMed.
- S. Dib, M. Boufatit, S. Chelouaou, F. Sadi-Hassaine, J. Croissant, J. Long, L. Raehm, C. Charnay and J. O. Durand, RSC Adv., 2014, 4, 24838–24841 RSC.
- M. Breugst and H.-U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
- A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 CrossRef CAS.
- J. L. Nyalosaso, E. Rascol, C. Pisani, C. Dorandeu, X. Dumail, M. Maynadier, M. Gary-Bobo, J. L. Kee Him, P. Bron, M. Garcia, J. M. Devoisselle, O. Prat, Y. Guari, C. Charnay and J. Chopineau, RSC Adv., 2016, 6, 57275–57283 RSC.
- M. Ménard, L. M. Ali, A. Vardanyan, C. Charnay, L. Raehm, G. Seisenbaeva, M. Gary Bobo, T. Theodossiou and J.-O. durand, J. Mater. Chem. B Search PubMed , to be submitted.
- K. Jobe, C. H. Brennan, M. Motevalli, S. M. Goldup and M. Watkinson, Chem. Commun., 2011, 47, 6036–6038 RSC.
- S. Hwang, W. Cha and M. E. Meyerhoff, Angew. Chem., Int. Ed., 2006, 45, 2745–2748 CrossRef CAS PubMed.
- K. Bürglová, N. Moitra, J. Hodacová, X. Cattoën and M. Wong Chi Man, J. Org. Chem., 2011, 76, 7326–7333 CrossRef PubMed.
- B. Jagadish, G. L. Brickert-Albrecht, G. S. Nichol, E. A. Mash and N. Raghunand, Tetrahedron Lett., 2011, 52, 2058–2061 CrossRef CAS PubMed.
- A. K. R. Junker, M. Tropiano, S. Faulkner and T. J. Sørensen, Inorg. Chem., 2016, 55, 12299–12308 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Syntheses of cyclen precursors, 1H, 13C, 29Si NMR spectra, DLS, zeta potentials, TGA, FTIR of functionalized MMSN, EDS analyses. See DOI: 10.1039/d1ra01581h |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |