
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective Pd-catalyzed α-alkylation of furans using alkyl iodides†
Jiaqi Yuanab,
Xiaofei Zhang*a and
Chunhao Yang *ab
*ab
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai, 201203, China
bSchool of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China. E-mail: xiaofeizhang@simm.ac.cn; chyang@simm.ac.cn
First published on 14th April 2021
Abstract
Herein, direct alkylation of the C–H bond at the α-position of furans catalyzed by palladium catalyst is reported. This protocol targets α-alkylfurans, achieving moderate to good yields under very practical reaction conditions. With a broad scope of substrates and good functional group tolerance, this method will has promising utility in medicinal chemistry.
Introduction
Alkyl-substituted heteroarene scaffolds are commonly found in drugs and other materials. Among these heteroarenes, α-alkyl-substituted furans are attractive for marketed drugs or active compounds, for example: Lapatinib,1 a tyrosine kinase inhibitor targeting HER2, was approved for the treatment of breast cancer in 2013; Lafutidine2 and Ranitidine,3 classic histamine H2-receptor antagonists, are clinically used for peptic ulcers; Navarixin,4 a selective CXC chemokine receptor 1/2 antagonist, is currently being used in a phase 2 clinical trial for treatment of chronic obstructive pulmonary disease (Fig. 1). Clearly, as important structures in drug research, the efficient synthesis and construction of α-alkylfurans deserves more attention. | ||
| Fig. 1 Drugs with α-alkylfuran scaffolds. | ||
While most direct alkylation reactions of furans fall into two types: (1) Friedel–Crafts alkylation, which usually requires Lewis acids/environmentally harmful solvents5–7 and has low selectivity and yields for furans; (2) nucleophilic substitution reactions, which need extremely low temperature conditions and strong base to form and stabilize the furan anion.8–10 Both types of reactions suffer from the limitation of functional group compatibility. Recently, transition-metal-catalyzed C–H functionalization has become a powerful and reliable tool for the direct alkylation of arenes, but most efficient procedures are designed for electron-deficient heteroarenes11–14 and acidic C–H bonds of azoles.15–18 As a typical electron-rich heteroarene, transition-metal-catalyzed C–H functionalization for the direct alkylation of furan has been rarely reported. In 2014, Zhou reported Pd-catalyzed alkylations of neutral heteroarenes (like benzoxazole) with alkyl halides,19 but few furan derivatives were employed as substrates. In 2015, Hartwig accomplished anti-Markovnikov hydroheteroarylation of unactivated alkenes with furans,20 but this transformation used a highly air-sensitive Ni(0)-N-heterocyclic carbene complex and gave only primary and secondary alkyl-substituted products. As far as we know, from 2015 to the present, only a few research papers involving transition-metal-catalyzed C–H alkylation of furans have been published. Evano and Nishikata achieved Cu21-and Fe22-catalyzed alkylation of heteroarenes with alkyl halides, respectively, via a radical pathway; however the coupling partners were limited to tertiary alkyl halides and only a few furan substrates were reported. Furthermore, Bao and Yin employed an Fe catalyst to achieve the alkylation of furans with alkyl diacyl peroxides23 and aliphatic aldehydes,24 respectively, but Bao's protocol is restricted because it involves dangerous peroxides and far in excess usage of furans, and Yin's method is strictly confined to acyl-substituted furan substrates and oxidative conditions. Therefore, general and systemic research focused on developing a convenient, practical and, most importantly, a functional-group-tolerant method for direct alkylation of furans is in high demand.
During research on the structure–activity relationship (SAR) of a furan-containing leading compound in our group, the introduction of different alkyl substituents at the α-position of furan became quite a challenge. So, herein, a general protocol for Pd-catalyzed regioselective α-alkylation of furans with alkyl iodides was developed.
Results and discussion
Tetrahydro-2H-pyran is a very familiar pharmacophore in bio-active compounds owing to its unique chemical and physical properties.25–28 Following screening in previous work, commercially available and easily accessible methyl furan-2-carboxylate (1a) and 4-iodotetrahydro-2H-pyran (2a) were used as substrates for the first synthesis of methyl 5-(tetrahydro-2H-pyran-4-yl)furan-2-carboxylate (3a) via direct C–H alkylation.Initially, we employed Zhou's optimal conditions for this reaction, but the desired product 3a was obtained with a low yield (Table 1, entry 1). By increasing the amount of ligand to 10 mol%, the yield of 3a was slightly increased to 36% (Table 1, entry 2). Several solvents with appropriate boiling points were tested and PhCF3 turned out to be the best choice (Table 1, entries 2–4). Later, the optimization of the reaction conditions focused on the screening of ligands (Table 1, entries 5–12) and Pd catalysts (Table 1, entries 13–16). As a result, Pd(PPh3)4 and Xantphos were found to be the best combination for this transformation. Subsequently, the replacement of Cs2CO3 with K2CO3/LiOH/K3PO4 failed to give better yields (Table 1, entry 17). Eventually, by adding 10 mol% Pd(PPh3)4, 20 mol% Xantphos, 2 equiv. (0.6 mmol) Cs2CO3 and 3 equiv. (0.9 mmol) 2a, and extending the reaction time to 48 h, the target compound 3a was obtained with 74% isolated yield (Table 1, entries 18 and 19).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Solvent | Yield (%)b |
| a Reaction conditions: methyl furan-2-carboxylate (1a, 0.3 mmol), 4-iodotetrahydro-2H-pyran (2a, 0.6 mmol), Pd catalyst (5 mol%), ligand (10 mol%) and Cs2CO3 (0.6 mmol) in 5 mL solvent under Ar at 110 °C for 24 h.b Isolated yields.c By employing Zhou's optimized conditions.d Cs2CO3 was replaced with K2CO3/LiOH/K3PO4, respectively (0.6 mmol).e By adding 10 mol% Pd(PPh3)4 and 20 mol% Xantphos.f By adding 10 mol% Pd(PPh3)4, 20 mol% Xantphos and 0.9 mmol 2a, extending the reaction time to 48 h. dppp = 1,3-bis(diphenylphosphino)propane; dppe = 1,2-bis(diphenylphosphino)ethane; dppb = 1,4-bis(diphenylphosphino)butane; dppf = 1,1′-bis(diphenylphosphino)ferrocene; XPhos = 2-(dicyclohexylphosphino)-2′,4′,6′-tri-i-propyl-1,1′-biphenyl; Xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene; BINAP = 1.1′-binaphthyl-2.2′-diphemyl phosphine; Johnphos = 2-(di-tert-butylphosphino)biphenyl. | ||||
| 1 | Pd(PPh3)4 | dppp | PhCF3 | 28c |
| 2 | Pd(PPh3)4 | dppp | PhCF3 | 36 |
| 3 | Pd(PPh3)4 | dppp | 1,4-dioxane | 23 |
| 4 | Pd(PPh3)4 | dppp | PhCH3 | 12 |
| 5 | Pd(PPh3)4 | dppe | PhCF3 | 27 |
| 6 | Pd(PPh3)4 | dppb | PhCF3 | 36 |
| 7 | Pd(PPh3)4 | dppf | PhCF3 | 45 |
| 8 | Pd(PPh3)4 | XPhos | PhCF3 | <10% |
| 9 | Pd(PPh3)4 | Xantphos | PhCF3 | 47 |
| 10 | Pd(PPh3)4 | BINAP | PhCF3 | 31 |
| 11 | Pd(PPh3)4 | Johnphos | PhCF3 | 11 |
| 12 | Pd(PPh3)4 | — | PhCF3 | <10% |
| 13 | Pd(OAc)2 | Xantphos | PhCF3 | ND |
| 14 | Pd2dba3 | Xantphos | PhCF3 | <10% |
| 15 | PdCl2dppf | Xantphos | PhCF3 | 28 |
| 16 | Pd2dba3·CHCl3 | Xantphos | PhCF3 | <10% |
| 17 | Pd(PPh3)4 | Xantphos | PhCF3 | <10%d |
| 18 | Pd(PPh3)4 | Xantphos | PhCF3 | 58e |
| 19 | Pd(PPh3)4 | Xantphos | PhCF3 | 74f |
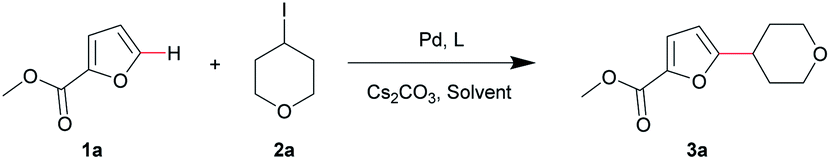
With the optimal conditions in hand, we first explored the substrate scope of furans (Table 2). This Pd-catalyzed direct C–H alkylation of furans was tolerant of a broad range of functional groups, such as ester, aldehyde, cyano, acetyl, amide and Boc groups. The target compounds were successfully synthesized with moderate to good yields (Table 2, 3a–f and 3h). It is noteworthy that no regio-isomers were detected in these reactions even for the preparation of 3c. 2-Phenylfuran and benzofuran also performed well to give products with good yields (Table 2, 3i and 3j). In contrast, from the experimental results, when the 2- or 3-position of furan was substituted with electron-donating groups, the yields were significantly decreased (Table 2, 3g, 3k and 3l). The relatively low yields for electron-donating substituents were due to the low conversion rate: most of the unreacted starting furan could be recovered after the reaction. An electron-withdrawing substituent on the furan ring clearly benefitted the reaction process.

The substrate scope of the alkyl iodides was then examined. Various unactivated/functionalized alkyl iodides (2) were treated with 1a under the optimal conditions (Table 3). Most secondary alkyl iodides afforded products with good yields, except for 3-iodooxetane (Table 3, 3m–3t). Interestingly, methyl furan-2-carboxylate (1a) reacted with iodocyclohexane gave an unsatisfactory result (yield <30%), while 2-acetylfuran performed well with iodocyclohexane to produce 3x in 78% yield. Furthermore, for tertiary alkyl iodides, 2-iodo-2-methylpropane gave the coupling product 3u in 81% yield. To our delight, although primary alkyl iodides like 1-iodohexane failed to afford the alkylated product (yield <10%), the first C–H α-difluoroethylation and α-trifluoroethylation of furan using ICH2CF2H and ICH2CF3 were accomplished with acceptable yields (Table 3, 3v and 3w). It is necessary to note that methyl furan-2-carboxylate 1a did not react with 4-bromotetrahydro-2H-pyran, even with 3 equiv. (0.09 mmol) NaI or KI under the optimal conditions.
To further demonstrate the synthetic utility of this method, we employed 3β-iodo-5-androsten-17-one as coupling substrate (Scheme 1). The desired compound 3y was produced with an acceptable yield of 42% under the optimal conditions. This late-stage modification strongly highlights the importance of this protocol and demonstrates potential applications in medicinal chemistry.
 | ||
| Scheme 1 The reaction of methyl furan-2-carboxylate and 3β-iodo-5-androsten-17-one. | ||
A proposed mechanism for the Pd-catalyzed radical alkylation of furans is outlined in Scheme 2. It starts with single electron transfer from [Pd0Ln] to the alkyl iodides, generating an alkyl radical A and [PdIILn]. The addition of alkyl radical A to 1a affords a delocalized radical intermediate B, which then produces the corresponding carbocation C and [Pd0Ln] through single electron transfer. Finally, deprotonation driven by aromatization furnishes the alkylated furans. To test the possibility of single electron transfers, control experiments with our standard conditions were performed in the presence of TEMPO (oxidanyl, (CH₂)₃(CMe₂)₂NO). The reaction of 1a and 2a with 1 equiv. TEMPO afforded the alkylated product 3a in low yield (<10%), but it did not give the corresponding product when 3 equiv. TEMPO was employed. This result indicates that the reaction may proceed via the described radical pathway.
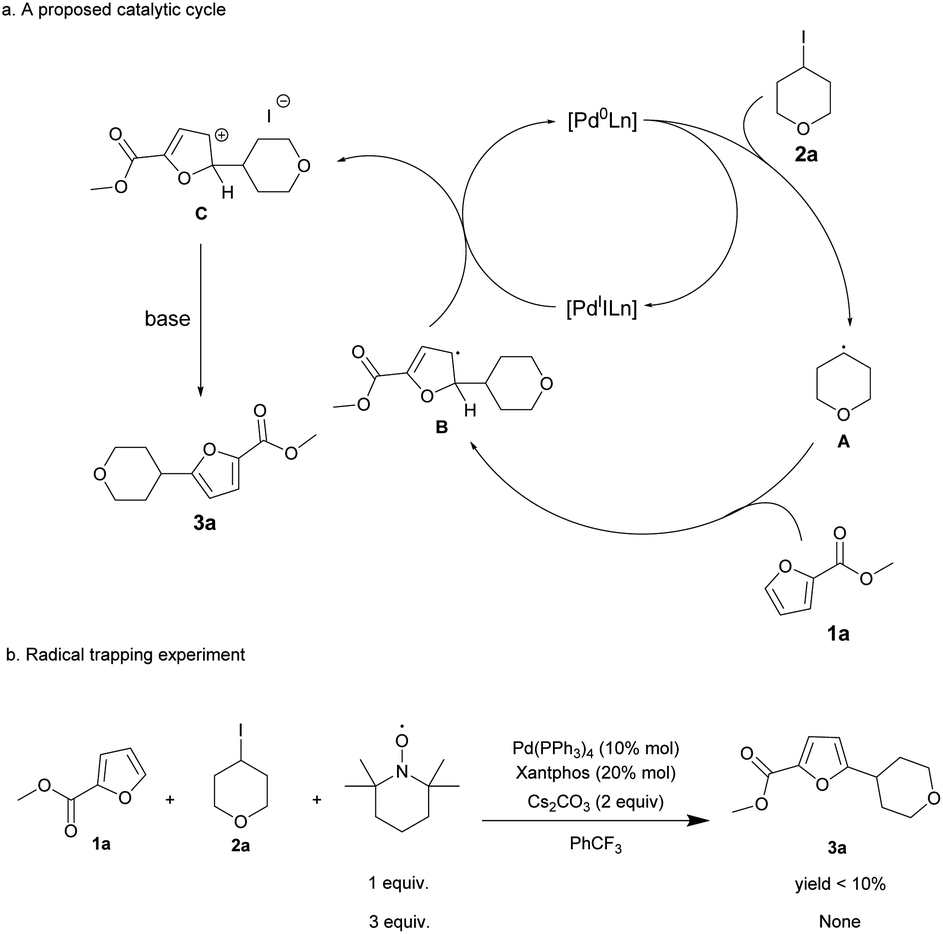 | ||
| Scheme 2 Possible mechanism and radical trapping experiment. | ||
Conclusions
In conclusion, a regioselective procedure for α-alkylation of furans using alkyl iodides via a simple Pd catalyst has been developed. This protocol provides a convenient and practical route to α-alkylfurans with good functional group compatibility and broad substrate scope. After this systemic research for alkylation of furans, the diversification of furan-containing compounds could be more easily achieved. The applications of this method for drug discovery from our group will be published in the near future.Experimental section
General information
All reagents were purchased from commercial suppliers and used without further purification. The progress of all of the reactions was monitored by thin layer chromatography (TLC) with standard TLC silica gel plates, and the developed plates were visualized under UV light. All of the compounds were purified by column chromatography. Chromatography was performed on silica gel (100–200 mesh). Nuclear magnetic resonance spectra (1H, 13C NMR) were recorded on Varian Mercury-400 and Bruker Avance III-500/600 spectrometers and CDCl3-d and CD3OD-d were used as solvent. NMR peaks were calibrated by reference to standard peaks of CDCl3 at 7.26 ppm for 1H and 77.16 ppm for 13C and standard peaks of CD3OD at 3.31 ppm for 1H and 49.00 ppm for 13C. For peak descriptions, the following abbreviations are used: s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), m (multiplet). Electron ionization high-resolution mass spectrometry (EI-HRMS) data were recorded using a Thermo DFS mass spectrometer. ESI-HRMS were recorded using an Agilent QTOF mass spectrometer.General experimental methods
A 15 mL Schlenk tube equipped with a stirrer bar was charged with Pd(PPh3)4 (0.03 mmol, 0.1 equiv.), Xantphos (0.06 mmol, 0.2 equiv.), Cs2CO3 (0.6 mmol, 2.0 equiv.), furan derivatives (0.3 mmol, 1.0 equiv.), alkyl iodides (0.9 mmol, 3.0 equiv.), and PhCF3 (5 mL). The tube was evacuated and backfilled with Ar 5 times, and the mixture was vigorously stirred in a pre-heated 110 °C oil bath for 48 h. The mixture was diluted with water (90 mL), and extracted three times with ethyl acetate (30 mL). The organic phase was dried over Na2SO4, concentrated in vacuo, and purified by column chromatography on silica gel.Characterization data of compounds
Author contributions
Conceptualization, C. Y.; methodology, J. Y. and X. Z.; chemical experiments, J. Y. and X. Z.; inspiration and discussions, J. Y., X. Z. and C. Y.; writing—original draft preparation, J. Y. and X. Z.; writing—review and editing, X. Z. and C. Y.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Science and Technology Commission of Shanghai Municipality (18431907100), the Institutes for Drug Discovery and Development, Chinese Academy of Sciences (CASIMM0120185003), and the State Key Laboratory of Drug Research Program (SIMM1903ZZ-03).Notes and references
- J.-C. Xuhong, X.-W. Qi, Y. Zhang and J. Jiang, Am. J. Cancer Res., 2019, 9, 2103–2119 CAS.
- M. Nakano, S. Kitano, M. Nanri and M. Kiniwa, Eur. J. Pharmacol., 2011, 658, 236–241 CrossRef CAS PubMed.
- S. M. Grant, H. D. Langtry and R. N. Brogden, Drugs, 1989, 37, 801–870 CrossRef CAS PubMed.
- S. I. Rennard, D. C. Dale, J. F. Donohue, F. Kanniess, H. Magnussen, E. R. Sutherland, H. Watz, S. Lu, P. Stryszak, E. Rosenberg and H. Staudinger, Am. J. Respir. Crit. Care Med., 2015, 191, 1001–1011 CrossRef CAS PubMed.
- J. E. Fitzpatrick, D. J. Milner and P. White, Synth. Commun., 1982, 12, 489–494 CrossRef CAS.
- S. Hadjikyriacou and R. Faust, Macromolecules, 1999, 32, 6393–6399 CrossRef CAS.
- F. Mohanazadeh and H. Amini, Bull. Korean Chem. Soc., 2010, 31, 3038–3040 CrossRef CAS.
- I. Chambrier, G. F. White and M. J. Cook, Chem. - Eur. J., 2007, 13, 7608–7618 CrossRef CAS PubMed.
- D. Kalaitzakis, E. Antonatou and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 400–402 RSC.
- D. Kalaitzakis, A. Kouridaki, D. Noutsias, T. Montagnon and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2015, 54, 6283–6287 CrossRef CAS PubMed.
- J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332–5333 CrossRef CAS PubMed.
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS PubMed.
- B. Xiao, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 616–619 CrossRef CAS PubMed.
- T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758 RSC.
- K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 13964–13965 CrossRef CAS PubMed.
- Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4451–4454 CrossRef CAS PubMed.
- O. Vechorkin, V. Proust and X. Hu, Angew. Chem., Int. Ed., 2010, 49, 3061–3064 CrossRef CAS PubMed.
- T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2012, 51, 775–779 CrossRef CAS PubMed.
- X. Wu, J. W. T. See, K. Xu, H. Hirao, J. Roger, J.-C. Hierso and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 13573–13577 CrossRef CAS PubMed.
- Y. Schramm, M. Takeuchi, K. Semba, Y. Nakao and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 12215–12218 CrossRef CAS PubMed.
- C. Theunissen, J. Wang and G. Evano, Chem. Sci., 2017, 8, 3465–3470 RSC.
- Y. Yamane, K. Yoshinaga, M. Sumimoto and T. Nishikata, ACS Catal., 2019, 9, 1757–1762 CrossRef CAS.
- F. Yu, T. Wang, H. Zhou, Y. Li, X. Zhang and H. Bao, Org. Lett., 2017, 19, 6538–6541 CrossRef CAS PubMed.
- W. Luo, Y. Yang, B. Liu and B. Yin, J. Org. Chem., 2020, 85, 9396–9404 CrossRef CAS PubMed.
- F. W. Goldberg, M. R. V. Finlay, A. K. T. Ting, D. Beattie, G. M. Lamont, C. Fallan, G. L. Wrigley, M. Schimpl, M. R. Howard, B. Williamson, M. Vazquez-Chantada, D. G. Barratt, B. R. Davies, E. B. Cadogan, A. Ramos-Montoya and E. Dean, J. Med. Chem., 2020, 63, 3461–3471 CrossRef CAS PubMed.
- B. Wang, J. Wu, Y. Wu, C. Chen, F. Zou, A. Wang, H. Wu, Z. Hu, Z. Jiang, Q. Liu, W. Wang, Y. Zhang, F. Liu, M. Zhao, J. Hu, T. Huang, J. Ge, L. Wang, T. Ren, Y. Wang, J. Liu and Q. Liu, Eur. J. Med. Chem., 2018, 158, 896–916 CrossRef CAS PubMed.
- G. A. Nishiguchi, A. Rico, H. Tanner, R. J. Aversa, B. R. Taft, S. Subramanian, L. Setti, M. T. Burger, L. Wan, V. Tamez, A. Smith, Y. Lou, P. A. Barsanti, B. A. Appleton, M. Mamo, L. Tandeske, I. Dix, J. E. Tellew, S. Huang, L. A. Mathews Griner, V. G. Cooke, A. Van Abbema, H. Merritt, S. Ma, K. Gampa, F. Feng, J. Yuan, Y. Wang, J. R. Haling, S. Vaziri, M. Hekmat-Nejad, J. M. Jansen, V. Polyakov, R. Zang, V. Sethuraman, P. Amiri, M. Singh, E. Lees, W. Shao, D. D. Stuart, M. P. Dillon and S. Ramurthy, J. Med. Chem., 2017, 60, 4869–4881 CrossRef CAS PubMed.
- A. Dietrich, T. Mueller, R. Paschke, B. Kalinowski, T. Behlendorf, F. Reipsch, A. Fruehauf, H.-J. Schmoll, C. Kloft and W. Voigt, J. Med. Chem., 2008, 51, 5413–5422 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01522b |
| This journal is © The Royal Society of Chemistry 2021 |