
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDemonstration of pH-controlled DNA–surfactant manipulation for biomolecules
Na Li,
Zijuan Liao,
Shupeng He,
Xiao Chen,
Shenhao Huang,
Yanwei Wang* and
Guangcan Yang *
*
Department of Physics, Wenzhou University, Wenzhou, 325035, China. E-mail: wangyw@wzu.edu.cn; yanggc@wzu.edu.cn; Fax: +86-577-86689010; Tel: +86-577-86689033
First published on 22nd April 2021
Abstract
The understanding of DNA–surfactant interactions is important for fundamental physical biology and developing biomedical applications. In the present study, we demonstrated a DNA–surfactant nano-machine model by modulating the compaction of DNA in dodecyldimethylamine oxide (DDAO) solutions. By controlling DDAO concentration and pH of solution, we are able to adjust the compacting force of DNA so as to pull biomolecular subunits connected to it. The pulling force of the machine depends on DDAO concentration and pH of solution, ranging from near zero to about 4.6 pN for 10 mM DDAO concentration at pH = 4. The response time of the machine is about 3 minutes for contracting and 2 minutes for releasing in 5 mM DDAO solution. We found that DDAO has no significant influence on DNA under basic conditions, but compacts DNA under acidic conditions, which is enhanced with decreasing pH of solution. Meanwhile, we found the accompanying charge inversion of DNA in the process of DNA compaction by DDAO.
1. Introduction
The interactions between polymers and surfactants in aqueous solutions have been intensively investigated in recent decades because of the fundamental interest in soft matter and their widespread applications.1–4 Among various polymer–surfactant systems, the interaction between DNA and cationic surfactants attracts special attention because of its significance in molecular biology and potential biomedical applications, such as genetic modification and control of gene transmission.5–7 Cationic surfactants can induce DNA to undergo a conformational transition from stretched strands to compact spheres through electrostatic attraction and further stabilized by hydrophobic interactions of the hydrocarbon tails.8,9 However, their biomedical applications of cationic surfactants are hindered by the detergent-related cytotoxicity from their high water solubility and poor efficiency in vitro in gene transfer.7 Amphoteric surfactants are potential candidates to overcome these disadvantages.10Amphoteric surfactants can achieve non-ionic to cationic transition under different pH conditions. Dodecyldimethylamine oxide (DDAO) is a typical amphoteric surfactant and suitable for investigating the interaction between DNA and surfactants. DDAO can easily bind DNA and induce its conformational change by hydrophobic and electrostatic interactions. In the interaction between DDAO and DNA, pH plays an important role due to its effect on the protonation of amphoteric surfactant in acidic solution. Under acidic conditions, DDAO becomes a cationic surfactant due to protonation of its amine oxide head group. However, it becomes a zwitterionic surfactant under alkaline conditions due to the net zero charge of the amine oxide head group. Therefore, the solution properties of amine oxide surfactants strongly depend on pH.9 In general, weak hydrophobic interaction dominates in a DDAO–DNA complex in basic solutions while electrostatic force plays a more significant role in the attraction between them due to the protonation of DDAO.11–13 However, the detail of the interaction mechanism of DNA–surfactant is not fully understood yet.
In present study,we used three experimental tools: dynamic light scattering (DLS), magnetic tweezers (MT) and atomic force microscopy (AFM),14–16 to study pH effect on the interaction between DDAO and DNA and then apply the DNA condensing feature of amphoteric surfactants to construct a DNA–surfactant nano-machine model. In the model, the condensing force is modulated by the concentration of DDAO and pH of solution. It is shown that the interaction between non-ionic DDAO and DNA is weak due to hydrophobic interaction, and is unable to cause DNA compaction, while the electrostatic force between DDAO and DNA is enhanced by lowering pH in solution leading to the protonation of amine oxide head group. For example, at fixing pH = 4, the strength of interaction between DDAO and DNA can be modulated by DDAO concentration (1 to 10 mM) from 1.8 to 4.6 pN, corresponding typical forces of noncovalent interaction in biomacromolecular systems. The model may find some potential applications in manipulation of biomolecules such as DNA and proteins.17
2. Materials and methods
2.1 Materials
Double strands λ-phage DNA (48502 bp) was purchased from New England Biolabs company, and its initial concentration of 500 ng μL−1. DDAO were purchased from Sigma at high-purity grade (>99%) and was used without further purification. Phosphate buffer saline (PBS), bovine serum albumin (BSA, 1 mg L−1) and other chemical reagents were also purchased from Sigma. Purified water was obtained from a Milli-Q system (Millipore, Billerica, MA, USA), and TRIS buffer (10 mM) was used as both stock solution and experimental buffer solution. All chemical agents were used as received and all measurements were repeated at least twice for consistency.2.2 Tethering DNA by magnetic tweezers (MT)
A transverse magnetic tweezers system (Fig. 1) is set up on an inverted microscope (Nikon TE2000U, Japan) monitoring the dynamic process of tethering DNA. The detail of setup can be found in previous work.14,24 The procedure can be described briefly as follows: a DNA chain is bound at one end to an immobile support (the glass sidewalls) and at the other end to a paramagnetic bead.18–21 A permanent magnet controlled by a micromanipulator system (MP-285), Sutter Instruments (Novato, CA, USA) is used to exert a force on the paramagnetic bead to pull tethered DNA. The movement of paramagnetic bead was recorded by a CCD camera in real-time. The sample cell is built as follows, A 0.17 mm thick coverslip with one side polished and coated with silane reagent, then it is sandwiched in between two glass slides. We use the polydimethylsiloxan to block out the open side of the structure. Next, two holes with a diameter of 1 mm each were created on the top glass slide and linked with a glass capillary to facilitate buffer out or in. In the Fig. 2 the drawing in the dashed square is zoom-in image of sidewall–DNA–bead structure in the sample cell. The entire motion of the small magnetic spheres was video-taped by Fast Fourier Transform-based software, and the video results were analyzed by Mathcad Professional software, and the critical cohesion of DNA molecules was obtained by the Worm-Like-Chain (WLC) model.19 The DNA–beads–support complex is constructed as the procedure as follows: one end of DNA is modified by digoxigenin so as to attach the anti-digoxigenin-coated glass sidewall; the other end is modified with biotin to bind avidin-coated magnetic beads. To form DNA–bead constructs, we gently mixed 0.5 μL stock solution of magnetic beads coated with streptavidin (M-280, Dynal Biotech, Oslo, Norway) with 0.5 μL modified DNA for 30 minutes in 200 μL buffer solution. The coverslips were polished, washed, dried, and then the polished sides were treated with a sigmacote solution. A cover glass slide is glued on a glass slide to serve as the sidewall to anchor an end of DNA. A syringe pump is used to control the inflow and outflow of the sample cell connecting with the soft silicone tube in the glass capillary of the cell. So the location is not fixed. The cell with a polished sidewall was dealt with anti-digoxygenin at first and then was rinsed with PBS containing 5 mg mL−1 BSA at pH 8.0. BSA is the blocking buffer to decrease the nonspecific interaction. DNA–bead constructs were flushed into the cell, then a side wall–DNA–paramagnetic bead structure was formed, as shown in Fig. 1. After the attachment of DNA–bead to the sidewall, the PBS buffer was eluted by injecting Tris buffer. | ||
| Fig. 1 A schematic diagram of magnetic tweezers. | ||
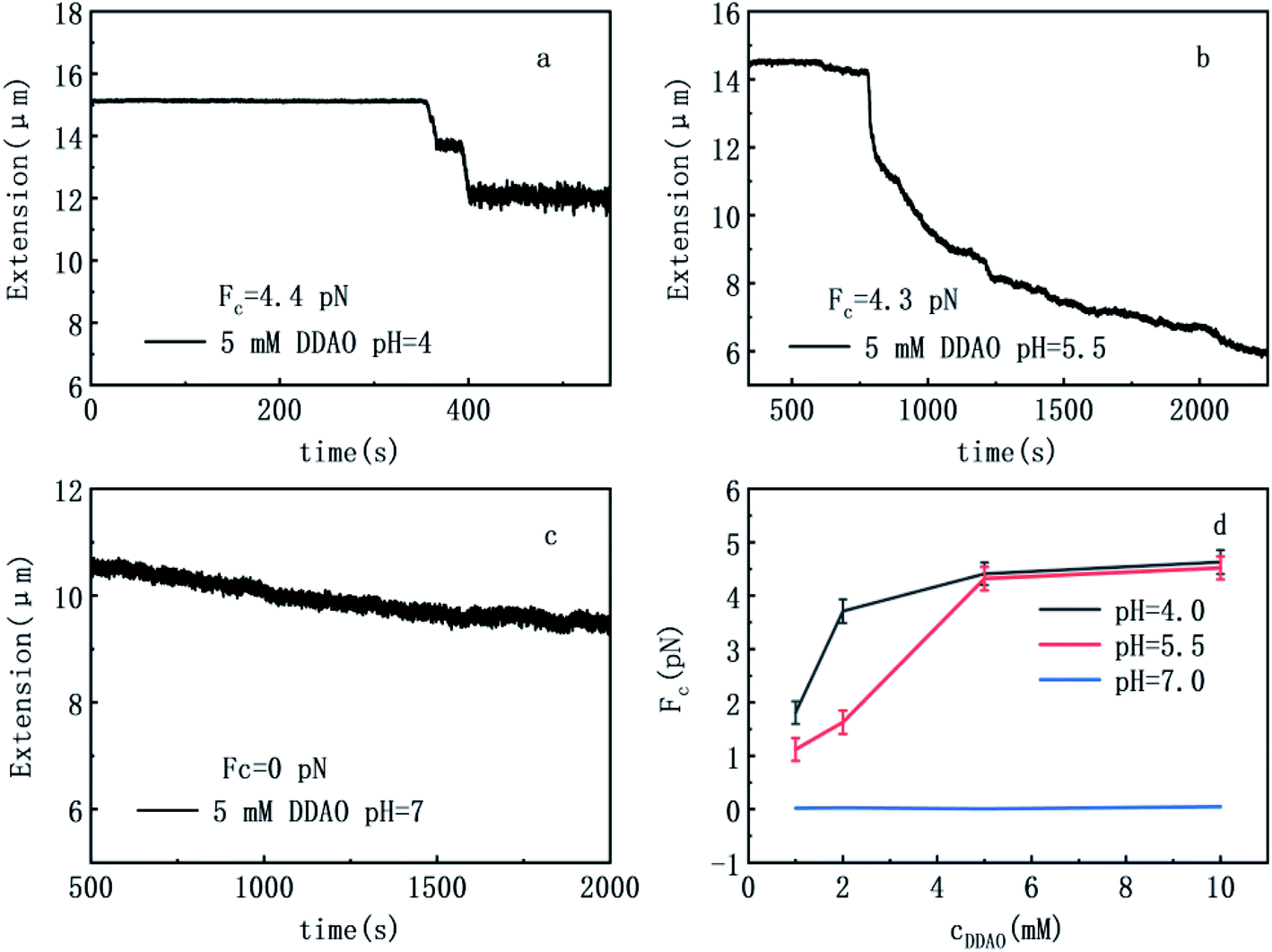 | ||
| Fig. 2 DNA extension as a function of pulling time. ((a) 5 mM DDAO pH = 4; (b) 5 mM DDAO pH = 5.5; (c) 5 mM DDAO pH = 7) DNA condensing force of DNA–DDAO complexes as a function of DDAO concentrations of solutions at various pH (d: 1 mM, 2 mM, 5 mM, 10 mM). | ||
The DDAO solution with specified pH and concentration for DNA tethering was prepared by the protocol described in DLS measurement. We flushed the DDAO solution into the flow cell by a syringe pump. In a typical measurement, we pull DNA to its maximal length (about 16 μm) by moving the magnet close to the paramagnetic bead to apply a force of more than 10 pN. Then, we moved the magnet slowly away to lower the force to a needed value and monitor the conformational change of DNA influenced by the added agents.
2.3 Dynamic light scattering (DLS)
The electrophoresis-mobility measurements were achieved in a dynamic light scattering (DLS)22–29 device of Malvern Zetasizer nano ZS90 equipped with the patented M3-PALS technique. The light scattering from a He–Ne gas laser (λ = 633 nm) is collected by an avalanche photodiode mounted on the goniometer arm in the perpendicular direction to the incident light. The DNA samples were diluted to a concentration of 1 ng μL−1 in a buffer solution containing 10 mM Tris (pH = 7.2) and, then different concentrations of DDAO were added. All measurements were carried out after 5 minutes incubation at room temperature, and 1 mL volume sample cell for DNA solution was used for each measurement.2.4 Atomic force microscopy (AFM)
The sample preparation process for atomic force microscopy (AFM) is similar to previous published work by ours and other groups,30–33 and can be briefly described as follows: mica disks were used as substrates for DNA adsorption, and their surfaces were always freshly cleaved before use. The disks are about 1 cm2 square or circular pieces and attached to glass slides. At first, a drop of mixture solution of DDAO–DNA about 50 μL was deposited on a freshly cleaved mica surface for 5 min. Then, the surface was rinsed with distilled water and dried with a gentle flow of nitrogen gas. All chemical agents were used as received and all measurements were repeated at least three times to obtain consistent results. All the prepared samples were scanned by AFM (JPK Instruments AG, Berlin, Germany) in AC mode. A silicon AFM probe (NCHR-50, NanoWorld Corporation, Neuchatel, Switzerland) with aluminium coating was used and its spring coefficient and the resonance frequency are 42 N m−1 and 320 kHz, respectively. The imaging area is 5 μm × 5 μm and the scan rate is 1.0 Hz. Each image obtained has a resolution of 512 × 512 pixels (4–6 nm per pixel).3. Results and discussions
3.1 Tethering DNA–DDAO complex
To explore the interaction mechanism between DDAO and DNA, we measured condensing forces of DNA–DDAO complexes by magnetic tweezers at various pH and DDAO concentrations. The DNA compaction is the process of one DNA chain going from free extensible state to a more compactly ordered structure. We used magnetic tweezers to pull DNA–DDAO complexes in a flow cell under different pH and DDAO conditions. In the setup, we can see the tethered DNA compaction and measure the tethering force simultaneously when flushing DDAO solution into the flow cell.DNA compaction causes a continuous stepwise shrinking of extension when we decreases magnetic force (F) below a critical condensing force Fc. The results are summarized in Fig. 2. From (a) to (c) in Fig. 2, pH of solution increases from 4, then to 5, finally to 7, but keeping DDAO concentration at 5 mM. In Fig. 2(a) (pH = 4), we can see that the extension curve of the DNA contains some stepwise leap in the shrinking, and the corresponding critical condensing force is about 4.4 pN. Then, we increase pH of solution to 5.5, as shown in Fig. 3(b), in which the DNA critical condensing force is 4.3 pN. The condensing force basically is almost unchanged. In the case, the extension curve of DNA shrinks continuously, and no stepwise leaps does not appear. However, if we increase pH of solution to 7, a neutral value, as shown in Fig. 2(c), the condensing force is almost approaching 0 pN, and DNA extension curve shows very little or no shrinking. We also explored the force spectra of the system at other concentrations (1 mM, 2 mM, 10 mM) of DDAO with the different pH (pH = 4, 5.5, 7). The results are summarized in Fig. 2(d). We made the control experiment to measure the change of DNA extension at different PH value (7, 5.5, 4) in which concentration of DDAO is 0 mM in Fig. 3, so the pH value can change a little persistence length of DNA, but has no influence on the compaction of DNA. This result is consistent with the experiments absent of ions at lower ph = 3 in our previous work.27
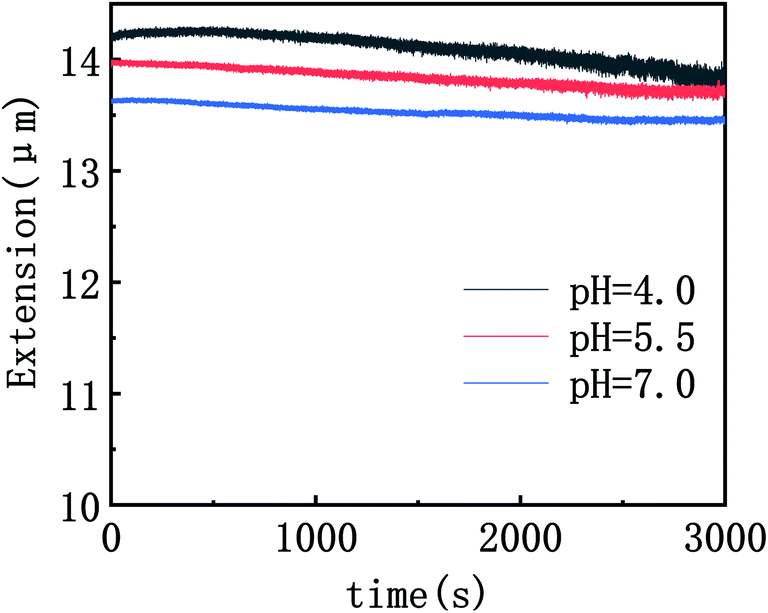 | ||
| Fig. 3 DNA extension as a function of pulling time. (10 mM Tris;0 mM DDAO). | ||
To verify the attraction between segments of single DNA chain, we changed pH of solution up and down by flushing two buffers into the sample cell alternately while keeping the pulling of tethered DNA. Fig. 4(a) and (b) show the cyclic process of shrinking and releasing of a single tethered DNA at various DDAO concentrations by switching pH of solution between 4 and 8 with the same buffer condition of 10 mM Tris. For example, as shown in Fig. 4(a), we initially pulled a tethered single DNA at a force in a solution of 2 mM DDAO (pH = 4), and the DNA shrunk discontinuously. Then, we injected the Tris (10 mM, pH = 8) with the same concentration of DDAO into the cell, the extension of tethered DNA recovered to its contour length immediately following the flush, appearing as a one-jump process. In Fig. 4(a), it has the delayed response in second cycle. It may be the non-uniformity of liquid flow in cell. So the change of PH value in cell had the delayed time. Fig. 4(b) shows the similar shrinking and releasing procedures but with 5 mM concentrations of DDAO in solution. In other words, when the solution is alkaline, DNA–DDAO is relaxed while it is compacted under acidic conditions. Therefore, we can construct a DNA nano-machine based on this cyclic process controlled by pH of solution while fixing DDAO concentration at some value. For example, at fixing pH = 4, the maximal working force of the DNA machine can reach 1.8 pN for DDAO concentration 1.0 mM, and 4.6 pN for DDAO concentration 10 mM. The force strength is very useful in molecular manipulation for biomacromolecules such as proteins.
 | ||
| Fig. 4 The cyclic process of DNA extension–time curve measured by MT in releasing process with different concentration of DDAO in 10 mM Tris (pH = 3) and stretching process with different concentration of DDAO in 10 mM Tris, pH = 8. (a) 2 mM DDAO. (b) 5 mM DDAO. | ||
3.2 DNA–DDAO morphology by atomic force microscopy
In order to further analyse the interaction between DNA and DDAO, we investigated the morphology change of DNA–DDAO complex by atomic force microscopy (AFM), as shown in Fig. 5. Fig. 5(a–l) show the images of DDAO-DNA complex at different pH values (pH = 4, pH = 5.5, pH = 7) with increasing DDAO concentrations, from 1 mM, 2 mM, 5 mM, 10 mM. Fig. 5(a–c) correspond to the case 1 mM DDAO concentration at pH = 4, 5.5 and 7 respectively. In Fig. 5(a), we can see the dense floral DNA on mica at pH = 4. If we increase pH from 4 to 5.5, many DNA network structures are observed as in Fig. 5(b). When pH value is increased further to 7 as in Fig. 5(c), the networks become a free loose forms of DNA. When the concentration of DDAO is 2 mM, as shown in Fig. 5(d–f), the DNA conformation changes from dense flowery shape under acidic conditions to grid shape and then to free loose shape with the increase of pH value. When the concentration is 5 mM, as shown in Fig. 5(g–i), the conformation of DNA varies from a compact small sphere to a free loose shape with increasing pH of incubation solution. If we increase DDAO concentration to 10 mM, the DNA morphology change very slightly, is quite similar to the case of 5 mM, as shown in Fig. 5(j–l) for different pH. From these AFM images, we can see that increasing pH of incubation solution for DNA deposition hinders the ability of DDAO compacting DNA, as shown in Fig. 5 by row such as from (a) to (c) or from (d) to (f). On the other hand, the ability of DDAO compacting DNA increases with DDAO concentration when pH of solution is fixed, corresponding to that DNA–DDAO complex becomes more and more compact, as shown in Fig. 5 by column, such as from (a) to (j) or from (b) to (k). These AFM results are consistent to the measurement of condensing forces by MT, where more compact DNA structure corresponds to higher unraveling force. | ||
| Fig. 5 Atomic force images of DNA–DDAO complexes at different DDAO concentrations and pHs. ((a) 1 mM DDAO pH = 4; (b) 1 mM DDAO pH = 5.5; (c) 1 mM DDAO pH = 7; (d) 2 mM DDAO pH = 4; (e) 2 mM DDAO pH = 5.5; (f) 2 mM DDAO pH = 7; (g) 5 mM DDAO pH = 4; (h) 5 mM DDAO pH = 5.5; (i) 5 mM DDAO pH = 7; (j) 10 mM DDAO pH = 4; (k) 10 mM DDAO pH = 5.5; (l) 10 mM DDAO pH = 7). | ||
3.3 Electrophoretic mobility of DNA–DDAO complex
DNA shows a negative zeta potential and electrophoretic mobility in aqueous solution due to ionization of its phosphate backbone, so it is often regarded as a negatively charged biological polyelectrolyte (or polyanion). When cationic agents such as surfactants are added to the solution, they electrostatically interact with DNA and the cations compensate or neutralizes DNA surface negative charge, result in that the zeta potential of DNA approaches zero or even positive value in some cases.34The electrophoretic mobilities of DNA–DDAO complexes at various pH and DDAO concentrations in solutions are shown in Fig. 6. The electrophoretic mobility of complex changed from −0.32 × 10−4 cm2 V−1 s−1 to 3.01 × 10−4 cm2 V−1 s−1 when the complex system pH = 3 and the concentration increased from 0.5 mM to 10 mM. We can see that the electrophoretic mobility of DDAO–DNA complex increases with the concentration of DDAO, corresponding the upper shifting of mobility curves in Fig. 6. Meanwhile it decreases with increasing pH of solution, the experimental results showed that the electrophoretic mobility of the complex decreased from 1.24 × 10−4 cm2 V−1 s−1 to −1.04 × 10−4 cm2 V−1 s−1 when the concentration of the system was 2 mM and the pH value increased from 3 to 8, corresponding the negative slopes of all the mobility curves in the figure. DDAO cannot completely neutralize the negative charge of the DNA, even when the pH reaches 3 at lower concentration of DDAO (<1 mM), the electrophoretic mobility of the complex is still negative,It is known experimentally that the electrophoretic mobility is −0.32 × 10−4 cm2 V−1 s−1 at the complex concentration of 0.5 mM and pH = 3. This is different from the strong electrostatic effect shown by the CTAB. The electrophoretic mobility of the complex is still negative in pH = 3 with high DDAO protonation, indicating that hydrophobic interaction plays a role in the binding process between DDAO and DNA. DDAO the system with higher concentration than 1 mM, the electrophoretic mobility of the complex increased sharply in the range of pH = 8–5.
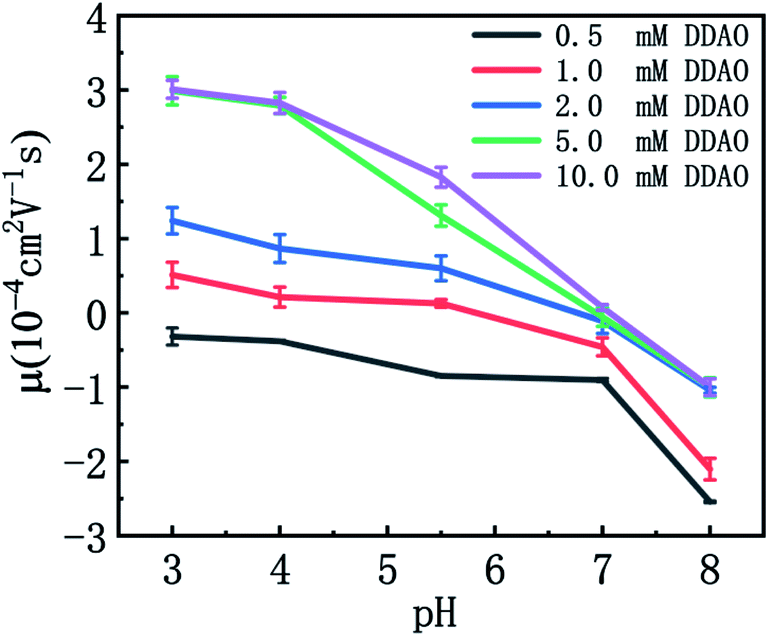 | ||
| Fig. 6 Electrophoretic mobility of DNA–DDAO complexes as a function of pH of solutions at various DDAO concentrations (0.5 mM, 1 mM, 2.0 mM, 5.0 mM, 10.0 mM). The concentration of DNA is 1 ng μL−1. | ||
3.4 Microscopic mechanism of DNA–DDAO interaction
The mechanical and electrokinetic properties of DNA–DDAO complex observed in previous sections can be ascribed to the amphoteric feature of DDAO. It is reported in the literature that the critical micelle concentration of DDAO increases with the degree of protonation,35 which is 1 mmol L−1 for the nonionic state and 2.2 mmol L−1 for the fully cationic state,36 and the protonation of DDAO micelles is easier than that of monomers.35 We present a feasible and consistent microscopic mechanism for DNA–DDAO interaction to explain the working process of our machine model and other related observation. In neutral or alkaline solution (pH ≥ 7), DDAO behaves as a non-ionic surfactant, and its net charge is zero due to its neutral amine oxide head group. In the case, the main attraction between DNA and DDAO is the hydrophobic interaction, which is quite weak and not able to induce DNA compaction. This is corresponding the case shown in Fig. 2(c), in which we can see that the DNA extension curve slopes down slightly but no strong shrinking can be observed, and the condensing force of DNA is close to 0 pN. When pH of solution is low, corresponding the acidic case, DDAO becomes cationic surfactant from charge neutral zwitterionic surfactant. In the case, DDAO concentration plays a very important role for DNA charge neutralization and compaction. When DDAO concentration is low such as less than 2 mM, the surfactant molecules in solution behave as monomers or dimers, are not able to form many micelles. Monomer and dimer have only charged one or two elementary charges, and behave like monovalent or divalent cations. They can neutralize part of negative charge of DNA, but cannot induce DNA compaction. When DDAO concentration goes up, they form more and more micelles in solution, DNA attracts more monomer, dimers and micelles. When the concentration is beyond some critical value, most of DDAO molecules in solution form into micelles carrying many positive charges, corresponding multivalent cations. They are capable to induce DNA compaction and neutralize DNA surface charge or even lead to charge inversion of DDAO–DNA complex. In the case, DNA segments can overcome coulombic repulsive barrier and become attractive for each other due to the conformational entropic effect. The attraction depends on DDAO concentration and its charge by protonation relating with pH in solution. Finally, the attraction of DNA to DDAO micelles is saturated and no more surfactant micelles bind to DNA chains.Because of the saturation of micelles binding to DNA, the condensing force shows the same saturated feature shown in Fig. 2(d). For example, DNA condensing forces at 5 mM and 10 mM DDAO concentration are almost same at pH = 5.5 and 4 respectively. The reason is that DDAO concentration is above the critical concentration forming micelles and the surfactant has been in the ionic state in these acidic environments. However, the condensing force alters dramatically with pH of solution when DDAO concentration is around the critical micelle concentration. For example, DNA condensing force decrease rapidly from 3.8 pN to 1.6 pN when we increase pH of solution from 4 to 5.5 while fixing DDAO concentration at 2 mM. At this DDAO concentration higher pH corresponds to many monomer and dimers, but less micelles in solution, while lower pH means that most of DDAO molecules in solution form micelles and few of them are monomers and dimers.
The same scenario appears in the process of DNA charge compensation by DDAO. We can see that the electrophoretic mobility curves are very close for the cases of high DDAO concentration, such as 5 and 10 mM shown in Fig. 6. At high concentration of DDAO, most of the surfactant molecules form cationic micelles in low pH solution while non-ionic micelles in neutral or alkaline solution. The cationic micelles carry many positive charges resulting in the charge inversion of DNA–DDAO complex when pH of solution is just below 7. In solution of low concentration of DDAO, the surfactant molecules exist in forms of monomer or dimers, carrying only one or two positive charges, which can neutralize part of DNA surface charge but cannot induce its charge inversion. The result is shown in Fig. 6 at low part corresponding to 0.5 mM DDAO concentration. In the case of middle DDAO concentration, such as 1.0 mM, the electrophoretic mobility of DNA–DDAO can be adjusted by pH of solution in a quite large range from negative to positive values, implying the process of charge inversion.
4. Conclusions
In summary, we investigated the interaction between DDAO and DNA, and examined the effects of pH and DDAO concentration on DNA compaction. A pH-controlled DNA–surfactant nano-machine model was constructed by applying DNA condensing feature of amphoteric surfactants. More importantly, the condensing force can be controlled by pH in solution. The maximal contracting force reaches about 5 pN at 10 mM DDAO concentration. The force results from the electrostatic interaction between DDAO and DNA, which is enhanced by lowering pH in solution leading to the protonation of amine oxide head group of DDAO.The model may find some potential applications in manipulation of biomolecules such as DNA and proteins. A possible application is to applying force between different subunits of a protein or different proteins. As shown in Fig. 7(a), we can pull the two components of a protein by altering pH in DDAO solution when a DNA chain has been cross-linked to the two parts.17 If we need to know how close the two parts are, some fluorescence modification can be done in advance, and then measure the signal of fluorescence resonance transfer (FRET) for their distance. Fig. 7(b) shows a similar application but applying force between two proteins. For biomolecule manipulation, we have to maintain their native structures in the procedure. Indeed, proteins might denature at very low pH such as less than 4. However, the most proteins could maintain their native structures in mild acidic conditions.37 On the other hand, DNA is only denatured at very high pH. Thus, the present method for manipulating biomolecules can work in mild acidic and basic condition, being suitable for most biochemical and biophysical applications.
 | ||
| Fig. 7 Schematic diagram of a possible application of DNA–DDAO manipulation for (a) intra- and (b) inter-biomolecules. This is a very primary and tentative proposal. | ||
Author contributions
Yanwei Wang and Guangcan Yang conceived and designed the experiments; Na Li, Zijuan Liao, Shupeng He, Shenhao Huang and Xiao Chen performed the experiments; Yanwei Wang, Na Li and Guangcan Yang analyzed the data; Guangcan Yang, Yanwei Wang and Na Li wrote the paper.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (12074289, 11574232) and Graduate Innovation Fund Project of Wenzhou University (No. 3162019011).Notes and references
- E. Grueso, C. Cerrillos, J. Hidalgo and P. Lopez-Cornejo, Langmuir, 2012, 28, 10968 CrossRef CAS PubMed.
- L. Xu, L. Feng, J. Hao and S. Dong, Colloids Surf., B, 2015, 134, 105 CrossRef CAS PubMed.
- L. Feng, L. Xu, J. Hao and S. Dong, Colloids Surf., A, 2016, 501, 65 CrossRef CAS.
- X. Li, D. Sun, Y. Chen, K. Wang, Q. He and G. Wang, Biochem. Biophys. Res. Commun., 2018, 495, 2559 CrossRef CAS PubMed.
- D. D. Lasic, H. Strey, M. C. Stuart, R. Podgornik and P. M. Frederik, J. Am. Chem. Soc., 1997, 119, 832 CrossRef CAS.
- J. Teychené, D. Didacus-Prins, N. Chouini-Lalanne, V. Sartor and C. Déjugnat, J. Mol. Liq., 2019, 295, 111712 CrossRef.
- J. Clamme, S. Bernacchi, C. Vuilleumier, G. Duportail and Y. Mély, Biochim. Biophys. Acta, Biomembr., 2000, 1467, 347 CrossRef CAS.
- F. Rios, M. Lechuga, M. Fernandez-Serrano and A. Fernandez-Arteaga, Chemosphere, 2017, 171, 324 CrossRef CAS PubMed.
- Y. S. Mel'nikova and B. Lindman, Langmuir, 2000, 16, 5871 CrossRef.
- G. Uphues, Chemische Revue über die Fett- und Harz-Industrie., 1998, 100, 490 CAS.
- P. M. Macdonald, Colloids Surf., A, 1999, 147, 115 CrossRef CAS.
- Y. Wang, P. L. Dubin and H. Zhang, Langmuir, 2001, 17, 1670 CrossRef CAS.
- V. Mengarelli, L. Auvray, D. Pastre and M. Zeghal, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 127 CrossRef CAS PubMed.
- J. Yan, D. Skoko and J. F. Marko, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 011905 CrossRef PubMed.
- H. G. Hansma, R. Golan, W. Hsieh, C. P. Lollo, P. Mullen-Ley and D. Kwoh, Nucleic Acids Res., 1998, 26, 2481 CrossRef CAS PubMed.
- C. Gosse and V. Croquette, Biophys. J., 2002, 82, 3314 CrossRef CAS PubMed.
- N. Y. Tretyakova, A. t. Groehler and S. Ji, Acc. Chem. Res., 2015, 48, 1631 CrossRef CAS PubMed.
- W. B. Fu, X. L. Wang, X. H. Zhang, S. Y. Ran, J. Yan and M. Li, J. Am. Chem. Soc., 2006, 128, 15040 CrossRef CAS PubMed.
- Y. Wang, X. Zhang and G. Yang, RSC Adv., 2015, 5, 29594 RSC.
- Y. Wang, S. Ran and B. Man, Colloids Surf., B, 2011, 83, 61 CrossRef CAS PubMed.
- B. Chen, Y. Wang and G. Yang, RSC Adv., 2019, 9, 41050 RSC.
- K. Besteman, M. A. Zevenbergen, H. A. Heering and S. G. Lemay, Phys. Rev. Lett., 2004, 93, 170802 CrossRef CAS PubMed.
- K. Besteman, K. Van Eijk and S. G. Lemay, Nat. Phys., 2007, 3, 641 Search PubMed.
- Z. Guo, Y. Wang, A. Yang and G. Yang, Soft Matter, 2016, 12, 6669 RSC.
- S. Marchetti, G. Onori and C. Cametti, J. Phys. Chem. B, 2006, 110, 24761 CrossRef CAS PubMed.
- S. Qiu, Y. Wang and B. Cao, Soft Matter, 2015, 11, 4099 RSC.
- T. Gao, W. Zhang and Y. Wang, Polymers, 2019, 11, 337 CrossRef PubMed.
- Y. Wang, R. Wang and B. Cao, Sci. Rep., 2016, 6, 1 CrossRef CAS PubMed.
- Y. Wang, R. Wang and T. Gao, Polymers, 2018, 10, 244 CrossRef PubMed.
- P. Hinterdorfer and Y. F. Dufrene, Nat. Methods, 2006, 3, 347 CrossRef CAS PubMed.
- A. Japaridze, A. Benke, S. Renevey, C. Benadiba and G. Dietler, Macromolecules, 2015, 48, 1860 CrossRef CAS.
- Y. Wang, S. Ran, B. Man and G. Yang, Soft Matter, 2011, 7, 4425 RSC.
- Y. Wang, S. Ran and G. Yang, Sci. Rep., 2014, 4, 1 CAS.
- D. Jing, J. Zhang, L. Ma and G. Zhang, Colloid Polym. Sci., 2004, 282, 1089 CrossRef CAS.
- Y. Imaishi, R. Kakehashi and T. Nezu, J. Colloid Interface Sci., 1998, 197, 309 CrossRef CAS PubMed.
- H. Maeda and R. Kakehashi, Adv. Colloid Interface Sci., 2000, 88, 275 CrossRef CAS PubMed.
- E. P. O'Brien, B. R. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2012, 134, 979 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |