
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReadily-fabricated supported MgO catalysts for efficient and green synthesis of diethyl carbonate from ethyl carbamate and ethanol
Fengjiao Li *ab,
Liguo Wang*c,
Shuang Xuc,
Shuting Liangd and
Ningning Zhanga
*ab,
Liguo Wang*c,
Shuang Xuc,
Shuting Liangd and
Ningning Zhanga
aShenzhen Automotive Research Institute, Beijing Institute of Technology, Shenzhen 518118, Guangdong, China. E-mail: lifengjiao@szari.ac.cn
bCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, Guangdong, China
cKey Laboratory for Green Process and Engineering, National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: lgwang@ipe.ac.cn
dCollege of Chemical and Environmental Engineering, Chongqing University of Arts and Sciences, Yongchuan, Chongqing 402160, China
First published on 26th April 2021
Abstract
Developing cost-effective, high-efficiency and heterogeneous catalysts is of prime importance for the green synthesis of diethyl carbonate (DEC) from ethyl carbamate (EC) and ethanol. Herein, a series of MgO/γ-Al2O3 catalysts were readily fabricated by an impregnation method for DEC synthesis from EC and ethanol. The activities of the as-prepared MgO/γ-Al2O3 catalysts as well as the individual MgO or γ-Al2O3 were first tested in the batch reactor. Among the investigated samples, the MgO/γ-Al2O3 with a MgO loading of 10 wt% (denoted as 10% MgO/γ-Al2O3) exhibited the largest amount of stronger basic sites, and the highest activities with EC conversion of 41.8% and DEC yield of 30.4%, respectively. Furthermore, the DEC yield was greatly boosted to 52.1% with a high DEC selectivity of 93.8% over the 10% MgO/γ-Al2O3 catalyst under the optimized reaction conditions in the fixed bed reactor, outperforming most of the reported catalysts.
1. Introduction
Diethyl carbonate (DEC) is an important linear carbonate in the 21st century, which can be widely used as a carbonylating reagent, alkylating reagent, solvent, electrolyte, fuel additive, etc.1–3 Various methods have been adopted to produce DEC. The earliest one is the phosgenation of ethanol to synthesize DEC in 1941, which involved an extremely hazardous raw material such as phosgene.4 To avoid the usage of the poisonous phosgene, a number of phosgene-free routes such as transesterification of carbonate,5–8 carbonation of ethanol,9,10 coupling reaction from CO2, epoxides and ethanol,11 ethanolysis of CO2,12–16 ethanolysis of urea,17–24 and ethanolysis of ethyl carbamate (EC)24–33 have been developed in the past years. It is worth noting that ethanolysis of urea or EC to produce DEC, which is considered as indirect utilization of CO2 to high value-added products, has been attracting wide attention and interest in the recent years.As shown in eqn (1) and (2), ethanolysis of urea to DEC actually occurs in two steps: (i) ethanolysis of urea to EC intermediate and NH3 by-product, (ii) ethanolysis of EC intermediate to DEC product and NH3 by-product. The latter one is well known as the rate-determining step, which has dominant impacts on DEC synthesis and strongly calls for the development of efficient catalysts. Urea is easy to decompose at high reaction temperatures, usually causing unsatisfactory DEC yield and selectivity via direct ethanolysis of urea. However, the more stable EC intermediate can be easily prepared by ethanolysis of urea with high yield and selectivity even without any catalysts.17 Therefore, ethanolysis of EC is a more economical and promising route for large-scale production of DEC when compared with the direct ethanolysis of urea to DEC.
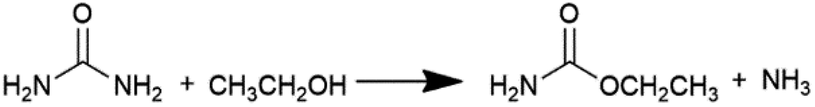 | (1) |
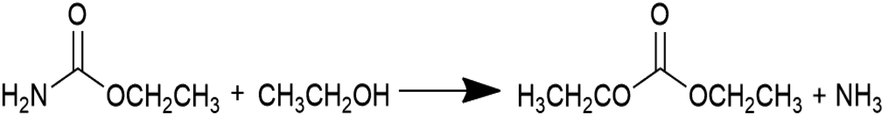 | (2) |
The ethanolysis of EC to DEC is very sluggish, and the obtained DEC yield under conventional conditions is usually less than 5% without the presence of catalysts.26–28,31–33 Actually, the DEC yield could be improved to 22.9% with a DEC selectivity of 48.0% from EC in supercritical ethanol, but the extremely harsh conditions of 573 K and 13.2 MPa will inevitably increase the investment costs on reactors.12 Therefore, the research work is more focused on developing efficient catalysts for DEC synthesis from EC and ethanol. For instance, Zhao et al. applied different transition metal chlorides to investigate the catalysis effects in the batch reactor, and found that the activities followed the sequence of MnCl2 > CoCl2 > ZnCl2 > CdCl2 > NiCl2.33 However, the EC conversion and DEC yield over the highest MnCl2 is only 23% and 11%, respectively. In addition, these transition metal chlorides were homogeneous and normally suffered from the difficulties in separating from the products. In contrast, single or mixed metal oxides such as PbO,25 ZnO24 and zinc-based mixed oxides (e.g., ZnO–PbO,26 and Zn–Al–M based oxides (M = Ca, Mg, La and Y)30), MgO28 and magnesium-based mixed oxides (e.g., waste slag derived oxides,27 transition metal-modified mesoporous Mg–Al mixed oxides,29 and Mg–Al–Y mixed oxides31), and CeO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 32 were more frequently employed in the batch reactors. In general, the DEC yields over the Pb-containing oxides are lower than 21%.25,26 In addition, the post-treatment of the non-environment-friendly Pb element is another challenge for green and large-scale synthesis of DEC. As for the single ZnO or ZnO-containing mixed oxides, the ZnO component was converted to the homogenous Zn(NH3)2(NCO)2 during the reaction, which functioned as the real active composition for DEC synthesis,24,26 and thus would also lead to the problems of separating catalysts. Recently, the Mg-containing oxides have shown great potentials as heterogenous catalysts for efficient DEC synthesis in the batch reactors.27–29,31 It was discovered that the activity of MgO is strongly dependent on the preparation method, and the MgO nanosheet (MgO-SC-450) exhibited the highest catalytic activities among the investigated MgO samples prepared by different methods.28 However, these methods to obtain MgO-based materials appear to be expensive and complicated for practical applications of DEC synthesis. Furthermore, the mechanical strength of MgO is relatively low, and shaping of MgO-based materials is also complex. It can be also noted that the above-mentioned catalysts were used in the batch reactors. Therefore, it still remains a formidable challenge to develop highly effective, low-cost, and readily-fabricated MgO-based catalysts with strong mechanical strength for continuous synthesis of DEC via ethanolysis of EC in the fixed bed reactors.
32 were more frequently employed in the batch reactors. In general, the DEC yields over the Pb-containing oxides are lower than 21%.25,26 In addition, the post-treatment of the non-environment-friendly Pb element is another challenge for green and large-scale synthesis of DEC. As for the single ZnO or ZnO-containing mixed oxides, the ZnO component was converted to the homogenous Zn(NH3)2(NCO)2 during the reaction, which functioned as the real active composition for DEC synthesis,24,26 and thus would also lead to the problems of separating catalysts. Recently, the Mg-containing oxides have shown great potentials as heterogenous catalysts for efficient DEC synthesis in the batch reactors.27–29,31 It was discovered that the activity of MgO is strongly dependent on the preparation method, and the MgO nanosheet (MgO-SC-450) exhibited the highest catalytic activities among the investigated MgO samples prepared by different methods.28 However, these methods to obtain MgO-based materials appear to be expensive and complicated for practical applications of DEC synthesis. Furthermore, the mechanical strength of MgO is relatively low, and shaping of MgO-based materials is also complex. It can be also noted that the above-mentioned catalysts were used in the batch reactors. Therefore, it still remains a formidable challenge to develop highly effective, low-cost, and readily-fabricated MgO-based catalysts with strong mechanical strength for continuous synthesis of DEC via ethanolysis of EC in the fixed bed reactors.
In this work, using the common γ-Al2O3 with strong mechanical strength as the support, a series of MgO/γ-Al2O3 catalysts with different MgO loadings (e.g., 5 wt%, 10 wt% and 15 wt%) were readily prepared by a facile impregnation method for the synthesis of DEC from EC and ethanol. The structures and physiochemical properties of the above supported catalysts, the γ-Al2O3 support and the pure MgO sample were systematically characterized by X-ray diffraction (XRD), X-ray photoelectron spectra (XPS), field-emission scanning electron microscope (FE-SEM), Fourier transform infrared (FTIR) spectra, N2 sorption, and temperature programmed desorption of CO2 (CO2-TPD). The screening and reusability of catalysts was first evaluated in the batch reactor. Afterwards, the effects of reaction conditions on the performances over the most active catalyst were investigated in the fixed bed reactor for continuous synthesis of DEC from EC and ethanol. This work provides a comprehensive insight into a very cost-effective and highly efficient heterogeneous catalyst system of MgO/γ-Al2O3 for continuously manufacturing DEC via the reaction of EC and ethanol.
2. Experimental
2.1 Materials
EC (≥98.0% purity), ethanol (99.7% purity), cyclohexanol (≥ 99.0% purity), Mg(NO3)2·6H2O (≥99.0% purity) and quartz sand (20–40 mesh) were commercially purchased from Sinopharm Chemical Reagent Co., Ltd., China. γ-Al2O3 (20–40 mesh) was obtained from Aladdin Industrial Corporation, China.2.2 Catalyst preparation
The γ-Al2O3 was pre-treated at 873 K for 2 h prior to use. Various MgO supported on γ-Al2O3 catalysts with different MgO loadings of 5 wt%, 10 wt%, and 15 wt% were prepared by an impregnation method. First, the γ-Al2O3 support was impregnated with a desired amount of magnesium nitrate solution for 18 h at room temperature, followed by drying at 383 K for 12 h and calcination in air at 673 K for 3 h. The obtained catalysts were denoted as 5% MgO/γ-Al2O3, 10% MgO/γ-Al2O3 and 15% MgO/γ-Al2O3, respectively. Pure MgO employed in this paper was also prepared by calcining Mg(NO3)2·6H2O at 673 K in air for 3 h for comparison.2.3 Catalyst characterization
X-ray diffraction (XRD) patterns were acquired with a PANalytical Empyrean diffractometer using Cu Kα radiation at 40 kV, 40 mA.Field-emission scanning electron microscopy (FE-SEM) images were collected on an FEI Quanta 250 FEG microscope.
X-ray photoelectron spectra (XPS) tests were conducted on an ESCALAB 250Xi spectrometer with Al Kα radiation (1486.6 eV). The C 1 s peak at 284.8 eV was applied as a reference to calibrate all the peaks.
Fourier transform infrared (FTIR) spectra were recorded on a Tensor 27 Fourier-transform infrared spectrometer from 4000 to 400 cm−1.
N2 sorption analysis experiments were carried out on a Quantachrome Autosorb-1 instrument at 77 K. Prior to the measurements, all the samples were outgassed in vacuum at 573 K for 3 h. The specific surface areas (SBET) of the samples were determined using the N2 adsorption data at a relative pressure (P/P0) from 0.05 to 0.30 according to the Barrett–Emmett–Teller (BET) method. The total pore volume (VP) and the average pore diameter (Dp) of the samples were obtained by analyzing the desorption branch of the isotherms based on the Barrett–Joyner–Halenda (BJH) model.
The CO2 temperature program desorption (CO2-TPD) experiments were performed on an AutoChem II 2920 with a thermal conductivity detector (TCD). Typically, the sample was preheated in helium (25 mL min−1) at 393 K for 60 min, and then cooled to 323 K. Afterwards, the sample was saturated with a mixture of 10 vol% CO2/He (25 mL min−1) for 1 h. The physically-adsorbed CO2 was removed by flushing with helium (25 mL min−1) at 323 K for 30 min. Finally, the temperature was increased to 923 K at a heating rate of 10 K min−1.
2.4 Catalytic tests
Typically, 2.0 g catalyst (20–40 mesh, about 2.6 mL) was charged into the fixed bed reactor and the residue zone of the reactor was filled with quartz sand (20–40 mesh). Then, the reactor was heated to the expected reaction temperature after being pressured with N2 to the desired pressure. Afterwards, the solution containing EC and ethanol was continuously introduced into the reactor after being preheated to 333 K. After the reaction was finished, the products were cooled in a product condenser at 323 K, in order to avoid the formation of ammonium carbonate from the reaction of carbon dioxide and ammonia. Simultaneously, at the top of the condenser, the gas products were released through the pipeline. The liquid products were introduced into the product storage vessel at the bottom of the condenser. Liquid samples were analyzed by the method similar to those in the batch reactor.
3. Results and discussion
3.1 Catalyst characterization
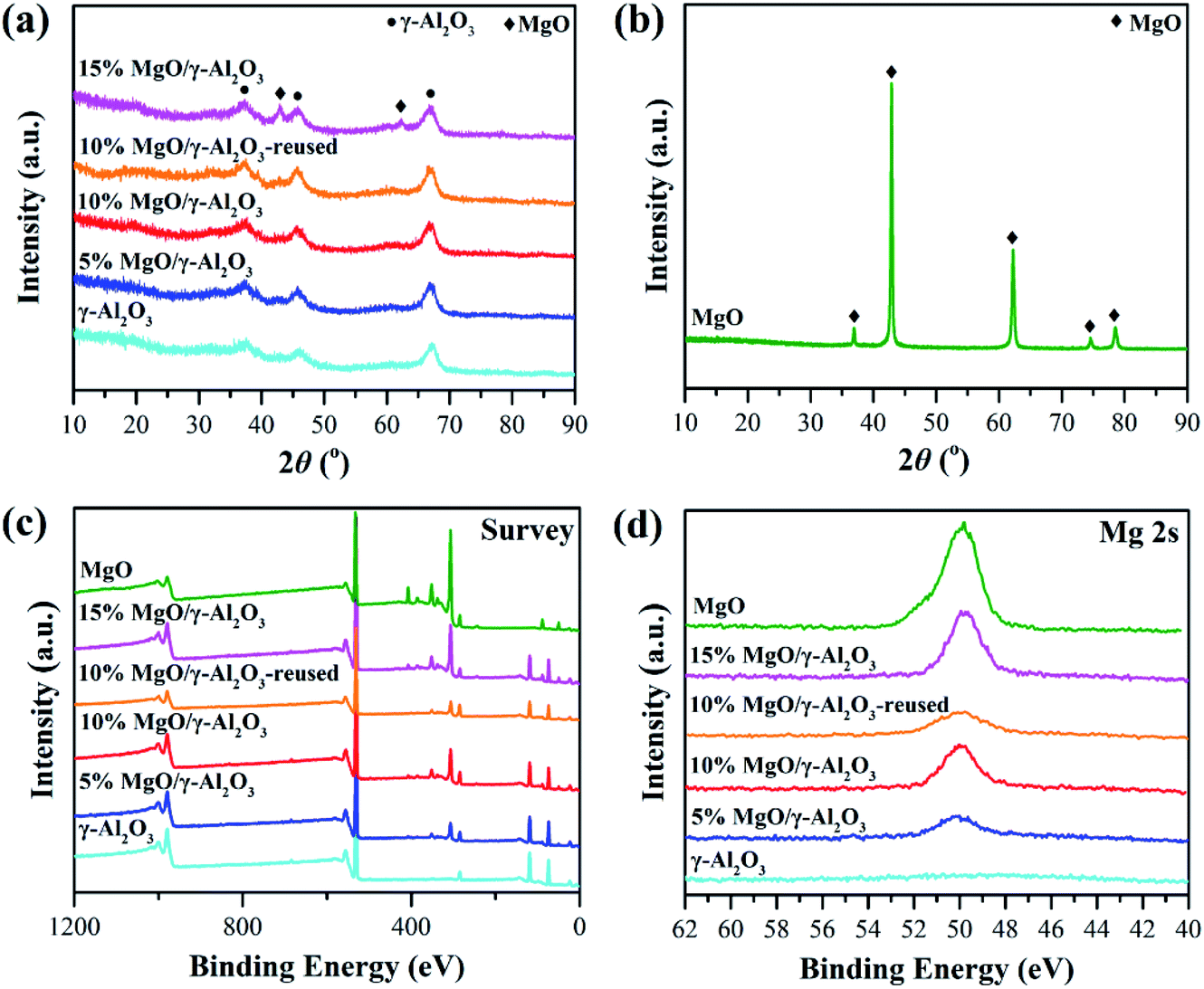 | ||
| Fig. 1 XRD patterns (a and b), XPS survey spectra (c) and Mg 2s high resolution XPS spectra (d) of the γ-Al2O3, 5% MgO/γ-Al2O3, 10% MgO/γ-Al2O3, 15% MgO/γ-Al2O3, MgO and 10% MgO/γ-Al2O3-reused samples. | ||
 | ||
| Fig. 2 SEM images of (a) MgO, (b) γ-Al2O3, (c) 5% MgO/γ-Al2O3, (d) 10% MgO/γ-Al2O3, (e) 15% MgO/γ-Al2O3 and (f) 10% MgO/γ-Al2O3-reused samples. | ||
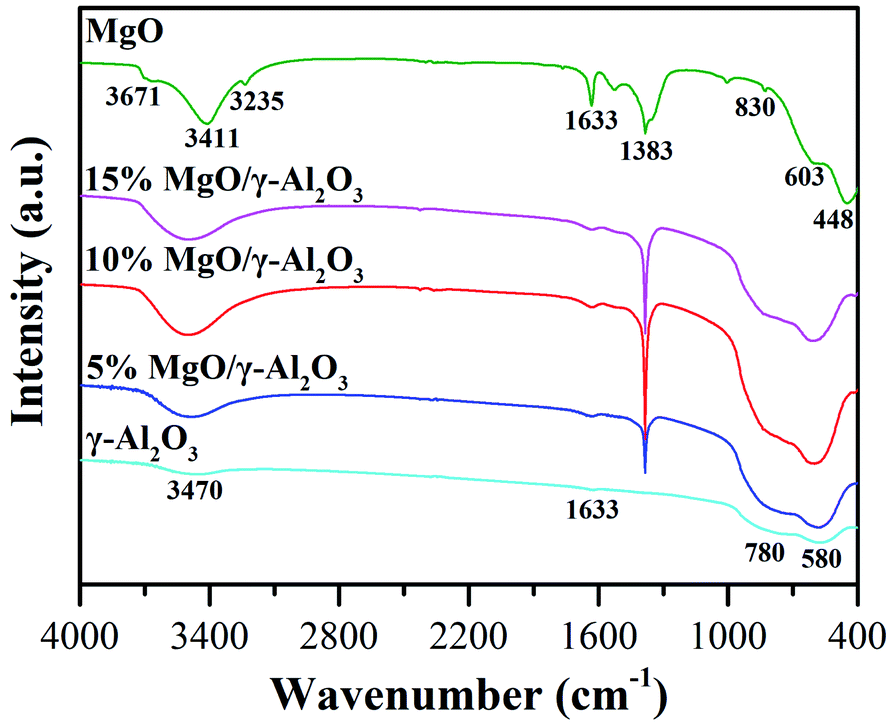 | ||
| Fig. 3 FTIR of the γ-Al2O3, 5% MgO/γ-Al2O3, 10% MgO/γ-Al2O3, 15% MgO/γ-Al2O3 and MgO samples. | ||
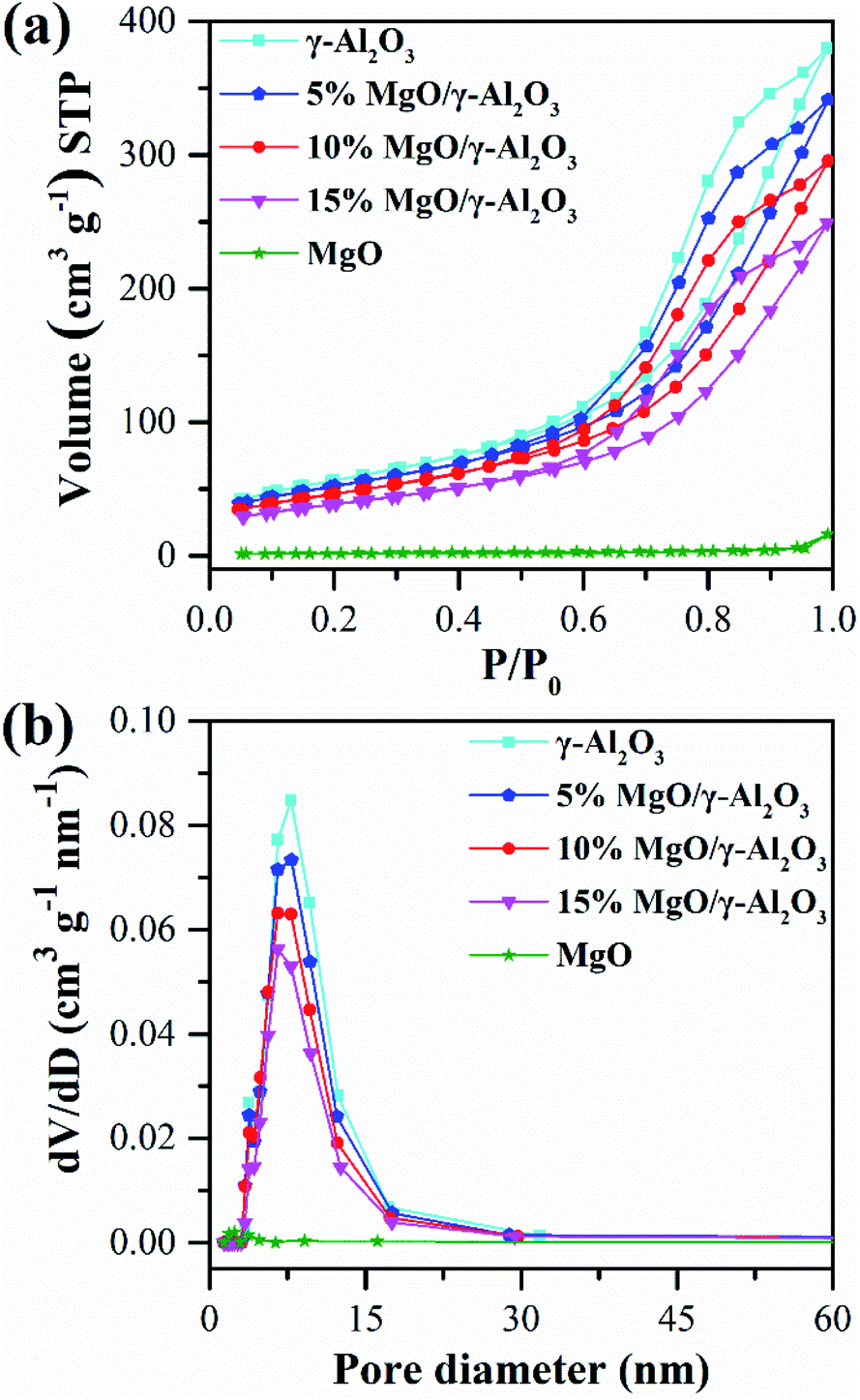 | ||
| Fig. 4 N2 adsorption and desorption isotherms (a) and the corresponding pore size distributions (b) of the γ-Al2O3, 5% MgO/γ-Al2O3, 10% MgO/γ-Al2O3, 15% MgO/γ-Al2O3 and MgO samples. | ||
| Sample | SBET (m2 g−1) | VP (cm3 g−1) | Dp (nm) |
|---|---|---|---|
| γ-Al2O3 | 203.0 | 0.59 | 11.6 |
| 5% MgO/γ-Al2O3 | 187.5 | 0.53 | 11.3 |
| 10% MgO/γ-Al2O3 | 167.3 | 0.46 | 11.0 |
| 15% MgO/γ-Al2O3 | 138.3 | 0.39 | 11.2 |
| MgO | 5.5 | 0.02 | — |
Specifically, the 10% MgO/γ-Al2O3 sample possesses a moderate surface area of 167.3 m2 g−1 with a pore volume of 0.46 cm3 g−1 and an average pore diameter of 11.0 nm. The enlarged specific surface areas of the supported MgO/γ-Al2O3 samples were beneficial for the active sites to access to the reactants, thus facilitating the enhancement of the EC conversion and DEC yield over the supported MgO/γ-Al2O3 samples.
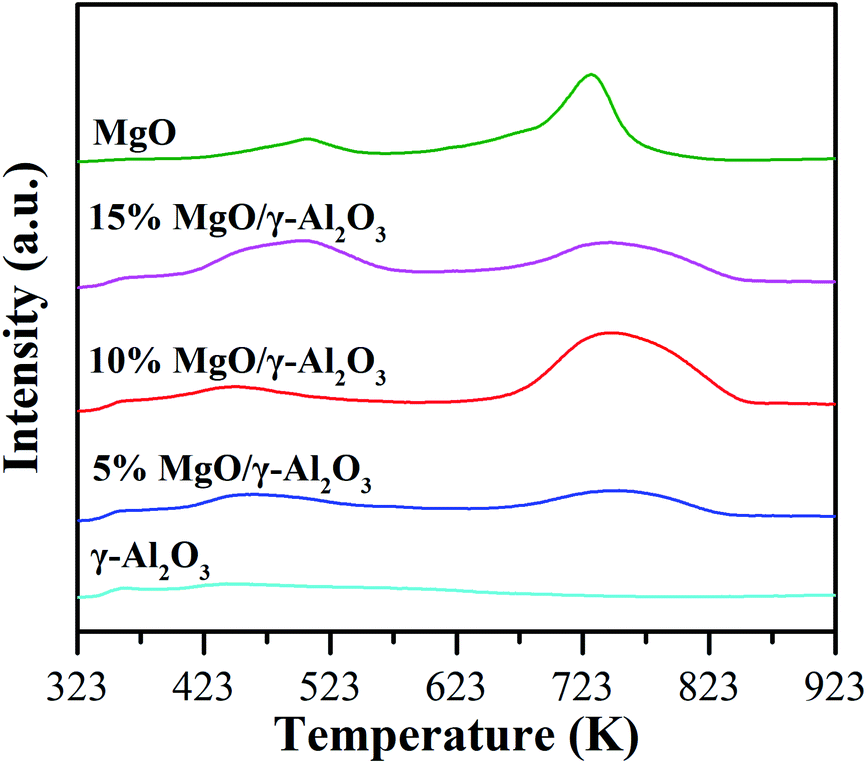 | ||
| Fig. 5 CO2-TPD profiles of the γ-Al2O3, 5% MgO/γ-Al2O3, 10% MgO/γ-Al2O3, 15% MgO/γ-Al2O3 and MgO samples. | ||
| Sample | Number of basic sites (μmol g−1) | |||
|---|---|---|---|---|
| Weak | Medium | Strong | Total | |
| γ-Al2O3 | 221.5 | 276.9 | 22.1 | 520.5 |
| 5% MgO/γ-Al2O3 | 380.4 | 538.1 | 617.1 | 1535.6 |
| 10% MgO/γ-Al2O3 | 270.5 | 301.9 | 1054.3 | 1626.7 |
| 15% MgO/γ-Al2O3 | 426.6 | 881.7 | 888.4 | 2196.7 |
| MgO | 72.6 | 334.5 | 593.2 | 1000.3 |
3.2 DEC synthesis in the batch reactor
The performances of the γ-Al2O3 support, the three supported MgO catalysts, and the individual MgO sample for DEC synthesis from EC and ethanol were first evaluated in the batch reactor, and the results are summarized in Table 3. The EC conversion and DEC yield over the γ-Al2O3 support are 8.3% and 3.6%, respectively (entry 2), which are very close to those in the blank experiment (entry 1), indicating that the γ-Al2O3 support was quite inert for DEC synthesis from EC and ethanol. In addition, the pure MgO sample obtained by simple calcination of Mg(NO3)2·6H2O at 673 K in air also showed inferior activities towards this reaction (9.5% of EC conversion and 4.0% of DEC yield, entry 6), which is worse than the reported MgO catalysts by thermal decomposition (i.e., MgO-MCBP-450 and MgO-MH-450) and precipitation methods (i.e., MgO-SC-450, MgO-SH-450 and MgO-NH-450),28 also suggesting that the preparation method had a significant impact on the activities of MgO. However, the three supported MgO/γ-Al2O3 catalysts obtained by the facile impregnation method exhibited much higher catalytic abilities than the γ-Al2O3 support or the pure MgO sample. As shown in entries 3–5, both the EC conversion and the DEC yield increased with the MgO loading increasing to 10 wt%, and then dropped when the MgO loading was further increased to 15 wt%. It can be clearly found that in the batch reactor, the activities of the supported MgO catalysts follow the similar order with the total amount of strong basic sites. Specifically, the 10% MgO/γ-Al2O3 catalyst with the largest total amount of strong basic sites exhibited the highest EC conversion of 41.8% and the highest DEC yield of 30.4%, respectively, while the 5% MgO/γ-Al2O3 catalyst with the smallest total amount of strong basic sites showed the lowest activities. The total amount of strong basic sites over the pure MgO sample is close to that over the 5% MgO/γ-Al2O3 catalyst, but the activities of the former one is much lower than the latter one, indicating that the high dispersion of MgO on the γ-Al2O3 support and the enlarged surface area were also beneficial for the enhanced activities of the supported MgO catalysts. More importantly, the supported MgO catalysts obviously possessed stronger basic sites than the pure MgO sample in the region of desorption temperature higher than 673 K (Fig. 5), therefore, it can be concluded that larger amount of stronger basic sites of the supported MgO catalysts actually played the very crucial role in the DEC synthesis from EC and ethanol.| Entry | Catalyst | EC conv. (%) | DEC yield (%) | DEC sel. (%) | NEEC yield (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 473 K, 34 mmol of EC, 0.34 mol of ethanol, 10 wt% of catalyst amount (based on the weight of EC), and 3 h. | |||||
| 1 | None | 6.1 | 4.4 | 72.1 | — |
| 2 | γ-Al2O3 | 8.3 | 3.6 | 43.6 | 1.4 |
| 3 | 5% MgO/γ-Al2O3 | 25.0 | 10.6 | 42.5 | 2.5 |
| 4 | 10% MgO/γ-Al2O3 | 41.8 | 30.4 | 72.6 | 1.6 |
| 5 | 15% MgO/γ-Al2O3 | 32.2 | 19.3 | 59.9 | 2.5 |
| 6 | MgO | 9.5 | 4.0 | 41.5 | 1.3 |
In a word, the above results suggested that the outstanding activities of the supported MgO catalysts could be attributed to the co-contribution of the high dispersion of MgO on the γ-Al2O3 support, the enlarged surface area, and larger amount of stronger basic sites.
The reusability of the 10% MgO/γ-Al2O3 catalyst for DEC synthesis in the batch reactor was also investigated for practical application. After the reaction, the catalyst was centrifuged, washed, and directly reused for the next cycle, and the results are shown in Fig. 6. Obviously, the 10% MgO/γ-Al2O3 catalyst could be recycled and reused at least five times without significant variation in the activity. Furthermore, XRD, XPS and SEM studies of the 10% MgO/γ-Al2O3 catalyst after the fifth cycle (denoted as 10% MgO/γ-Al2O3-reused) were also performed, in order to gain a deep insight into the stability of the 10% MgO/γ-Al2O3 catalyst. As exhibited in Fig. 1a, compared with the fresh 10% MgO/γ-Al2O3 catalyst, there were no obvious changes to the crystallite size and crystal phase after the fifth reuse.
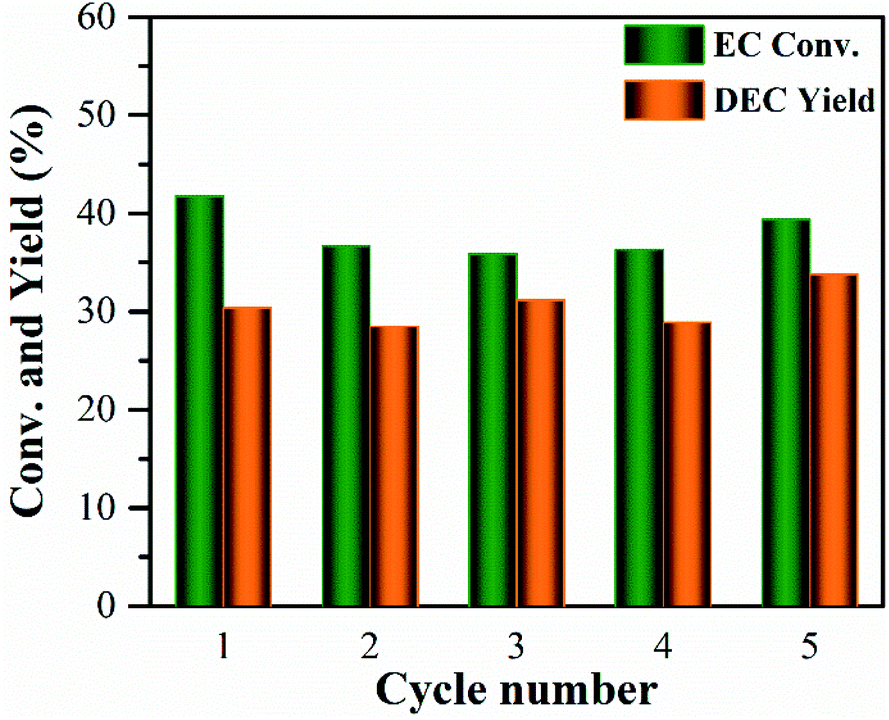 | ||
| Fig. 6 The reusability of the 10% MgO/γ-Al2O3 catalyst for DEC synthesis from EC and ethanol in the batch reactor. Reaction conditions: 473 K, 34 mmol of EC, 0.34 mol of ethanol, 10 wt% of catalyst amount (based on the weight of EC), and 3 h. | ||
XPS also shows no distinct changes between the fresh and the reused 10% MgO/γ-Al2O3 catalyst, indicating that no new species were formed after reuse (Fig. 1c and d). In addition, the 10% MgO/γ-Al2O3-reused catalyst showed similar morphologies (Fig. 3f) in comparison with the fresh one. Therefore, it could be concluded that the 10% MgO/γ-Al2O3 catalyst has a remarkable structure stability and reusability for DEC synthesis from EC and ethanol.
3.3 DEC synthesis in the fixed-bed reactor
Despite of the high activities of the 10% MgO/γ-Al2O3 catalyst in the batch reactor, the NH3 by-product was accumulated in the batch reactor and would restrict the shift of the reaction equilibrium to DEC synthesis, which is disadvantageous to the improvement of DEC yield. Therefore, the performances of the 10% MgO/γ-Al2O3 catalyst for continuous synthesis of DEC from EC and ethanol were further investigated in the fixed-bed reactor, which could efficiently shift the reaction equilibrium to the side of DEC synthesis by removing the ammonia by-product simultaneously from the reactor. In addition, this reactor would also alleviate the problems of the undesirable side reactions, thus helping to improve the DEC yield and selectivity.At first, the experiment only with quartz sand filled into the whole tube of the fixed bed reactor was conducted under the reaction condition of 463 K, 1.6 MPa, EC solution liquid hourly space velocity (LHSV) of 2.02 h−1, and the molar ratio of ethanol to EC (shorted as n(EtOH)/n(EC)) of 10. As shown in Fig. 7a, in the absence of the 10% MgO/γ-Al2O3 catalyst, both the EC conversion and the DEC yield are extremely low, which are 3.7% and 1.9%, respectively, demonstrating that the quartz sand is inactive towards this reaction. However, after the 10% MgO/γ-Al2O3 catalyst was loaded into the reaction zone in the fixed bed reactor, the EC conversion and the DEC yield were greatly boosted to 35.6% and 30.3% under the same reaction conditions, also re-confirming the superior activities of the 10% MgO/γ-Al2O3 catalyst (Fig. 7a). Furthermore, the effects of reaction conditions such as reaction pressure, reaction temperature, and EC solution LHSV on the catalytic performances of the 10% MgO/γ-Al2O3 catalyst for continuous synthesis of DEC from EC and ethanol in the fixed-bed reactor were also investigated.
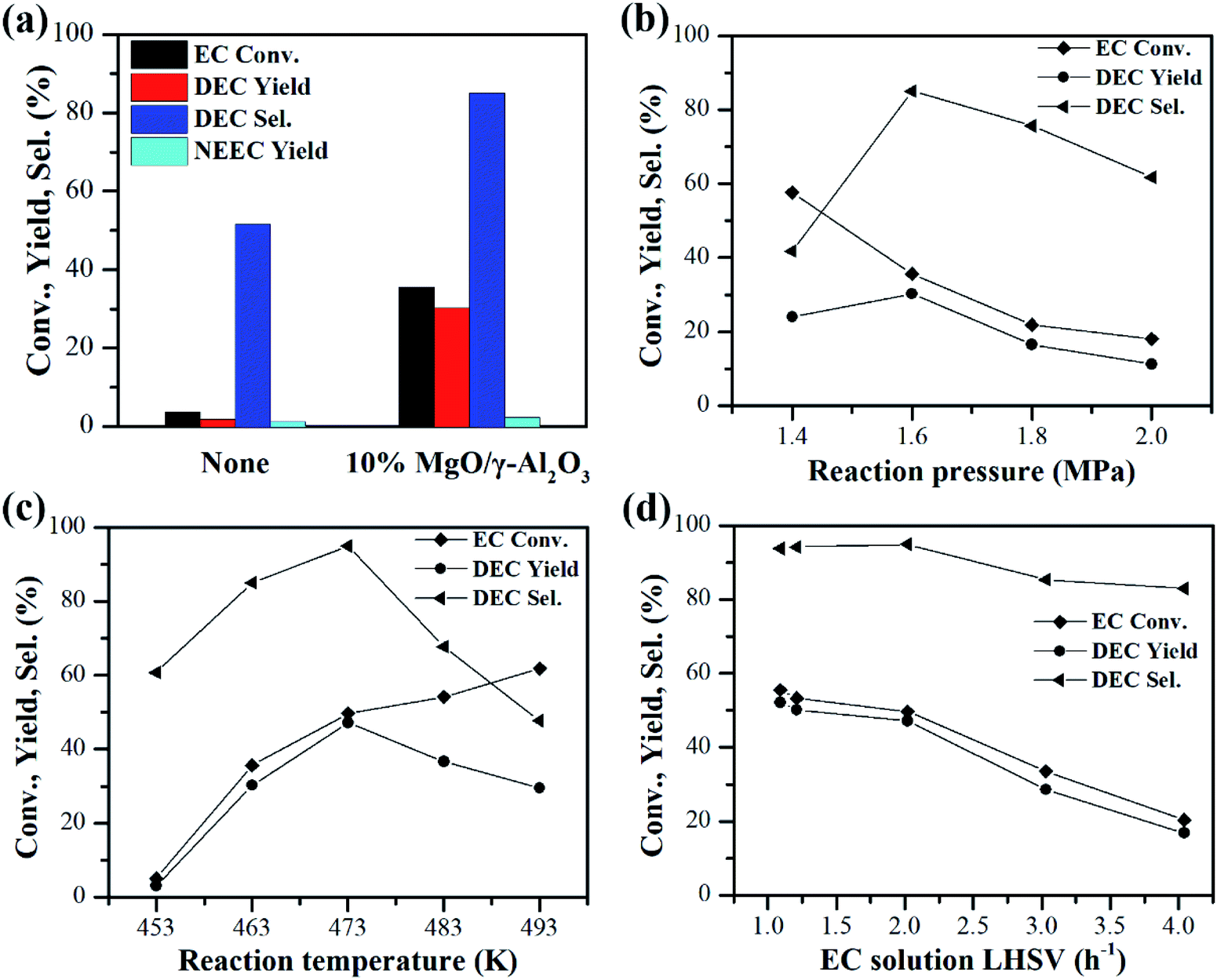 | ||
| Fig. 7 Effects of reaction conditions on the performances of the 10% MgO/γ-Al2O3 catalyst for continuous synthesis of DEC from EC and ethanol in the fixed bed reactor, 2 g of catalyst, n(EtOH)/n(EC) of 10. (a) Comparison before and after the addition of the 10% MgO/γ-Al2O3 catalyst. Reaction conditions: 463 K, 1.6 MPa, EC solution LHSV of 2.02 h−1; (b) effect of reaction pressure. Reaction conditions: 463 K, EC solution LHSV of 2.02 h−1; (c) effect of reaction temperature. Reaction conditions: 1.6 MPa, EC solution LHSV of 2.02 h−1; (d) effect of EC solution LHSV. Reaction conditions: 473 K, 1.6 MPa. | ||
The effect of reaction pressure is shown in Fig. 7b. Theoretically, low pressure is beneficial for the ammonia by-product to escape from the reaction system.40 As a result, the EC conversion gradually increased with the reaction pressure decreasing from 2.0 MPa to 1.4 MPa. However, the DEC yield first increased with the reaction pressure increasing from 1.4 MPa to 1.6 MPa, and then dropped from 1.6 MPa to 2.0 MPa. In addition, 85.0% of DEC selectivity was obtained at 1.6 MPa, which was higher than that in the batch reactor at a higher reaction temperature of 473 K (72.6% of DEC selectivity, entry 4 in Table 3). Therefore, the suitable reaction pressure for the DEC synthesis in the fixed bed reactor should be 1.6 MPa.
The effect of reaction temperature is displayed in Fig. 7c. The EC conversion increased with the reaction temperature increasing from 453 K to 493 K. In contrast, the DEC yield first increased with the reaction temperature increasing from 453 K to 473 K, and then dropped after 473 K. As disclosed by previous work, DEC could be decomposed into CO2 and diethyl ether at temperatures of 453–513 K over catalysts.41 Therefore, the rapid drop of DEC yields from 483 to 493 K is largely ascribed to the decomposition of the DEC to carbon dioxide and diethyl ether at too high temperatures. It should be noted that the maximal DEC yield of 47.1% with a DEC selectivity higher than 90% was achieved at 473 K. Therefore, the optimized reaction temperature for the DEC synthesis in the fixed-bed reactor was 473 K.
The effect of EC solution LHSV on the continuous synthesis of DEC from EC and ethanol over the 10% MgO/γ-Al2O3 catalyst was displayed in Fig. 7d. Both the EC conversion and the DEC yield decreased with the EC solution LHSV increasing from 1.09 h−1 to 4.04 h−1. A higher EC solution LHSV indicated that a larger amount of EC was used. As the amount of catalyst is constant, the low EC solution LHSV of 1.09 h−1 was advantageous to obtain high DEC yield.
It should be noted that under the optimized reaction conditions of 473 K, 1.6 MPa, EC solution LHSV of 1.09 h−1, n(EtOH)/n(EC) of 10, 2 g of 10% MgO/γ-Al2O3 catalyst, EC conversion of 55.5%, a maximum DEC yield of 52.1% and a high DEC selectivity of 93.8% were achieved in the fixed bed reactor, which are superior to most of the reported catalysts.24–27,30,32,33
The catalytic stability of the 10% MgO/γ-Al2O3 catalyst was also tested in the fixed-bed reactor. As presented in Fig. 8, with the reactants continuously pumping into the fixed bed reactor, no obvious changes in EC conversion and DEC yield were observed after 7 h of test over the 10% MgO/γ-Al2O3 catalyst, also indicative of the outstanding stability of the 10% MgO/γ-Al2O3 catalyst for DEC synthesis from EC and ethanol.
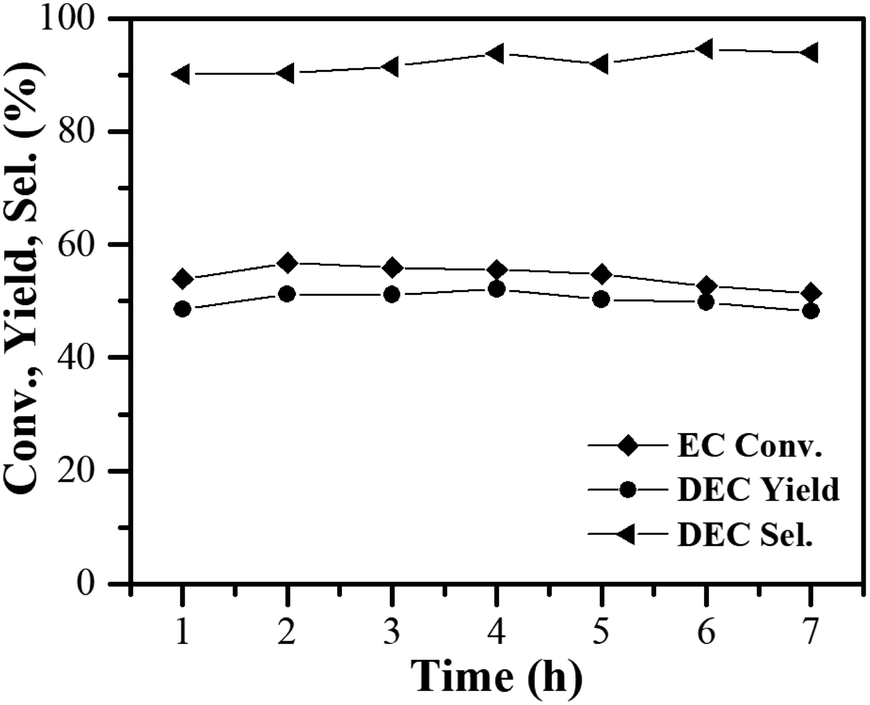 | ||
| Fig. 8 Continuous synthesis of DEC from EC and ethanol over time using the 10% MgO/γ-Al2O3 catalyst. Reaction conditions: 473 K, 1.6 MPa, EC solution LHSV of 1.09 h−1, n(EtOH)/n(EC) of 10, 2 g catalyst. | ||
Based on the above results, it could be concluded that the readily-fabricated 10% MgO/γ-Al2O3 catalyst was a very promising candidate as a highly-efficient, cost-effective and heterogenous catalyst for green and continuous synthesis of DEC using low-cost and environmental-benign EC and ethanol as raw materials, which is also an efficient way to indirectly utilize CO2 to synthesize high value-added chemicals.
4. Conclusions
In this work, low-cost and effective supported MgO catalysts readily fabricated by an impregnation method were employed for green synthesis of DEC from EC and ethanol. The 10% MgO/γ-Al2O3 catalyst presented the highest activities in the fixed bed reactor with a maximum DEC yield of 52.1% and a high DEC selectivity of 93.8%, respectively, which are superior to the individual γ-Al2O3 or MgO, and most of the reported catalysts. The outstanding performances of the 10% MgO/γ-Al2O3 catalyst could be attributed to the co-contribution of the high dispersion of MgO on the γ-Al2O3 support, the enlarged surface area, and larger amount of stronger basic sites. This work has provided a readily-fabricated and economical catalyst system to efficiently synthesize high value-added DEC indirectly from CO2.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by National Natural Science Foundation of China (No. 21706162).Notes and references
- K. Shukla and V. C. Srivastava, RSC Adv., 2016, 6, 32624–32645 RSC.
- K. Shukla and V. C. Srivastava, Catal. Rev., 2017, 59, 1–43 CrossRef CAS.
- S. Huang, B. Yan, S. Wang and X. Ma, Chem. Soc. Rev., 2015, 44, 3079–3116 RSC.
- I. E. Muskat, F. Strain, US Pat., 2379250, 1941 Search PubMed.
- H. Iida, R. Kawaguchi and K. Okumura, Catal. Commun., 2018, 108, 7–11 CrossRef CAS.
- S.-J. Wang, S.-H. Cheng, P.-H. Chiu and K. Huang, Ind. Eng. Chem. Res., 2014, 53, 5982–5995 CrossRef CAS.
- C. Murugan and H. C. Bajaj, Fuel Process. Technol., 2011, 92, 77–82 CrossRef CAS.
- K. Shukla and V. C. Srivastava, Ind. Eng. Chem. Res., 2018, 57, 12726–12735 CrossRef CAS.
- H. Wang, Y. Xiang, M. Guo, J. Su, G. Wang, W. Cui and Z. Deng, Energy Fuels, 2020, 34, 8697–8706 CrossRef CAS.
- S. Huang, P. Chen, B. Yan, S. Wang, Y. Shen and X. Ma, Ind. Eng. Chem. Res., 2013, 52, 6349–6356 CrossRef CAS.
- L. Wang, H. Li, S. Xin, P. He, Y. Cao, F. Li and X. Hou, Appl. Catal., A, 2014, 471, 19–27 CrossRef CAS.
- L.-C. Zhao, Z.-Q. Hou, C.-Z. Liu, Y.-Y. Wang and L.-Y. Dai, Chin. Chem. Lett., 2014, 25, 1395–1398 CrossRef CAS.
- B.-Y. Yu, P.-J. Wu, C.-C. Tsai and S.-T. Lin, J. CO2 Util., 2020, 41, 101254 CrossRef CAS.
- O. Arbeláez, E. Hernández, L.-M. González, F. Bustamante and A.-L. Villa, Chem. Eng. Technol., 2019, 42, 1135–1143 CrossRef.
- M. Buchmann, M. Lucas and M. Rose, Catal. Sci. Technol., 2021, 11, 1940–1948 RSC.
- J. Wang, Z. Hao and S. Wohlrab, Green Chem., 2017, 19, 3595–3600 RSC.
- S. Xin, L. Wang, H. Li, K. Huang and F. Li, Fuel Process. Technol., 2014, 126, 453–459 CrossRef CAS.
- P. Wang, S. Liu, F. Zhou, B. Yang, A. S. Alshammari and Y. Deng, RSC Adv., 2015, 5, 19534–19540 RSC.
- K. Shukla and V. C. Srivastava, Fuel Process. Technol., 2017, 161, 116–124 CrossRef CAS.
- J. Zhang, T. Yan, Y. Yang, J. Sun, Y. Lin and M. Wei, Chin. J. Catal., 2019, 40, 515–522 CrossRef CAS.
- A. Angelini, A. Dibenedetto, D. Curulla-Ferré and M. Aresta, RSC Adv., 2015, 5, 88401–88408 RSC.
- A. Dibenedetto, A. Angelini, M. Aresta, S. Fasciano, M. E. Cucciolito, F. Ruffo, B. M. Aresta, D. Curulla-Ferré and E. De Giglio, Appl. Catal., A, 2015, 493, 1–7 CrossRef CAS.
- T. Yan, W. Bing, M. Xu, Y. Li, Y. Yang, G. Cui, L. Yang and M. Wei, RSC Adv., 2018, 8, 4695–4702 RSC.
- W. Zhao, W. Peng, D. Wang, N. Zhao, J. Li, F. Xiao, W. Wei and Y. Sun, Catal. Commun., 2009, 10, 655–658 CrossRef CAS.
- L. Guo, X. Zhao, H. An and Y. Wang, Chin. J. Catal., 2012, 33, 595–600 CrossRef CAS.
- H. An, X. Zhao, L. Guo, C. Jia, B. Yuan and Y. Wang, Appl. Catal., A, 2012, 433, 229–235 CrossRef.
- L. Wang, H. Li, S. Xin and F. Li, Catal. Commun., 2014, 50, 49–53 CrossRef CAS.
- F. Li, H. Li, L. Wang, P. He and Y. Cao, Catal. Sci. Technol., 2015, 5, 1021–1034 RSC.
- D. Wang, X. Zhang, C. Liu, T. Cheng, W. Wei and Y. Sun, Appl. Catal., A, 2015, 505, 478–486 CrossRef CAS.
- K. Shukla and V. C. Srivastava, Catal. Lett., 2017, 147, 1891–1902 CrossRef CAS.
- D. Wang, X. Zhang, D. Zhou, S. Liu and W. Wei, Fuel Process. Technol., 2017, 167, 404–415 CrossRef CAS.
- Y. Ma, H. Wang, L. Wang and H. Li, J. Environ. Manage., 2019, 232, 952–956 CrossRef CAS PubMed.
- W. Zhao, D. Feng, J. Nong, G. Cao, X. Liu, Z. Tang and Y. Chen, React. Kinet., Mech. Catal., 2016, 117, 639–654 CrossRef CAS.
- D.-e. Jiang, B. Zhao, Y. Xie, G. Pan, G. Ran and E. Min, Appl. Catal., A, 2001, 219, 69–78 CrossRef CAS.
- J. Gangwar, B. K. Gupta, S. K. Tripathi and A. K. Srivastava, Nanoscale, 2015, 7, 13313–13344 RSC.
- M. Aazza, H. Ahlafi, H. Moussout, C. Mounir, A. Fadel and A. Addad, J. Environ. Chem. Eng., 2020, 8, 103707–103718 CrossRef CAS.
- A. Ansari, A. Ali, M. Asif and Shamsuzzaman, New J. Chem., 2018, 42, 184–197 RSC.
- J. P. Dhal, M. Sethi, B. G. Mishra and G. Hota, Mater. Lett., 2015, 141, 267–271 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2015, 504, 103–109 CrossRef CAS.
- D. Wang, X. Zhang, W. Wei and Y. Sun, Chem. Eng. Technol., 2012, 35, 2183–2188 CrossRef CAS.
- M. Selva, M. Fabris and A. Perosa, Green Chem., 2011, 13, 863–872 RSC.
| This journal is © The Royal Society of Chemistry 2021 |