
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA biocompatible polypyrrole membrane for biomedical applications†
Shujun Cuiabc,
Jifu Mao d,
Mahmoud Rouabhiaa,
Saïd Elkoune and
Ze Zhang*bc
d,
Mahmoud Rouabhiaa,
Saïd Elkoune and
Ze Zhang*bc
aResearch Group on Oral Ecology, Faculty of Dentistry, Université Laval, Québec (QC), Canada. E-mail: Mahmoud.rouabhia@fmd.ulaval.ca
bDepartment of Surgery, Faculty of Medicine, Université Laval, Québec (QC), Canada. E-mail: Ze.Zhang@chg.ulaval.ca
cDivision of Regenerative Medicine, Research Center of CHU de Québec-Université Laval, Québec (QC), Canada
dKey Laboratory of Textile Science & Technology of Ministry of Education and College of Textiles, Donghua University, Shanghai, China
eDepartment of Mechanical Engineering, Université de Sherbrooke, Sherbrooke (QC), Canada
First published on 10th May 2021
Abstract
Polypyrrole (PPy) is the most widely investigated electrically conductive biomaterial. However, because of its intrinsic rigidity, PPy has only been used either in the form of a composite or a thin coating. This work presents a pure and soft PPy membrane that is synergically reinforced with the electrospun polyurethane (PU) and poly-L-lactic acid (PLLA) fibers. This particular reinforcement not only renders the originally rather fragile PPy membrane easy to manipulate, it also prevents the membrane from deformation in an aqueous environment. Peel and mechanical tests confirmed the strong adhesion of the fibers and the significantly increased tensile strength of the reinforced membrane. Surface electrical conductivity and long-term electrical stability were tested, showing that these properties were not affected by the reinforcement. Surface morphology and chemistry were analyzed with scanning electron spectroscopy (SEM), X-ray photoelectron spectroscopy (XPS), and Fourier transform infrared spectroscopy (FTIR). Material thermal stability was investigated with thermogravimetric analysis (TGA). Finally, the adhesion and proliferation of human skin keratinocytes on the membrane were assessed by Hoechst staining and the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay. In conclusion, this membrane proves to be the first PPy-based soft conductive biomaterial that can be practically used. Its electrical conductivity and cytocompatibility promise a wide range of biomedical applications.
1. Introduction
Polypyrrole (PPy) is an inherently conductive polymer formed through consecutive couplings of the oxidized heterocyclic pyrrole (Py) monomers and oligomers.1 Unlike polyacetylene, PPy is hardly crystalline; and instead of forming a planar structure, PPy grows and aggregates layer by layer,2,3 making it low in tensile strength, with a lack of plasticity and elasticity, and poor in processability.4Meanwhile, due to its electrical conductivity, environmental stability and excellent biocompatibility,5 PPy has been intensively investigated as a biomaterial.6 Considering its potential applications in electrical stimulation including in clinic therapies,7–11 PPy also becomes an interesting scaffolding material or an interface to mediate electrical stimulation to cells or tissues, such as in cancer therapy or tissue regeneration.12,13 All of these, however, are based on the promise to have a processable PPy scaffold with adequate mechanical strength.14
Abundant research work has concentrated on improving the mechanical properties of PPy by combining it with supporting polymers.6,15 A commonly used method is to blend PPy particles with supporting polymers including in situ polymerization of PPy in a polymer solution, followed by solution casting and solvent evaporation to obtain a composite PPy membrane. Polymers that are frequently used include polylactide (PLA),15 polyurethane (PU),16–18 chitosan19 and polycaprolactone (PCL).20 With this approach, normally only a small percentage of the PPy particles are dispersed in the supporting polymer, which should be adequate to make the composite conductive but not too high to make the mechanical property weak. The key point of this technique is to homogeneously distribute the PPy particles and, at the same time, form a PPy network to conduct electricity. Another widely used method is surface modification.21–23 For example, soaking a polyester fabric in pyrrole monomer solution followed by oxidation polymerization can lead to a conductive textile, with a thin layer of PPy coated on the individual fibers if pyrrole is not excessive to fill the microporous structure.
Other nonconventional technologies have also been used. For example, electrospinning a polymer/PPy solution can generate fibrous conductive scaffolds.24,25 In situ vapor-phase polymerization of pyrrole monomers in cellulose gels followed by supercritical drying can produce a conductive aerogel.26 A flexible PPy/cellulose composite membrane was prepared through electrochemical polymerization by firstly sputter-coating a paper with platinum.27 The work of PPy and its composites in biomedical applications have been extensively reviewed recently.28–30
However, the above-mentioned methods have two major limitations. In the approach of using PPy particles as fillers, conductivity of the composite is significantly lower than that of the pure PPy, for the supporting polymers are normally insulators. In the second approach, i.e., surface coating, although the thin PPy coating can be as conductive as the pure PPy, this conductivity deteriorates rapidly once used in an aqueous environment due to dedoping.3 The lowering of conductivity in either approach can be as significant as 3 to 4 orders.
The free standing soft PPy membrane synthesized via a template-assisted interfacial polymerization technique largely addressed these two problems.31 This PPy membrane has a distinctive physical structure that, for the first time, makes the pure PPy membrane soft and flexible. However, the tensile strength of this membrane is poor, with an elastic modulus of 3.4 MPa and an ultimate elongation 2.5%, making it easily damaged if not handled properly. Because free standing pure PPy membranes are hardly available and non-extendable, the tensile strength of pure PPy in literature is very rare. One group reported a PPy foam with a tensile strength of 18 kPa at 3.3% elongation.32
In this study, we aimed to improve the mechanical strength of the free-standing soft PPy membrane without losing its other properties. For that purpose, polyurethane (PU) and poly(D,L-lactide) (PLLA) were sequentially electrospun onto the PPy membrane. The final membrane displayed unchanged electrical conductivity, significantly improved tensile strength and handling property, and an excellent cytocompatibility.
2. Materials and methods
2.1 Materials
Pyrrole (98%, Alfa Aesar), ferric chloride hexahydrate (FeCl3·6H2O, 98%, Sigma-Aldrich), methyl orange (MO, ACS reagent, dye content 85%, Sigma-Aldrich), chloroform (CHCl3, HPLC grade, Fisher Scientific), PU (Tecoflex™ SG-80A, Thermedics), PLLA (intrinsic viscosity 1.3, Hycail). All chemicals except pyrrole were used as received. Pyrrole was distilled and kept at 4 °C before use.2.2 Synthesis of PPy membrane
PPy membranes were synthesized according to the technique published in our previous work.31 Briefly, 3 mL pyrrole was dissolved in 150 mL CHCl3 and maintained at 4 °C for at least 1 h before use. The amount of 18.2 g of FeCl3·6H2O and 5 mM of MO were simultaneously dissolved into 320 mL of deionized water, and stirred for 30 min to obtain a FeCl3/MO complex. The interfacial polymerization started when the FeCl3/MO complex was transferred onto the top of the cold pyrrole chloroform solution in a 15 cm in diameter beaker. After 48 h polymerization at 4 °C, the PPy membrane was collected and washed sequentially with ethanol and deionized water to colorless. A HCl solution (1.67 mol L−1) was dropped into the washing solution and the absence of pink color indicated the complete removal of FeCl2 and MO. Then the membrane was dried at ambient temperature and named PPy for later use.2.3 Strengthening PPy membrane with electrospun fibers
After trying 10, 12, 15 and 18 wt% concentrations, 15 wt% was found most appropriate to spin both PU and PLLA fibres using a handheld electrospinning device (Bona, Qingdao, China). The PU and PLLA solutions of 15 wt% were separately prepared by adding solid polymer pellets to CHCl3, and then stirred at room temperature overnight. A volume of 1.5 mL of the polymer solution was then fed into a syringe that was then loaded to the handheld electrospinning device. Fibres were spun through a #25 gauge needle to cover 241 cm2 of PPy surface (e.g., 0.93 mg cm−2) at a feeding rate of about 2.25 mL h−1 under a DC voltage about 9 kV at 25 °C and 65% humidity. The bubble side of the PPy membranes31 was used as the collector, which was 25 cm away from the needle. To spin composite fibers, the PU fibers were firstly spun, followed by the PLLA fibers, as illustrated in Fig. 1. The fibre-reinforced PPy membrane was then placed in an oven at 40 °C overnight to evaporate CHCl3. The reinforced membranes herein were identified as PPy-PU, PPy-PLLA and PPy-PU/PLLA.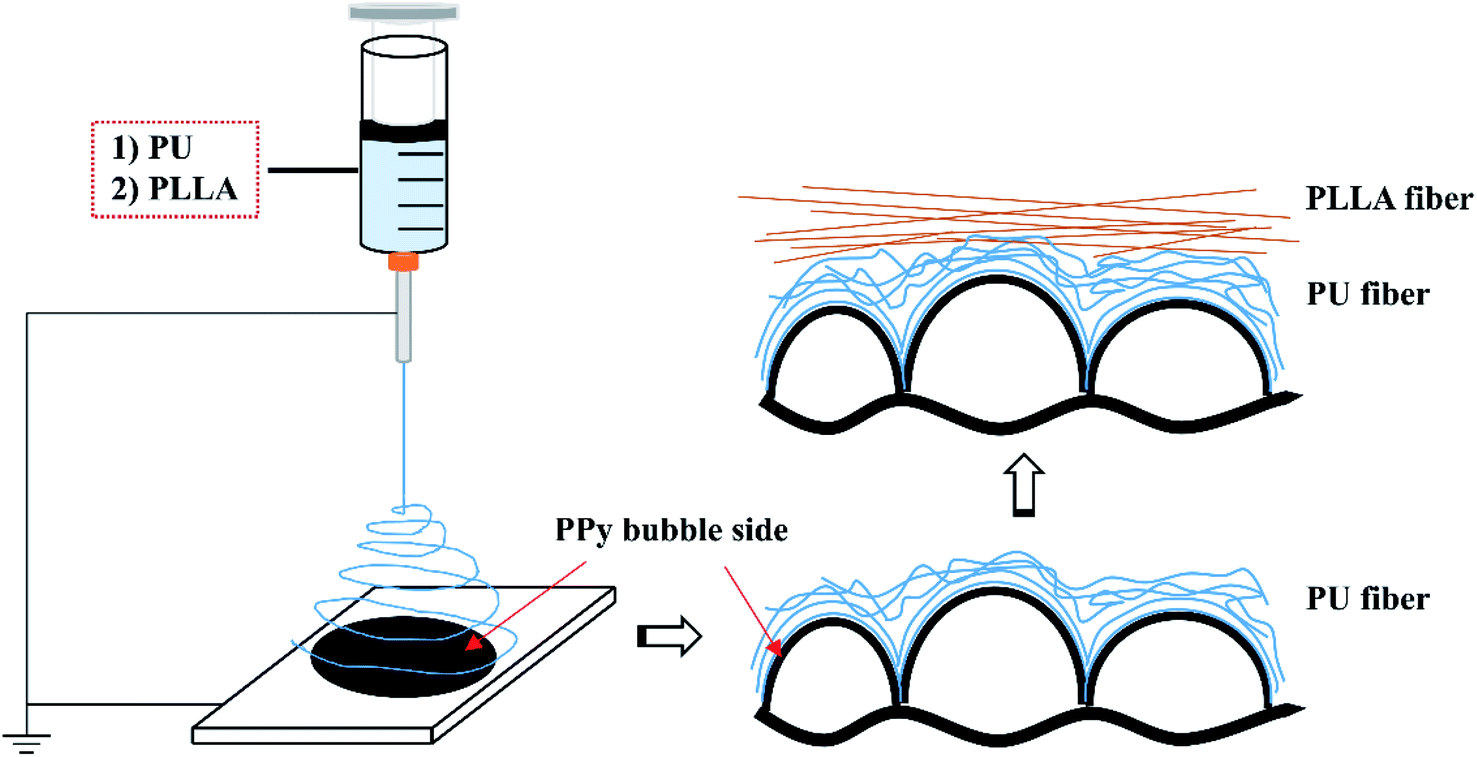 | ||
| Fig. 1 Schematic illustration of how the electrospun fibers are assembled on top of the bubble surface of PPy membrane. | ||
In addition, the dried original and reinforced PPy membranes were immersed in deionized water for 7 days with the water refreshed twice a day to further remove impurities and reduce cytotoxicity. The membranes were subsequently dried naturally in the fume hood. The membranes, after 7 day wash, were named as wPPy, wPPy-PU, wPPy-PLLA, and wPPy-PU/PLLA.
2.4 Characterizations
For Hoechst staining, the cells were first washed three times with PBS and then fixed with a mixture of methanol and acetone (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 10 min. After, the cells were incubated with a solution of 2 mg mL−1 of Hoechst 33342 in PBS for 15 min at room temperature and then washed with PBS. The cells were finally observed under an epifluorescence microscope (Axiophot, Zeiss, Oberkochen, Germany), and photographed. The cells cultured on glass slides were used as the control group.
:1) for 10 min. After, the cells were incubated with a solution of 2 mg mL−1 of Hoechst 33342 in PBS for 15 min at room temperature and then washed with PBS. The cells were finally observed under an epifluorescence microscope (Axiophot, Zeiss, Oberkochen, Germany), and photographed. The cells cultured on glass slides were used as the control group.
To do MTT assay, the prepared 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg mL−1) was stored at 4 °C prior to use. The cells were refreshed with the new medium containing 10% (v/v) MTT and cultivated in an incubator for 4 h without light. The supernatant was then carefully removed and 2 mL of 0.04 N HCl in isopropanol (lysis solution) was added. Fifteen minutes later, 200 mL of the solution was transferred in triplicate from each well to a 96-well flat bottom plate. Absorbance of the formazan at 550 nm was determined using an ELISA reader (Model 680, BioRad Laboratories, Mississauga, ON, Canada). Cells cultured in standard 24-well plate were used as control.
2.5 Statistical analysis
All data were presented as mean ± standard deviation (n ≥ 3) when appropriate. Student t-test and Two-Way Analysis of Variance (ANOVA) were performed to compare the variations between two experiments or among a group of experiments, respectively. p <0.05 was considered statistically significant.
3. Results and discussion
3.1 The synergy between PU and PLLA fibers
The surface of the soft PPy membrane has two distinct topographies: a flat nanotube side and a porous bubble side (Fig. S1, ESI† document). Since the flat surface after removal of the nanotubes was used for cell culture, the reinforcement could only be done on the bubble side. The size and shape of the bubbles vary, ranging from 1 mm to 0.1 mm. The bubbles can be closed or open (Fig. S1B, ESI† document). Such a surface topography significantly reduced the contact area between the PPy bubbles and any non-compliant fibers. Consequently, in this work we used two types of polymer fibers, i.e., PU and PLLA. The PU fibers are soft and compliant as compared with the PLLA fibres, so can better follow the surface topography of the PPy bubbles to achieve a large adhesion area. However, the PU fibers can absorb a small amount of water and became slightly swollen in culture medium, forcing the soft PPy membrane to deform. Therefore, the rigid PLLA fibers were added to strengthen and stabilize the PU reinforced PPy membrane. As shown in Fig. 2, the PU fibers landed on the bubble surface and deformed according to the surface topography (Fig. 2, F&H), forming an intimate contact to PPy. In comparison, the PLLA fibers were straight and spanned the neighboring bubbles without following surface contour (Fig. 2C and G). When electrospinning PU and then PLLA on the membrane, the composite fibers showed a strong adhesion to the PPy membrane, where the compliant PU fibers acted as an adhesive primer between the PPy and PLLA (Fig. 2I, S1D–E†). The thickness of either PU or PLLA layer was estimated to be between 50 to 70 μm. | ||
| Fig. 2 SEM images of the fibers and the cross section of the reinforced membranes. (A– C) PLLA reinforced membrane, showing the PLLA fibers before (A) and after (B) wash, and the cross-section of the washed membrane. (D–F) PU reinforced membrane, showing PU fibers before (D) and after (E) wash, and the cross-section of the washed membrane. (G–I) PLLA/PU reinforced and washed membrane wPPy-PU/PLLA, showing the straight PLLA fibers (G), compliant PU fibers (H), and the cross-section of the membrane (I). | ||
Safety must be adequately ensured for biomaterials. Reducing cytotoxicity through washing is a critical step for the as-prepared membranes. After wash, tiny dents appeared over the surface of the PLLA and PU fibers, as shown in Fig. 2B and E. The probable causes include the extraction of residual chloroform that was not completely evaporated through conventional drying, and the hydrolytic degradation of the PLLA fiber during washing.34
Peel test showed that the PLLA fibers were very easily peeled off the PPy membrane, whereas the PU fibers adhered strongly to the PPy membrane making it impossible to peel off the PU fibers without breaking the PPy membrane (Fig. 3B and C). These results are consistent with the SEM observations, i.e., the PU fibers were better integrated with the PPy bubbles. The Tecoflex™ SG-80A is a polyether-based aliphatic thermoplastic polyurethane, of which the electrospun fibers were highly elastic and compliant. The PU fibers were also very sticky to the PPy probably because that the urethane groups might have formed hydrogen bonds with the PPy. The PLLA, on the other hand, is a polymer with high crystallinity, which imposes rigidity and is compatible with the PU fibers. Consequently, the PU fibers stuck to both PPy and PLLA firmly, like a glue.
 | ||
| Fig. 3 Peel test of the electrospun fibers on PPy, showing the weak adhesion of the PLLA fibers and the strong adhesion of the PU fibers. (A) illustration of the peel test; (B) PLLA fibers were easily peeled off without damaging the PPy membrane; (C) PU fibers cannot be peeled off without breaking the PPy membrane. | ||
To test the usability, the PU/PLLA reinforced PPy membranes and the non-reinforced (naked) PPy membranes were pressed with screws to the bottom of a home-made cell culture device (Fig. S2a, ESI† document) and then released, mimicking the manipulations in a real cell culture experiment. The naked PPy membranes were found broken while the reinforced membranes were intact (Fig. S2b and c, ESI† document). Because of the flexibility of the PU fibers and rigidity of the PLLA fibers, the reinforced PPy membrane exhibited a sufficient resistance to deformation and pressure.
3.2 Mechanical and handling properties
In general, the stress–strain curves of the PPy membranes are linear and show very low stress and strain at failure, which is expectable for such a brittle material (Fig. 4). Interestingly, the washing procedure significantly reduced the ultimate strain (4.13 vs. 2.78%, p < 0.01) and stress (693 vs. 377 kPa, p < 0.05) of the PPy (Table 1). Knowing that PPy is not vulnerable to hydrolysis, we believe that such a drop in mechanical property is likely because of the leaching of dopants and possibly other small molecules such as oligomers out of the PPy, causing the PPy less compact and generating weak points. These small molecules also could have acted as plasticizers that render the PPy slightly more ductile. The differences in the strain and stress at break of the PPy are insignificant between the PPy and PPy-PU groups before wash (p > 0.5), indicating that the electrospinning of PU did not change the ductility of the PPy membrane. However, after wash, the PPy-PU membranes showed the largest decline in stress at break, i.e., 544.09 vs. 229.25 kPa (p < 0.01). This is because that the PU fibers absorbed water and expanded, which stretched and damaged the PPy membrane. In fact, the PPy-PU membranes were found deformed during washing. Importantly, such a PU fiber-caused membrane deformation was not observed among the PPy-PU/PLLA membranes during washing. Apparently, the rigid PLLA fibers prevented such a deformation as well as the damage to the PPy.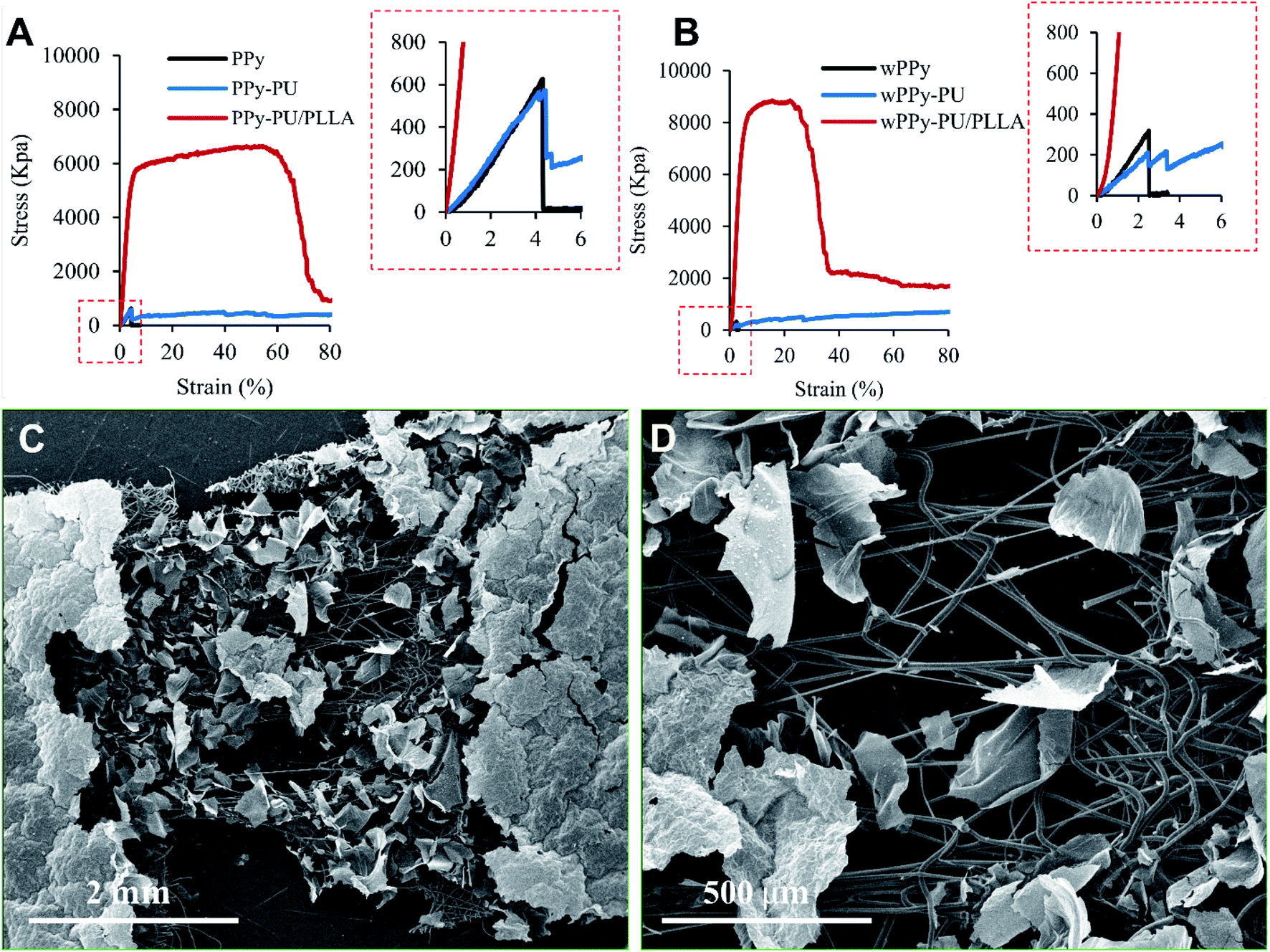 | ||
| Fig. 4 Stress–strain curves of the membranes, before (A) and after (B) 7 day wash, and the SEM photos of the stretched and broken wPPy-PU/PLLA specimen at low (C) and high (D) magnifications. | ||
For the reinforced membranes, the high stress and strain reflect the property of the electrospun fibers rather than that of the PPy. Even the reinforced membranes recorded a much higher stress and strain, the PPy membrane still broke at a low strain. As shown in Fig. 4, the failure of PPy can be clearly identified for the PPy and PPy-PU membranes. For the PPy-PU/PLLA membrane, the break point of the PPy was shield by the strong PLLA fibers and became undetectable. Table 1 only lists the stress and strain of the PPy at failure because these are the data that really mater. In this work, the role of the fibers is to prevent the strain of the PPy membrane from reaching the strain of failure during manipulation, not to make a strong and elastic composite membrane. The PLLA fibers showed a yield point followed by a large plastic deformation before failure, which is normal. They became stiffer after wash likely because of the better organized crystalline structures and the complete removal of the residual chloroform, which is supported by the SEM results. The PU fibers on the other hand were very elastic and recorded a large deformation at low stress.
3.3 Electrical conductivity
The surface conductivity of the membranes is presented in Table 2. The influence of wash and electrospinning on surface conductivity was analyzed through two-way ANOVAs. The 7 day wash significantly reduced the surface conductivity of both PPy (p < 0.01) and PPy-PU/PLLA (p < 0.01) membranes. In fact, the surface conductivity dropped by three orders of magnitude after wash. The reason is the leaching out of the dopants from PPy, resulting in the decay of conductivity. The difference between the PPy and PPy-PU/PLLA groups before wash was significant (p < 0.01), meaning that chloroform in the electrospinning process can affect PPy conductivity. However, this small difference is negligible in terms of conductivity, particularly for conductive polymers. This difference disappeared after wash (p > 0.05), which marks the effectiveness of washing in comparison of conventional drying in removing trace chloroform from polymers.| Condition | Membrane | Surface electrical conductivity (S cm−1) |
|---|---|---|
| Before 7 day wash | PPy | 0.161 ± 0.016 |
| PPy-PU/PLLA | 0.109 ± 0.012 | |
| After 7 day wash | wPPY | 2.41 × 10−4 ± 2.58× 10−5 |
| wPPy-PU/PLLA | 2.17 × 10−4 ± 1.73× 10−5 |
The peak voltage gradient at the wound edge was reported 140 mV mm−1 in skin and 42 mV mm−1 in cornea.35,36 So the electrical stability of the wPPy-PU/PLLA membranes was tested in a 200 mV mm−1 electrical field. As shown in Fig. 5, after a sharp decline in the first 24 h, which was about 65% of the initial conductivity, the conductivity continued to decrease but in a slow and linear fashion, with 13% of the initial conductivity retained after 10 days. The resistivity at this point was at the level of 103 ohm cm, which is still similar to that of the living animal tissues.37 The first exponential decrease was due to the de-doping of anions from the PPy to the aqueous environment, a process enhanced by the reductive electrical potential. However, at same time the anions in the medium could diffuse back to re-dope the PPy. The second relatively stable stage in Fig. 5 is attributed to the dynamic de-doping and re-doping processes. Knowing the electrical behaviors of the reinforced PPy membranes in aqueous environment, researchers are better guided about how to use such PPy membranes in biomedicine.
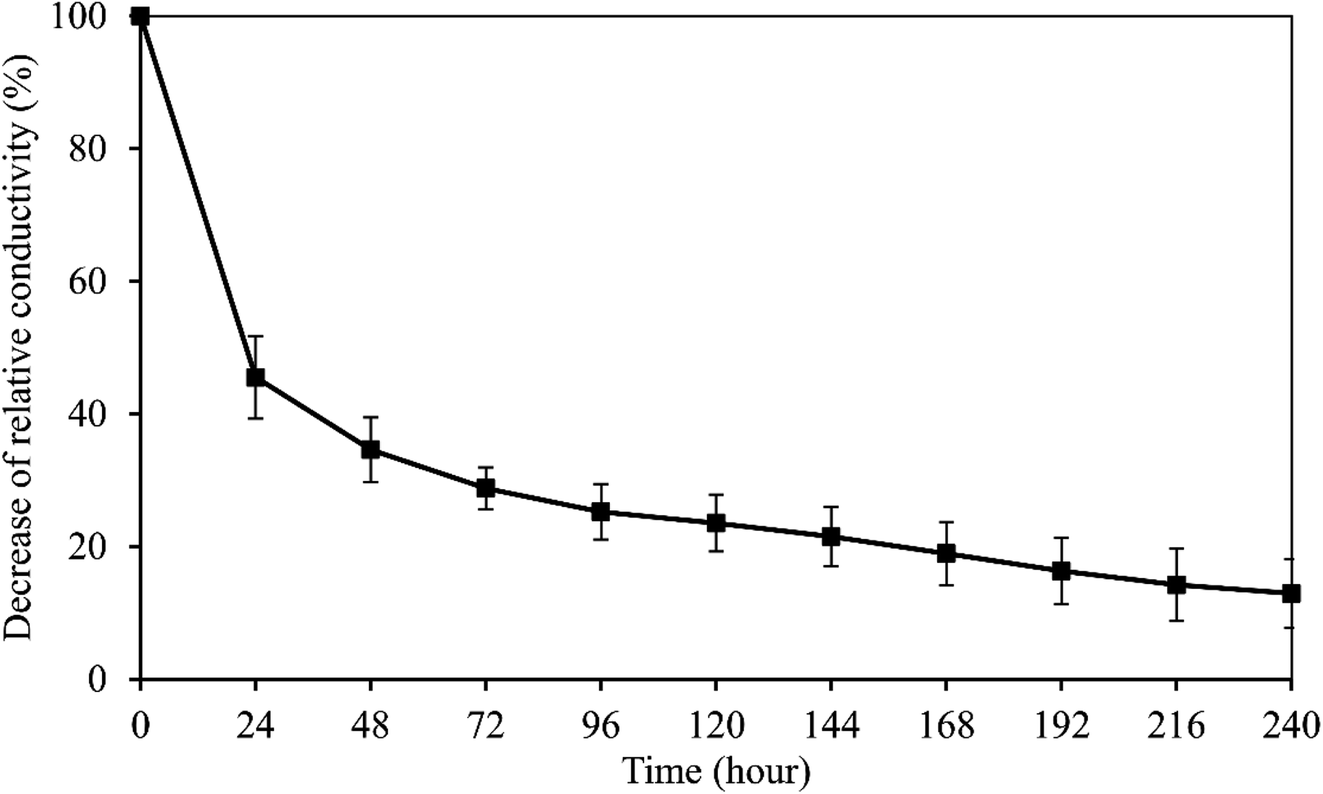 | ||
| Fig. 5 Electrical stability of the wPPy-PU/PLLA membrane. | ||
3.4 XPS and FTIR
The influence of wash on surface elemental composition of PPy (Table 3) was analyzed through Student t-test. The difference in elemental composition is significant for Cl2p (p < 0.001), but insignificant for C1s (p > 0.5), N1s (p > 0.5) and O1s (p > 0.05). This means that the chemical structure of the PPy was not affected after 7 days in water except the loss of chlorine anions, as shown in Fig. S3.† The presence of a significant amount of oxygen testifies the oxidation in PPy, which nevertheless is normal considering that the surface tested (nanotube side, with the nanotubes removed) was synthesized in water phase that inevitably contained dissolved oxygen. The loss of chlorine anions is supported by Fig. 6, showing clearly that the amount of oxidized pyrrole rings (positively charged N) was significantly reduced from 30.44% before wash to 22.13% after wash, explaining the deterioration in conductivity before and after wash.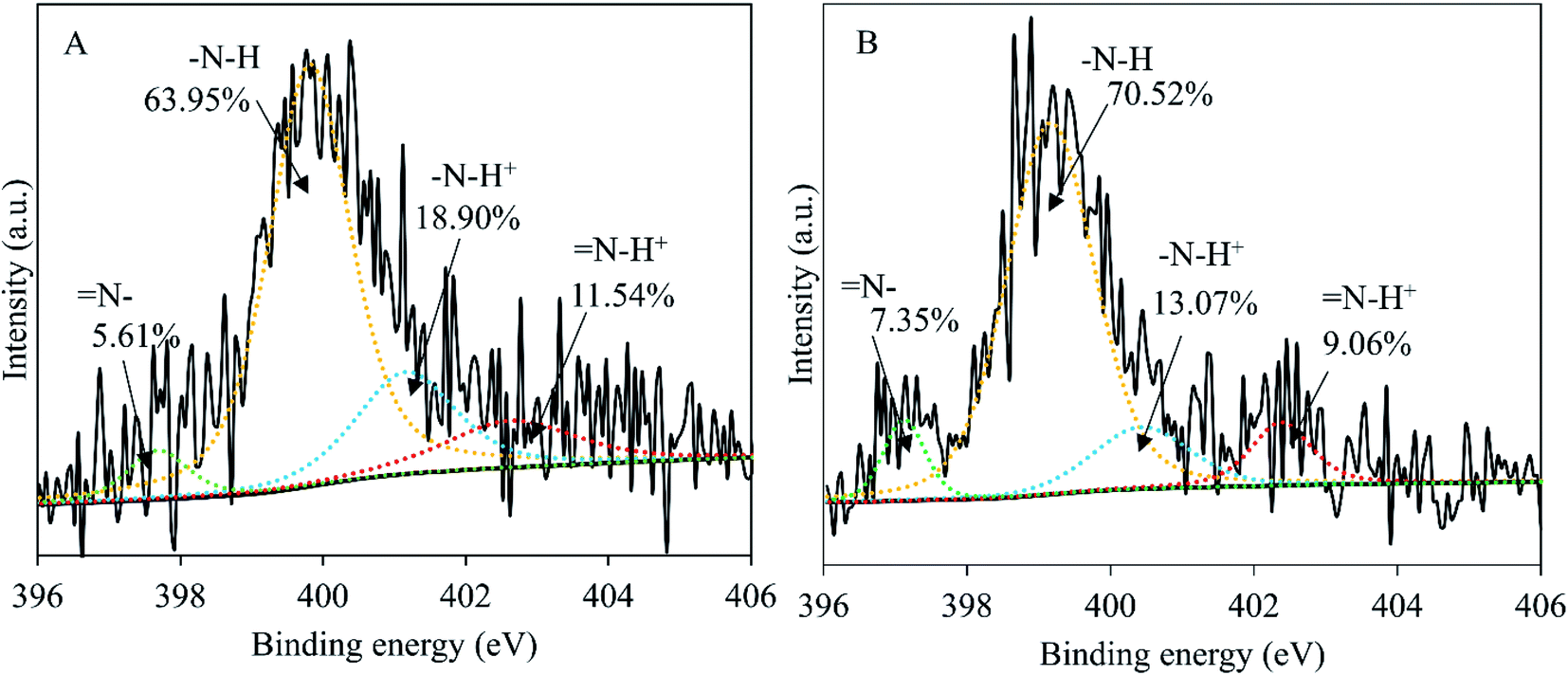 | ||
| Fig. 6 Curve fittings of the high resolution XPS spectra of N1s, PPy membrane before 7 day wash (A) and after 7 day wash (B). | ||
The characteristic absorptions of the PPy and MO identified from the FTIR spectra in Fig. 7 are summarized in Table 4. The MO absorptions do not exist in the spectrum of any membrane, proving that MO was completely washed out. MO is cytotoxic and its elimination is essential to insure membrane cytocompatibility. It was found that the spectra of the naked and the fiber reinforced membranes are basically identical before and after wash (Fig. 7b vs. c, d vs. e), further proving that the electrospinning did not alter PPy chemistry. The peaks in highlight in Fig. 7 identify the absorptions that changed before and after wash. According to literature, the absorption at 1450 cm−1 was due to C–N stretching in pyrrole aromatic rings.38 The band at 1299 cm−1 is attributed to C–N in-plane deformation.39 Thus after washing, pyrrole aromatic ring vibrations became weak, possibly because of the low doping ratio. After wash, the absorptions at 961 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and 850 cm−1 also became weaker and the ones at 900 cm−1 and 780 cm−1 became stronger. The absorption at 961 cm−1 is attributed to CC–C in-plane and out-of-plane deformation, and those at 900 cm−1, 850 cm−1 and 780 cm−1 are caused by C–H vibrations.39 We conclude that the electrospinning did not bring detectable changes to PPy chemistry, and that the changes caused by washing were likely because of the dedoping and changes in oxidation states, which however requires further investigation.
C) and 850 cm−1 also became weaker and the ones at 900 cm−1 and 780 cm−1 became stronger. The absorption at 961 cm−1 is attributed to CC–C in-plane and out-of-plane deformation, and those at 900 cm−1, 850 cm−1 and 780 cm−1 are caused by C–H vibrations.39 We conclude that the electrospinning did not bring detectable changes to PPy chemistry, and that the changes caused by washing were likely because of the dedoping and changes in oxidation states, which however requires further investigation.
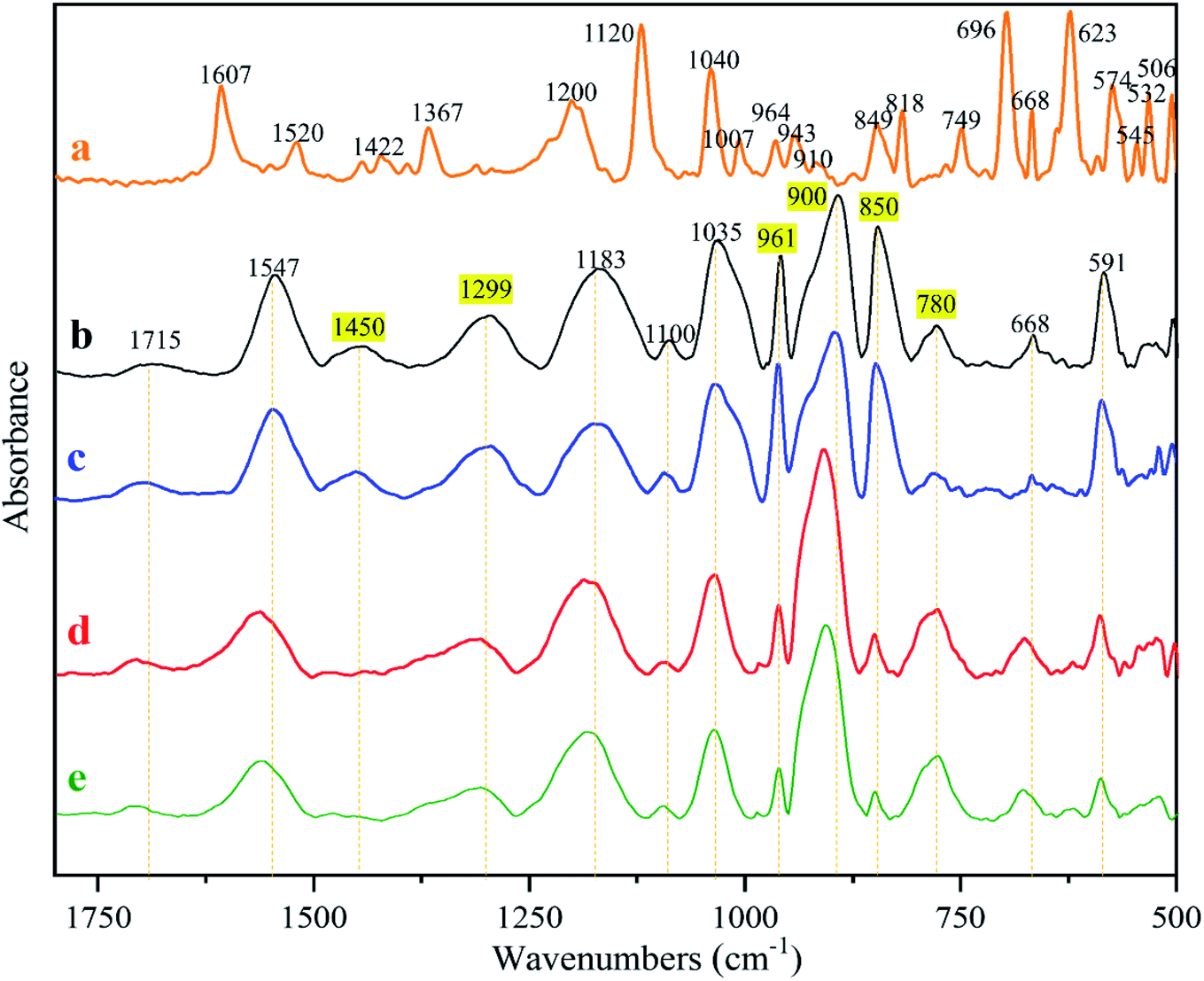 | ||
| Fig. 7 Infrared spectra of (a) MO; (b) PPy; (c) PPy-PU/PLLA; (d) wPPy; (e) wPPy-PU/PLLA. | ||
| Materials | Peak assignment | Absorptions (cm−1) | Ref. |
|---|---|---|---|
| PPy | Stretching of C–C and CC |
1565, 1547 | 39 and 40 |
| C–N and ring stretching vibration | 1450, 1310 | ||
| Out of plane bending of C–H and ring modes | 700 to 1000 | ||
| MO | CC vibrations in the aromatic rings | 1607, 1520 | 41 |
| CH3 vibrations | 1422 | ||
| Azo group NN vibration |
1367 | ||
| Sulfate groups from sulfonate species | 1000 to 1200 |
3.5 Thermal stability
As presented in Fig. 8A and C, the thermogravimetric curves of the naked PPy show three stages of weight loss. The first stage is from 25 to 125 °C where the weight loss is mainly from water and low molecular weight volatiles absorbed in the membranes. In the second stage, from 125 to 300 °C, the weight loss remains small meaning a low degradation rate, which should come from the less stable components in PPy such as the structures with oxidation. The third and most significant degradation starts around 300 °C and does not stop at 600 °C, which accounts for 60% to 67% of the weight loss. The residue at 600 °C is 20.4% for the original PPy and 12.6% for the PPy after the 7 day wash, indicating that when chlorine anions were used as dopant the PPy with a higher doping ratio was more stable.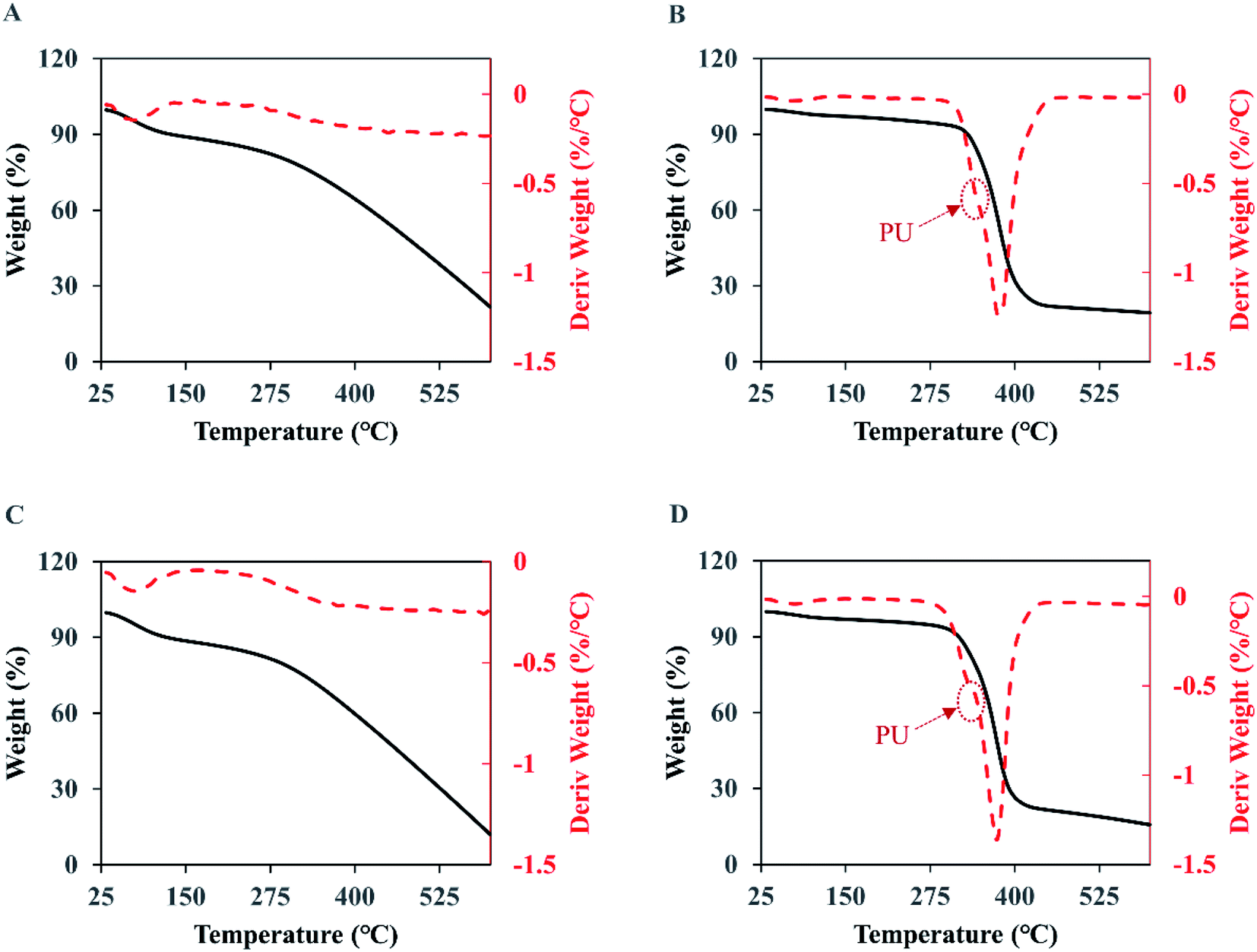 | ||
| Fig. 8 The TGA (solid black curve) and DTG (dash red curve) analyses of the membranes. (A) PPy; (B) PPy-PU/PLLA; (C) wPPy; (D) wPPy-PU/PLLA. | ||
For the PU/PLLA reinforced PPy membranes (Fig. 8B and D), the weight loss happened in four stages, with an initial loss of volatile maters below 125 °C, followed by a slow weight loss up to 300 °C, then a significant drop of weight from 300 to 450 °C, and finally a slow weight loss up to 600 °C. Clearly, the relatively narrow degradation temperature of PU and PLLA is largely responsible for the sharp weight loss between 300 and 450 °C. The DTG curves show a shoulder at 340 °C (arrows), which should be due to PU that is less stable than PLLA. Student t-test revealed no difference (p > 0.1) in weight loss at the 3rd stage (72.6 vs. 71.1), meaning that the wash did not have impact on the thermal stability of the fibers. The significant differences at the 4th stage and in final residue are due to the dedoped and less stable PPy. To conclude, the thermal stability of PPy was affected starting from 300 °C because of the dedoping caused by the intensive wash; and the electrospinning did not affect PPy stability (Table 5).
| Weight loss (%) | Residue (%) | ||||
|---|---|---|---|---|---|
| 25–125 °C | 125–300 °C | 300–600 °C | @600 °C | ||
| PPy | 9.4 ± 0.4 | 9.6 ± 1.7 | 60.7 ± 5.0 | 20.4 ± 3.0 | |
| wPPY | 9.3 ± 1.3 | 10.7 ± 0.4 | 67.5 ± 0.7 | 12.6 ± 1.5 | |
| 25–125 °C | 125–300 °C | 300–450 °C | 450–600 °C | @600 °C | |
| PPy-PU/PLLA | 2.5 ± 0.2 | 3.7 ± 0.1 | 72.6 ± 2.3 | 2.5 ± 0.2 | 18.7 ± 1.9 |
| wPPy-PU/PLLA | 2.8 ± 0.3 | 5.5 ± 2.8 | 71.1 ± 4.6 | 6.8 ± 3.4 | 14.0 ± 2.2 |
3.6 Cytocompatibility
Fig. 9 presents the cell proliferation after 24, 48 and 72 h of culture. It can be seen that the amount of cells on the membranes (A, B and C) were comparable to that on the glass slide (D, E and F). Many cells on the membrane were out of focus because of the uneven surface morphology. The quantitative data depicted by the histograms also show a similar number of cells at 24 h and 48 h. At 72 h, however, a significantly higher number of cells was found on the membranes (0.58 vs. 0.48, p < 0.01), which was probably because of the larger surface area of the PPy-PU/PLLA membrane allowing more space to proliferate. These results indicate that the reinforced membrane has an excellent cytocompatibility and can support keratinocyte growth, which indicates the potential of using such conductive membranes in skin wound care.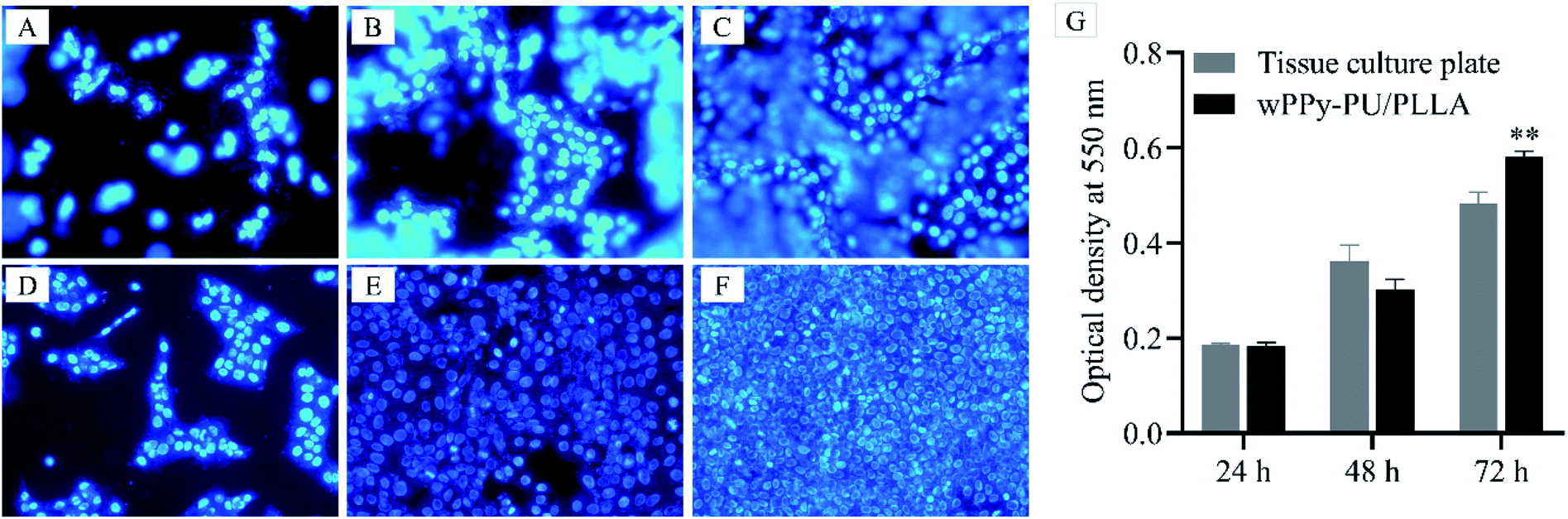 | ||
| Fig. 9 Adhesion of human skin keratinocytes on wPPy-PU/PLLA membrane at 24 h (A), 48 h (B) and 72 h (C), showing a comparable cell density to the controls on glass slide (D–F). The histograms show the proliferation of the keratinocytes, showing a comparable or higher number of cells on the wPPy-PU/PLLA membrane compared to that in the tissue culture plate. **p < 0.01. | ||
4. Conclusions
Electrospun PU and PLLA fibers were successfully utilized to strengthen the soft PPy membrane without sacrificing its electrical conductivity, electrical stability, thermal stability, and surface chemistry, which for the first time makes the soft PPy membrane practically usable. The unique combination of PU and PLLA fibers ensured their strong attachment to the PPy surface, no membrane deformation in aqueous solution despite the multi-layered structure, and the sufficient mechanical strength to stand normal manipulation without damaging the membrane. The reinforced membrane remained highly flexible and light weight, and supported the proliferation of human skin keratinocytes. Such a microporous, flexible, high surface area and easy to use conductive PPy membrane is very useful for a variety of biomedical applications such as electrically stimulated cell culture and reconstruction of conductive tissues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was funded by the Canadian Institutes of Health Research CIHR Project Grant 148523. The first author acknowledges the studentships from the Projet Intégrateurs of the Research Center for High Performance Polymer and Composite Systems (CREPEC) and La Fondation du CHU de Québec. The technical assistance of Pascale Chevallier in XPS measurement is greatly appreciated.References
- B. A. Bolto, R. McNeill and D. E. Weiss, Aust. J. Chem., 1963, 16, 1090–1103 CrossRef CAS.
- G. Street, S. Lindsey, A. Nazzal and K. Wynne, Mol. Cryst. Liq. Cryst., 1985, 118, 137–148 CrossRef CAS.
- T. V. Vernitskaya and O. N. Efimov, Russ. Chem. Rev., 1997, 66, 443 CrossRef.
- F. Yilmaz, Conducting polymers, 2016 Search PubMed.
- Z. Zhang, M. Rouabhia and S. E. Moulton, Conductive Polymers: Electrical Interactions in Cell Biology and Medicine, CRC Press, 2017 Search PubMed.
- J. F. Mao and Z. Zhang, Polypyrrole as electrically conductive biomaterials: Synthesis, biofunctionalization, potential applications and challenges, in Cutting-Edge Enabling Technologies for Regenerative Medicine, ed. H. J. Chun, C. H. Park, I. K. Kwon and G. Khang, Springer, 2018, pp. 347–370 Search PubMed.
- M. Levin, Mol. Biol. Cell, 2014, 25, 3835–3850 CrossRef PubMed.
- S. Meng, Z. Zhang and M. Rouabhia, J. Bone Miner. Metab., 2011, 29, 535–544 CrossRef CAS PubMed.
- G. Tai, M. Tai and M. Zhao, Burns Trauma, 2018, 6, 20 Search PubMed.
- P. Waldauf, J. Gojda, T. Urban, N. Hruskova, B. Blahutova, M. Hejnova, K. Jiroutkova, M. Fric, P. Jansky, J. Kukulova, F. Stephens, K. Rasova and F. Duska, Trials, 2019, 20, 1–11 CrossRef PubMed.
- S. Meng, M. Rouabhia and Z. Zhang, J. Appl. Bioeng., 2011, 37–62 Search PubMed.
- D. O'Connor and B. Caulfield, Support Care Cancer, 2018, 26, 3661–3663 CrossRef PubMed.
- R. Balint, N. J. Cassidy and S. H. Cartmell, Tissue Eng., Part B, 2013, 19, 48–57 CrossRef CAS PubMed.
- C. F. Guimarães, L. Gasperini, A. P. Marques and R. L. Reis, Nat. Rev. Mater., 2020, 5, 351–370 CrossRef.
- G. Shi, M. Rouabhia, Z. Wang, L. H. Dao and Z. Zhang, Biomaterials, 2004, 25, 2477–2488 CrossRef CAS PubMed.
- H. H. Wu, D. K. Sheng, X. D. Liu, Y. Zhou, D. Li, F. Ji, S. B. Xu and Y. M. Yang, Polymer, 2020, 189, 122–181 CrossRef.
- Y. X. Zhao, Y. F. Li, W. M. Kang, Y. He, W. Liu, H. Liu and B. W. Cheng, RSC Adv., 2017, 7, 49576–49585 RSC.
- C. R. Broda, J. Y. Lee, S. Sirivisoot, C. E. Schmidt and B. S. Harrison, J. Biomed. Mater. Res., Part A, 2011, 98, 509–516 CrossRef PubMed.
- F. Y. Qi, Y. Q. Wang, T. Ma, S. Zhu, W. Zeng, X. Y. Hu, Z. Y. Liu, J. H. Huang and Z. J. Luo, Biomaterials, 2013, 34, 1799–1809 CrossRef CAS PubMed.
- J. G. Hardy, R. C. Sukhavasi, D. Aguilar, M. K. Villancio-Wolter, D. J. Mouser, S. A. Geissler, L. Nguy, J. K. Chow, D. L. Kaplan and C. E. Schmidt, J. Mater. Chem. B, 2015, 3, 8059–8064 RSC.
- J. Pelto, M. Bjorninen, A. Palli, E. Talvitie, J. Hyttinen, B. Mannerstrom, R. Suuronen Seppanen, M. Kellomaki, S. Miettinen and S. Haimi, Tissue Eng., Part A, 2013, 19, 882–892 CrossRef CAS PubMed.
- Y. L. Wang, M. Rouabhia and Z. Zhang, J. Mater. Chem. B, 2013, 1, 3789–3796 RSC.
- W. Jing, Q. Ao, L. Wang, Z. Huang, Q. Cai, G. Chen, X. Yang and W. Zhong, Chem. Eng., 2018, 345, 566–577 CrossRef CAS.
- J. F. Zhou, Y. G. Wang, L. Cheng, Z. Wu, X. D. Sun and J. Peng, Neural Regen. Res., 2016, 11, 1644 CrossRef CAS PubMed.
- B. Shu, X. B. Liu, J. F. Zhou, H. Huang, J. Y. Wang, X. D. Sun, C. Qin and Y. H. An, CNS Neurosci. Ther., 2019, 25, 951–964 CrossRef PubMed.
- C. Y. Foo, N. M. Huang, H. N. Lim, Z. T. Jiang and M. Altarawneh, Eur. Polym. J., 2019, 117, 227–235 CrossRef CAS.
- X. Fu, J. K. Wang, A. C. Ramírez-Pérez, C. Choong and G. Lisak, Flexible conducting polymer-based cellulose substrates for on-skin applications, Mater. Sci. Eng., C, 2020, 108, 110392 CrossRef CAS PubMed.
- M. Talikowska, X. Fu and G. Lisak, Application of conducting polymers to wound care and skin tissue engineering: A review, Biosens. Bioelectron., 2019, 135, 50–63 CrossRef CAS PubMed.
- B. Guo and P. X. Ma, Conducting polymers for tissue engineering, Biomacromolecules, 2018, 19, 1764–1782 CrossRef CAS PubMed.
- M. Talikowska, X. Fu and G. Lisak, Application of conducting polymers to wound care and skin tissue engineering: A review, Biosens. Bioelectron., 2019, 35, 50–63 CrossRef PubMed.
- J. F. Mao, C. J. Li, H. J. Park, M. Rouabhia and Z. Zhang, ACS Nano, 2017, 11, 10409–10416 CrossRef CAS PubMed.
- C. H. Wang, Y. J. Ding, Y. Yuan, A. Y. Cao, X. D. He, Q. Y. Peng and Y. B. Li, Small, 2016, 12, 4070–4076 CrossRef CAS PubMed.
- F. M. Smits, Bell Syst. Tech. J., 1958, 37, 711–718 CrossRef.
- G. Gorrasi and R. Pantani, in Synthesis, Structure and Properties of Poly (lactic acid), ed. D. Lorenzo, M. AndroschRené Laura, Springer, 2018, pp. 119–151 Search PubMed.
- A. T. Barker, L. F. Jaffe and J. W. Vanable Jr, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 1982, 242, R358–R366 CrossRef CAS PubMed.
- M. C. Chiang, K. R. Robinson and J. W. Vanable Jr, Exp. Eye Res., 1992, 54, 999–1003 CrossRef CAS PubMed.
- C. F. Kay and H. P. Schwan, Circ. Res., 1956, 4, 664–670 CrossRef CAS PubMed.
- B. Tian and G. Zerbi, J. Chem. Phys., 1990, 92, 3886–3891 CrossRef CAS.
- F. N. Ajjan, M. J. Jafari, T. Rębiś, T. Ederth and O. Inganäs, J. Mater. Chem. A, 2015, 3, 12927–12937 RSC.
- Z. Ahmad, M. A. Choudhary, A. Mehmood, R. Wakeel, T. Akhtar and M. A. Rafiq, Macromol. Res., 2016, 24, 596–601 CrossRef CAS.
- J. J. Murcia, M. C. Hidalgo, J. A. Navío, J. Araña and J. M. Doña-Rodríguez, Appl. Catal. B, 2014, 150–151, 107–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01338f |
| This journal is © The Royal Society of Chemistry 2021 |