
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer-supported synthesis of N-substituted anthranilates as the building blocks for preparation of N-arylated 3-hydroxyquinolin-4(1H)-ones†
Soňa Krajčovičováa,
Jan Hlaváč a and
Kristýna Vychodilová*b
a and
Kristýna Vychodilová*b
aDepartment of Organic Chemistry, Faculty of Science, Palacký University, 17. Listopadu 12, 77146 Olomouc, Czech Republic
bInstitute of Molecular and Translational Medicine, Faculty of Medicine and Dentistry, Palacký University, Hněvotínská 5, 77900 Olomouc, Czech Republic. E-mail: kristyna.vychodilova@upol.cz
First published on 2nd March 2021
Abstract
Fast and simple access to N-arylated 3-hydroxyquinolin-4(1H)-ones starting from easily available 1-methyl-2-iodoterephthalate and variously substituted anilines is presented. N-Alkylated anthranilic acid derivatives represent important intermediates. They can be advantageously prepared by solid-phase synthesis, by Buchwald–Hartwig amination or reductive amination with wide substrate scope and with excellent crude purities.
3-Hydroxyquinolin-4(1H)-ones (3-HQs), flavones aza-analogues, represent an interesting family of biologically active compounds.1–3 The methodology for 3-HQ's synthesis is well established, starting from anthranilic acid derivatives and bromoacetophenones.4 The formed phenacylesters cyclize upon heating in various acids or N-methyl-2-pyrrolidone (NMP) (Scheme 1). The synthetic pathway was later successfully adapted to using a solid support which enabled simple, fast and high-throughput organic synthesis of larger collections of 3-HQs for biological screening (Scheme 1).5
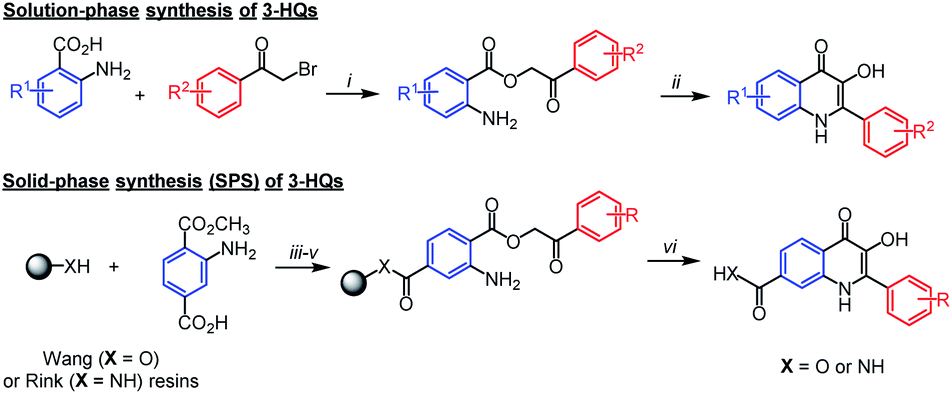 | ||
Scheme 1 Routinely used synthesis of 3-HQs in solution and on a solid support. Reagents and conditions: (i) K2CO3, DMF; (ii) refluxing acid or NMP; (iii) 1-hydroxybenzotriazole (HOBt), N,N′-diisopropylcarbodiimide (DIC), CH2Cl2/DMF; (iv) potassium trimethylsilanolate (TMSOK), DMF; (v) 2-bromoacetophenone, triethylamine, DMF; (vi) CH2Cl2/TFA 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, then reflux in acid. :1, then reflux in acid. | ||
Although the synthesis of 3-HQs substituted on 2-phenyl as well as the quinoline benzene ring is relatively widely explored,6,7 preparation of N-substituted 3-HQs was yet barely described. Hradil et al.8 studied the effect of substituents at the nitrogen atom in the course of cyclization of phenacyl anthranilates. N-methyl and N-phenyl derivatives were studied; however, the desired N-methyl-3-HQ was obtained in very low yield and N-phenyl-3-HQ was even not isolated. Spacilova et al.9 managed to prepare N-amino-3-HQ by the cyclization of 2-hydrazinobenzoic acid phenacyl ester, however, the procedure lacks general application. Recently Wang et al.10 reported synthesis of aza-rocaglates involving N-methyl-3-HQ as one of the intermediates. Interestingly, they used basic conditions for the cyclization of N-methylphenacyl ester and obtained the product in 85% yield. Nevertheless, a wider application of the method was not studied. It is worth mentioning that direct N-alkylation of the 3-HQ scaffold is not feasible as it leads to a mixture of mono- and dialkylated products due to the presence of reactive hydroxy group.4
3-HQs have been reported as strong cytotoxic agents, they act as inhibitors of microtubule formation in tubulin polymerization1 or the species able to target translation elongation factor.2 Furthermore, 3-HQs were recognized to exhibit antiprotozoal and immunosuppressive effects.3 In the field of physico-chemical properties, compounds bearing 3-HQ scaffold have been identified as promising fluorescent agents.11,12 In this regard they exhibit dual fluorescence spectra with sufficiently separated emission bands, with the intensities' ratio independent on the molecule concentration, thus enable the 3-HQs to be e.g. fluorescent probes in the biological system.
However, due to unavailability of the general method applicable for the preparation of N-substituted-3-HQs, the properties of such compounds are unknown.
With respect to well-known advantages of solid-phase synthesis (SPS) in the preparation of drug-like molecules we decided to modify the previously reported SPS synthesis of 3HQs5 towards their N-alkylated analogues. Rink AM resin 1 was acylated with 1-methyl-2-aminoterephthalate and subjected to N-alkylation (Scheme 2). Although N-substituted anthranilic acids belong to pharmaceutically relevant intermediates,13–15 their preparation on solid support has not been reported yet. Consequently we adopted conditions from traditional solution-phase chemistry describing N-arylation by Ullmann type reactions16,17 and copper or nickel catalyzed Chan–Evans–Lam cross-coupling.18,19 However, we were not able to convert 2 to corresponding N-substituted derivatives (Scheme 2, reaction conditions iii–iv) and the starting material only was isolated. For this reason, we prepared immobilized 1-methyl-2-iodoterephthalate 3 and subjected this intermediate to amination by aromatic nucleophilic substitution, Buchwald reaction, Ullmann reaction and Miyaura coupling. Either no conversion of the starting material to the product was observed (Scheme 2, reaction conditions vi–vii) or we detected only dehalogenation of the starting iodo-derivative (Scheme 2, reaction conditions viii–ix).
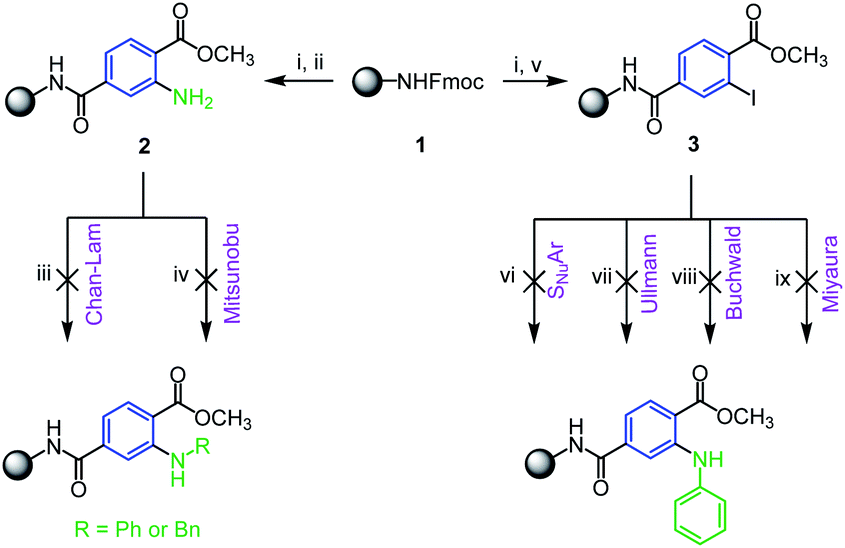 | ||
| Scheme 2 Attempted preparation of N-substituted aminoterephthalates on solid support. Reagents and conditions (per 100 mg of 1): (i) DBU/CH2Cl2 1:1, r.t., 10 min (ii) 1-methyl-2-aminoterephthalate (0.3 M), HOBt (0.3 M), DIC (0.3 M), CH2Cl2/DMF 1:1, r.t., 16 h; (iii) phenylboronic acid (0.2 M), Cu(OAc)2 (0.2 M), triethylamine (0.4 M), CH3CN, 80 °C, 16 h; (iv) benzyl alcohol (0.3 M), PPh3 (0.3 M), diisopropyl azodicarboxylate (DIAD) (0.3 M), THF, 0 °C to r.t., 16 h; (v) 1-methyl-2-iodoterephthalate (0.2 M), HOBt (0.2 M), DIC (0.2 M), DMF, r.t. 4 h; (vii) aniline (0.3 M), K3PO4 (0.3 M), CuI (0.03 M), ethylene glycol (0.3 M), DMSO, 100 °C, 16 h; (viii) aniline (0.4 M), LiHMDS (1 M in THF, 0.4 M) Pd2dba3 (0.02 M), XPhos (0.04 M), THF, 60 °C, 16 h; (ix) bis(pinacolato)diboron (0.3 M), KBr (0.3 M), PhOK (0.3 M), Pd(PPh3)Cl2 (0.03 M), PPh3 (0.06 M), toluene, reflux, 16 h, then (viii). | ||
Luckily, we finally succeeded in optimizing the Buchwald–Hartwig amination20–22 (Table 1) with aniline using the second generation of XPhos palladium as the pre-catalyst and K3PO4 as a base in the mixture of toluene/DMF at 100 °C. Under these conditions the desired intermediate 4 was received in an excellent crude purity of 94% (calculated from UHPLC-UV traces). Motivated by this result we subsequently tested variously substituted anilines, benzylamines and other aliphatic amines. The reaction worked smoothly with all tested anilines, with both electron-withdrawing (entries 2–8, Table 1) and electron-donating (entries 9–14, Table 1) substituents as it afforded the products in high crude purities. It is worth mentioning that only use of SPS allows to isolate the corresponding products in such high crude purities because the residual catalyst and solution-phase reagents were simply removed by common washing procedure. In contrast, Buchwald–Hartwig amination in solution phase requires removal of the catalyst by tedious column chromatography. Compared to anilines no reaction was observed for aliphatic amines including benzylic, cyclic or acyclic ones (entries 15–18, Table 1), which was caused presumably due to the inefficient formation of active complex with palladium catalyst.
|
|||
|---|---|---|---|
| Entry | RNH2 | Cmp. no. | Crude purity (%) |
| a Calculated from UHPLC-UV traces, n.d. = not detectable. | |||
| 1 | Aniline | 4 | 94 |
| 2 | 2,4-Dinitroaniline | 5 | 99 |
| 3 | 4-Chloraniline | 6 | 90 |
| 4 | 3-Chloraniline | 7 | 95 |
| 5 | 2-Fluorphenyl | 8 | 95 |
| 6 | 2-Trifluormethylaniline | 9 | 70 |
| 7 | 4-Aminobenzoic acid | 10 | 70 |
| 8 | 3-Aminobenzoic acid | 11 | 60 |
| 9 | 4-Methylaniline | 12 | 90 |
| 10 | 3-Methylaniline | 13 | 81 |
| 11 | 4-Aminoindan | 14 | 95 |
| 12 | 4-Morpholinoaniline | 15 | 95 |
| 13 | 4-Methoxyaniline | 16 | 85 |
| 14 | 2,4-Dimethoxyaniline | 17 | 95 |
| 15 | Benzylamine | 18 | n.d. |
| 16 | 4-Methoxybenzylamine | 19 | n.d. |
| 17 | Cyclohexylamine | 20 | <10 |
| 18 | Octylamine | 21 | n.d. |
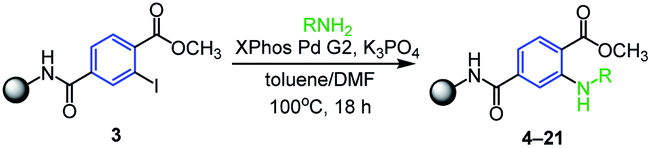
The successfully prepared N-arylated methyl esters 4–17 were then hydrolyzed by typical procedure for SPS chemistry23 employing potassium trimethylsilanolate (TMSOK) in THF (Scheme 3). All intermediates 18–29 were obtained in high crude purities (up to 90%) and were further reacting with 2-bromoacetophenone which yielded phenacyl esters 30–41 again with high crude purities (Scheme 3).
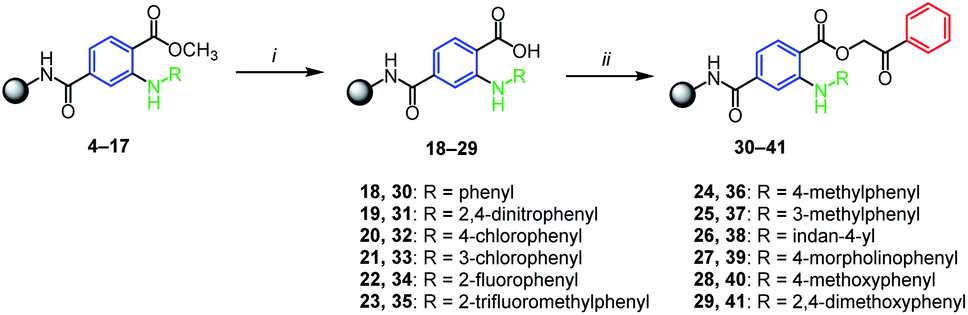 | ||
| Scheme 3 Hydrolysis of methyl esters and alkylation to phenacyl esters. Reagents and conditions: (i) TMSOK, THF, r.t., 16 h; (ii) 2-bromoacetophenone, triethylamine, DMF, r.t., 16 h. | ||
As the Buchwald–Hartwig coupling with aliphatic amines failed, we considered the possible alkylation using the reductive amination of immobilized aminoterephthalate 2. Interestingly, we observed no conversion to desired products when using conditions typical for solid support, i.e. sodium triacetoxyborohydride or sodium cyanoborohydride as the reductive agents.6,24 Thus we adopted the solution-phase protocol for catalytic silane-based direct reductive amination in refluxing THF reported by Apodaca et al.25 (Scheme 4). Using this procedure, benzyl moiety was successfully introduced and derivative 42 was obtained in 90% crude purity (according to UHPLC-UV traces). Intermediate 42 was hydrolyzed to carboxylic acid 43 and alkylated with 2-bromoacetophenone but an equimolar mixture of compounds 44 and 45 was formed (Scheme 4). Formation of 45 was caused by an excess of solution-phase reagents which is a typical feature of solid-phase synthesis. Therefore, we changed the reaction sequence and performed the reductive amination in the stage of phenacyl ester 46. The reaction proceeded well with benzaldehyde, cyclohexanecarboxaldehyde and pentanal (Scheme 4, compounds 47–49).
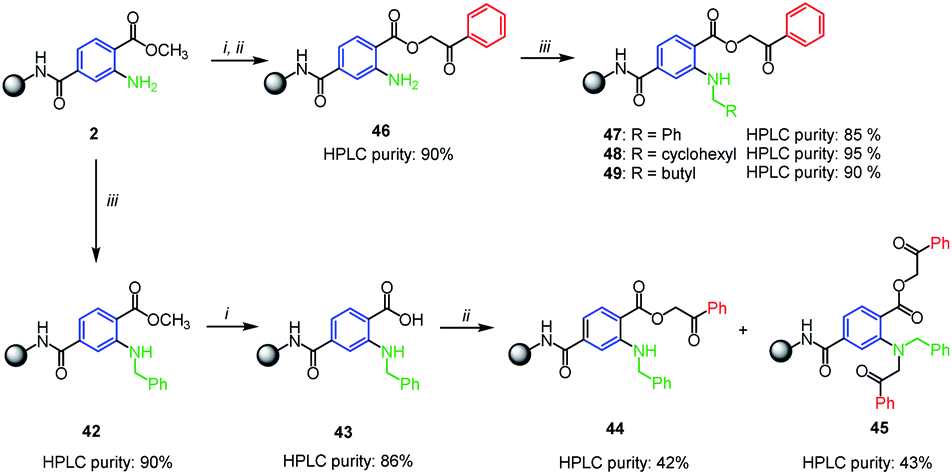 | ||
| Scheme 4 Silane-based reductive amination on solid phase. Reagents and conditions: (i) TMSOK, THF, r.t., 16 h; (ii) 2-bromoacetophenone, triethylamine, DMF, r.t., 16 h; (iii) benzaldehyde, Bu2SnCl2, phenylsilane, THF, reflux, 16 h. | ||
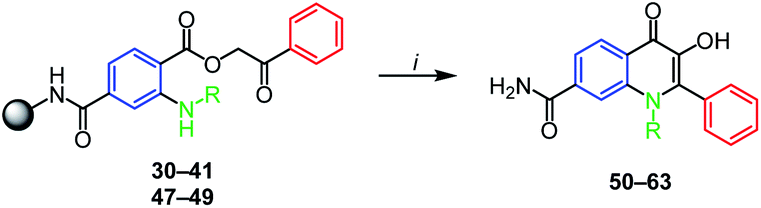
The final step leading to targeted N-substituted 3-HQs 50–63 was performed by traditional acid-catalyzed cyclization. Several acids (PPA, AcOH, H2SO4) were employed, however the successful cyclization occurred only when neat refluxing trifluoracetic acid (TFA) was used. Due to acid-lability of the Rink amide linker these conditions led to cleavage of phenacyl esters from the resin and their subsequent cyclization. The cyclization was feasible for phenacyl esters 36–38 and 40–41 prepared from electron-rich anilines (products 56–58, 60–61, Table 2). Interestingly, although sterically demanding, N-indanyl 3-HQ 58 was obtained smoothly and with excellent purity. Similarly, phenacyl esters with halogen atoms on N-phenyl ring 32–34 can be cyclized as well (products 52–54, Table 2). In this case the mesomeric effect of halogens probably prevails the inductive effect. Going down with nitrogen nucleophilicity the reaction gets problematic. Derivatization of N-phenyl ring by nitro groups or trifluormethyl group (compounds 51 and 55 respectively, Table 2) led to low reactivity of phenacyl esters towards cyclization. 4-Morpholinophenyl derivative 39 did not react to expected product 59, probably due to the protonation of morpholine nitrogen under acidic conditions causing electron-withdrawal from the aniline NH. The cyclization to 3-HQs did not work with N-aliphatic substitution (benzyl derivatives as well as aliphatic ones), no matter if cyclized or acyclic (compounds 62–63, Table 2). When compared to N-arylated esters, this result can be explained by the protonation of more basic N-alkylated intermediates by TFA which breaks down the cyclization. It is worth mentioning that in contrast to high crude purities (Table 2) the limited yields of final compounds were caused by problematic purification and not by the cyclization itself. Significant peak broadening and tailing were observed using reverse phase sorbents and for some derivatives, the RP HPLC, were not applicable.26 Interestingly, in our case, the purification method strongly depended on N-phenyl substitution, where alkyl groups such as methyl or indanyl were feasible to purify by reversed-phase semipreparative HPLC, opposite to chloro- and methoxy moieties that required column chromatography using silica gel.
:1, r.t., 30 min to 2 h, then neat TFA, reflux, 24 h to 5 days
|
|||
|---|---|---|---|
| Cmp. no. | R– | Crude purity of products (%) | Overall yieldb (%) |
| a No conversion to product.b Yield is calculated after purification from the starting loading of the resin.c Purified via column chromatography on silica gel.d Reversed-phase semipreparative HPLC purification. | |||
| 50 | Phenyl- | 78 | 80c |
| 51 | 2,4-Dinitrophenyl | n.da | — |
| 52 | 4-Chlorphenyl | 85 | 14d |
| 53 | 3-Chlorphenyl | 70 | 24d |
| 54 | 2-Fluorphenyl | 50 | — |
| 55 | 2-Trifluormehtylphenyl | n.da | — |
| 56 | 4-Methylphenyl | 90 | 8d |
| 57 | 3-Methylphenyl | 90 | 14d |
| 58 | Indan-4-yl | 85 | 8d |
| 59 | 4-Morpholinophenyl | n.da | — |
| 60 | 4-Methoxyphenyl | 85 | 42c |
| 61 | 2,4-Dimethoxyphenyl | 70 | 88c |
| 62 | Benzyl | n.da | — |
| 63 | Cyclopentanemethyl | n.da | — |
Due to unreactivity of some phenacyl esters towards TFA-mediated cyclization, other conditions27 were screened on the representative examples: N-benzyl-phenacyl ester 47, N-cyclopentanemethyl phenacyl ester 48 and N-2,4-dinitrophenyl-phenacyl ester 31 were heated in sulphuric acid, acetic acid, polyphosphoric acid, NMP or simply melted. Unfortunately, no conversion to desired products was observed.
Finally, as mentioned earlier, Wang et al.10 managed to convert N-methyl phenacyl ester to corresponding 3-HQ by sodium hydride in THF. Thus, we tested this procedure, however, we only observed very rapid hydrolysis of ester to the carboxylic acid, without traces of desired product.
We successfully developed the solid-phase synthetic approach applicable for the preparation of collection of N-substituted anthranilic acid derivatives. The methodology covers wide range of substrate scope; aromatic substitution can be introduced by Buchwald–Hartwig amination, while aliphatic and benzylic moieties can be incorporated by reductive amination. With respect to use of immobilized starting materials the protocol enables to easily remove the catalysts by simple filtration which provides the intermediates in high crude purities. The resulting immobilized N-substituted anthranilic acid derivatives represent compounds of biological interest28,29 or can be theoretically used as precursors for the preparation of e.g. acridine/acridones derivatives with antimalarial,30 antiviral,31,32 antibacterial,33 and other34–36 activities. In our case we applied them for the preparation of N-substituted 3-HQs. The synthetic route is feasible for compounds bearing N-aryl moiety with electron donating substituents. Due to number of readily available electron rich anilines the developed strategy can be used, in combination with previously developed procedures,6,7 to easily and rapidly prepare collections of novel 3-HQ's for determination of their biological and physicochemical properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (reg. no. 18-26557Y).References
- J. Rehulka, K. Vychodilová, P. Krejci, S. Gurska, P. Hradil, M. Hajduch, P. Dzubak and J. Hlavac, Eur. J. Med. Chem., 2020, 192, 112176 CrossRef CAS
.
- K. Burglova, G. Rylova, A. Markos, H. Prichystalova, M. Soural, M. Petracek, M. Medvedikova, G. Tejral, B. Sopko, P. Hradil, P. Dzubak, M. Hajduch and J. Hlavac, J. Med. Chem., 2018, 61, 3027–3036 CrossRef CAS
- P. Hradil, J. Hlavac, M. Soural, M. Hajduch, M. Kolar and R. Vecerova, Mini-Rev. Med. Chem., 2009, 9, 696–702 CrossRef CAS
- P. Hradil and J. Jirman, Collect. Czech. Chem. Commun., 1995, 60, 1357–1366 CrossRef CAS
- M. Soural and V. Krchnak, J. Comb. Chem., 2007, 9, 793–796 CrossRef CAS
- M. Soural, J. Hlavac, P. Funk, P. Dzubak and M. Hajduch, ACS Comb. Sci., 2011, 13, 39–44 CrossRef CAS
- M. Soural, J. Hlavac, P. Hradil, I. Frysova, M. Hajduch, V. Bertolasi and M. Malon, Eur. J. Med. Chem., 2006, 41, 467–474 CrossRef CAS
- P. Hradil, J. Hlavac and K. Lemr, J. Heterocycl. Chem., 1999, 36, 141–144 CrossRef CAS
- L. Spacilova, J. Hlavac, P. Hradil, I. Frysova, M. Soural, P. Krejci and M. Malon, J. Heterocycl. Chem., 2006, 43, 1065–1070 CrossRef CAS
- W. Wang, R. Cencic, L. Whitesell, J. Pelletier and J. Porco, Chem.–Eur. J., 2016, 22, 12006–12010 CrossRef CAS
- P. Funk, K. Motyka, P. Dzubak, P. Znojek, S. Gurska, J. Kusz, C. McMaster, M. Hajduch and M. Soural, RSC Adv., 2015, 5, 48861–48867 RSC
- K. Motyka, J. Hlavac, M. Soural, P. Hradil, P. Krejci, L. Kvapil and M. Weiss, Tetrahedron Lett., 2011, 52, 715–717 CrossRef CAS
- S. Sharma, V. K. Srivastava and A. Kumar, Eur. J. Med. Chem., 2002, 37, 689–697 CrossRef CAS
- A. Varnavas, L. Lassiani, V. Valenta, F. Berti, L. Mennuni and F. Makovec, Bioorg. Med. Chem., 2003, 11, 741–751 CrossRef CAS
- J. F. Sun, Y. J. Xu, X. H. Kong, Y. Su and Z. Y. Wang, Neurosci. Lett., 2019, 696, 67–73 CrossRef CAS
- H. Rao, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2005, 70, 8107–8109 CrossRef CAS
- H. Rao, Y. Jin, H. Fu, Y. Jiang and Y. Zhao, Chem.–Eur. J., 2006, 12, 3636–3646 CrossRef CAS
- S. Liu and L. Xu, Asian J. Org. Chem., 2018, 7, 1856–1863 CrossRef CAS
- S. Ando, Y. Hirota, H. Matsunaga and T. Ishizuka, Tetrahedron Lett., 2019, 60, 1277–1280 CrossRef CAS
- A. S. Guram, R. A. Rennels and S. L. Buchwald, Angew. Chem.,
Int. Ed. Engl., 1995, 34, 1348–1350 CrossRef CAS
- J. Louie and J. F. Hartwig, Tetrahedron Lett., 1995, 36, 3609–3612 CrossRef CAS
- V. Zimmermann and S. Brase, J. Comb. Chem., 2007, 9, 1114–1137 CrossRef CAS
- R. C. D. Brown, J. Keily and R. Karim, Tetrahedron Lett., 2000, 41, 3247–3251 CrossRef CAS
- S. Krajcovicova, J. Stankova, P. Dzubak, M. Hajduch, M. Soural and M. Urban, Chem.–Eur. J., 2018, 24, 4957–4966 CrossRef CAS
- R. Apodaca and W. Xiao, Org. Lett., 2001, 3, 1745–1748 CrossRef CAS
- T. Volná, K. Motyka and J. Hlaváč, Chromatographia, 2016, 79, 1153–1163 CrossRef
- M. Soural, P. Hradil, S. Krupkova and J. Hlavac, Mini-Rev. Org. Chem., 2012, 9, 426–432 CrossRef CAS
- V. B. Oza, H. M. Petrassi, H. E. Purkey and J. W. Kelly, Bioorg. Med. Chem. Lett., 1999, 9, 1–6 CrossRef CAS
- D. Tiwari, S. Haque, S. Misra and R. Chandra, Int. J. Drug Dev. Res., 2011, 3, 265–271 CAS
- J. X. Kelly, M. J. Smilkstein, R. Brun, S. Wittlin, R. A. Cooper, K. D. Lane, A. Janowsky, R. A. Johnson, R. A. Dodean, R. Winter, D. J. Hinrichs and M. K. Riscoe, Nature, 2009, 459, 270–273 CrossRef CAS
- J. R. Goodell, A. A. Madhok, H. Hiasa and D. M. Ferguson, Bioorg. Med. Chem., 2006, 14, 5467–5480 CrossRef CAS
- J. R. Goodell, F. Puig-Basagoiti, B. M. Forshey, P. Y. Shi and D. M. Ferguson, J. Med. Chem., 2006, 49, 2127–2137 CrossRef CAS
- A. R. Benoit, C. Schiaffo, C. E. Salomon, J. R. Goodell, H. Hiasa and D. M. Ferguson, Bioorg. Med. Chem. Lett., 2014, 24, 3014–3017 CrossRef CAS
- S. H. Watterson, P. Chen, Y. Zhao, H. H. Gu, T. G. M. Dhar, Z. Xiao, S. K. Ballentine, Z. Shen, C. A. Fleener, K. A. Rouleau, M. Obermeier, Z. Yang, K. W. McIntyre, D. J. Shuster, M. Witmer, D. Dambach, S. Chao, A. Mathur, B. C. Chen, J. C. Barrish, J. A. Robl, R. Townsend and E. J. Iwanowicz, J. Med. Chem., 2007, 50, 3730–3742 CrossRef CAS
- L. A. Howell, A. Howman, M. A. O'Connell, A. Mueller and M. Searcey, Bioorg. Med. Chem. Lett., 2009, 19, 5880–5883 CrossRef CAS
- L. I. James, V. K. Korboukh, L. Krichevsky, B. M. Baughman, J. M. Herold, J. L. Norris, J. Jin, D. B. Kireev, W. P. Janzen, C. H. Arrowsmith and S. V. Frye, J. Med. Chem., 2013, 56, 7358–7371 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01308d |
| This journal is © The Royal Society of Chemistry 2021 |