
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbazole sulfonamide-based macrocyclic receptors capable of selective complexation of fluoride ion†
Na Luoa,
Junhong Lia,
Tao Sunb,
Suran Wana,
Peijia Lia,
Nan Wua,
Ya Yana and
Xiaoping Bao *a
*a
aState Key Laboratory Breeding Base of Green Pesticide and Agricultural Bioengineering, Key Laboratory of Green Pesticide and Agricultural Bioengineering, Ministry of Education, Center for Research and Development of Fine Chemicals, Guizhou University, Guiyang 550025, China. E-mail: baoxp_1980@aliyun.com
bCollege of Chemistry and Chemical Engineering, Guizhou Key Laboratory of High Performance Computational Chemistry, Guizhou University, Guiyang 550025, China
First published on 10th March 2021
Abstract
Two carbazole sulfonamide-based macrocycles 1 and 2 were facilely synthesized and carefully evaluated for their anion recognition properties. The obtained results revealed that macrocycle 1 with a 1,3-xylyl linker was able to bind fluoride ion more strongly and selectively in acetonitrile medium than its strong competitors (like acetate and dihydrogen phosphate anions), with a large binding constant (Ka) of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 878 M−1. More importantly, an exclusive fluoride recognition was achieved for macrocycle 1 in the more polar DMSO-d6 solution, albeit with a moderate affinity of Ka = 147 M−1. Compared with macrocycle 1, macrocycle 2 bearing a 2,6-lutidinyl linkage exhibited a remarkable change not only in the anion affinity but also in the anion selectivity, although with only a slight difference in their molecular structures.
878 M−1. More importantly, an exclusive fluoride recognition was achieved for macrocycle 1 in the more polar DMSO-d6 solution, albeit with a moderate affinity of Ka = 147 M−1. Compared with macrocycle 1, macrocycle 2 bearing a 2,6-lutidinyl linkage exhibited a remarkable change not only in the anion affinity but also in the anion selectivity, although with only a slight difference in their molecular structures.
Introduction
Fluoride ions play crucial roles in many chemical, biological, and environmental processes, thus the development of fluoride-selective receptors and/or sensors has become a research focus in the field of supramolecular chemistry.1,2 However, only a handful of examples of anion receptors enabling the simultaneous discrimination of fluoride from its strong competitors (like acetate and dihydrogen phosphate anions) by means of their remarkably different affinities have been reported so far,3–12 if ruling out some reports of specific reactions-based detection of fluoride. As we all know, the backbone of carbazole is an ideal platform for constructing efficient anion receptors since it is easy to install various anion binding groups onto this backbone and then cooperatively bind anions with the assistance of the 9-position NH unit.13 Moreover, it has been shown that macrocyclic anion receptors are capable of pre-organizing relevant recognition sites, thus being favorable for achieving strong and selective complexation towards specific anions based on size and/or shape complementarity to the macrocycles.14,15 Nonetheless, the preparation of such macrocycles generally requires lengthy and tedious synthetic procedures due to their sophisticated structures. Among the commonly-used anion-binding groups, sulfonamide NH can form a stronger N–H⋯anion hydrogen bond than its amide NH analogue.16,17 In addition, the utilization of aromatic CH group as hydrogen-bond donor for constructing anion receptors was hitherto less studied,18,19 although they had some unique advantages over the conventional NH/OH hydrogen-bond donors including a remarkable resistance towards the deprotonation event20 and a larger affinity towards softer anions in some cases.21 In 2016, we synthesized 1,8-disulfonamidocarbazole-dipyrromethane Schiff-base macrocycle & its reduced derivative and evaluated their anion binding properties by UV-vis and 1H NMR titration methods.22 The results indicated that two macrocyclic receptors exhibited strong binding interactions (Ka > 104 M−1) with fluoride, acetate and dihydrogen phosphate ions over other anions in the highly competitive DMSO medium, but both macrocycles were poorly selective among these three anions.Based on all of the above considerations, we herein reported a carbazole sulfonamide-based macrocycle 1 containing four hydrogen-bond donors as anion-binding sites, namely one carbazole NH, two sulfonamide NHs and one aromatic CH groups. In order to clarify the effects of aromatic CH group of the 1,3-xylyl linker in 1 on the anion-recognition behaviors, macrocycle 2 with a 2,6-lutidinyl linker was also synthesized for comparison.
Results and discussion
Preparation of macrocycles 1 and 2
As shown in Scheme 1, macrocycles 1 and 2 were facilely synthesized by condensation of carbazole-1,8-disulfonyl chloride 322,23 with the appropriate diamines in dry CH2Cl2 using TEA as a catalyst, giving reasonable yields of 30% and 25%, respectively. Both macrocycles were fully characterized by 1H NMR, 13C NMR, and HRMS (high resolution mass spectrometry) techniques. Additionally, single crystals of 1 and 2 suitable for X-ray diffraction analysis were obtained by slow evaporation of their CH3CN solution at room temperature. As shown in Fig. 1, two macrocycles both adopted a folded conformation in the solid state. More specifically, two sulfonamide NH protons in 1 pointed away from the macrocyclic cavity and the dihedral angle between carbazole plane and phenyl ring was measured to be 12.49°. In other words, NH and CH recognition units of 1 displayed a divergent orientation in the anion-free state. Unlike 1, carbazole NH and one of the two sulfonamide NHs of 2 pointed in the same direction. Relative to 1, the dihedral angle between two aromatic rings of 2 was slightly reduced to 11.20°.
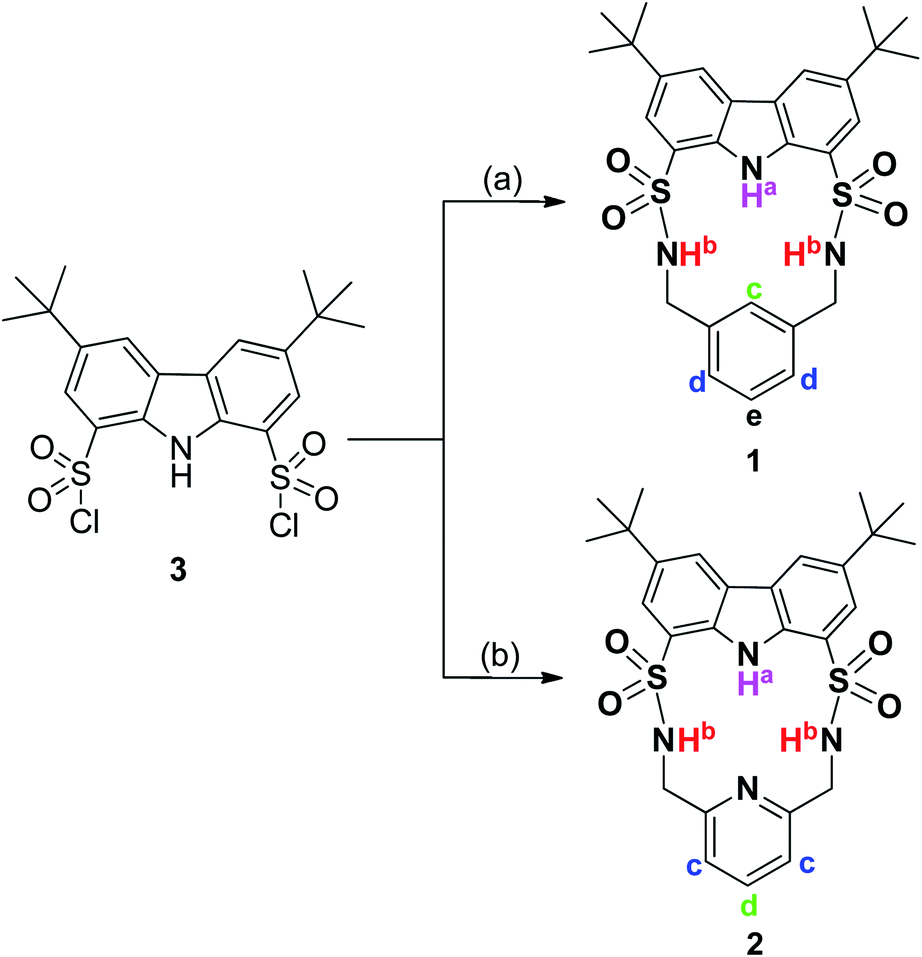 | ||
| Scheme 1 Synthesis of macrocycles 1 and 2. Reaction conditions: (a) 1,3-xylenediamine/dry CH2Cl2/dry TEA/rt/30%; (b) 2,6-lutidinylenediamine/dry CH2Cl2/dry TEA/rt/25%. | ||
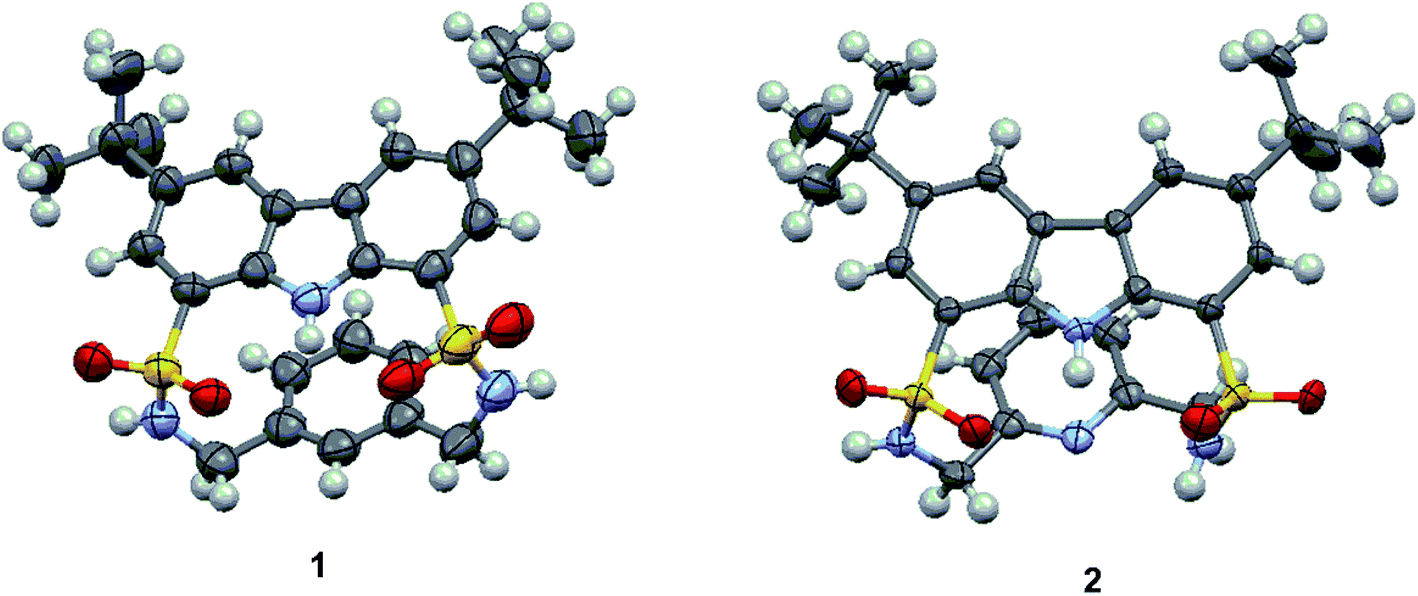 | ||
| Fig. 1 Crystal structures of macrocycles 1 and 2. | ||
Anion recognition studies
The binding interactions between macrocyclic receptors and a variety of anions (including spherical F−/Cl−/Br−, tetrahedral H2PO4−/HSO4−/ClO4−, and trigonal planar PhCOO−/CH3COO−/NO3−) were initially investigated by 1H NMR titration method in CD3CN. Upon addition of tetra-n-butylammonium fluoride (TBAF) into a CD3CN solution of 1 (Fig. 2a), the proton signals assigned to carbazole NH (Ha) and sulfonamide NHs (Hb) rapidly vanished and then reappeared (after 1.0 equiv. of F−). Notably, remarkable downfield shifts were observed for these two signals throughout the whole titration process (+4.56 and +4.99 ppm, respectively), indicative of the formation of strong intermolecular hydrogen bonds between these NH protons and the bound F−. As expected, the signal of aromatic CH (Hc) also underwent a relatively small but well-defined downfield shift of +0.58 ppm, proving its effective participation in the stabilization of the resultant 1·F− complex. Both molar ratio (Fig. 2b) and Job's plot (Fig. S1†) methods provided vigorous supports for the formation of 1:1 1/F− complex in CD3CN solution. However, the binding interaction of 1 with F− in this medium was too strong to be accurately determined by 1H NMR titration method (Ka > 10000 M−1), as inferred from the presence of a sharp titration isotherm in Fig. 2b.24,25
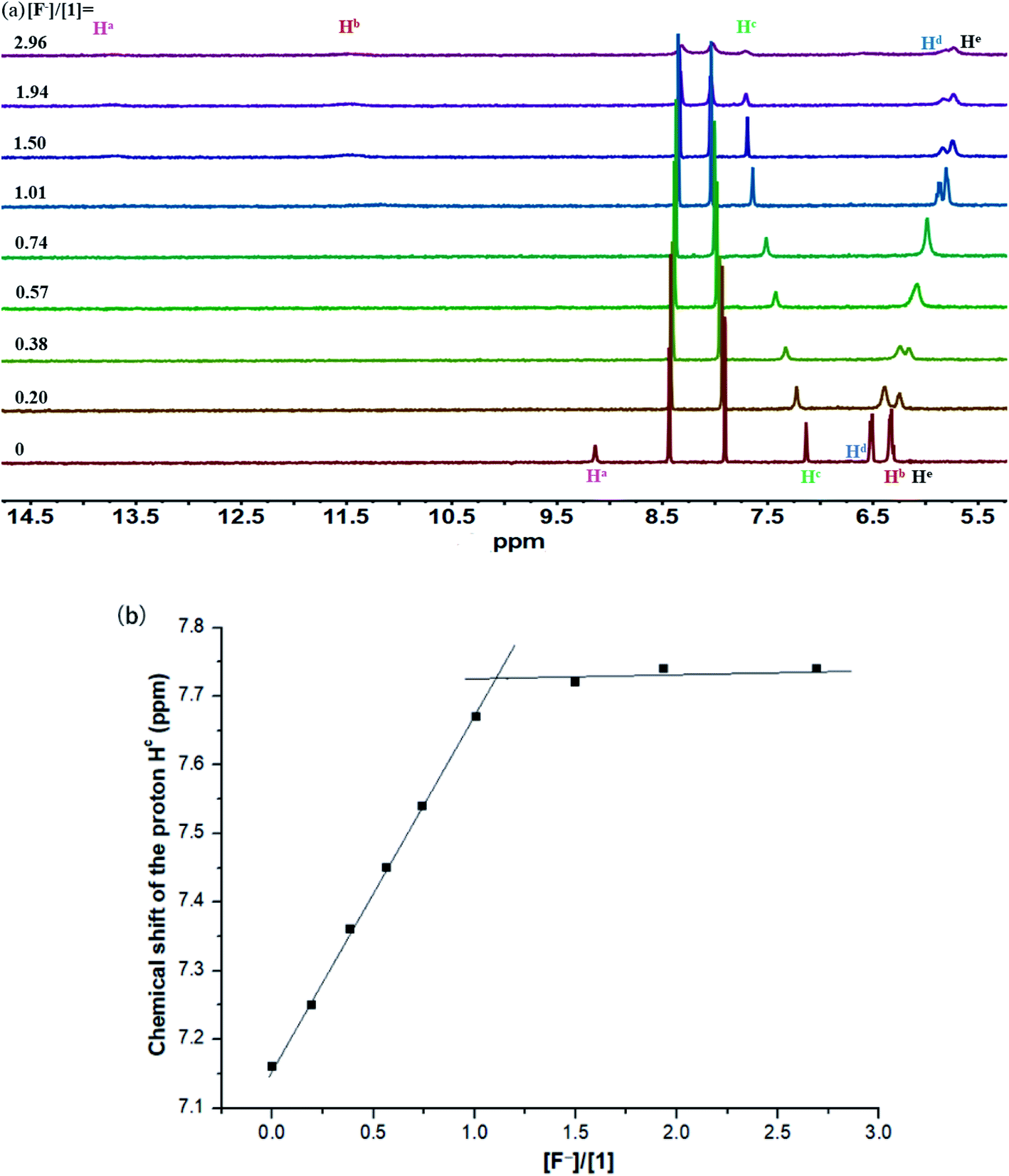 | ||
| Fig. 2 (a) Stack plot of 1H NMR titration of macrocycle 1 (1.6 mM) with TBAF in CD3CN at 298 K. (b) Chemical shift changes of the proton Hc in 1 upon addition of TBAF in CD3CN (Ka > 10000 M−1). | ||
Unlike fluoride, the signal of carbazole NH in 1 was found to be traceable during the overall titration process and move downfield consistently (from 9.14 to 9.84 ppm) after addition of H2PO4− (Fig. 3a). Likewise, the signal of Hc also underwent a clear downfield shift from 7.14 to 7.49 ppm. Notably, a strong binding with H2PO4− by macrocycle 1 (Ka = 4920 M−1) was observed in acetonitrile (Fig. 3b).
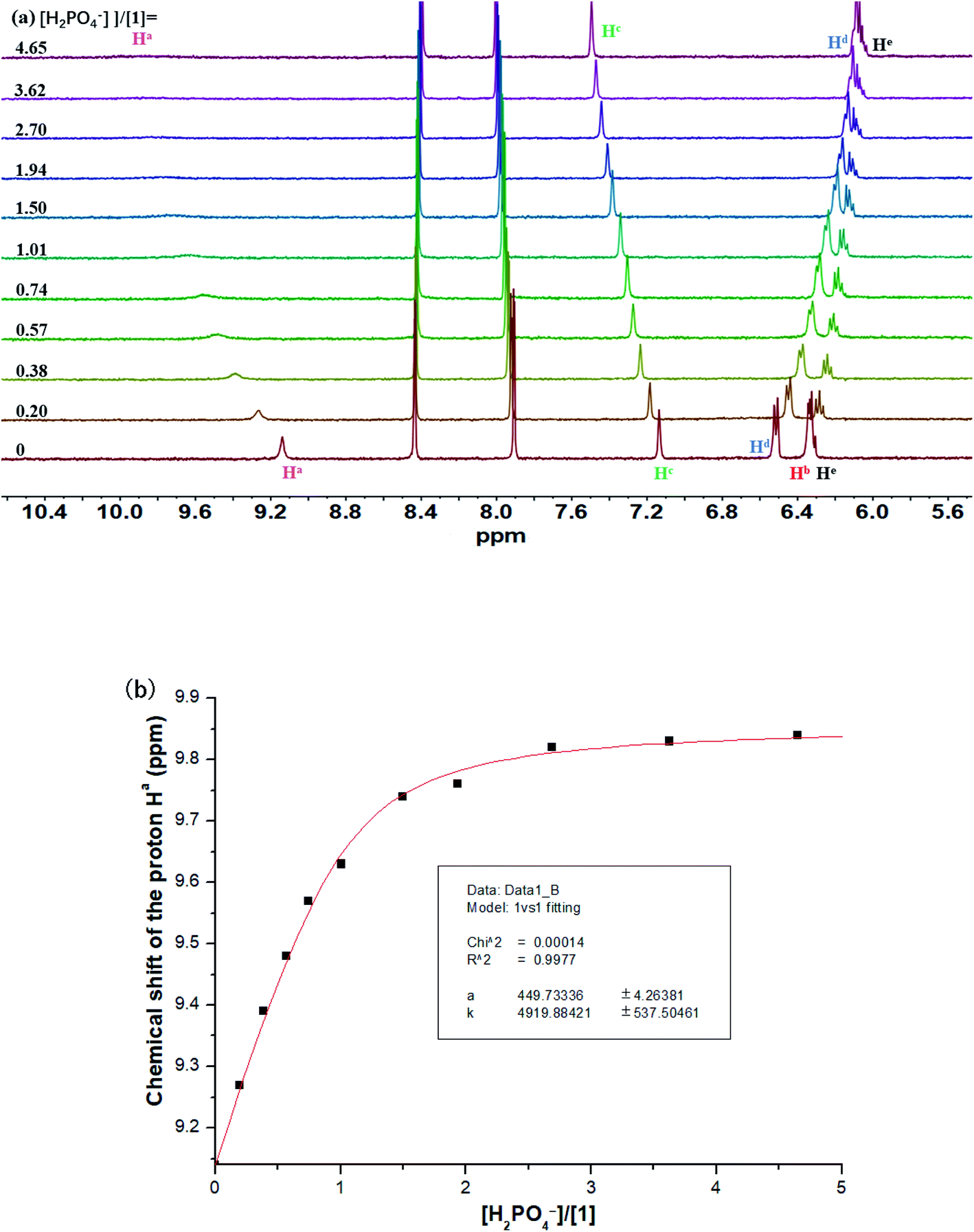 | ||
| Fig. 3 (a) Stack plot of 1H NMR titration of macrocycle 1 (1.6 mM) with TBAH2PO4 in CD3CN at 298 K. (b) Fitting binding isotherms of macrocycle 1 with TBAH2PO4 in CD3CN at 298 K, showing chemical shift changes of the proton Ha based on a 1:1 binding model. | ||
After addition of other competitive anions like PhCOO− and CH3COO− (Fig. S2 and S4†), two NH signals of 1 disappeared rapidly. Meanwhile, the signal of Hc experienced a continuous downfield shift of ca. +0.78 and +0.44 ppm, respectively. In the case of chloride ion (Fig. S6†), sulfonamide NH of 1 exhibited a pronounced downfield shift of +0.61 ppm. At the same time, a small downfield shift of +0.26 ppm was also observed for aromatic CH. In contrast, carbazole NH only underwent a slight downfield shift of +0.05 ppm during the titration process. These findings clearly indicated that chloride ion was bound mainly by three intermolecular hydrogen bonds, contributing from two SO2NH and one CH units.
In all cases except for fluoride ion, the 1H NMR titration profiles gave the best fitting results using a 1:1 binding model (Table 1). For macrocycle 1, the observed binding trend followed the order F− (>10000 M−1) > H2PO4− (4920 M−1) > PhCOO− (1013 M−1) > CH3COO− (279 M−1) > Cl− (91 M−1) > Br− (24 M−1) > NO3−/HSO4− (<10 M−1) > ClO4− (no binding), which was inconsistent with the Hofmeister order of related anions (H2PO4− > CH3COO− > F− > Cl− > Br−).26 Of note, an unusual preference of 1 for tetrahedral H2PO4− over more basic & trigonal planar PhCOO− and CH3COO− was seen. The same binding trend was also found for macrocycle 2, with the exception of a reversed order involving HSO4− and Br−. It was noteworthy that two types of protons on the pyridine ring of 2 (Hc and Hd) both experienced a visible upfield shift upon exposure to F−, H2PO4−, CH3COO−, PhCOO−, and Cl− ions (Fig. S22, S24, S26, S28, and S32†), which was presumably caused by the proximity effect between these anions and the pyridine moiety after the complexation.
| 1 | 2 | Ka(1)/Ka(2) | |
|---|---|---|---|
| a Determined by the 1H NMR titration experiments in CD3CN, by monitoring the proton signal of the receptors showing the most significant chemical shift changes over the titration process. The errors in the Ka values were within 15%. The R2 values for the 1:1 non-linear fitting to determine Ka values ranged between 0.981 and 0.999.b All the anions were used as their tetrabutylammonium salts.c Determined by the UV-vis titration experiments in CH3CN, due to the inability to measure Ka value accurately by 1H NMR titration method.d NB = no binding. |
|||
| F− | 50878 ± 4721c |
12885 ± 1962c |
3.9 |
| H2PO4− | 4920 ± 538 | 2428 ± 309 | 2.0 |
| PhCOO− | 1013 ± 91 | 2005 ± 75 | 0.5 |
| CH3COO− | 279 ± 21 | 940 ± 46 | 0.3 |
| Cl− | 91 ± 2 | 335 ± 7 | 0.3 |
| Br− | 24 ± 1 | <10 | >2.4 |
| HSO4− | <10 | 42 ± 6 | <0.2 |
| NO3− | <10 | <10 | — |
| ClO4− | NBd | NBd | — |
| Ka(F−)/Ka(H2PO4−) | 10.3 | 5.3 | 1.9 |
| Ka(F−)/Ka(PhCOO−) | 50.2 | 6.4 | 7.8 |
| Ka(F−)/Ka(CH3COO−) | 182.4 | 13.7 | 13.3 |
| Ka(F−)/Ka(Cl−) | 559.1 | 38.6 | 14.5 |
To accurately quantify binding affinities of both macrocycles towards F− in acetonitrile solution, UV-vis titration studies were subsequently carried out. As shown in Fig. 4, a gradual increase in the absorption peak at 294 nm was detected for 1, in the presence of increasing concentrations of TBAF. Meanwhile, two pseudo-isosbestic points were discovered at 305 and 355 nm. A 1:1 1/F− binding model generated an excellent fitting result with Ka = 50878 M−1 (consistent with the affinity range determined by 1H NMR titration experiments), which was at least ten times higher than its nearest competitor H2PO4−. To validate the nature of fluoride-induced spectroscopic changes of 1 (intermolecular hydrogen-bonding interactions or NH deprotonation effect), the titration of 1 with strong base TBAOH was also performed. As shown in Fig. S38,† the absorption peak at 294 nm of 1 was considerably weakened upon addition of OH−, along with a noticeable enhancement in the absorption bands centered at 275 and 325 nm. The above phenomena were completely different with those caused by F−, which confirmed that multiple hydrogen-bonding interactions between 1 and fluoride accounted for the observed spectral changes in Fig. 4. In addition, the affinity of macrocycle 2 with F− was determined to be 12885 M−1 under the same experimental conditions (Fig. S39 and S40†).
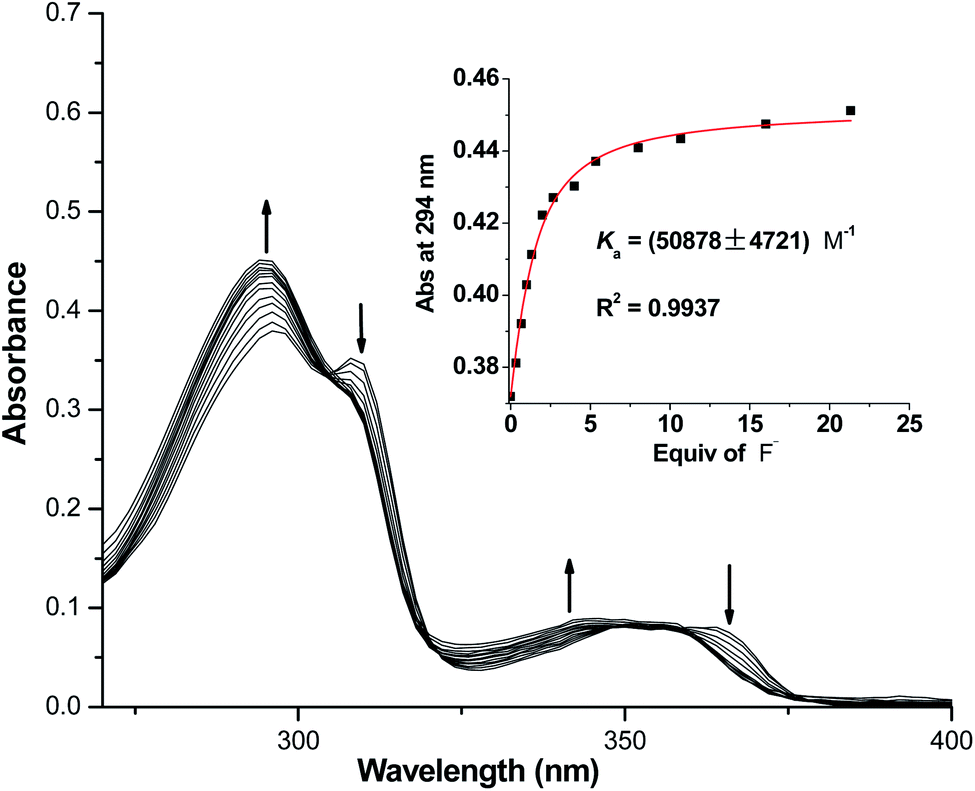 | ||
| Fig. 4 UV-vis titration of macrocycle 1 (20 μM) with TBAF in CH3CN (Inset: A 1:1 non-linear curve fitting of the absorbance at 294 nm of macrocycle 1 against the added F−). | ||
After a close inspection of the data from Table 1, some preliminary conclusions could be drawn as follows: compared with 1, a decrease in the affinity was detected for 2 with F−, H2PO4−, and Br−, with a value of 3.9-, 2.0- and >2.4-fold, respectively. On the contrary, a higher affinity of 2 than 1 was discovered towards other anions like PhCOO−, CH3COO−, Cl−, and HSO4− (ranging from 2.0- to >4.2-fold). For macrocycle 1 bearing a 1,3-xylyl linker, the presence of an additional aromatic CH group as the potential anion-binding site was beneficial to anion complexation. After replacing the 1,3-xylyl linker with a 2,6-lutidinyl spacer, the resultant 2 was probably endowed with a certain degree of preorganization for its SO2NH protons in solution (via intramolecular hydrogen-bonding interactions with the pyridine nitrogen atom), which was also conducive to anion binding. Simultaneously, this replacement meant the loss of an additional anion-binding site. Therefore, different change trends in the affinity towards various anions were observed for two macrocycles, depending on comprehensive outcomes of the aforementioned factors. Among all the tested anions, the affinity of 2 for HSO4− was most significantly enhanced (with respect to 1), which may result from the existence of an additional hydrogen bond between bisulfate hydroxyl proton and pyridine nitrogen atom of 2.27
It should be noted that macrocycle 1 displayed a remarkable improvement in the binding selectivity for fluoride over chloride, acetate, benzoate, and dihydrogenphosphate anions (relative to macrocycle 2), as revealed by selectivity factors expressed as ratios of the related binding constants Ka(F−)/Ka(Cl−), Ka(F−)/Ka(CH3COO−), Ka(F−)/Ka(PhCOO−), and Ka(F−)/Ka(H2PO4−). For example, a 5.3-fold preference for F− over H2PO4− in 2 was improved moderately to 10.3-fold in the case of 1. Moreover, the binding selectivity of 1 for F− over PhCOO− was pronouncedly improved from 6.4 (in the case of 2) to 50.2. In addition, a 13.7:1 selectivity for F− over CH3COO− in 2 was increased sharply to 182.4:1 as for 1. More significantly, the binding selectivity for F− over Cl− was dramatically enhanced from 38.6 to 559.1, while changing the macrocycles from 2 to 1. In a word, a slight structural change in these macrocyclic receptors gave rise to profound influences not only on the binding affinity but also on the binding selectivity.
Previous studies have documented that solvent effects in supramolecular systems played a significant role in the affinity and selectivity of anion receptors.28–30 Thus, we subsequently studied recognition properties of 1 in the more polar DMSO-d6 medium. As displayed in Fig. 5a, the addition of 0.2 equiv. of F− firstly triggered a significant broadening of the signals assigned to carbazole NH and sulfonamide NHs, and then they experienced a thorough disappearance while continually increasing the concentration of fluoride ion. Meanwhile, the resonance of aromatic CH proton was found to progressively move downfield from 7.17 to 7.59 ppm. Noteworthily, no NH deprotonation event took place for 1 before the addition of 2.8 equiv. of F−. In other words, a small amount of F− firstly formed hydrogen-bonded complex with 1, and then more F− (≥2.8 equiv.) induced NH deprotonation in 1 (as evidenced by the appearance of a characteristic triplet of the HF2− species centered at 16.20 ppm).31,32 Based on the titration data extracted from hydrogen-bonding interaction stage, a rough Ka of 147 M−1 (Fig. 5b) was determined (a 1:1 binding stoichiometry between 1 and F− in DMSO-d6 supported by Job plot method in Fig. S21†). Interestingly, upon addition of other competitive anions like H2PO4− and CH3COO− (Fig. S13 and S15†), only the disappearance of two NH signals was observed for 1 (probably due to a rapid proton exchange process),33 without meaningful chemical shift changes for other proton signals. These observations demonstrated no or very weak binding of 1 with these anions in this medium.34,35 Taken together, macrocycle 1 achieved an exclusive recognition towards fluoride ion in the strongly polar DMSO-d6 medium, albeit with a moderate affinity.
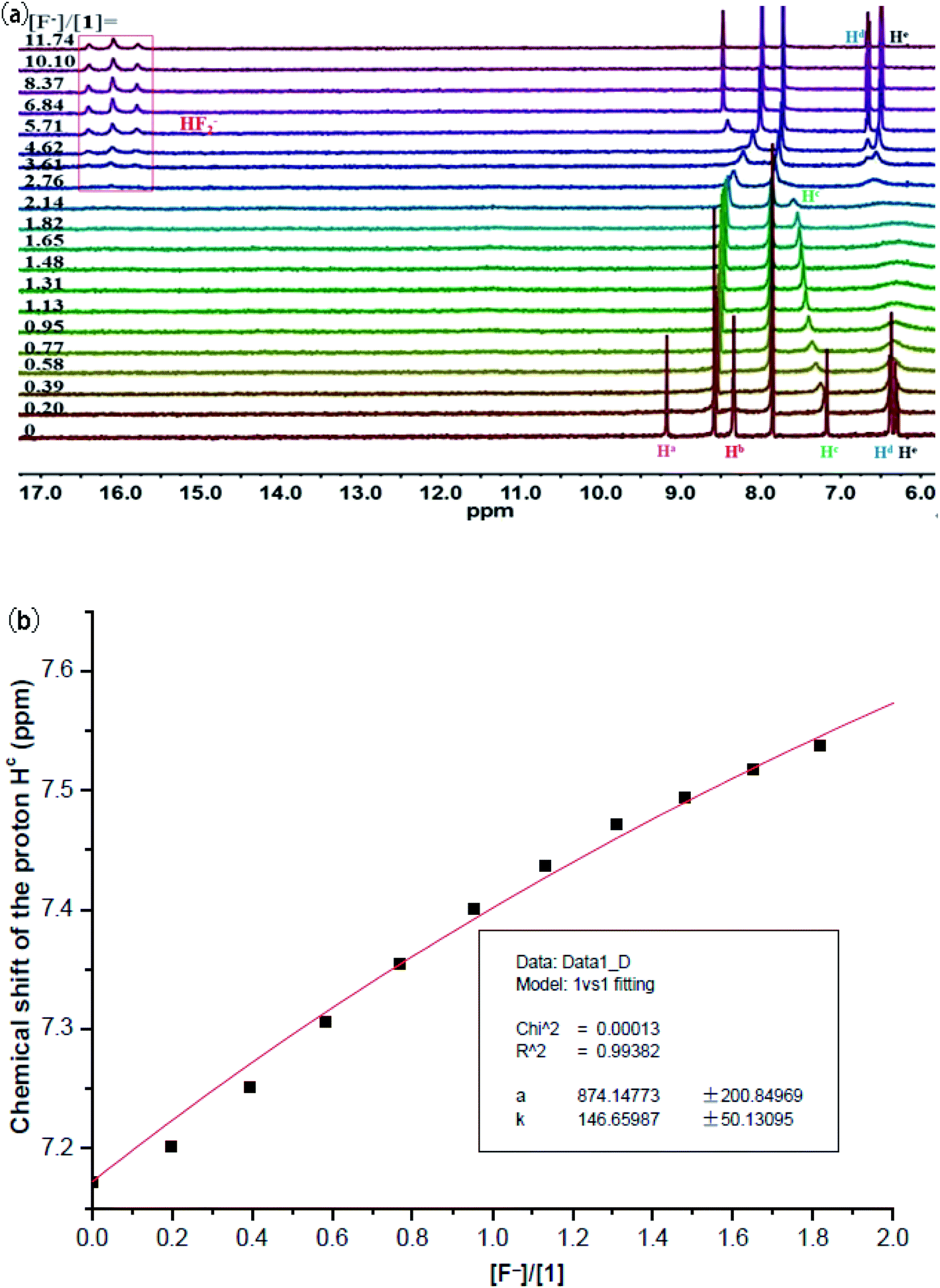 | ||
| Fig. 5 (a) 1H NMR titration of macrocycle 1 (1.6 mM) with TBAF in DMSO-d6. (b) Fitting binding isotherms of macrocycle 1 with TBAF in DMSO-d6 at 298 K, showing chemical shift changes of the proton Hc based on a 1:1 binding model. | ||
Single crystals of fluoride complex of macrocycle 1 were obtained by slow evaporation of an acetonitrile solution of 1 in the presence of excess fluoride ion. Unexpectedly, the binding model in the solid state (1/F− = 2:1) turned out to be different from that observed in solution (1/F− = 1:1). As seen from Fig. 6, one fluoride ion was captured by two sulfonamide NH groups (N⋯F distances of 2.650 and 2.641 Å, respectively, and the corresponding N–H⋯F angles of 160.4 and 169.1°) from two adjacent host molecules. This disparity in the binding stoichiometry between solution phase and solid state had been previously reported,36–40 likely resulting from molecular packing effect in the latter case.37
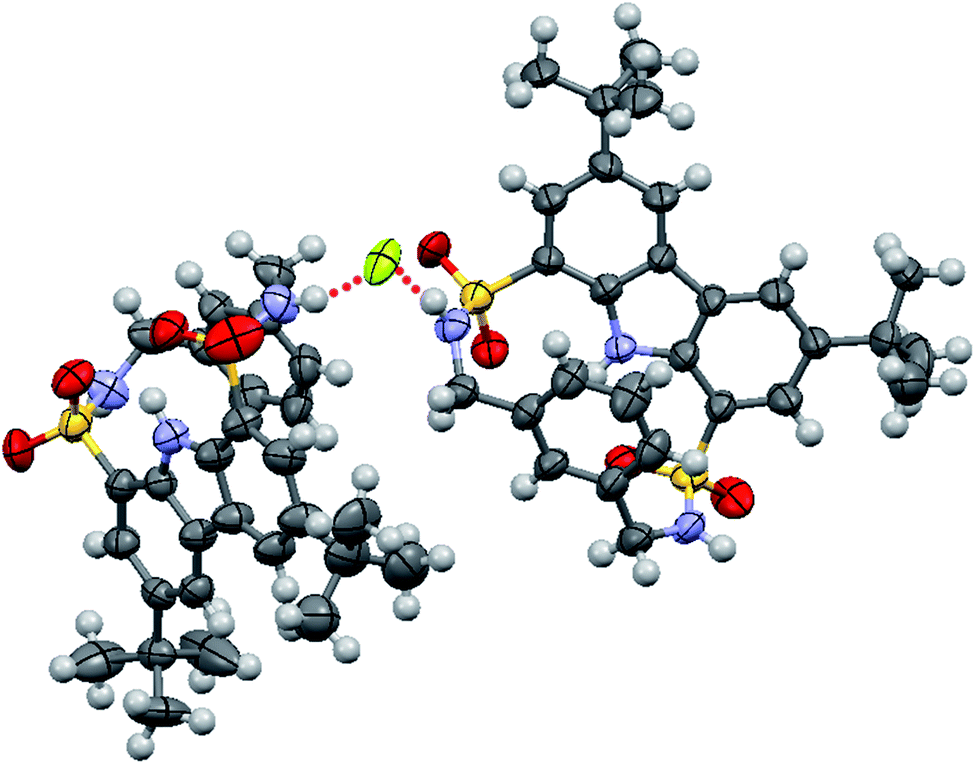 | ||
| Fig. 6 Crystal structure of the formed complex between macrocycle 1 and fluoride ion. The counterion (TBA+) was omitted for clarity. | ||
In order to shed more light on the nature of the formed complex between macrocycle 1 and fluoride ion, density functional theory (DFT) calculations were performed at the B3LYP/6-311++G(d,p) level under the polarizable continuum model (PCM) using CH3CN as a solvent in the Gaussian 16 package. As shown in Fig. 7, an energy-minimized structure of the 1:1 1/F− complex confirmed the presence of four intermolecular hydrogen bonds involving 1 and the bound F− ion. More specifically, the F⋯N/C distances between F− and four hydrogen-bond donors (one carbazole NH, two sulfonamide NHs and one aromatic CH units) were determined to be 2.567, 2.768, 2.814, and 2.964 Å, respectively. Combined with their bond angles (172.74, 171.68, 167.28, and 90.41°, respectively), a conclusion could be drawn that carbazole NH and sulfonamide NHs made a main contribution to the stabilization of the 1/F− complex. In the meanwhile, aromatic CH group from the 1,3-xylyl linker also played a role in the hydrogen-bonding interaction with fluoride, albeit a relatively smaller contribution. These results of molecular optimization were in good agreement with the aforementioned 1H NMR titration experiments in CD3CN (Fig. 2). Of note, macrocycle 1 had to undergo a conformational change (from a divergent pattern to a convergent style as for the NH/CH binding units) for cooperative utilization of four hydrogen-bond donors for the fluoride binding. Interestingly, the dihedral angle between carbazole plane and phenyl ring remained nearly unchanged (12.21° in the resulting complex), that is, a near 180-degree rotation took place for the benzene ring of 1 upon fluoride binding.
 | ||
| Fig. 7 DFT-optimized structure of macrocycle 1/F− complex. | ||
Conclusions
In summary, two carbazole sulfonamide-based macrocycles 1 and 2 were facilely prepared and assessed as anion receptors by 1H NMR, UV-vis titration, solid-state X-ray, and DFT calculation methods. The obtained results indicated that macrocycle 1 had a powerful and selective binding towards fluoride ion in acetonitrile solution, over its strong competitors including CH3COO− and H2PO4− anions. More importantly, macrocycle 1 was found to recognize F− exclusively in the more polar DMSO-d6 solution. As a well-performed anion receptor, macrocycle 1 may have the potential for future applications in fluoride sensing, transmembrane fluoride transport and fluoride extraction, after a further structural modification.Experimental
Materials and general methods
All the chemicals were purchased from commercial suppliers and used without further purification (unless stated otherwise). Melting points were measured on a XT-4 binocular microscope (Beijing Tech Instrument Co., China) and uncorrected. All the 1H and 13C NMR data were determined on a JEOL-ECX 500 NMR spectrometer at 298 K using TMS as an internal standard, and chemical shift (δ) was expressed in parts per million (ppm). The following abbreviations were utilized in expressing the multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. High resolution mass spectra (HRMS) were recorded on a Thermo Scientific Q Exactive Hybrid Quadrupole-Orbitrap mass spectrometer. UV-vis spectra were collected on a Beijing PGENERAL TU-1900 spectrometer. The X-ray crystallographic data were collected using a Bruker D8 Venture diffractometer. All the non-linear curve fitting analysis were conducted using the previously-reported equations41 and the software of Origin 6.0.Synthesis
:1, v/v) as an eluent, yielding 1 as a white solid (40.4 mg, 30% yield), mp > 250 °C. 1H NMR (500 MHz, CD3CN) δ: 9.14 (s, 1H), 8.44 (d, J = 5.0 Hz, 2H), 7.91 (d, J = 5.0 Hz, 2H), 7.14 (s, 1H), 6.52 (d, J = 5.0 Hz, 2H), 6.34–6.30 (m, 3H), 4.18 (d, J = 5.0 Hz, 4H), 1.47 (s, 18H); 1H NMR (500 MHz, DMSO-d6) δ: 9.18 (s, 1H), 8.58 (d, J = 5.0 Hz, 2H), 8.34 (t, J = 5.0 Hz, 2H), 7.86 (d, J = 5.0 Hz, 2H), 7.17 (s, 1H), 6.38 (d, J = 5.0 Hz, 2H), 6.31–6.28 (m, 1H), 4.07 (d, J = 5.0 Hz, 4H), 1.45 (s, 18H); 13C NMR (125 MHz, DMSO-d6) δ: 142.4, 136.7, 133.4, 130.6, 127.0, 126.4, 123.5, 122.8, 122.4, 122.3, 46.3, 34.7, 31.6; HRMS (ESI) m/z: [M + H]+ calcd for C28H34N3O4S2: 540.1985, found: 540.1978.1H NMR titration studies
1H NMR titrations were performed on a JEOL-ECX 500 NMR spectrometer, working at a frequency of 500 MHz with the probe temperature at 298 K. All the tested anions were added as their tetra-n-butylammonium (TBA) salts. In all cases, the 1H NMR titrations were carried out while keeping the concentration of the macrocycles 1 or 2 (1.6 mM in CD3CN or DMSO-d6) constant through dissolving guest anions with the same macrocycle solution to prepare guest solution. The guest solution was then progressively added to the macrocycle solution using an appropriate pipette, and the resulting 1H NMR spectrum was recorded after each addition. The above operations ensured that the concentration of the macrocycle remained constant, whilst the concentration of the added anions varied. Finally, a non-linear curve fitting analysis using a 1:1 binding model was performed to fit experimental data and gave the corresponding binding constants (Ka).41
UV-vis titration studies
UV-vis titration studies were conducted on a TU-1900 UV-visible spectrophotometer, by adding an acetonitrile solution containing guest anions F− or OH− (20 mM, using their tetrabutylammonium salts) into an acetonitrile solution of the macrocycle (20 μM) in a quartz cuvette at 298 K. The resultant UV-vis spectra were recorded after each addition. Finally, the absorbance values at the specific wavelength were plotted against equivalents of the added anions and fitted based on a 1:1 binding stoichiometry.41
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully thank the National Natural Science Foundation of China (No. 21161005) for financial support.Notes and references
- Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511–5571 CrossRef CAS PubMed.
- M. Cametti and K. Rissanen, Chem. Soc. Rev., 2013, 42, 2016–2038 RSC.
- M. Cametti and K. Rissanen, Chem. Commun., 2009, 2809–2829 RSC.
- M. Vázquez, L. Fabbrizzi, A. Taglietti, R. M. Pedrido, A. M. González-Noya and M. R. Bermejo, Angew. Chem., Int. Ed., 2004, 43, 1962–1965 CrossRef.
- K. J. Chang, D. Moon, M. S. Lah and K. S. Jeong, Angew. Chem., Int. Ed., 2005, 44, 7926–7929 CrossRef CAS PubMed.
- S. O. Kang, J. M. Llinares, D. Powell, D. VanderVelde and K. Bowman-James, J. Am. Chem. Soc., 2003, 125, 10152–10153 CrossRef CAS.
- R. Nishiyabu and P. Anzenbacher, J. Am. Chem. Soc., 2005, 127, 8270–8271 CrossRef CAS PubMed.
- R. Nishiyabu, M. A. Palacios, W. Dehaen and P. Anzenbacher, J. Am. Chem. Soc., 2006, 128, 11496–11504 CrossRef CAS PubMed.
- R. Nishiyabu and P. Anzenbacher, Org. Lett., 2006, 8, 359–362 CrossRef CAS.
- G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2007, 2121–2123 RSC.
- J. Yoo, M. S. Kim, S. J. Hong, J. L. Sessler and C. H. Lee, J. Org. Chem., 2009, 74, 1065–1069 CrossRef CAS PubMed.
- R. Samanta, B. S. Kumar and P. K. Panda, Org. Lett., 2015, 17, 4140–4143 CrossRef CAS PubMed.
- P. A. Gale, Chem. Commun., 2008, 4525–4540 RSC.
- K. Choi and A. D. Hamilton, Coord. Chem. Rev., 2003, 240, 101–110 CrossRef CAS.
- M. Chmielewski and J. Jurczak, Tetrahedron Lett., 2004, 45, 6007–6010 CrossRef CAS.
- C. Caltagirone, G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2008, 61–63 RSC.
- D. Mondal, A. Sathyan, S. V. Shinde, K. K. Mishra and P. Talukdar, Org. Biomol. Chem., 2018, 16, 8690–8694 RSC.
- Y. C. He, Y. M. Yan, H. B. Tong, Z. X. Ren, J. H. Wang, Y. B. Zhang, J. B. Chao and M. L. Wang, Chem. Commun., 2020, 56, 9364–9367 RSC.
- Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262–1271 RSC.
- Z. Li, C. Rao, L. Chen, C. Fu, T. Zhu, X. Chen and C. Liu, J. Org. Chem., 2019, 84, 7518–7522 CrossRef CAS PubMed.
- L. M. Eytel, H. A. Fargher, M. M. Haley and D. W. Johnson, Chem. Commun., 2019, 55, 5195–5206 RSC.
- F. Zhang, Y. H. Zhou and X. P. Bao, Supramol. Chem., 2016, 28, 305–313 CrossRef CAS.
- Á. L. Fuentes de Arriba, M. G. Turiel, L. Simón, F. Sanz, J. F. Boyero, F. M. Muñiz, J. R. Morán and V. Alcázar, Org. Biomol. Chem., 2011, 9, 8321–8327 RSC.
- X. P. Bao, X. Wu, S. N. Berry, E. N. W. Howe, Y. T. Chang and P. A. Gale, Chem. Commun., 2018, 54, 1363–1366 RSC.
- N. Busschaert, M. Wenzel, M. E. Light, P. Iglesias-Hernández, R. Pérez-Tomás and P. A. Gale, J. Am. Chem. Soc., 2011, 133, 14136–14148 CrossRef CAS PubMed.
- V. Mazzini and V. S. J. Craig, Chem. Sci., 2017, 8, 7052–7065 RSC.
- M. J. Chmielewski and J. Jurczak, Tetrahedron Lett., 2005, 46, 3085–3088 CrossRef CAS.
- C. Caltagirone, J. R. Hiscock, M. B. Hursthouse, M. E. Light and P. A. Gale, Chem.–Eur. J., 2008, 14, 10236–10243 CrossRef CAS PubMed.
- C. Caltagirone, P. A. Gale, J. R. Hiscock, S. J. Brooks, M. B. Hursthouse and M. E. Light, Chem. Commun., 2008, 3007–3009 RSC.
- X. P. Bao, Y. H. Zhou and B. A. Song, Sens. Actuators, B, 2012, 171–172, 550–555 CrossRef CAS.
- L. T. Wang, X. M. He, Y. Guo, J. Xu and S. J. Shao, Org. Biomol. Chem., 2011, 9, 752–757 RSC.
- S. O. Kang, D. Powell, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 1921–1925 CrossRef CAS.
- Z. A. Tabasi, E. A. Younes, J. C. Walsh, D. W. Thompson, G. J. Bodwell and Y. M. Zhao, ACS Omega, 2018, 3, 16387–16397 CrossRef CAS.
- S. J. Choi, B. Yoon, J. D. Ray, A. Netchaev, L. C. Moores and T. M. Swager, Adv. Funct. Mater., 2020, 30, 1907087 CrossRef CAS.
- G. Picci, M. Kubicki, A. Garau, V. Lippolis, R. Mocci, A. Porcheddu, R. Quesada, P. C. Ricci, M. A. Scorciapino and C. Caltagirone, Chem. Commun., 2020, 56, 11066–11069 RSC.
- M. D. Gu, Y. Lu and M. X. Wang, J. Org. Chem., 2020, 85, 2312–2320 CrossRef CAS PubMed.
- S. Sugiura, Y. Kobayashi, N. Yasuda and H. Maeda, Chem. Commun., 2019, 55, 8242–8245 RSC.
- S. Xiong, F. Chen, T. Zhao, A. Li, G. Xu, J. L. Sessler and Q. He, Org. Lett., 2020, 22, 4451–4455 CrossRef CAS PubMed.
- C. J. Serpell, A. Y. Park, C. V. Robinson and P. D. Beer, Chem. Commun., 2021, 57, 101–104 RSC.
- C. Guo, H. Wang, V. M. Lynch, X. Ji, Z. A. Page and J. L. Sessler, Chem. Sci., 2020, 11, 5650–5657 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of 1, 2 & the complex (1)2·F−, 1H NMR titration spectra, UV-vis titration spectra as well as 1H NMR, 13C NMR & HRMS spectral files of macrocycles 1 and 2. CCDC 1976672, 2021529 and 1976667. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01285a |
| This journal is © The Royal Society of Chemistry 2021 |