
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Synthesis, characterization and cytotoxicity evaluation of a novel magnetic nanocomposite with iron oxide deposited on cellulose nanofibers with nickel (Fe3O4@NFC@ONSM-Ni)†
Pouya Ghamari Kargar‡
 a,
Maryam Noorian‡b,
Elham Chamanic,
Ghodsieh Bagherzade*a and
Zahra Kiani*d
a,
Maryam Noorian‡b,
Elham Chamanic,
Ghodsieh Bagherzade*a and
Zahra Kiani*d
aDepartment of Chemistry, Faculty of Sciences, University of Birjand, Birjand, 97175- 615, Iran. E-mail: P.ghamari71@gmail.com; Fax: +98 56 32345192; Tel: +98 56 32345192
bStudent Research Committee, Birjand University of Medical Sciences, Birjand, Iran
cDepartment of Clinical Biochemistry, Birjand University of Medical Sciences, Birjand, Iran
dDepartment of Pharmacology, Birjand University of Medical Sciences, Birjand, Iran. E-mail: Kiani.za@gmail.com; gbagherzade@gmail.com; bagherzade@birjand.ac.ir; Tel: +985632381920
First published on 12th May 2021
Abstract
A heterogeneous, magnetically recoverable nanocomposite, Fe3O4@NFC@ONSM-Ni(II) was prepared by immobilization of a novel Ni(II) Schiff base complex on Fe3O4@NFC nanoparticles followed by treatment with melamine. This trinuclear catalyst has been characterized using several analytical techniques including FT-IR, TEM, Fe-SEM, EDX, DLS, ICP, TGA, VSM, and XRD. It was used as an efficient catalyst for one-pot solvent-free synthesis of 1,4-dihydropyridine and poly-hydro quinoline derivatives through Hantzsch reaction. This catalyst showed remarkable advantage over previously reported catalysts due to suitable conditions, short reaction time, high efficiency and lower catalyst load and timely recovery of the magnetic catalyst. Moreover, the effects of Fe3O4@NFC@ONSM-Ni(II) nanoparticles on the in vitro proliferation of human leukemia cell line (k562) and human breast cancer cells (MDA-MB-231) were investigated. The results of MTT and Hochest assays suggested that the nanoparticles could effectively inhibit the proliferation of these cancer cells in a time- and concentration-dependent manner.
Introduction
Schiff base metal complexes are one of the most interesting topics studied in coordination chemistry. They play a significant role in chemistry, biology, and medical imaging, and also have enzymatic, anti-viral, anti-coagulant and antitumor properties.1,2 The di and trinuclear ligands can form Schiff base complexes by bonding to other metal ions. Multinuclear transition metal complexes have attracted great attention because of their potentially useful properties, such as notable catalytic activity, modelling the metal binding sites of metalloproteins, and their recent applications in the area of nanoscale materials. One of the synthetic strategies to prepare polynuclear transition metal complexes is the use of simple metal ion complexes which have the appropriate functionality to act as ligands for another metal ion.3 There is currently a great deal of interest in the synthesis and characterization of polynuclear nickel, due to its wide-ranging potential applications such as catalysts, electron transfer mediators in dye-sensitized solar cells, antiviral agents, and molecular nanomagnets.4–7Magnetic nanoparticles (MNPs) as one of the best-known nanomaterials have been investigated in a wide variety of potential medical diagnostic and therapeutic applications such as imaging, magnetic hyperthermia, magnetic particle resonance,8 anti-cancer drug delivery and catalysis.9–12 Ling et al. (2011) studied the effect of iron oxide nanocrystals with the docetaxel as an anticancer drug. Their results indicated that nanoparticles have an antiproliferative effect on PC3 prostate cancer cells.13–15 Khan et al. (2012) investigated the effects of Fe3O4 MNPs on the human lung epithelial cancer cells (A549) and normal human lung fibroblasts (IMR-90). Their results showed that Fe3O4 MNPs has significant cytotoxicity effects on cancer cell lines but not on the normal cells.16 But, one of the disadvantages of Fe3O4 MNPS is magnetic diminution over time that along with the encapsulation of Fe3O4 nanoparticle nucleus with a shell-layer, is an easy way to overcome the above-mentioned shortcoming.17,18 Recently, scientists have made a great effort to synthesize and regenerate of environmentally-friendly nanoparticles by using biocompatible polymers which have unique features such as increasing colloidal stability, avoiding RES absorption of nanoparticles, and providing a surface for ligand composition like peptides19
Cellulose due to its good biocompatibility, biological degradation, and non-toxicity is considered as one of the most important natural renewable polymers, which can be used as a promising organic material for magnetic nanoparticles.20
Multi component reactions (MCRs) have become a promising tool for rapid preparation of compound libraries of small molecules. They are a valuable, efficient, time-saving, atom-economic, and environmentally friendly resource. They are a suitable method for the preparation of compounds with biological properties. In recent years, a lot of attention has been paid to the three and four component reactions, for example, synthesis of the 1,4-dihydropyridine (1,4-DHP) and poly-hydro quinolines (PHQ) derivatives via Hantzsch reaction,21 as important nitrogen heterocyclic compounds with valuable pharmaceutical and biological properties. The recent studies have revealed that 1,4-DHP and PHQ derivatives have several activities including neuroprotection, bronchodilation,22 seroprotective,23 anti-tumor,24 anti-hypertensive,25 platelet anti-aggregatory,26 and cerebral anti-ischemic effects.27 These examples clearly illustrate the notable potential of 1,4-DHP derivatives in novel drugs design.28 In this regard,4-aryl-1,4-DHPs are the most important subclass of Ca2+ channel blocker drugs.29,30 Such as felodipine, nifedipine, nimodipine, nicardipine, amlodipine, etc.31,32 Due to the above-mentioned importance of 1,4-DHPs and PHQs, one of the amazing research challenges that have caught the attention of chemists is their synthesis. However, some drawbacks including utilization of volatile and toxic solvents and expensive catalysts, large energy-wasting,35–38 long reaction times, and boring workup procedures may still be pre-determined as some important drawbacks that should be overcome by using a more efficient and green synthesis method to further improve reaction conditions and increase yields.33,34
During our ongoing research into the activity of trinuclear nickel catalysts finally, after a successful study of trinuclear catalysts, their application to the 1,4-dihydropyridines and polyhydroquinolines to produce corresponding pyridine compounds with high conversion and good selectivity has been investigated. In this study, a tri-nuclear magnetic nanocomposite with iron oxide deposited on cellulose nanofibers with nickel Fe3O4@NFC@ONSM-Ni(II) was prepared and characterized using FT-IR, TEM, Fe-SEM, EDX, DLS, ICP, TGA, VSM, and XRD. We then explored its applicability for solvent-free one-pot synthesis of 1,4-DHPs and PHQs derivatives. To the best of our knowledge, this is the first report of using a trinuclear catalyst based on magnetic nanofiber cellulose to prepare these Hantzsch derivatives and also a new approach to such combinations. Finally, as part of our ongoing efforts to develop new catalytic methods and to find a wider potential of Fe3O4 in clinical applications and knowing that cell culture is considered as the first-line therapy for screening therapeutic efficacy and safety of drugs before transplantation into the body, the in vitro cytotoxic effects of Fe3O4@NFC@ONSM-Ni(II) nanoparticle on the k562 and MDA-MB-231 cell lines were examined.
Results and discussion
Catalyst characterization
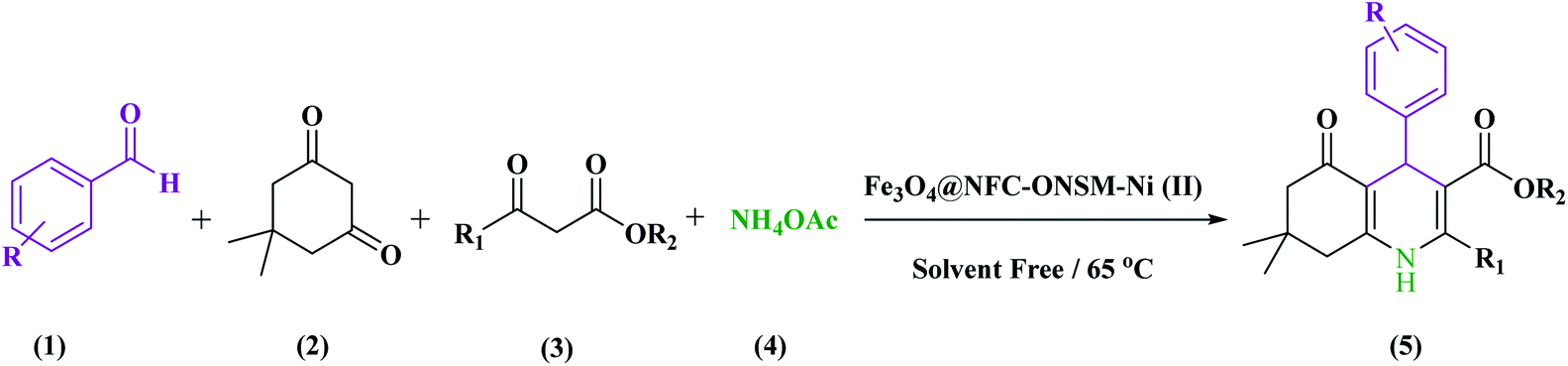
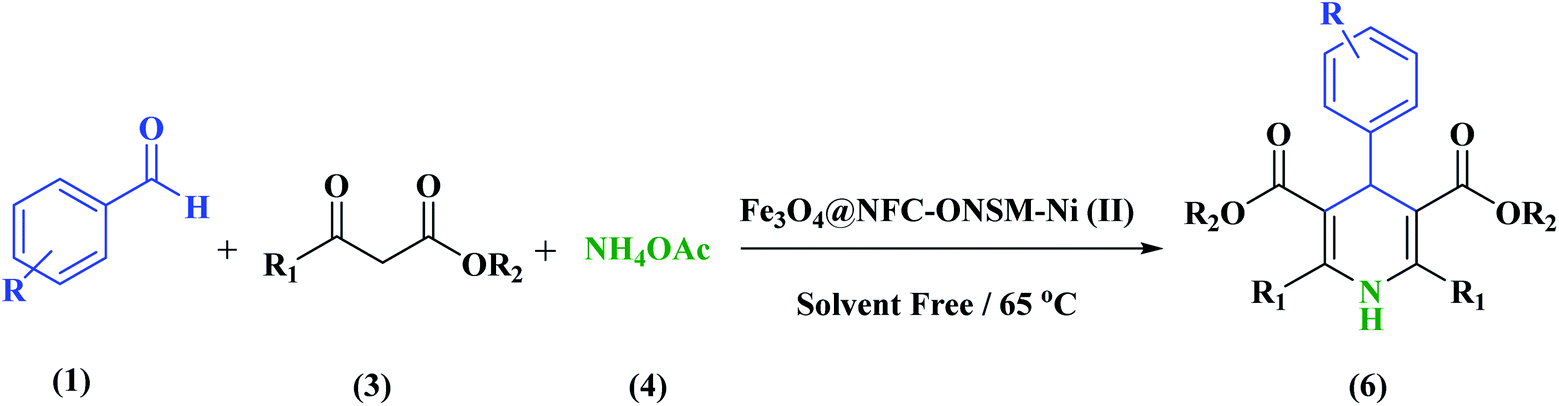
In this study, catalyst Fe3O4@NFC@ONSM-Ni(II) was prepared in several steps. At the first step, the oxygen atom in the magnetite (Fe3O4) attacked the cellulose nanofibers as a nucleophile and a C–O bond was then formed by the interaction between the Fe3O4 and the cellulose. Thereafter, silane, nickel–metal complex was added to the mixture consisting of Fe3O4@NFC. Fe3O4, Fe3O4@NFC, Schiff Base, and Fe3O4@NFC@ONSM-Ni(II) (Scheme 1). Characterization of the Fe3O4@NFC@ONSM-Ni(II) structure was performed using various techniques such as FT-IR, FESEM, TEM, XRD, DLS, EDX, and VSM. Spectra of (a) Schiff base, (b) Fe3O4@NFC, (c) Fe3O4@NFC-Cl, (d) Fe3O4@NFC@Schiff base, and (e) Fe3O4@NFC@ONSM-Ni(II) are shown in (Fig. 1). The FT-IR spectrum of Schiff base (Fig. 1a) showed the stretching vibrations of the C–Cl, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group at 760 and 1632 cm−1 and the stretching vibrations of the C–O, CC aromatic, and O–H group at around 1059 cm−1, 1415–1530 cm−1, and 3259 cm−1, respectively. As presented (Fig. 1b), FT-IR spectrum of the Fe3O4 stretching vibrations Fe–O and O–H were at 574 cm−1 and 3414 cm−1 that approved the formation of Fe3O4 nanoparticles, respectively. Besides the peaks Fe3O4 (Fig. 1c), the stretching vibrations confirmed the OH group at 3339 cm−1 and the stretching vibrations of the C–O group at around 1059 cm−1 in the presence of cellulose. Moreover (Fig. 1d), shows the FT-IR spectrum as a new sharp peak at 713 and 1156 cm−1 that was assigned to C–Cl and Si–O stretching vibration. Additionally, the spectrum (Fig. 1e) indicated that the Schiff base is supported on the Fe3O4@nanofiber cellulose. Besides the above-mentioned bands, the complexion of nickel-metal to ligand IV leads the imine bond sorption 1632 cm−1 to lower wavenumbers 1612 cm−1 by about 17 cm−1, denoting the participation of nitrogen in bonding with metal ion and supporting the coordination of ν(CV) stretch to the metal via a nitrogen atom. Also, the main sorption band at 790 cm−1 (C–Cl) in the FT-IR spectrum of ligand, represents the functionalization of Fe3O4@NFC-CPTMS nanoparticles with Ni(II) complex VI (Fig. 1e).
N group at 760 and 1632 cm−1 and the stretching vibrations of the C–O, CC aromatic, and O–H group at around 1059 cm−1, 1415–1530 cm−1, and 3259 cm−1, respectively. As presented (Fig. 1b), FT-IR spectrum of the Fe3O4 stretching vibrations Fe–O and O–H were at 574 cm−1 and 3414 cm−1 that approved the formation of Fe3O4 nanoparticles, respectively. Besides the peaks Fe3O4 (Fig. 1c), the stretching vibrations confirmed the OH group at 3339 cm−1 and the stretching vibrations of the C–O group at around 1059 cm−1 in the presence of cellulose. Moreover (Fig. 1d), shows the FT-IR spectrum as a new sharp peak at 713 and 1156 cm−1 that was assigned to C–Cl and Si–O stretching vibration. Additionally, the spectrum (Fig. 1e) indicated that the Schiff base is supported on the Fe3O4@nanofiber cellulose. Besides the above-mentioned bands, the complexion of nickel-metal to ligand IV leads the imine bond sorption 1632 cm−1 to lower wavenumbers 1612 cm−1 by about 17 cm−1, denoting the participation of nitrogen in bonding with metal ion and supporting the coordination of ν(CV) stretch to the metal via a nitrogen atom. Also, the main sorption band at 790 cm−1 (C–Cl) in the FT-IR spectrum of ligand, represents the functionalization of Fe3O4@NFC-CPTMS nanoparticles with Ni(II) complex VI (Fig. 1e).
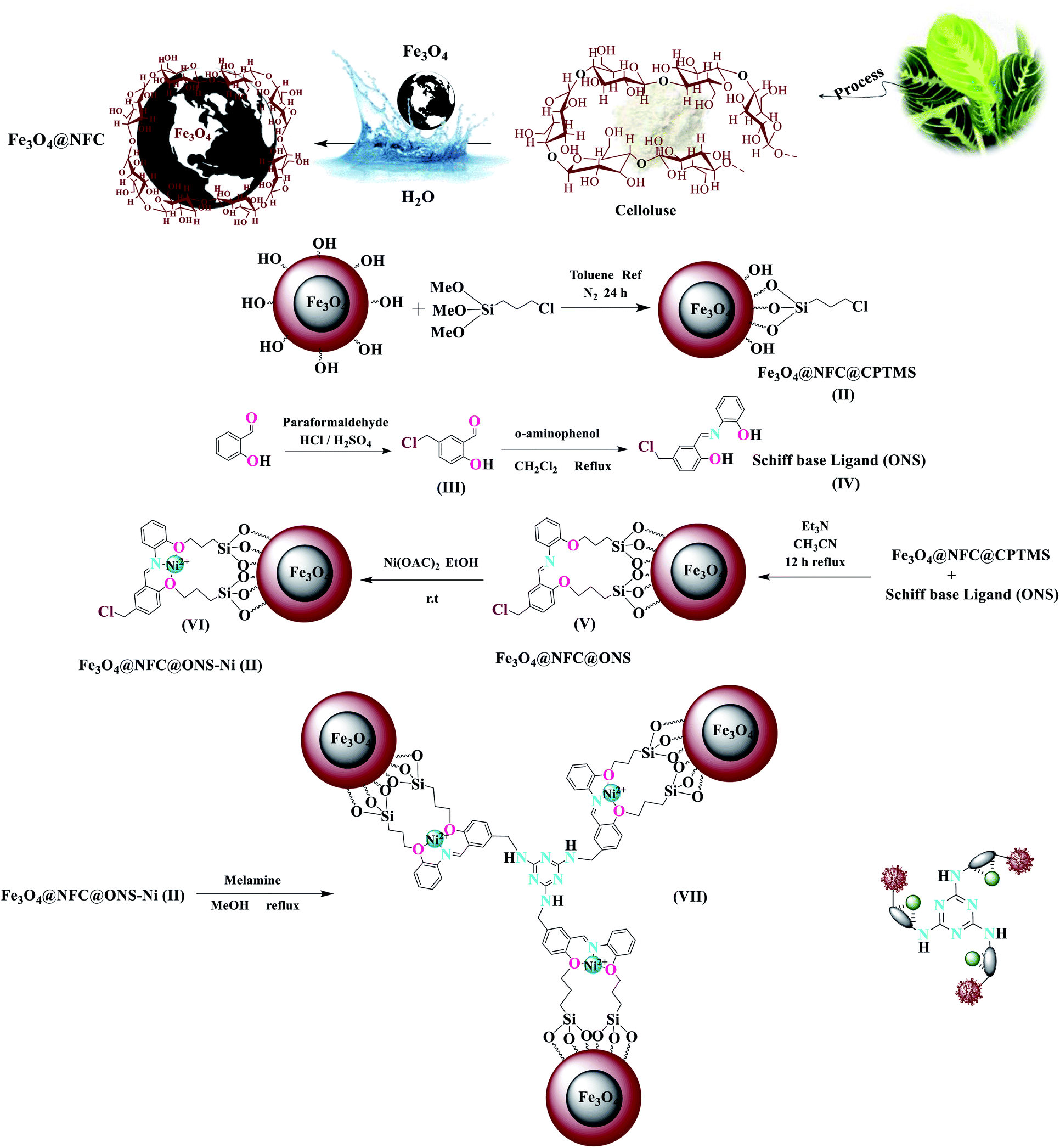 | ||
| Scheme 1 The synthetic route for Fe3O4@NFC@ONSM-Ni(II). | ||
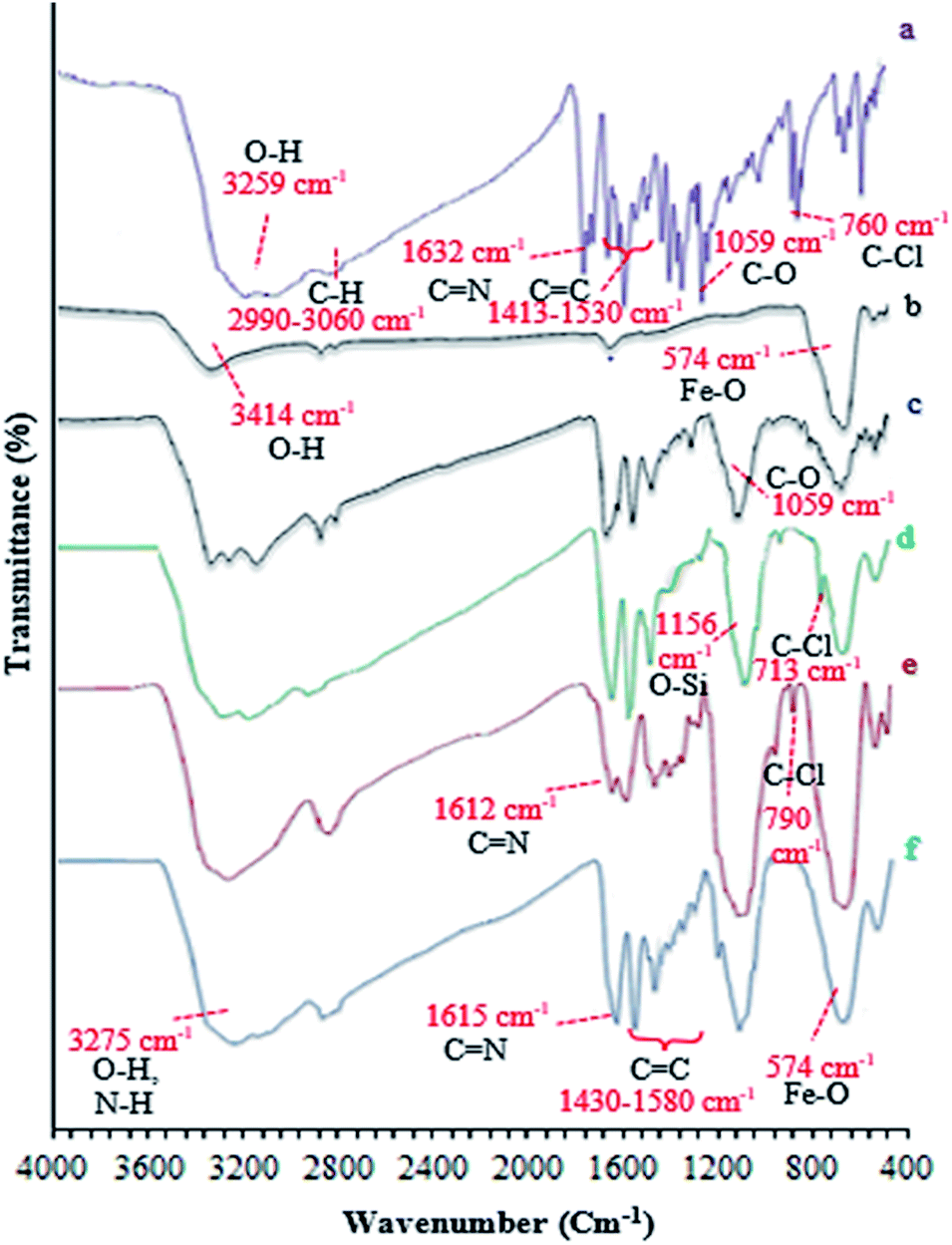 | ||
| Fig. 1 FT-IR spectra of (a) Schiff base (ONS), (b) Fe3O4, (c) Fe3O4@NFC, (d) Fe3O4@NFC–Cl, (e) Fe3O4@NFC@ONS, (f) Fe3O4@NFC@ONSM-Ni(II). | ||
Finally, the main sorption bands at 578 cm−1 (Fe–O), 1135 cm−1 (Si–O), 1434 cm−1 (CC), 1615 cm−1 (CN), and 3275 cm−1 (O–H and N–H) in the FT-IR spectrum of Fe3O4@NFC@ONSM-Ni(II), represent the functionalization of Fe3O4@NFC@ONS-Ni(II) nanoparticles with melamine VII (Fig. 1f). The Fe-SEM images illustrate in Fig. 2a, b, and (Fig. 2a) present that the Fe3O4 have an average diameter between 18 and 20 nm as well as an approximately spherical shape by the SEM in (Fig. 2b) micrographs presentation that Fe3O4@NFC@ONSM-Ni(II) have a larger particle size and a smoother surface. Fe-SEM analysis was executed for consecutive synthetization steps of catalyst (Fig. 2a–d). According to FE-SEM images, the synthesized Fe3O4 and Fe3O4@NFC are almost spherical in shape and well dispersed, howbeit in some area's larger structures with non-spherical morphology are perceived (Fig. 2a and c). The Fe-SEM images also corroborate the spherical structure of the Fe3O4 and show that the Fe3O4 have a homogeneous distribution and are similar in size in accordance with the TEM image. The Fe-SEM images show an increment in the size of the Fe3O4 at each step, in the match with the dynamic light scattering (DLS) results. It is interesting that the resulting spherical morphology of Fe3O4@NFC and Fe3O4@NFC@ ONSM-Ni(II) catalyst shows that the functionalization of the nanofiber cellulose, silica, and ligand are monotone coated on the Fe3O4 nanoparticles to form a shell. Fe-SEM images were used for further research of the surface morphology of the prepared catalyst. Of the nanoparticles by the silica and nickel complexes, respectively, was accomplished regularly and harmoniously, with no aberration in shape or aggregation in the particles. The Fe3O4 and Fe3O4@NFC particles have an average diameter of 13–15 and 20–22 nm (Fig. 2a and c), respectively, consistent with their corresponding DLS analyses (Fig. 2b and d). As shown in Fig. 2e, the Fe-SEM image, clearly show a catalyst of organic and inorganic different ingredients in a homogeneous network of Fe3O4@NFC@ONSM-Ni(II) which have an average diameter of 23–26 nm (Fig. 2f). In EDX analysis For the Fe3O4 (Fig. 3a: magnetic), Fe3O4@NFC (Fig. 3b: core–shell), and Fe3O4@NFC-ONSM-Ni(II) (Fig. 3c: salen complex nanocatalyst), the results of this analysis displayed the presence of Ni, Fe, Si, O, N, and C elements which could be acceptable evidence of the modification of the Fe3O4 surface by the nanocatalyst.
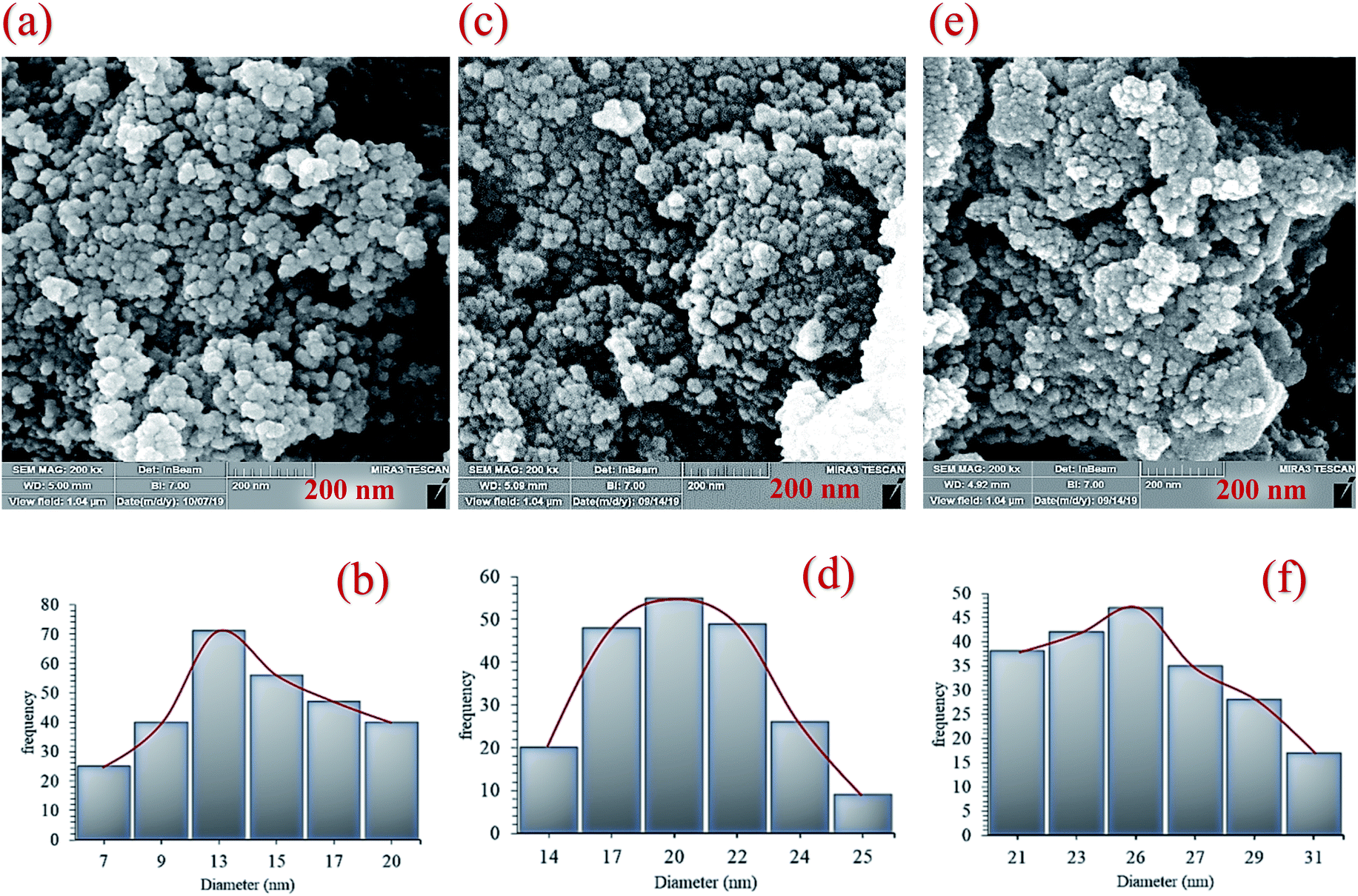 | ||
| Fig. 2 Field emission-scanning electron microscopy images and dynamic light scattering results of (a and b) Fe3O4, (c and d) Fe3O4@NFC, and (e and f) Fe3O4@NFC-ONSM-Ni(II) complex nanocomposite. | ||
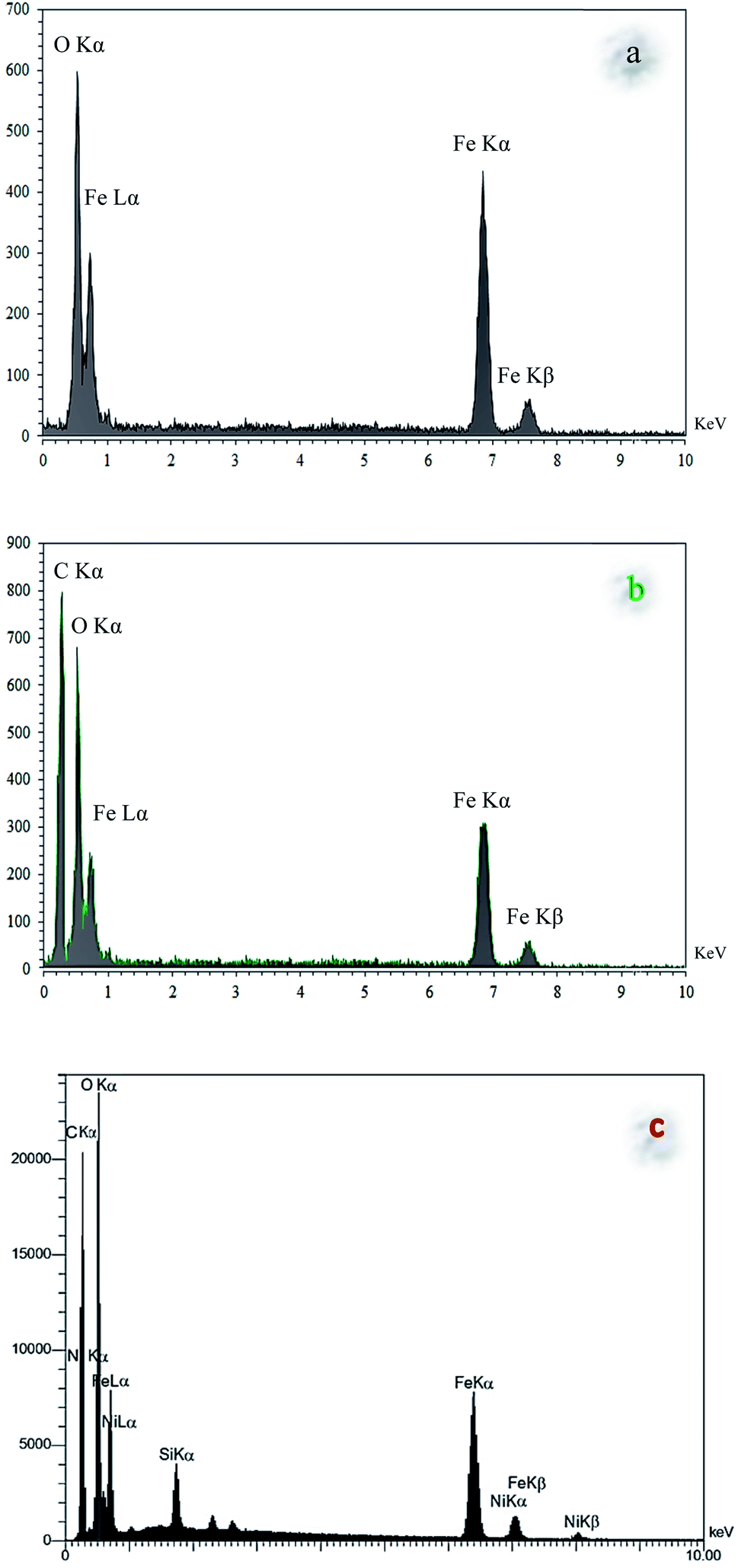 | ||
| Fig. 3 The EDS spectra of (a) Fe3O4, (b) Fe3O4@NFC, (c) Fe3O4@NFC@ONSM-Ni(II). | ||
Therefore, it can be inferred that the full elements were loaded onto the magnetic surface (Fe3O4) of Fe3O4@NFC-ONSM-Ni(II) (Fig. 3c). According to the results from ICP and EDX, the amount of copper in Fe3O4@NFC-ONSM-Ni(II) nanocatalyst was calculated at 1.52 mmol g−1.
The X-ray diffraction patterns of the Fe3O4, Fe3O4@NFC, and Fe3O4@NFC@ONSM-Ni(II) are shown in (Fig. 4). According to Fig. 4a, size of the magnetic NPs was determined by X-ray line broadening using the Debye–Scherrer formula (D = 0.9/ˇ cos), where D is the average crystalline size. Several prominent Bragg reflections by their indices (220), (311), (400), (422), (511), and (440) revealed that the resultant magnetic NP was Fe3O4 with the structure of an inverse spinel.39 XRD pattern in (Fig. 4b) shows that peaks at 2θ = 35.87°, 43.97°, 62.92°, 71.72°, and 74.77° correspond to the (111), (200), (220), (311), and (222) crystallographic phases in XRD pattern that are related to Ni(II).5 As a result, the high-angle XRD pattern of Fe3O4@NFC@ONSM-Ni(II) catalyst, as shown in Fig. 4c, fractured peaks corresponded to the standard Fe3O4 and Ni(II) have also illustrated that the surface rectification of MNPs does not detriment the structure of Fe3O4 core. While two peaks at 2θ = 16.77° and 20.71° corresponded to the (101) and (002) XRD pattern were confirmed to be related to cellulose.40
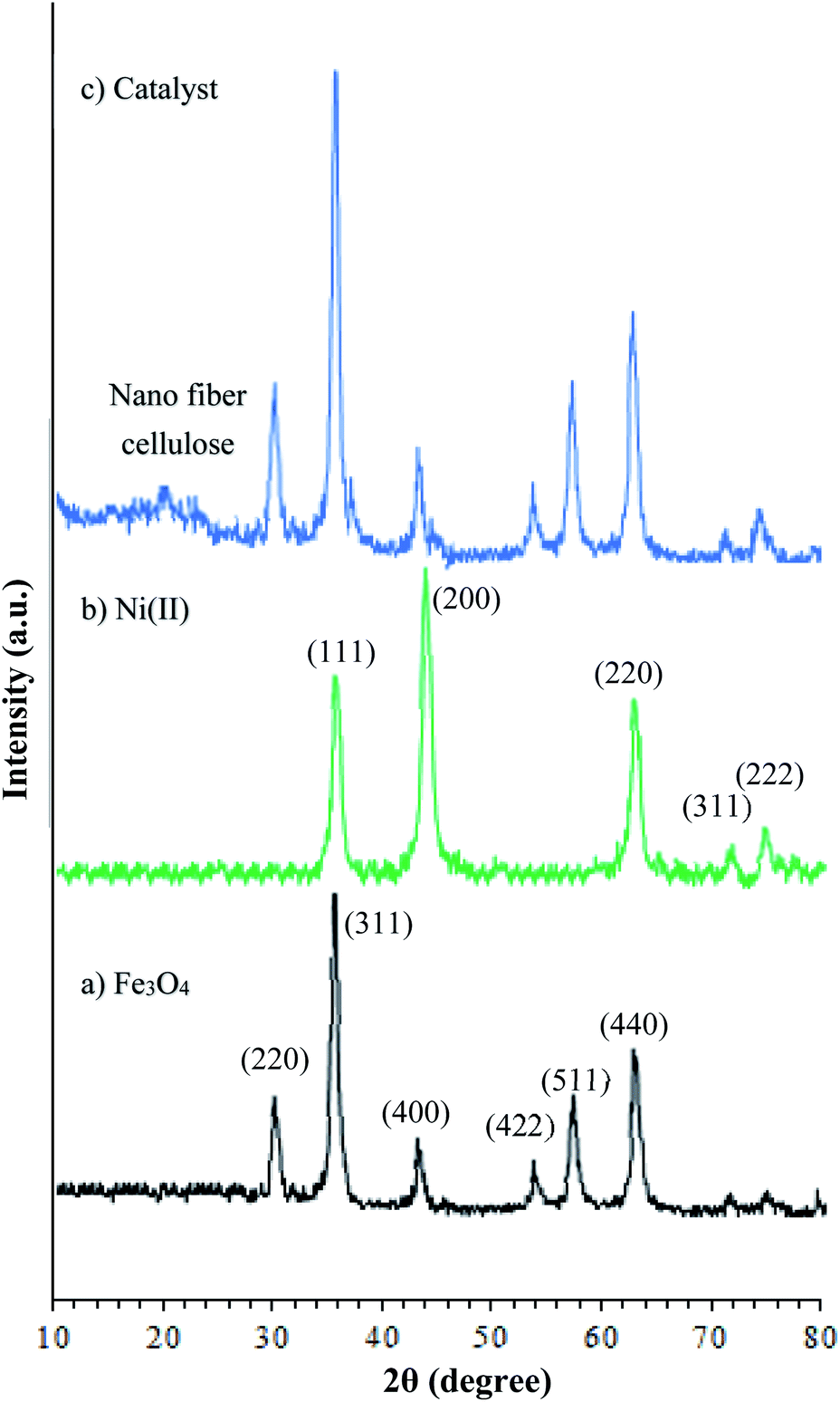 | ||
| Fig. 4 XRD spectra of (a) Fe3O4, (b) nickel(II), (c) Fe3O4@NFC@ONSM-Ni(II). | ||
Magnetic properties of different steps of the catalyst preparation were characterized by VSM. The magnetic curves are shown in Fig. 5. These curves showed that the approximate saturation magnetization value of Fe3O4@NFC and trinuclear nickel catalyst is 51 and 30 emu g−1. The reduction in the saturation magnetization of this compound compared to that in the pure MNPs (62 emu g−1) is due to coated nanofiber cellulose-shell and Schiff base Ni complex and the synergistic effect with melamine on its surface (Fig. 5a–c). Nevertheless, Fe3O4@NFC@ONSM-Ni(II) nanocatalyst exhibited the superparamagnetic characteristic and a high magnetization value, which can be readily separated from the mixture by a simple external magnet. These NPs exhibited high permeability in magnetization and good magnetic responsiveness, which their magnetization was sufficient in order to separation with an external magnetic field. The magnetization and demagnetization curves are coincident and did not any hysteresis phenomenon was found. As shown in Fig. 5, the remanent magnetization is equal to zero for all NPs.
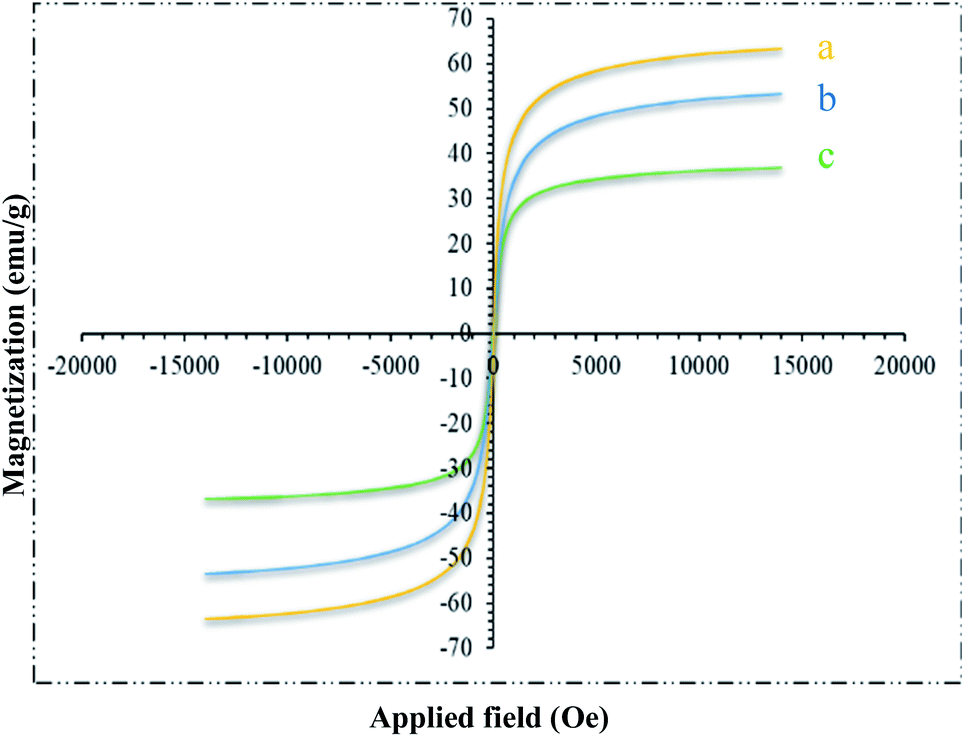 | ||
| Fig. 5 VSM pattern (a) Fe3O4, (b) Fe3O4@NFC and (c) Fe3O4@NFC@ONSM-Ni(II). | ||
The presence of appropriate magnetic properties allows Fe3O4@NFC@ONSM-Ni(II) to be completely, efficiently, and quickly separated from the reaction mixture by an external magnet. The structure of Fe3O4@NFC-ONSM-Ni(II) was studied using the transmission electron microscopy (TEM) (Fig. 6). These images are considered as a suitable tool for determining the size and structure of particles. The TEM images and histogram of Fe3O4@NFC-ONSM-Ni(II) showed the small particles of 16–28 nm.
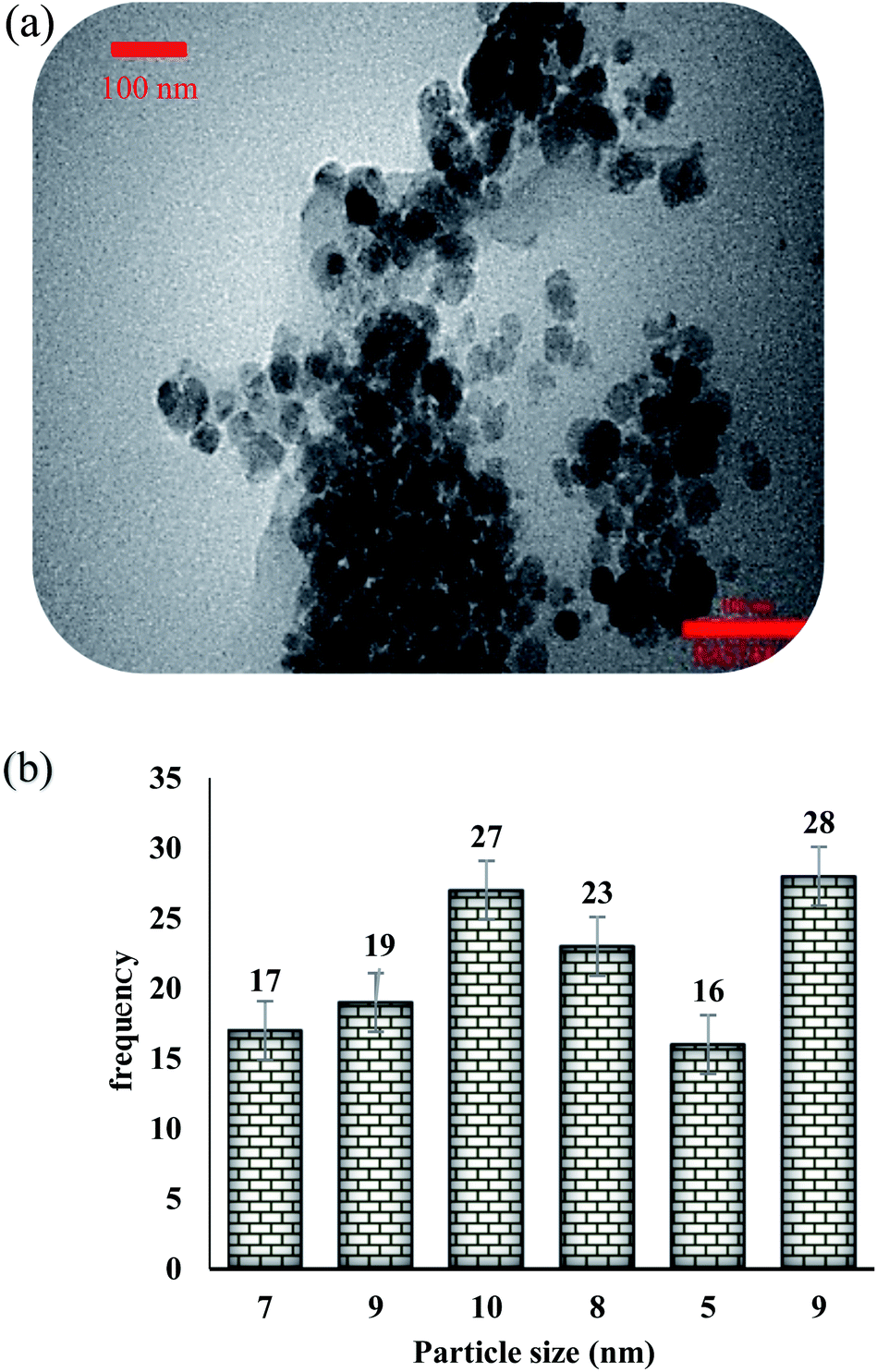 | ||
| Fig. 6 (a) TEM image; (b) particle size distribution histogram Fe3O4@NFC@ONSM-Ni(II). | ||
Electronic spectrum of Ni(OAc)2, Fe3O4, Fe3O4@NFC, and Fe3O4@NFC@ONSM-Ni(II) were carried out in H2O as a solvent at the region of 200–800 nm (Fig. 7). The synthesized Fe3O4 shows the maximum absorption peak at 360 nm. The spectrum of aqueous Fe3O4@cellulose solution exhibited a maximum at 250 and 275 nm, which was attributed to the absorption of cellulose in Fe3O4 NPs structure, which is masked after immobilization of Schiff base nickel complex by π–π* absorptions. In the UV-Vis spectrum the catalyst lmax and adsorption intensity appear in 355 nm, while the Ni(OAc)2 lmax is in region 400 nm, resulting in UV-Vis spectrum of Schiff base ligand to Ni(II) caused the reduction of absorption intensity of n–π* for CN bond and π–π* transitions for benzene ring, which confirmed the successful chelation of Ni(II) to the catalyst. Furthermore, the absence of resonant peak above 355 nm proved the metallic nature of Fe3O4@NFC NPs.
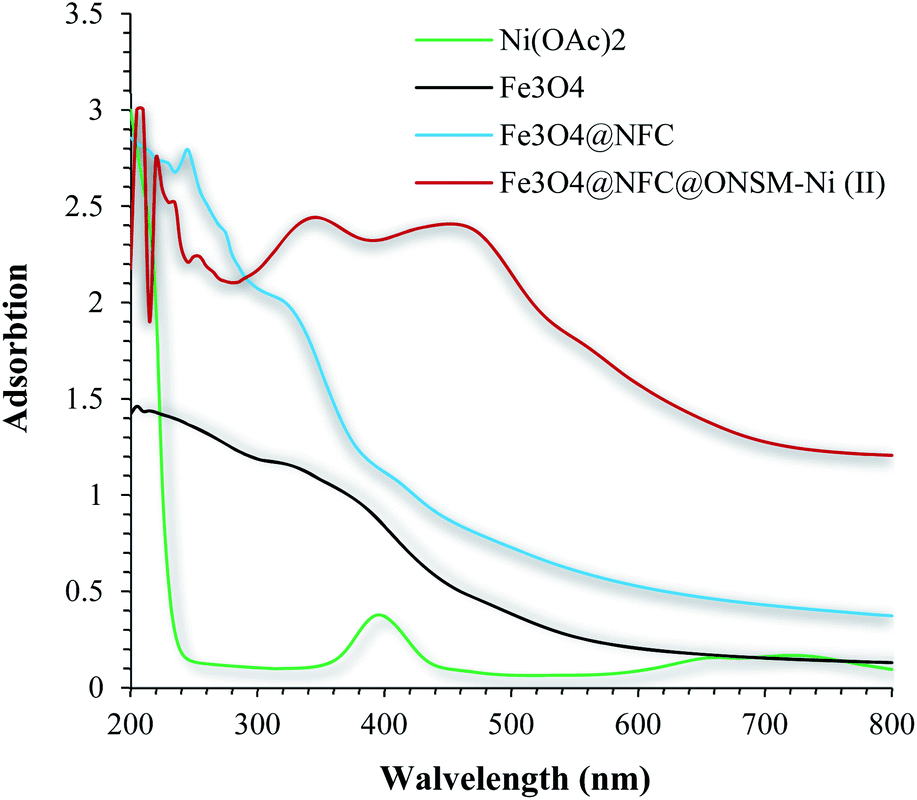 | ||
| Fig. 7 UV-Vis spectra of Ni(OAc)2, Fe3O4, Fe3O4@NFC, and Fe3O4@NFC@ONSM-Ni(II) in H2O solvent. | ||
Evaluation of the catalytic actuality of Fe3O4@NFC@ONSM-Ni (II) in the 1,4-dihydro pyridines and polyhydroquinolines reactions
A recently designed trinuclear catalyst was used to synthesize the polyhydroquinolines (PHQ) and 1,4-dihydropyridine (1,4-DHP) reaction and its activity was also evaluated (Scheme 2). Therefore, to optimize the polyhydroquinolines (PHQ) and 1,4-dihydropyridine (1,4-DHP) reaction parameters, we studied the effect of catalyst amount, solvent, temperature, and time were explored towards as a multi component reaction, and the results are shown in Table 1. The low yield of products was observed in H2O, EtOH, EtOAc, and CH3CN (Table 1, entry 1–4) even after 2 h stirring, respectively. In solvent-free situations, good yields of PHQ and 1,4-DHP derivatives were obtained within 20 and 30 min (97% and 96%, Table 1, entry 5 for PHQ and 1,4-DHP). After identifying the appropriate solvent, the next step was studying the role of the catalyst in the reaction rate and product performance. For this purpose, a series of equal reactions were conducted with the amounts of variant catalysts and the results are indicated in Table 1. According to these results, we piecemeal the enhancement of the amount of catalyst from 0 to 20 mg (Table 1, entries 6, 7, 8, and 9). In the absence of Fe3O4@NFC@ONSM-Ni(II), only a small amount of product was obtained, whereas the amount of catalyst increased to more than 15 mg, the reaction efficiency was almost constant and did not change much. It was observed that 15 mg was the most yield (Table 1, entry 5).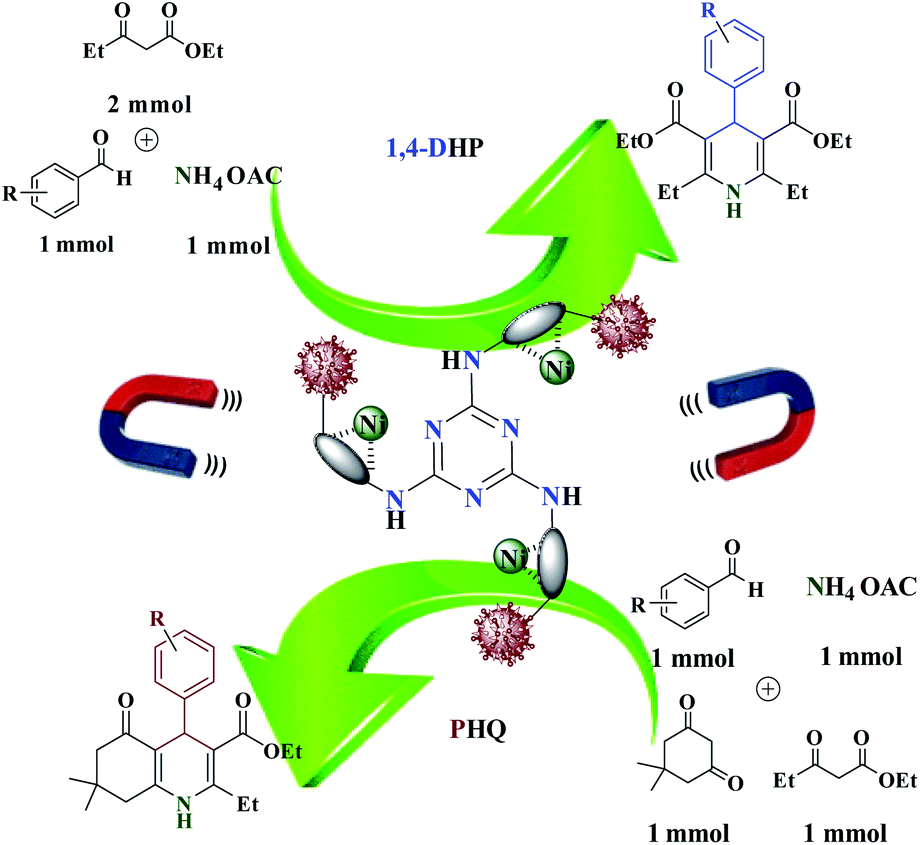 | ||
| Scheme 2 Solvent-free synthesis of 1,4-dihydropyridines and polyhydroquinolines catalysed by Fe3O4@NFC@ONSM-Ni(II). | ||
| Entry | Solvent | Catalyst (mg) | Temp (°C) | Time (min) | Yieldb (%) | ||
|---|---|---|---|---|---|---|---|
| PHQ | DHP | PHQ | DHP | ||||
| a (Reaction conditions) PHQ: benzaldehyde (1 mmol), dimedone (1 mmol), ethyl acetoacetate (1 mmol), and ammonium acetate (1.2 mmol) 1,4-DHP: benzaldehyde (1 mmol), ethyl acetoacetate (2 mmol), and ammonium acetate (1.2 mmol).b Yields of isolated product. | |||||||
| 1 | H2O | 15 | Reflux | 10 | 15 | 45 | 40 |
| 2 | EtOH | 15 | Reflux | 10 | 15 | 70 | 75 |
| 3 | EtOAc | 15 | Reflux | 10 | 15 | 40 | 30 |
| 4 | CH3CN | 15 | Reflux | 10 | 15 | 75 | 75 |
| 5 | Neat | 15 | 65 | 10 | 15 | 97 | 96 |
| 6 | Neat | — | 65 | 60 | 75 | Trace | Trace |
| 7 | Neat | 5 | 65 | 40 | 50 | 75 | 70 |
| 8 | Neat | 10 | 65 | 30 | 30 | 88 | 85 |
| 9 | Neat | 20 | 65 | 10 | 15 | 98 | 97 |
| 10 | Neat | 15 | — | 60 | 60 | 45 | 40 |
| 11 | Neat | 15 | 40 | 10 | 15 | 70 | 65 |
| 12 | Neat | 15 | 50 | 10 | 15 | 85 | 85 |
| 13 | Neat | 15 | 60 | 10 | 15 | 93 | 91 |
| 14 | Neat | 15 | 70 | 10 | 15 | 97 | 96 |
| 15 | Neat | 15 | 65 | 5 | 5 | 85 | 83 |
| 16 | Neat | 15 | 65 | 15 | 10 | 95 | 92 |
| 17 | Neat | 15 | 65 | 20 | 20 | 95 | 95 |
| 18 | Neat | 15 | 65 | 30 | 30 | 90 | 89 |
After finding the appropriate solvent and amount catalyst for the reaction of the model, we examined the effects of temperature and time on the desired reaction. To identify the appropriate temperature, at first, a series of reactions with equal conditions and equal amounts of catalyst for the model reaction at different temperatures was evaluated and the results are shown in Table 1 (entries 10–14). Notably, the most appropriate temperature is 65 °C for these reactions. In the final step, after optimizing the catalyst, solvent and temperature, the best time for the model reaction was selected (entries 15–18). As you can see, the best result among these different times was 10 min for PHQ and also 15 min for 1,4-DHP. With the optimized reaction situation in hand, the reaction was performed with different benzaldehydes with 15 mg Fe3O4@NFC@ONSM-Ni(II) NPs to prospect the scope and the present protocol, and accordingly, the results of these observations are summarized in Table 1. From the obtained results, polyhydroquinolines (PHQ) and 1,4-dihydropyridine (1,4-DHP) reaction was synthesized with various aldehydes containing lethal and donor electron groups with good yields, while lethal electron groups gave products slightly better those of donor electron groups.
The structures of the synthesized polyhydroquinolines (PHQ) and 1,4-dihydropyridine (1,4-DHP) derivatives were confirmed by melting point analysis (Tables 2 and 3). Notably, the spectral data for the selected compounds are discussed in the following (see Experimental for more details). The efficiency of Fe3O4, Fe3O4@NFC, Fe3O4@NFC@ONS-Ni(II) (mononuclear), and Fe3O4@NFC@ ONSM-Ni(II) (trinuclear) were separately studied in the model reaction (Fig. 8). As shown in Fig. 8, no product was gained by using Fe3O4 and Fe3O4@NFC species. However in the presence of Fe3O4@NFC@ONS-Ni(II), the reaction yields was far from satisfactory. As can be seen in Fig. 8, no product was gained by using Fe3O4 and Fe3O4@NFC species. However, when the reaction was carried out in the presence of Fe3O4@NFC@ONS-Ni(II), the result was far from satisfactory. These findings indicated that the enhanced magnetic catalytic activity of Fe3O4@NFC@ONSM-Ni(II) could be attributed to the synergistic effect of melamine and Fe3O4@NFC@ONS-Ni(II) towards the PHQ and 1,4-DHP reaction as a high-efficiency trinuclear catalyst (see the magnetic catalytic mechanism synthesis part for more details). With these interpretations, it can be inferred that the proper reaction of melamine and Fe3O4@NFC@ONS-Ni(II) in the synthesis of magnetic catalyst not only increases the power of the catalyst but also multiplies the activity of the magnetic catalyst. This property expedites and facilitates the reaction process in terms of time and other reaction conditions. These observations well proved the significant influence of the trinuclear catalyst to advance the PHQ and 1,4-DHP multi-component reactions. These observations well proved the significant influence of the trinuclear catalyst to advance the PHQ and 1,4-DHP multi-component reactions.
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | R1 | R2 | Product | Time (min) | Yieldb (%) | TON | TOF | Mp | Reported Mp (ref.) |
| a Reaction conditions: aryl aldehyde (1 mmol), ethyl acetoacetate (1 mmol), dimedone (1 mmol) and ammonium acetate (1 mmol), Fe3O4@NFC@ONSM-Ni(II) (15 mg), under solvent-free conditions at 65 °C.b Isolated yield. | ||||||||||
| 1 | H | Me | OEt | 5a | 10 | 97 | 194 | 1168 | 251–252 | 250–252 (ref. 41) |
| 2 | 2-Cl | Me | OEt | 5b | 15 | 95 | 190 | 760 | 210–211 | 210–212 (ref. 42) |
| 3 | 4-Cl | Me | OEt | 5c | 10 | 97 | 194 | 1168 | 249 | 248–250 (ref. 42) |
| 4 | 4-Br | Me | OEt | 5d | 5 | 97 | 194 | 2337 | 248–249 | 248–250 (ref. 43) |
| 5 | 4-Me | Me | OEt | 5e | 20 | 90 | 180 | 542 | 270 | 270–272 (ref. 42) |
| 6 | 4-MeO | Me | OEt | 5f | 15 | 92 | 182 | 728 | 249 | 247–249 (ref. 42) |
| 7 | 4-OH | Me | OEt | 5g | 20 | 97 | 194 | 584 | 246–247 | 245–247 (ref. 42) |
| 8 | 2-NO2 | Me | OEt | 5h | 15 | 95 | 190 | 760 | 212 | 210–212 (ref. 42) |
| 9 | 3-NO2 | Me | OEt | 5i | 25 | 92 | 182 | 437.5 | 240–241 | 240–242 (ref. 42) |
| 10 | 4-NO2 | Me | OEt | 5j | 10 | 97 | 194 | 1168 | 239–241 | 239–241 (ref. 42) |
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | R1 | R2 | Product | Time (min) | Yieldb (%) | TON | TOF | Mp | Reported Mp (ref.) |
| a Reaction conditions: aryl aldehyde (1 mmol), ethyl acetoacetate (2 mmol) and ammonium acetate (1 mmol), Fe3O4@NFC@ONSM-Ni(II) (15 mg), under solvent-free conditions at 65 °C.b Isolated yields. | ||||||||||
| 1 | H | Me | OEt | 6a | 10 | 97 | 192 | 768 | 159 | 158–160 (ref. 44) |
| 2 | 2-Cl | Me | OEt | 6b | 15 | 95 | 194 | 776 | 130 | 129–131 (ref. 45) |
| 3 | 4-Cl | Me | OEt | 6c | 10 | 97 | 190 | 1144 | 147 | 145–147 (ref. 44) |
| 4 | 4-Br | Me | OEt | 6d | 5 | 97 | 194 | 388 | 160–162 | 161–162 (ref. 44) |
| 5 | 4-Me | Me | OEt | 5e | 20 | 90 | 176 | 704 | 134–135 | 133–135 (ref. 44) |
| 6 | 4-MeO | Me | OEt | 6f | 15 | 92 | 184 | 728 | 162 | 160–162 (ref. 44) |
| 7 | 4-OH | Me | OEt | 6g | 20 | 97 | 194 | 388 | 223–224 | 223–225 (ref. 44) |
| 8 | 2-NO2 | Me | OEt | 6h | 15 | 95 | 184 | 1108 | 129 | 128–130 (ref. 46) |
| 9 | 3-NO2 | Me | OEt | 6i | 25 | 92 | 192 | 1156 | 137 | 136 (ref. 44) |
| 10 | 4-NO2 | Me | OEt | 6j | 10 | 97 | 190 | 570.5 | 166–167 | 165–167 (ref. 45) |
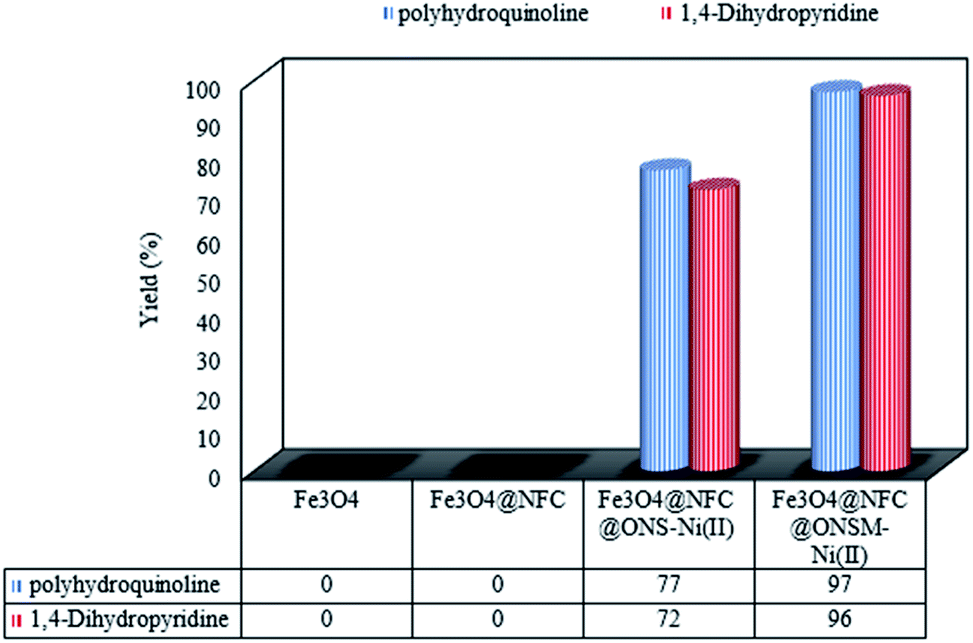 | ||
| Fig. 8 Comparison of the performance of catalysts with each other. Reaction conditions: PHQ: benzaldehyde (1 mmol), dimedone (1 mmol), ammonium acetate (1.5 mmol), ethyl acetoacetate (1 mmol), catalyst (15 mg), S. F., 65 °C. 1,4-DHP: benzaldehyde (1 mmol), ammonium acetate (1.5 mmol), ethyl acetoacetate (2 mmol), catalyst (15 mg), S. F., 65 °C. | ||
Reusability and stability of the Fe3O4@NFC@ONSM-Ni(II) catalyst as a tri-nuclear catalyst nickel in the 1,4-DHPs and PHQs reactions
One of the most important advantages which makes catalyst important for commercial applications, is the recycling property of the catalyst. In this method, to investigate the catalyst recyclability, experiments were performed for the model 1,4-DHPs and PHQs reactions, under the optimized reaction conditions. Interestingly, it was shown that the recovered catalyst, after each investigation, the catalyst was separated from the reaction mixture with a magnet. Then, the isolated catalyst was washed with water and EtOH, vacuum-dried at 50 °C for 3 h, and directly used in the next run of the reaction. Residual activation was analyzed by ICP for measurement.The amount of nickel that goes from the catalyst to the solution. As shown in Fig. 9, the catalyst was recovered and reused for at least five consecutive runs without notable loss of activity. The 1,4-DHPs and PHQs reactions yield reached to 90% and 90% for the 5th run. To show durability and structure of the catalyst, the recovered catalyst after 5th run was subjected to some analyses. Also, metal leaching of the catalyst was measured in each cycle. As shown in Fig. 8, a few leaching was observed for Fe3O4@NFC@ONSM-Ni(II), whereas only 3.2% for 1,4-DHPs reaction and 2.9% for PHQs reaction metal leaching was observed after the 5th run. Moreover, ICP analysis of the catalyst for each heavy metal demonstrated an insignificant change in their weight percentage than the corresponding fresh values: Fe 44.76, Si 2.45, Ni 5.8 w%. These results demonstrated insignificant changes in the percentages of the heavy metals and confirm the durability of the catalyst during recycling. One of the special properties of Fe3O4@NFC@ONSM-Ni(II) is the stability of the framework under reaction conditions, which leads to a decrease in the percentage of metal leaching into the organic solution and preservation of the heterogeneous nature of the system. In order to evaluate the heterogeneous nature of Fe3O4@NFC@ONSM-Ni(II), the hot leaching test was carried out for 1,4-DHPs and PHQs in CH3CN as solvent at 65 °C. For this experiment, the catalyst was separated after 50% of the reaction time and the remaining solution was stirred in the absence of Fe3O4@NFC@ONSM-Ni(II) for an additional 30 min. The result indicated that no further increase in either the conversion or selectivity occurred in the absence of the catalyst. For further confirmation, ICP analysis of the remaining solution exhibited a negligible amount of Ni in the reaction mixture. This finding established that Fe3O4@NFC@ONSM-Ni(II) is a typical heterogeneous catalyst (Fig. 10).
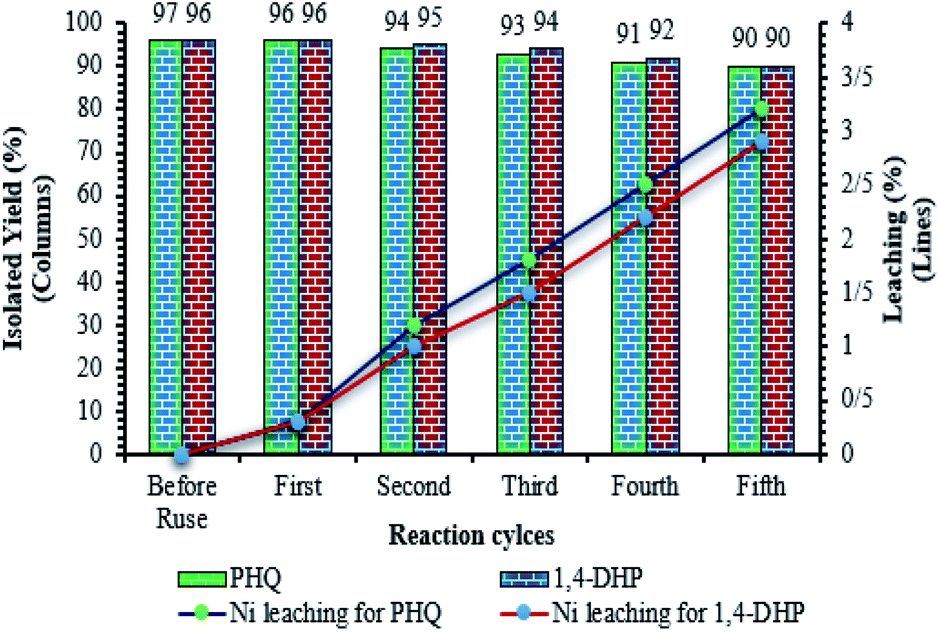 | ||
| Fig. 9 Recycling activity of the Fe3O4@NFC@ONSM-Ni(II) NPs. | ||
 | ||
| Fig. 10 Hot leaching test for (A) PHQ and (B) 1,4-DHP reactions using Fe3O4@NFC@ONSM-Ni(II) under optimized conditions. | ||
The recyclability tests of Fe3O4@NFC@ONSM-Ni(II)
One of the important factors for the heterogeneous catalyst in terms of sustainable chemistry is recyclability. FT-IR of the catalyst after five times reuse showed that the structure of the catalyst during the recycling process sustained unchanged. The spectrum related to the analysis of FT-IR functional groups is quite obvious in Fig. 11I. As presented (Fig. 11I(a–c)), FT-IR spectrum of the catalyst the stretching vibrations Fe–O and Si–O, C–O, CC, CN, C–H (aliphatic and aromatic), and O–H and N–H were at 574, 1059, 1156, 1430–1580, 1615, 2990–3050 and 3257 C m−1 that approved the formation of Fe3O4@NFC@ONSM-Ni(II) tri-nuclear catalyst respectively. Furthermore, the Fe-SEM and VSM analysis of the catalyst after five times of reuse showed that the design and morphology of the catalyst maintained unchanged during the recycling process (Fig. 11II). But the value of Fe3O4@NFC@ONSM-Ni(II) saturation has slightly decreased to 20 emu g−1 (Fig. 11III). These results proved that no substantial changes occurred in the chemical structure of the present catalyst.
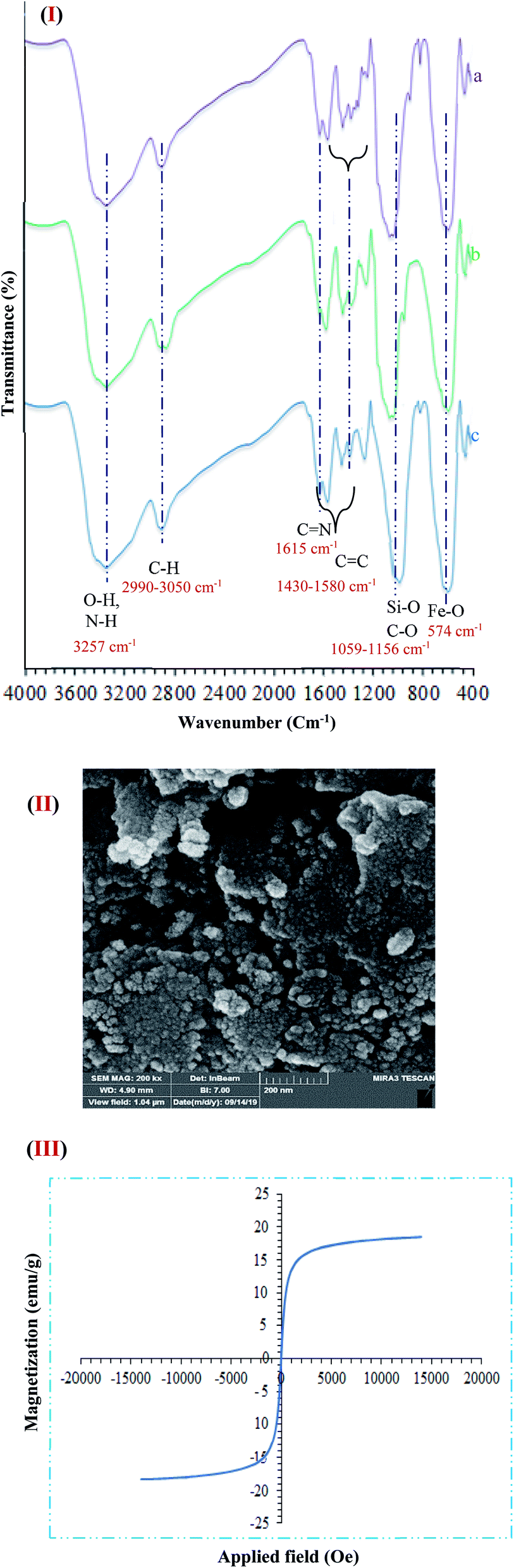 | ||
| Fig. 11 (I) FT-IR ((a) before recycling, (b) after recycling 1,4-Dihydropyridine, and (c) after polyhydroquinolines) (II) FE-SEM, and (III) VSM analysis of Fe3O4@NFC@ONSM-Ni(II) after five times. | ||
In order to evaluate the competency of the method for the syntheses of 1,4-dihydropyridine and poly-hydro quinolines, we compared the efficiency of Fe3O4@NFC@ONSM-Ni(II) with some recently reported catalysts for the multi-component reaction, as shown in Tables 4 and 5. These observations can be attributed to the strong interaction between the nodes of nickel and Schiff base ligand as an organic linker which maintains the integrity and structure of the Fe3O4@NFC@ONSM-Ni(II) during the reaction progress.
| Entry | Catalyst | Solvent | Temp/°C | Time/min | Yield | Reference |
|---|---|---|---|---|---|---|
| 1 | MCM-41 | EtOH | 90 | 15 | 90 | 48 |
| 2 | MBM-450 | EtOH | 90 | 40 | 90 | 49 |
| 3 | Ni(NO3)2-imine/thiophene-Fe3O4@SiO2 | Solvent-free | 100 | 20 | 96 | 50 |
| 4 | V-TiO2 | Solvent-free | 80 | 10 | 85 | 51 |
| 5 | PdRuNi@GO | DMF | 70 | 45 | 92 | 52 |
| 6 | Boehmite–SSA | EtOH | Reflux | 215 | 94 | 53 |
| 7 | BIL@MNP | Solvent-free | 70 | 15 | 92 | 54 |
| 8 | GSA@MNPs | EtOH | 80 | 240 | 90 | 55 |
| 9 | Fe3O4@NFC@ONSM-Ni(II) | Solvent free | 65 | 10 | 97 | This work |
| Entry | Catalyst | Solvent | Temp/°C | Time/min | Yield% | Reference |
|---|---|---|---|---|---|---|
| 1 | H5BW12O40 | EtOH | Reflux | 45 | 94 | 56 |
| 2 | SBA-SO3H | EtOH | R.T. | 25 | 86 | 57 |
| 3 | MIL-101-SO3H | EtOH | 60 | 8 h | 99 | 58 |
| 4 | Zr-SBA-16 | EtOH | 80 | 3 h | 77 | 59 |
| 5 | Alginic acid | EtOH | Reflux | 60 | 92 | 60 |
| 6 | Chitosan–CuSO4 | EtOH | Reflux | 65 | 95 | 61 |
| 7 | CeO2 | Solvent-free | 80 | 60 | 74 | 62 |
| 8 | NiFe2O4@SiO2@SO3H | H2O | 70 | 20 | 95 | 63 |
| 9 | Fe3O4@NFC@ONSM-Ni(II) | Solvent free | 65 | 15 | 96 | This work |
Mechanism studies
We proposed a plausible mechanism for the formation of polyhydroquinolines (PHQ) and 1,4-dihydropyridine (1,4-DHP) by using Fe3O4@NFC@ONSM-Ni(II) NPs is shown in Schemes 3 and 4.47 Accordingly, an acceptable mechanism for the synthesis of poly-hydro quinolines catalysed by Fe3O4@NFC@ONSM-Ni(II) is proposed in (Scheme 3). The reaction mechanism of four components of the pot can be predicted as ethyl acetoacetate, dimedone, aldehyde, and ammonium acetate along with Fe3O4@NFC@ONSM-Ni(II) catalyst. Dimedone reaction with the mediated aldehyde was obtained with Knoevenagel (I). In the next step, (I) reacted with ammonium acetate and the second molecule ethyl acetoacetate. Thereafter, two reactions were formed through Michael's addition and the mediator (II). Finally, by the reaction of intermediate with ammonia and ring-closing of intermediate (III), the next step was performed for the elimination of water from the desired product (IV). This mechanism follows the results shown in Table 2. Moreover, this proposed mechanism for the synthesis of 1,4-dihydropyridines involves Lewis acid catalysed cyclocondensation of intermediates A and B, causing knoevenagel condensation of one equivalent of ethyl acetoacetate with aldehyde and reaction of the second equivalent of ethyl acetoacetate with ammonia resulted from ammonium acetate, respectively. At the end, as a result of the reaction of intermediate C with ammonia and ring-closing of intermediate D, the formation of 1,4-DHP has occurred (Scheme 4). This mechanism follows the results shown in Table 3.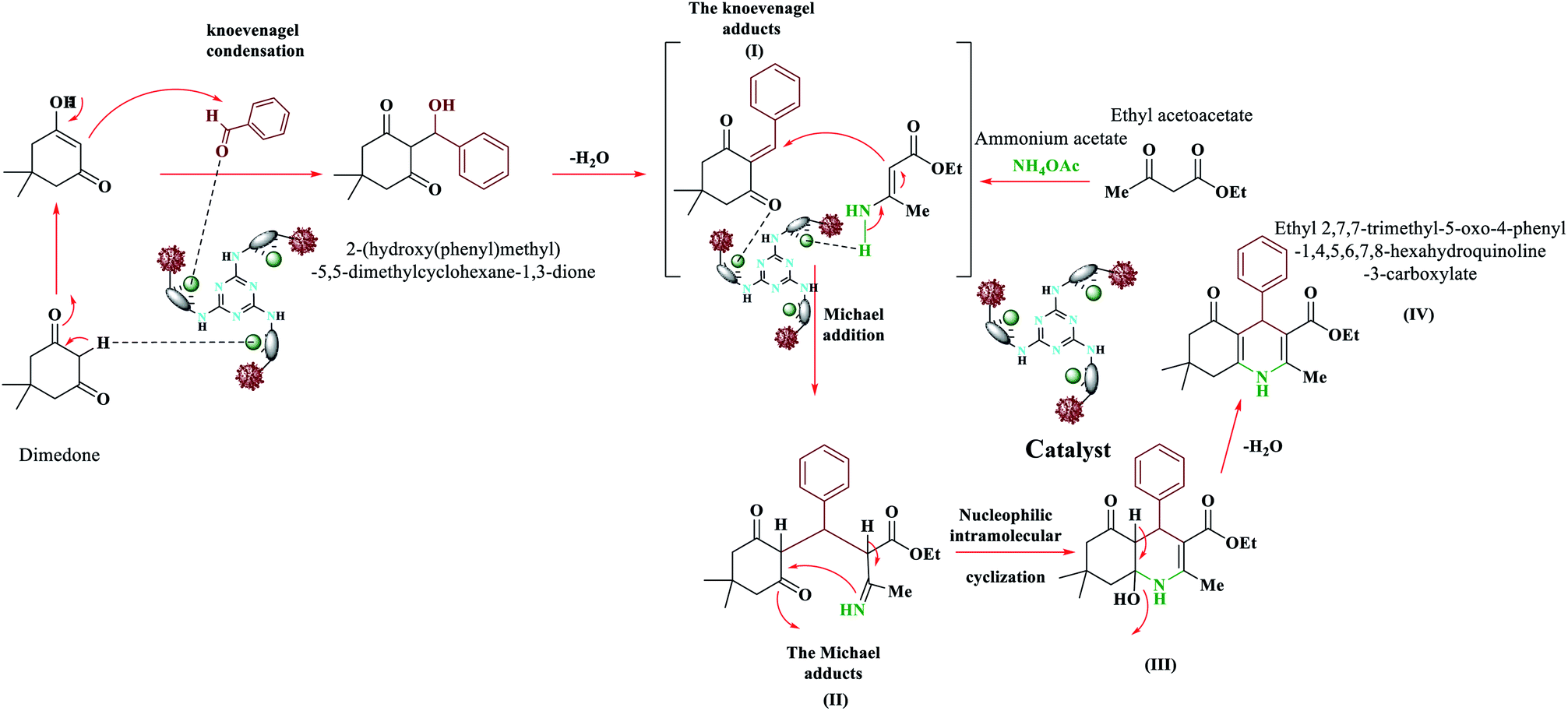 | ||
| Scheme 3 The proposed mechanism for the synthesis of compounds 1 to 10 polyhydroquinolines. | ||
 | ||
| Scheme 4 The proposed mechanism for the synthesis of compounds 1 to 10 1,4-dihydropyridine. | ||
Comparative study
To better clarify the merits of the competency tri-nuclear catalytic system over the reported metal-based other catalytic systems in the polyhydroquinolines and 1,4-dihydropyridine reactions, the comparing efficacy results were tabulated in Tables 4 and 5. It has been observed that the synthesized Fe3O4@NFC@ONSM-Ni(II) catalyst has almost a very good advantage over other reported catalysts which can be due to suitable reaction conditions, short reaction time, high reaction efficiency and lower catalyst load and on the other hand timely recovery of magnetic catalyst. In addition to the reusability of the catalyst, most importantly, the without use of organic and aqueous solvent that is environmentally friendly and does not require the use of any additives or toxic solvents supports the green chemistry approach well.Finally, a series of green metrics64 such as atom economy (AE), atom efficiency (AEF), carbon efficiency (CE), reaction mass efficiency (RME), optimum efficiency (OE), process mass intensity (PMI), E-factor (E), solvent intensity (SI), and water intensity (WI) were calculated to evaluate the greenness of the one-pot multi-component reaction of aldehydes, dimedone, ethyl acetoacetate and ammonium acetate for polyhydroquinolines, and aldehydes, ethyl acetoacetate and ammonium acetate for 1,4-dihydropyridines (Fig. 12 and 13, see ESI† for detailed calculations). To stable, the more greenness of the current catalyst over the reported catalysts in the one-pot multi-component reaction of aldehydes, dimedone, ethyl acetoacetate and ammonium acetate for polyhydroquinolines (Table 4, entries 7 and 8), and aldehydes, ethyl acetoacetate and ammonium acetate for 1,4-dihydropyridines (Table 5, entries 7 and 8), the current catalyst's green metrics was compared with those of two previously reported catalysts. As it is shown in Fig. 12a and 13a, the high values of the AE, AEF, CE, RME, and OE for the synthesis of PHQ and 1,4-DHP derivatives, illustrate well the greenness of the process. The lower the PMI, E, and SI, the more favourable is the process because of green chemistry. These values are less than 10 in the synthesis of the PHQ and 1,4-DHP mentioned above's (Fig. 12b and 13b).
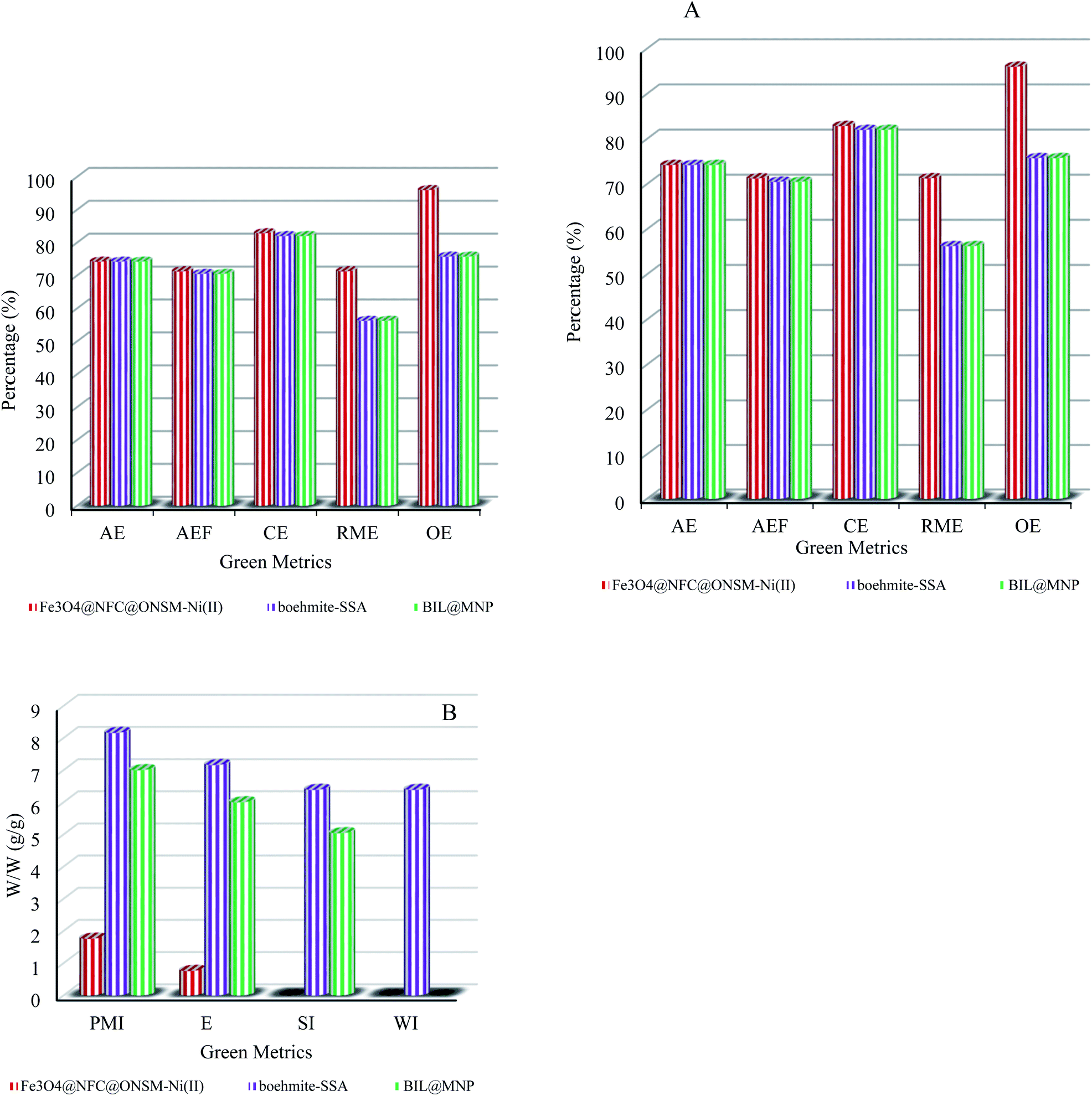 | ||
| Fig. 12 Green metrics including (a) AE, AEf, CE, RME, OE and (b) PMI, E, SI and WI for the one-pot four-component reaction (PHQ) of benzaldehyde, dimedone, ammonium acetate, ethyl acetoacetate, and catalysed by Fe3O4@NFC@ONSM-Ni(II) (this study), boehmite–SSA (Table 4, entry 7) and BIL@MNP (Table 4, entry 8); W/W = weight/weight (g/g). | ||
 | ||
| Fig. 13 Green metrics including (a) AE, AEf, CE, RME, OE and (b) PMI, E, SI and WI for the one-pot three-component reaction (1,4-DHP) of benzaldehyde, ammonium acetate, ethyl acetoacetate, and catalysed by Fe3O4@NFC@ONSM-Ni(II) (this study), NiFe2O4@SiO2@SO3H (Table 5, entry 9) and chitosan–CuSO4 (Table 5, entry 7); W/W = weight/weight (g/g). | ||
Hence, it can be concluded that about the high values of RME and low values of PMI, E, SI, and WI, this one-pot three and four-component process is an efficient and green protocol for synthesizing PHQ and 1,4-DHP (see ESI† for detailed calculations).
In vitro anticancer studies
In the next step, the synthesized Fe3O4@NFC@ONSM-Ni(II) nanoparticles were tested against K562 and MDA-MB-231 cell lines. According to MTT results, although the resistance of cancer cells to Fe3O4@NFC-ONSM-Ni(II) varied, the strong anti-cancer activity was observed in these tested cell lines. Fig. 14A shows the inhibitory effects of Fe3O4@NFC@ONSM-Ni(II) nanoparticles on the K562 cell growth. The IC50 for these cells at different times was calculated as 38.53, 34.11, and 27.58 at different times (24, 48, and 72 h), respectively. In order to investigate precisely the cytotoxicity of Fe3O4@NFC@ONSM-Ni(II) nanoparticles, we also investigated the cytotoxicity of compartment of Fe3O4@NFC@ONSM-Ni(II) nanoparticles separately on K562 cells and compared to Fe3O4@NFC@ONSM-Ni(II) nanoparticles (Fig. 14B). As it is shown in Fig. 14B, in the presence of Fe3O4, viability of K562 cells remains unchanged in comparison to control. Meanwhile, Fe3O4@NFC in low concentration lead to insignificant changes in cell viability vs. control, but at higher concentration (50 μg mL−1), reduced to 80% vs. control.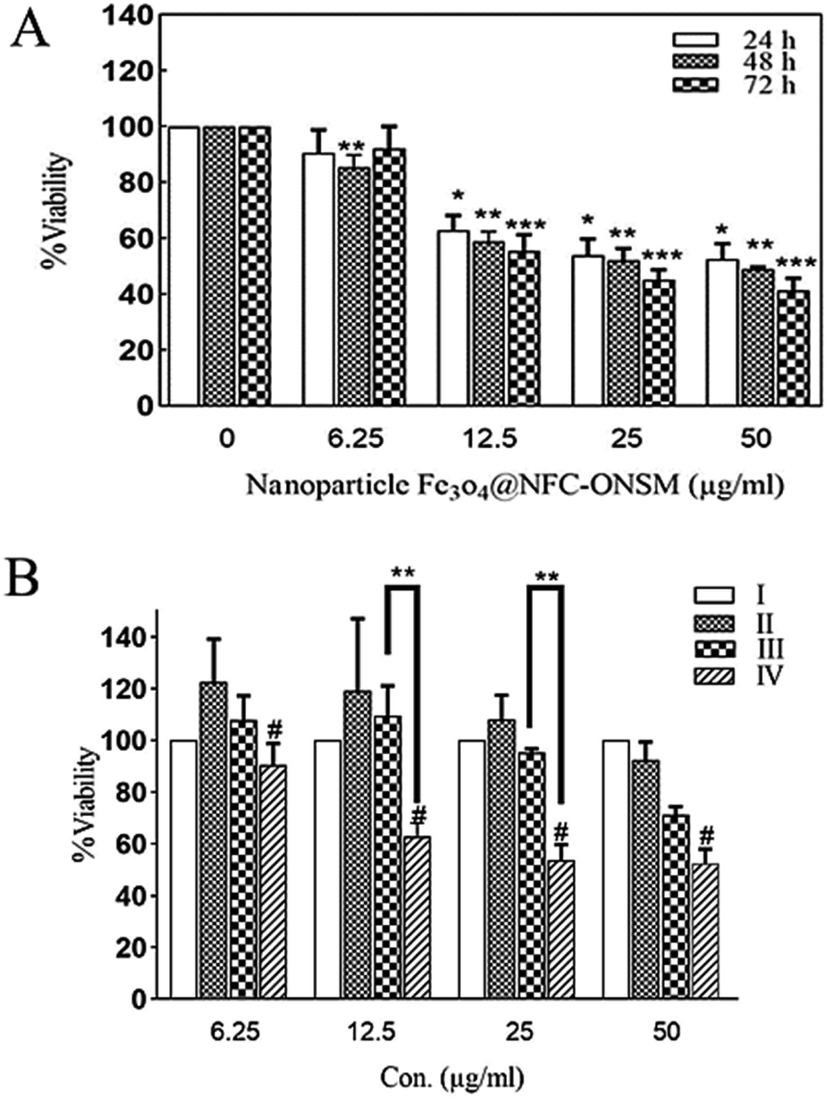 | ||
| Fig. 14 Dose–response effects on cell survival, after 24, 48, and 72 h of the incubation with Fe3O4@NFC@ONSM-Ni(II) nanoparticles (A), and percentage cell survival of control (I), Fe3O4 (II), Fe3O4@NFC(III) and Fe3O4 @NFC@ONSM-Ni(IV) (B), on K562 cell line. Significant differences (* time 24 h; ** time 48 h; and *** time 72 h), when compared with the control (P < 0.05). #: p < 0.05 compared to Fe3O4 the same concentration. | ||
As it was demonstrated, reduction of viability in the presence of Fe3O4@NFC@ONSM-Ni(II) nanoparticles (12.5 and 25 μg mL−1) significantly higher than Fe3O4@NFC. In addition, the survival of MDA-MB-231 cells in the presence of Fe3O4@NFC@ONSM-Ni(II) nanoparticles did not decrease after 24 hours compared to the control; however, it significantly decreased at 48 and 72 hours (Fig. 15). The IC50 for these cells at different times was calculated as 24, 48, and 72 (151.44, 68.15, and 54.2), respectively. According to the results, the inhibition of cell proliferation in both classes mostly depended on time and dose.
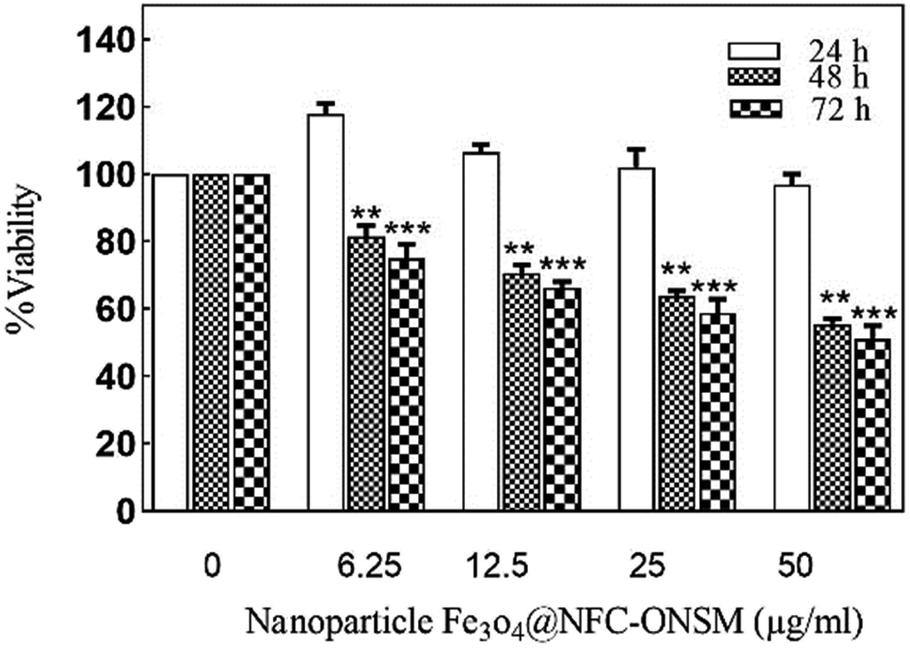 | ||
| Fig. 15 Dose–response effects on cell survival, after 24, 48, and 72 h of the incubation with Fe3O4@NFC@ONSM-Ni(II) nanoparticles in MDA-MB-231 cell line. Significant differences (* time 24 h; ** time 48 h; and *** time 72 h), when compared with the control (P < 0.05). | ||
In the next step, we evaluated the morphological changes of the cells by Hoechst staining. Accordingly, Hoechst staining is a method of differentiating apoptotic cells from living or necrotic cells [because apoptotic cells generally show dense DNA and fragmented nuclei, whereas living and necrotic cells do not]. Cells morphology after 48 h of exposure to Fe3O4@NFC@ONSM-Ni(II) nanoparticle clearly showed about 50–60% of the growth inhibition at a concentration of 50 μg mL−1 nanoparticle in the tested cell lines [Fig. 16A and B]. One of the studies performed in a similar manner was a study by Li et al. who investigated the viability of HeLa cells with Fe3O4@Au-HA NS nanoparticles by MTT assay. The results showed that after the incubation of HeLa cells with Fe3O4@Au-HA NSs at concentrations of 0.2, 0.4, 0.6, 0.8, 1.0, and 1.5 mm, when the gold concentration increased to 2.0 mm, FeSO4@Au-HA NS started to show toxicity. Based on the results of the homolytic process, it can be concluded that Fe3O4@ Au-HA NS has a strong biocompatibility in the studied concentration range, which can be consequently used for biomedical applications.65 Another study conducted on iron oxide nanoparticles (IONPs) tested the toxicity of INOPs on HeLa cells. The results of this nanoparticle's anti-cancer activity via MTT test have shown that the cell growth at a concentration of 150 μg mL−1 is inhibited by 50–60%. The Hoechst test results also showed that the cells were subjected to cytotoxicity caused by nanoparticles, which lost their nuclear density and membrane integrity and endured cell death or apoptosis.66
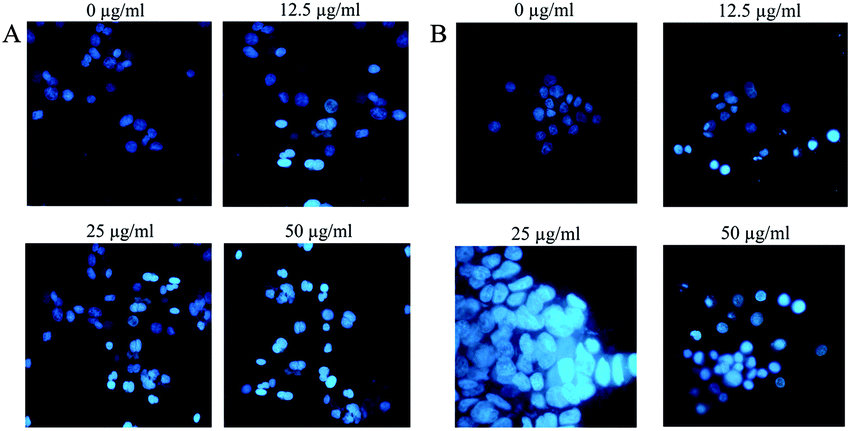 | ||
| Fig. 16 Morphology of K562 cells (A) and MDA-MB-231 cells (B) treated with different doses of Fe3O4@NFC@ONSM-Ni(II) nanoparticle after 48 hours. | ||
Experimental
Chemicals and physicochemical characterization
All reagents were obtained from joinery sources and then used as received with no further refinement. Thereafter, the purity of the synthesized compound was checked by TLC using aluminium plates, and then pre-coated with silica gel (60F, Merck). FT-IR spectra were recorded as KBr disks in the range of 400–4000 cm−1 on a JASCO FT/IR 4600 spectrophotometer, and Laguna Shore Cryotronics 7407 vibrating sample magnetometer (VSM) at room temperature. The elements in the samples were probed using energy-dispersive X-ray (EDX) spectroscopy accessory to the Philips scanning electron microscopy. Field emission scanning electron microscopy (FE-SEM) images were obtained on a FE-SEM Nanosem 450. Subsequently, ICP analysis was performed by VARIAN VISTA-PRO CCD simultaneous ICP-OES instrument. Transmission electron microscopy (TEM) was also performed on a Philips EM208 microscope operated at 100 kV. Finally, the X-ray diffraction (XRD) analysis was conducted using the Philips PW1730 automatic powder diffractometer for which Ni Kα radiation was employed.Synthesis of Fe3O4@NFC-CPTMS (I–II)
Core–shell (I) Fe3O4@NFC nanospheres were prepared in terms of the previously described method (Scheme 1).39 Initially, Fe3O4@NFC (1.5 g) was sonicated in dry toluene (15 mL) for 30 min, and then (CPTMS) 3-chloropropyl-trimethoxy silane (1 mL) was added slowly to the mixture. The mixture was then stirred for 24 h under N2 gas at reflux conditions. After performing the reaction, the catalyst was removed from the reaction mixture using an external magnet, washed with toluene and diethyl ether, and then dried for 8 h at 75 °C in a vacuum oven.Synthesis of 5-(chloromethyl)-2-hydroxy benzaldehyde (III)
We prepared (III) in terms of the above-mentioned procedure.40 Generally, for the preparation of 2-hydroxy 4-chloromethyl benzaldehyde, at first, salicylaldehyde (10 mmol), paraformaldehyde (0.49 g, 16.4 mmol), and HCl 37% (80 mmol) were mixed with several drops of the concentrated H2SO4 as a catalyst at 70 °C and stirred for 20 h (Scheme 1). At the second stage, the reaction mixture was cooled at room temperature, water (20 mL) was added to the mixture, and the product was then extracted into CH2Cl2 (20 mL). Notably, anhydrous Na2SO4 was used for drying the organic phase. The CH2Cl2 was removed using a rotary evaporator and dense yellow oil was put aside overnight. Eventually, a pale purple solid product III was then obtained (Scheme 1).FT-IR (KBr): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3151 (O–H), 3033, 2975, 2865 (C–H aldehyde), 1665 (CO), 1434 (CC), 673 (CH2–Cl) cm−1.
= 3151 (O–H), 3033, 2975, 2865 (C–H aldehyde), 1665 (CO), 1434 (CC), 673 (CH2–Cl) cm−1.
1HNMR (DMSO, 300 MHz): δ = 4.80 (s, 2H, CH2), 7.56–7.60 (d,d, 2H, H–Ar), 7.72 (s, 1H, H–Ar), 10.06 (s, 1H, CH of aldehyde), 11.30 (s, 1H, O–H) ppm.
13CNMR (DMSO, 75 MHz): δ = 47.0, 117.8, 124.8, 131.1, 136.1, 136.4, 148.4, 163.2, 197.7 ppm.
Synthesis of Schiff base ligand (ONS) (IV)
In the beginning, the solution of (2.0 mmol, 0.218 g) 2-aminophenol in 20 mL dichloromethane was added dropwise to the solution of 5-chloromethylsalicyaldehyde (5 mmol, 0.85 g) in 20 mL dichloromethane. Thereafter, this reaction mixture was stirred for 3 h at 40 °C. The 4-(chloromethyl)-2-(((2-hydroxyphenyl) imino) methyl) phenol solid was filtered off, and washed with CH2Cl2. Finally dried in an oven at 50 °C (yellowish orange powder, 98% yield).Synthesis of Fe3O4@NFC@ONS-Ni(II) nanocomposite (V–VI)
In a balloon containing acetonitrile (15 mL), the first 4-(chloromethyl) -2-(((2-hydroxyphenyl) imino) methyl) phenol (2 mmol) and triethylamine (4 mmol) was added and was stirred for 20 minutes. Then, Fe3O4@NFC@CPTMS (1 g) was added and the mixture is refluxed for 24 hours. Finally, Fe3O4@NFC@ONS(V) was filtered off, washed three times with acetonitrile and diethyl ether, and dried at 70 °C in the vacuum oven. The next step, Fe3O4@NFC@ONS (1.00 g) was mixed with Ni(OAc)2 (1.2 mmol) in ethanol (5 mL). The mixture was stirred for 12 hours at room temperature. After completion of the reaction, then it was filtered, and the solid obtained was washed with ethanol and dried at 70 °C overnight to give Fe3O4@NFC@ONS-Ni(II).Synthesis of Fe3O4@NFC@ONSM-Ni(II) nanocatalyst (VII)
The immobilized Fe3O4@NFC@ONS-Ni(II) (8) as well as a weighted amount of melamine (molar ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were transferred to a 25 mL oven round bottom flask. A solution containing triethylamine (3.0 mmol) in the MeOH (10.0 mL) was added to this mixture. Subsequently, the mixture was refluxed in N2 gas for 12 h. The catalyst Fe3O4@NFC@ONSM-Ni(II) was separated from the reaction mixture using a strong magnet, rinsed with water and ethanol, and was finally dried in a vacuum oven (6 h, 60 °C). Scheme 1 illustrates the perfect route for the preparation of VII. The nickel loading of Fe3O4@NFC@ONSM-Ni(II) was determined by the inductively coupled plasma (ICP) technique.
:1) were transferred to a 25 mL oven round bottom flask. A solution containing triethylamine (3.0 mmol) in the MeOH (10.0 mL) was added to this mixture. Subsequently, the mixture was refluxed in N2 gas for 12 h. The catalyst Fe3O4@NFC@ONSM-Ni(II) was separated from the reaction mixture using a strong magnet, rinsed with water and ethanol, and was finally dried in a vacuum oven (6 h, 60 °C). Scheme 1 illustrates the perfect route for the preparation of VII. The nickel loading of Fe3O4@NFC@ONSM-Ni(II) was determined by the inductively coupled plasma (ICP) technique.
General experimental procedure for the synthesis of polyhydroquinolines (PHQ)
In a round-bottomed flask aldehyde (1 mmol), dimedone (1 mmol), ammonium acetate (1.5 mmol), ethyl acetoacetate (1 mmol), and Fe3O4@NFC@ONSM-Ni(II) (15 mg, equal to 0.5 mol%) were thoroughly blended. The reaction mixture was heated up to 65 °C and the end of the reaction was confirmed with TLC (EtOAc:n-hexane). In the next step, hot ethanol (10 mL) was added to the reaction mixture and the product was then placed in the organic phase. Eventually, the green products crystallized in ethanol gave the pure products in 90–97% yields based on the starting aldehyde. The products were finally compared with the melting points of the previously reported products.
General experimental procedure for the synthesis of 1,4-dihydropyridine (1,4-DHP)
In a round-bottomed flask aldehyde (1 mmol), ammonium acetate (1.5 mmol), ethyl acetoacetate (2 mmol), and Fe3O4@NFC@ONSM-Ni(II) (15 mg, equal to 0.5 mol%) were thoroughly blended. The reaction mixture was heated up to 65 °C and the end of the reaction was confirmed with TLC (EtOAc:n-hexane). In the next step, hot ethanol (10 mL) was added to the reaction mixture and the product was then placed in the organic phase. Eventually, the green products crystallized in ethanol gave the pure products in 88–97% yields based on the starting aldehyde. The products were finally compared with the melting points of the previously reported products. The spectral data for the selected compounds (PHQ and 1,4-DHP) are as follows:
FT-IR (KBr): νmax = 3275 (NH-stretching), 3080 (H–Ar), 2963 (C–H aliphatic), 1700, 1610, 1450–1500, and 1158 (C–O) cm−1.
1HNMR (400 MHz, DMSO): δ = 0.87 (s, 6H, 2× CH3), 1.21–1.27 (s, 3H, CH3), 1.80 (s, 2H), 2.25–2.29 (s,s, 5H, CH3, CH2), 3.95–4.01 (q, 2H, O–CH2–CH3), 4.72 (s, 1H, CH), 6.92–7.09 (m, 5H, H–Ar), and 9.07 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 13.62, 18.51, 28.48, 31.18, 42.35, 43.13, 51.56, 62.35, 103.96, 111.90, 125.61, 128.42, 129.36, 144.40, 149.34, 150.43, 168.13, and 192.60 ppm.
FT-IR (KBr): νmax = 3275 (NH-stretching), 3196, 3080 (H–Ar), 2963 (C–H aliphatic), 1711, 1645, 1463–1605 (CC), and 1218 (C–O) cm−1.
1HNMR (400 MHz, DMSO): δ = 0.94 (s, 6H, 2× CH3), 1.25–1.31 (s, 3H, CH3), 1.81 (s, 2H), 2.29–2.33 (s,s, 5H, CH3, CH2), 4.01–4.06 (q, 2H, O–CH2–CH3), 4.72 (s, 1H, CH), 7.06–7.19 (d,d, 4H, H–Ar), and 9.07 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.59, 19.09, 27.02, 33.43, 43.13, 44.13, 51.56, 60.52, 103.96, 111.53, 129.36, 129.80, 131.16, 142.30, 150.29, 151.20, 168.13, and 194.48 ppm.
FT-IR (KBr): νmax = 3286 (NH-stretching), 3212, 3080 (H–Ar), 2963 (C–H aliphatic), 1695, 1615, 1498, and 1158 (C–O) cm−1.
1HNMR (400 MHz, DMSO): δ = 0.95 (s, 6H, 2× CH3), 1.21–1.29 (s, 3H, CH3), 1.81 (s, 2H), 2.29–2.33 (s,s, 5H, CH3, CH2), 3.90 (s, 3H, OCH3), 3.97–4.06 (q, 2H, O–CH2–CH3), 4.72 (s, 1H, CH), 6.92–7.09 (d,d, 4H, H–Ar), and 9.07 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.31, 19.37, 27.02, 31.18, 42.14, 43.41, 50.16, 55.53, 60.52, 102.08, 114.23, 119.01, 129.36, 137.98, 148.67, 150.94, 157.77, 167.46, and 194.54 ppm.
FT-IR (KBr): νmax = 3275 (NH-stretching), 3207, 3085 (H–Ar), 2963 (C–H aliphatic), 1700, 1610, 1494, and 1158 (C–O) cm−1.
1HNMR (400 MHz, DMSO): δ = 1.01 (s, 6H, 2× CH3), 1.12–1.18 (s, 3H, CH3), 1.64 (s, 2H), 2.21 (s, 2H, CH3 Ar), 2.24–2.33 (s,s, 5H, CH3, CH2), 4.12–4.19 (q, 2H, O–CH2–CH3), 4.72 (s, 1H, CH), 6.82–7.38 (d,d, 4H, H–Ar), and 9.02 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.31, 19.09, 21.18, 27.54, 32.78, 41.81, 42.14, 51.87, 61.51, 103.01, 111.53, 123.55, 128.42, 134.58, 141.82, 149.99, 150.59, 167.75, and 194.48 ppm.
FT-IR (KBr): νmax = 3342 (NH-stretching), 1689 (CO), 1651, 1491, 1218, and 1127 cm−1.
1H NMR (400 MHz, DMSO): δ = 1.31–1.40 (t, 6H, 2× CH3–CH2), 2.21 (s, 6H, 2× CH3), 3.89–4.02 (m, 4H, O–CH2–CH3), 5.84 (s, 1H, CH), 6.99–7.19 (m, 5H, H–Ar), and 9.06 (s, 1H, NH) ppm.
13C NMR (100 MHz, DMSO): δ = 16.86, 21.18, 43.13, 61.51, 102.70, 125.61, 128.42, 129.80, 144.81, 150.27, and 168.59 ppm.
1HNMR (400 MHz, DMSO): δ = 1.36–1.44 (t, 6H, 2× CH3–CH2), 2.25 (s, 6H, 2× CH3), 3.85–3.98 (s, 3H, CH3–O), 4.71 (s, 1H, CH), 7.06–7.39 (d,d, 2H, H–Ar), and 9.08 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.56, 19.24, 44.01, 61.36, 102.70, 128.34, 129.80, 131.16, 141.82, 150.29, and 168.13 ppm.
FT-IR (KBr): νmax = 3342 (NH-stretching), 2983 (C–H str of CH3), and 1694 (CO), 1491 cm−1.
1HNMR (400 MHz, DMSO): δ = 1.33–1.39 (t, 6H, 2× CH3–CH2), 2.53 (s, 6H, 2× CH3), 3.68 (s, 3H, CH3–O), 3.87–4.01 (m, 4H, O–CH2–CH3), 4.73 (s, 1H, CH), 7.08–7.13 (d of d, 4H, H–Ar), 8.17–8.23 (d, 2H, H–Ar), and 9.09 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.56, 19.24, 44.01, 53.30, 63.94, 113.93, 129.36, 137.98, 144.40, 150.27, 157.36, and 168.13 ppm.
FT-IR (KBr): νmax = 3326 (NH-stretching), 2978 (C–H str of CH3), 1694 (CO), 1646, 1523, 1218, and 1116 cm−1.
1HNMR (400 MHz, DMSO): δ = 1.36–1.43 (t, 6H, 2× CH3–CH2), 2.17 (s, 6H, 2× CH3), 3.85–3.97 (q, 4H, O–CH2–CH3), 4.71 (s, 1H, CH), 7.19–7.21 (d,d, 2H, H–Ar), 8.14–8.18 (d, 2H, H–Ar), and 8.98 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.56, 19.37, 42.14, 61.51, 101.98, 121.89, 125.61, 144.40, 150.94, 151.64, and 168.13 ppm.
FT-IR (KBr): νmax = 3331 (NH-stretching), 2983 (C–H str of CH3), 2234 (CN), 1694 (CO), 1485, and 1047 cm−1.
1HNMR (400 MHz, DMSO): δ = 1.37–1.44 (t, 6H, 2× CH3–CH2), 2.26 (s, 6H, 2× CH3), 3.88–3.95 (q, 4H, O–CH2–CH3), 4.71 (s, 1H, CH), 7.50–7.71 (d,d, 4H, H–Ar), and 8.98 (s, 1H, NH) ppm.
13CNMR (100 MHz, DMSO): δ = 14.31, 18.51, 44.01, 60.77, 102.54, 109.81, 118.22, 128.42, 133.48, 146.11, 150.27, and 167.91 ppm.
Cell culture
K562 and MDA-MB-231 cell lines were obtained from the Pasteur Institute of Iran and then enriched with RPMI1640 and DMEM high glucose, which were enriched with 10% fetal bovine serum and 5% carbon dioxide at 37 °C, respectively. In order to conduct the analyses, when the cells reached at least 70 percent of cell growth, MDA-MB-231 cells were separated from the flask by 1 percent trypsin, centrifuged, and the resulting cell pellet was then suspended in 1 mL of culture medium. K562 cells were also transferred to Falcon and after centrifugation, cell precipitate was suspended in 1 mL of culture medium. Those cells with high viability of 90 percent were used for the experiment after verifying the absence of contamination.Investigation of toxicity using MTT test
The toxicity of Fe3O4@NFC-ONSM-Ni(II) nanoparticles on cell lines was evaluated using MTT method. Cells (104 cells at the third passage, exponential phase) were separately treated with various concentrations of nanoparticles (6.25, 12.5, 25, and 50 μg mL−1) and then incubated in time periods of 24, 48, and 72 hours. Afterward, MTT (5 mg mL−1) was added to each well and the plate was incubated in an incubator for 4 hours (CO2: 5% and 37 °C). In the following step, the precipitate was thoroughly mixed in 100 μL of methyl sulfoxide (DMSO) to dissolve formazan crystal. Ultimately, the colour absorption of each treated well was read by the Eliza Reader at 570 nm. In this test, three repeats were considered for each concentration, and the cell survival rate was finally calculated using the following formula:| Cell survival rate = (Light absorption control/test light absorption) × 100 |
Morphological evaluation of the treated cells with Hoechst 33285 staining
For performing this staining, different concentrations of K562 and MDA-MB-231 cells (1.5 × 106 in each well) (12.5, 25, and 50 μg mL−1) as well as nanoparticles Fe3O4@NFC@ONSM-Ni(II) were treated for 48 hours on 6-well plates. Next, the cells were treated with Hoechst 33285 (1 mg mL−1). In this way, 1 μL of the coloured solution was mixed with 100 μL cell suspension (1.5 × 106 cells) and carefully placed on a microscopic slide with 10 μL of Hoechst stain, and a fluorescence Inverted Microscopy was then used to observe the slide.Statistical analysis
Data were analyzed using SPSS software version 16 by ANOVA analysis and Tukey check in one way. The statistics were drawn with the help of PRISM software. The significance level was considered as P < 0.05.Conclusion
In this study, we successfully synthesized a novel trinuclear nickel nanomagnetic catalyst, namely Fe3O4@NFC@ONSM-Ni(II) through a mechanical method using nanofiber cellulose as a substrate and a Schiff base ligand as an anchor. Fe3O4@NFC@ONSM-Ni(II) nanocatalyst was characterized using different techniques including FT-IR, TEM, FE-SEM, DLS, VSM, XRD, UV-Vis, EDX, and ICP, which emphasized the proposed structure, spherical morphology, being nano-scale, and metals loading. Also, good reactivity and recyclability, ease of testing, commercial availability and non-toxicity, cost-effectiveness, and compatibility with the climate as a green catalyst in controlling adverse reactions in the sensitive layer and supporting a wide range of reactions can be mentioned as good reasons for this catalyst. This novel trinuclear nickel catalyst used as a robust and magnetically recoverable catalyst for the one-pot synthesis of 1,4-dihydropyridine and polyhydroquinolines from derivatives via symmetrical and unsymmetrical Hantzsch reaction. The significant advantages of this method include a mild reaction, short time, high efficiency of reactions, and the absence of precious metals and green reaction conditions. Cytotoxicity results of Fe3O4@NFC@ONSM-Ni(II) against two different cancer cells clearly demonstrated in lymphoblastic cancer cells, low concentration increase cell death but at solid tumors (MDA-MB231) higher concentration at prolonged time incubation was needed (15 μg mL−1 and 25 μg mL−1 respectively IC50 for K562 cells at 24 h and MDA-MB 231 cell at 48 h). Although, the component of Fe3O4@NFC@ONSM-Ni (Fe3O4, Fe3O4@NFC) indicated low toxicity than Fe3O4@NFC@ONSM-Ni(II) on cancer cells. Morphological assessment revealed apoptotic cells increased than necrotic cell after treatment of Fe3O4@NFC@ONSM-Ni with K562 and MDA-MB231 cells. Although, further investigations require to investigate precise mechanisms of cell death of Fe3O4@NFC@ONSM-Ni.Conflicts of interest
There is no contrast to express.Acknowledgements
We gratefully acknowledge the financial support of the research Council of the University of Birjand and Birjand University of Medical Sciences.References
- P. Ghamari Kargar, S. Aryanejad and G. Bagherzade, Appl. Organomet. Chem., 2020, e5965 CAS.
- G. Kumar, D. Kumar, S. Devi, R. Johari and C. P. Singh, Eur. J. Med. Chem., 2010, 45, 3056–3062 CrossRef CAS PubMed.
- A. Shebi and S. Lisa, Carbohydr. Polym., 2018, 201, 39–47 CrossRef CAS PubMed.
- A. Y. Louie and T. J. Meade, Chem. Rev., 1999, 99, 2711–2734 CrossRef CAS PubMed.
- P. Ghamari Kargar, M. Bakherad, A. Keivanloo and A. H. Amin, Iran. J. Catal., 2018, 8(3), 179–187 Search PubMed.
- H. Khashei Siuki, G. Bagherzade and P. Ghamari Kargar, ChemistrySelect, 2020, 5, 13537–13544 CrossRef CAS.
- P. Mukherjee, C. Biswas, M. G. B. Drew and A. Ghosh, Polyhedron, 2007, 26, 3121–3128 CrossRef CAS.
- M. Suleman and S. Riaz, Med. Eng. Phys., 2020, 86, 128–137 CrossRef PubMed.
- N. Kohler, C. Sun, J. Wang and M. Zhang, Langmuir, 2005, 21, 8858–8864 CrossRef CAS PubMed.
- N. K. Sahu, J. Gupta and D. Bahadur, Dalt. Trans., 2015, 44, 9103–9113 RSC.
- J. Zhang, Y. Wang, H. Ji, Y. Wei, N. Wu, B. Zuo and Q. Wang, J. Catal., 2005, 229, 114–118 CrossRef CAS.
- A. Farzin, S. A. Etesami, J. Quint, A. Memic and A. Tamayol, Adv. Healthc. Mater., 2020, 9, 1901058 CrossRef CAS PubMed.
- P. H. Linh, N. V. Chien, D. D. Dung, P. H. Nam, D. T. Hoa, N. T. N. Anh, L. V. Hong, N. X. Phuc and P. T. Phong, J. Mater. Sci., 2018, 53, 8887–8900 CrossRef CAS.
- B. Lesiak, N. Rangam, P. Jiricek, I. Gordeev, J. Tóth, L. Kövér, M. Mohai and P. Borowicz, Front. Chem., 2019, 7, 642 CrossRef CAS PubMed.
- Y. Ling, K. Wei, Y. Luo, X. Gao and S. Zhong, Biomaterials, 2011, 32, 7139–7150 CrossRef CAS PubMed.
- M. I. Khan, A. Mohammad, G. Patil, S. A. H. Naqvi, L. K. S. Chauhan and I. Ahmad, Biomaterials, 2012, 33, 1477–1488 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40(7), 3941–3994 RSC.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- M. Calero, M. Chiappi, A. Lazaro-Carrillo, M. J. Rodríguez, F. J. Chichón, K. Crosbie-Staunton, A. Prina-Mello, Y. Volkov, A. Villanueva and J. L. Carrascosa, J. Nanobiotechnology, 2015, 13, 16 CrossRef.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2021, 11, 4339–4355 RSC.
- M. G. Dekamin, M. Ghanbari, M. R. Moghbeli, M. Barikani and S. Javanshir, Polym. Plast. Technol. Eng., 2013, 52, 1127–1132 CrossRef CAS.
- R. Mannhold, B. Jablonka, W. Voigt, K. Schönafinger and E. Schraven, Eur. J. Med. Chem., 1992, 27, 229–235 CrossRef CAS.
- B. Hemmateenejad, R. Miri, M. Akhond and M. Shamsipur, Arch. Pharm., 2002, 335, 472–480 CrossRef CAS.
- R. Eshaghi Malekshah, B. Fahimirad and A. Khaleghian, Int. J. Nanomedicine, 2020, 15, 2583–2603 CrossRef PubMed.
- F. Bossert, H. Meyer and E. Wehinger, Angew. Chem., Int. Ed. Engl., 1981, 20, 762–769 CrossRef.
- (a) R. A. Janis and D. J. Triggle, J. Med. Chem., 1983, 26, 775–785 CrossRef CAS PubMed; (b) R. S. Kumar, A. Idhayadhulla, A. J. Abdul Nasser and J. Selvin, Eur. J. Med. Chem., 2011, 46, 804–810 CrossRef CAS PubMed.
- R. G. Bretzel, C. C. Bollen, E. Maeser and K. F. Federlin, Am. J. Kidney Dis., 1993, 21, S53–S64 CrossRef.
- Q. Ren, R. Wang, H. Wang, J. Key, D. J. L. Brett, S. Ji, S. Yin and P. Kang Shen, J. Mater. Chem. A, 2016, 4, 7591–7595 RSC.
- S. M. Vahdat, F. Chekin, M. Hatami, M. Khavarpour, S. Baghery and Z. Roshan kouhi, Chinese J. Catal., 2013, 34, 758–763 CrossRef CAS.
- P. N. Kalaria, S. P. Satasia and D. K. Raval, Eur. J. Med. Chem., 2014, 78, 207–216 CrossRef CAS PubMed.
- G. Tenti, J. Egea, M. Villarroya, R. León, J. C. Fernández, J. F. Padín, V. Sridharan, M. T. Ramos and J. C. Menéndez, Medchemcomm, 2013, 4, 590 RSC.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chemie Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- J.-P. Jolivet, S. Cassaignon, C. Chanéac, D. Chiche, O. Durupthy and D. Portehault, Comptes Rendus Chim, 2010, 13, 40–51 CrossRef CAS.
- M. Nasr-Esfahani, S. J. Hoseini, M. Montazerozohori, R. Mehrabi and H. Nasrabadi, J. Mol. Catal. A: Chem., 2014, 382, 99–105 CrossRef CAS.
- J. Safari and Z. Zarnegar, J. Mol. Catal. A: Chem., 2013, 379, 269–276 CrossRef CAS.
- (a) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chemie Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed; (b) Q. Zhou, Z. Wan, X. Yuan and J. Luo, Appl. Organomet. Chem., 2016, 30, 215–220 CrossRef CAS.
- E. Doustkhah, S. Rostamnia and A. Hassankhani, J. Porous Mater., 2016, 23, 549–556 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 RSC.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 37086–37097 RSC.
- P. Ghamari kargar, G. Bagherzade and H. Eshghi, RSC Adv., 2020, 10, 32927–32937 RSC.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- S. Mondal, B. C. Patra and A. Bhaumik, ChemCatChem, 2017, 9, 1469–1475 CrossRef CAS.
- M. Rahimifard, G. Mohammadi Ziarani, A. Badiei, S. Asadi and A. Abolhasani Soorki, Res. Chem. Intermed., 2016, 42, 3847–3861 CrossRef CAS.
- Q. Tang, X. Gong, P. Zhao, Y. Chen and Y. Yang, Appl. Catal., A, 2010, 389, 101–107 CrossRef CAS.
- N. Koukabi, E. Kolvari, M. A. Zolfigol, A. Khazaei, B. S. Shaghasemi and B. Fasahati, Adv. Synth. Catal., 2012, 354, 2001–2008 CrossRef CAS.
- E. I. Stankevich, E. E. Grinshtein and G. Y. Dubur, Chem. Heterocycl. Compd., 1975, 11(2), 196–198 CrossRef.
- J. Safari and Z. Zarnegar, RSC Adv., 2013, 3, 26094–26101 RSC.
- L. Nagarapu, M. D. Kumari, N. V. Kumari and S. Kantevari, Catal. Commun., 2007, 8, 1871–1875 CrossRef CAS.
- M. Abdollahi-Alibeik and A. Rezaeipoor-Anari, J. Magn. Magn. Mater., 2016, 398, 205–214 CrossRef CAS.
- L. Shiri, L. Heidari and M. Kazemi, Appl. Organomet. Chem., 2018, 32, e3943 CrossRef.
- G. B. Dharma Rao, S. Nagakalyan and G. K. Prasad, RSC Adv., 2017, 7, 3611–3616 RSC.
- T. Demirci, B. Çelik, Y. Yıldız, S. Eriş, M. Arslan, F. Sen and B. Kilbas, RSC Adv., 2016, 6, 76948–76956 RSC.
- A. Ghorbani-Choghamarani and B. Tahmasbi, New J. Chem., 2016, 40, 1205–1212 RSC.
- Q. Zhang, X.-M. Ma, H.-X. Wei, X. Zhao and J. Luo, RSC Adv., 2017, 7, 53861–53870 RSC.
- M. Hajjami and B. Tahmasbi, RSC Adv., 2015, 5, 59194–59203 RSC.
- T. Momeni, M. M. Heravi, T. Hosseinnejad, M. Mirzaei and V. Zadsirjan, J. Mol. Struct., 2020, 1199, 127011 CrossRef CAS.
- E. Doustkhah, S. Rostamnia and A. Hassankhani, J. Porous Mater., 2016, 23, 549–556 CrossRef CAS.
- N. Devarajan and P. Suresh, New J. Chem., 2019, 43, 6806–6814 RSC.
- R. Maheswari, V. V. Srinivasan, A. Ramanathan, M. P. Pachamuthu, R. Rajalakshmi and G. Imran, J. Porous Mater., 2015, 22, 705–711 CrossRef CAS.
- M. G. Dekamin, S. Ilkhanizadeh, Z. Latifidoost, H. Daemi, Z. Karimi and M. Barikani, RSC Adv., 2014, 4, 56658–56664 RSC.
- M. G. Dekamin, E. Kazemi, Z. Karimi, M. Mohammadalipoor and M. R. Naimi-Jamal, Int. J. Biol. Macromol., 2016, 93, 767–774 CrossRef CAS PubMed.
- O. D'Alessandro, Á. G. Sathicq, J. E. Sambeth, H. J. Thomas and G. P. Romanelli, Catal. Commun., 2015, 60, 65–69 CrossRef.
- B. Zeynizadeh, S. Rahmani and E. Eghbali, Polyhedron, 2019, 168, 57–66 CrossRef CAS.
- F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752–768 RSC.
- J. Li, Y. Hu, J. Yang, P. Wei, W. Sun, M. Shen, G. Zhang and X. Shi, Biomaterials, 2015, 38, 10–21 CrossRef CAS PubMed.
- T. Shanmugasundaram, M. Radhakrishnan, A. Poongodi, K. Kadirvelu and R. Balagurunathan, MRS Commun, 2018, 8, 604–609 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01256h |
| ‡ These authors are co-first authors and contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |