DOI:
10.1039/D1RA01253C
(Paper)
RSC Adv., 2021,
11, 11536-11540
Peripherical thioester functionalization induces J-aggregation in bithiophene-DPP films and nanoparticles†
Received
15th February 2021
, Accepted 8th March 2021
First published on 19th March 2021
Abstract
In this work we demonstrated that the peripherical thioacetylation of a bithiophene–DPP molecule can greatly influence the solid-state properties triggering the formation of NIR emitting J-aggregates in both bithiophene–DPP films and nanoparticles. The morphology and the kinetic and thermal stability of the organic nanoparticles were also investigated.
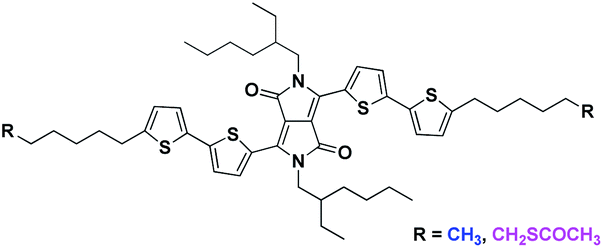
The control of supramolecular aggregation of organic semiconductors is a critical and fundamental achievement since aggregation strongly affects the solid-state optic, electronic and optoelectronic properties of conjugated systems. Although processing conditions play a key role in the solid-state arrangement of organic semiconductors in thin film,1 chemists are attracted by the possibility to control the organization of conjugated molecules by the structural design.2–5 In this context, it is particularly interesting the case of diketopyrrolo[3,4.c]pyrrole (DPP) derivatives. DPPs are gaining a prominent position among organic dyes for optics and electronics thanks to their simple synthesis, intense color, high photostability, and good charge transport properties.6,7 Several studies have been reported on the control of the DPP aggregation varying the conjugation of the core or the N-substitution,8–11 while less explored is the impact on the DPP self-assembly given by the presence of terminal non-conjugated flexible chains.12,13 In our previous works we observed that varying the length of terminal aliphatic side chains (C8 vs. C16) in a donor–acceptor–donor (DAD) triazole–thiophene–DDP derivative, it was possible to induce the formation of H- or J-aggregates in thin films and organic nanoparticles (ONPs).14,15 Especially, we found that, for both the C8 and C16 substituted molecules, H-aggregates were preferentially formed during the self-assembly, but in the case of C8 substituted species, the resulting ONPs were characterized by low colloidal stability with a remarkable tendency to form micrometric rod-like particles. This morphological evolution of ONPs was associated to an enhancement of the J-aggregate absorption, demonstrating the importance of terminal aliphatic chains in the supramolecular organization of the dye. The possibility to control the formation of H- or J-aggregates is a crucial aspect for the solid-state applications of DDPs. For instance, it has been reported that the increase in the DPP π–π stacking, with the consequent H-aggregation, improves carrier mobility in organic field-effect transistors16 while, in bulk heterojunction solar cells, DPPs that generate H-aggregates display distinctly lower performances than those forming J-aggregates.17 Furthermore, J-aggregates show NIR photoluminescence and they can be used as efficient solid state NIR emitters.18–20 In this work we report the effect of introduction of thioacetyl groups at the end of long alkyl chains on the self-assembly behavior of a bithiophene–DPP derivative (DPP4T). The two compared molecules share the same conjugated core, N-substitution (2-ethylhexyl) and terminal aliphatic moieties (hexyl chains), which are unsubstituted for DPP4T-CH3, or functionalized with terminal thioacetyl groups for DPP4T-CH2SAc (Fig. 1). DPP4T-CH3 represents the reference system for this study since it leads to the formation of both H- and J-aggregates once in the solid state.13,21,22 Hence, even small variations in the ratio between the two supramolecular species induced by the introduction of different substituents, can be easily detected and quantified. The interest about the correlations between the thioester functionalization and the supramolecular assembly was driven by recent findings on the organization of photoactive ternary blends in the presence of -SAc functionalized oligo(arylenethiophene)s.23 In fact, we showed that thioester polarizable group can modify the interactions with the solvent used for the processing, also impacting on the solid-state organization.
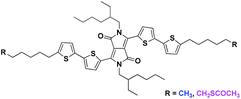 |
| | Fig. 1 Molecular structure of DPP4T-CH3 and of the thioacetate species DPP4T-CH2SAc. | |
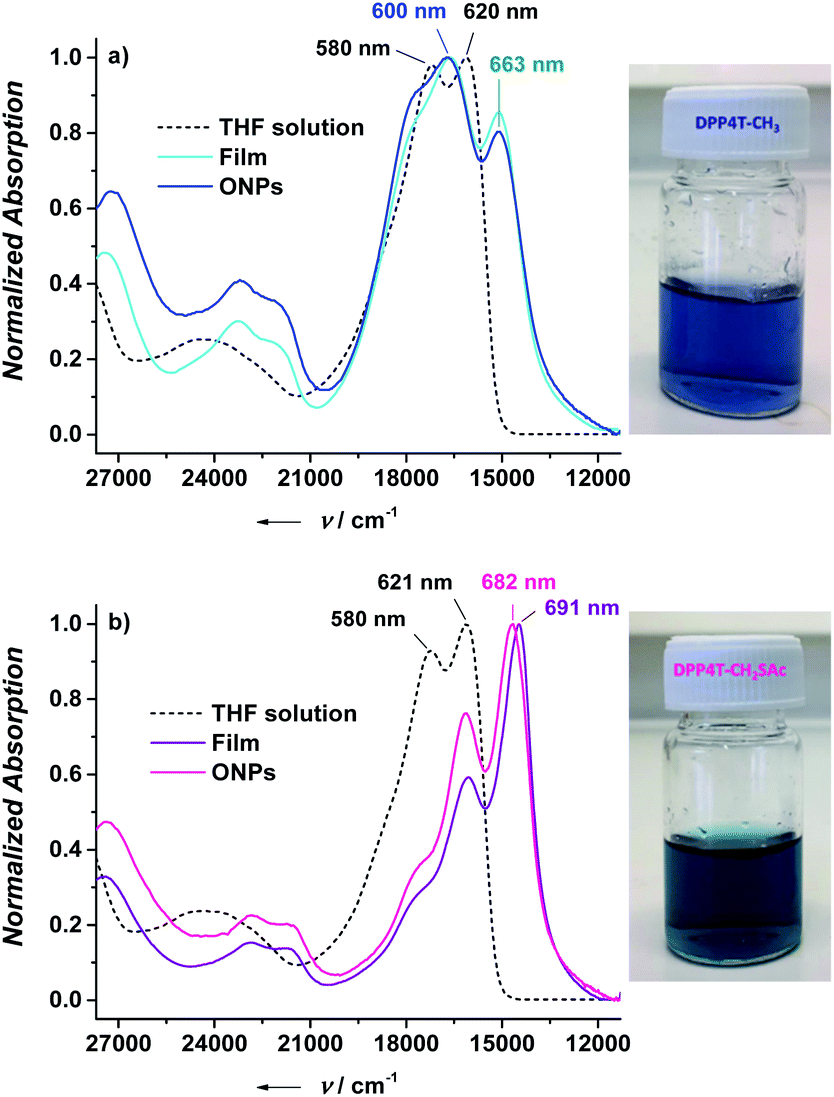
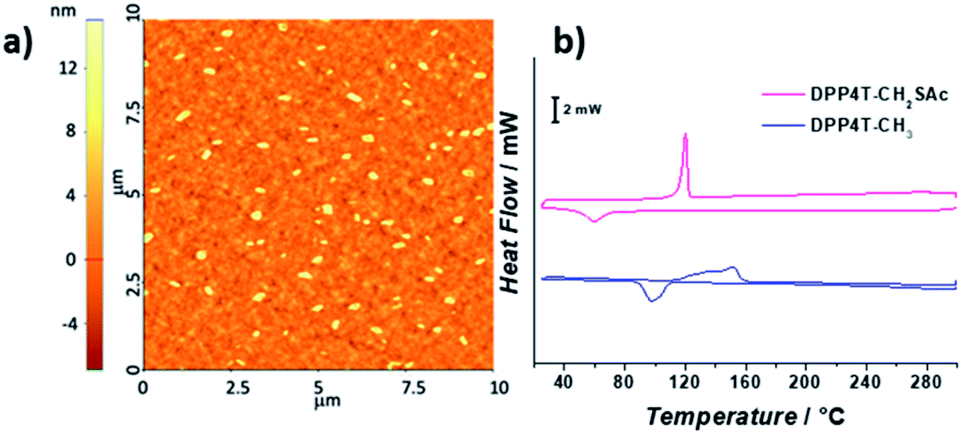
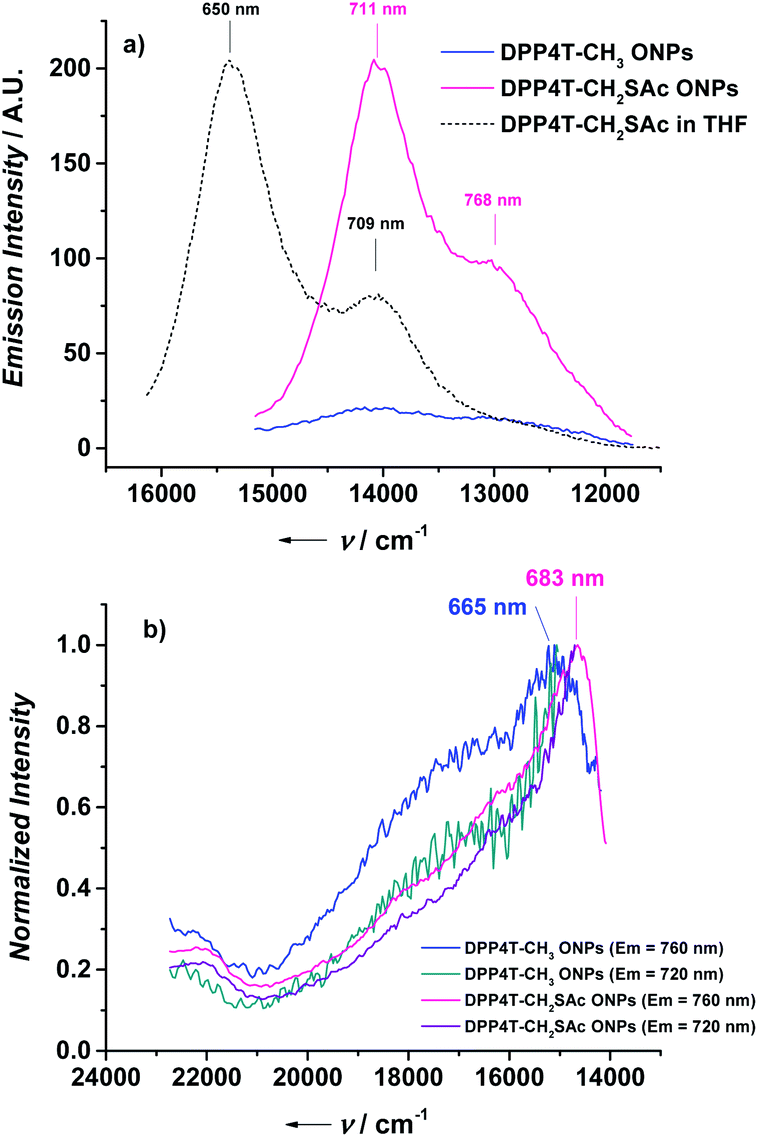
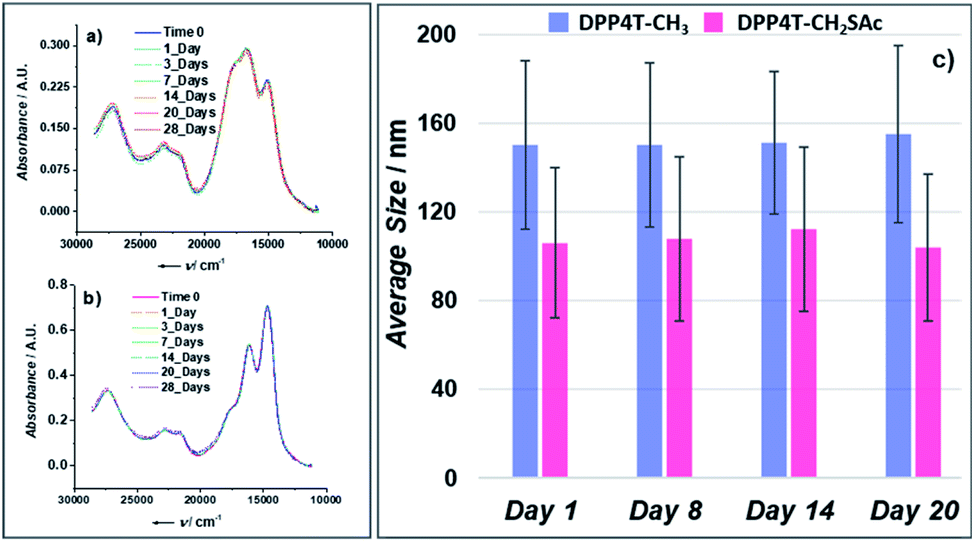
In the present study, in order to distinguish the two effects, DPP4T-CH3 and DPP4T-CH2SAc were processed with different techniques and in different conditions: as thin films by spin-coating from a chloroform solution, or as ONPs obtained via re-precipitation method from a tetrahydrofuran (THF) solution in water. The two DPP derivatives were synthesized through a palladium-catalysed Suzuki–Miyaura coupling, a straightforward metal-catalyzed cross-coupling reaction.24,25 Synthetic details are provided in the ESI.† First, we performed the electrochemical and the optical characterization of the two dyes in solution. From the comparison between the cyclic voltammograms of the two species (Fig. S1†) it is clear that the peripherical thioacylation has negligible effects on the electrochemical bandgap (Eg) of the conjugated molecules. Both the oligomers show HOMO and LUMO energies of −5.4 and −3.5 eV respectively, with a resulting bandgap of 1.9 eV (Table S1†). Results are in perfect agreement with the literature data available for DPP4T-CH3 21,22,26 and with the UV-VIS characterization in different solvents (CHCl3, THF, toluene). In fact, from the comparison between the normalized UV-vis spectra of the two dyes in THF (black dotted line in Fig. 2a and b), only minor differences in the relative intensities between the vibronic peaks at 620 and 580 nm can be detected. This difference in the relative intensity between the two absorption maxima can be observed also in other solvents (Fig. S2a and b†), suggesting a different solvation behaviour for two molecules due to the presence of the -SAc groups. The fluorescence is peaked in the red region at around 650 nm (Fig. S2c and d†), with a Stoke's shift of ≈30 nm. The main optical properties in solution are listed in Table S2.† The solid-state aggregation properties were investigated on thin films of both species on quartz glasses (details in ESI†). As already reported by Pron and co-workers, DPP4T-CH3 exhibits poor filming properties,22 and even testing different solvents it was not possible to obtain uniform depositions. Contrarily, DPP4T-CH2SAc can be deposited as thin film independently on the solvent used, indicating an easy processability of thioester-functionalized organic semiconductors.23 The high uniformity of DPP4T-CH2SAc film was confirmed by the AFM analysis (Fig. 3a), that revealed a film thickness of ≈137 nm and a surface roughness (Rg) of 2.42 ± 0.1 nm. The absorption behaviour of the two molecules as thin film completely differs as can be seen from Fig. 2. DPP4T-CH3 film presents an absorption maximum blue-shifted with respect to the maximum in solution and attributed to the formation of H-aggregates, and a further, red-shifted relative maximum, associated to the J-species. The blue-shifted absorption peak is at 600 nm with a H-shift of 540 cm−1 and a shoulder at 566 nm, while the peak of J-aggregates is red-shifted of 1020 cm−1 at 663 nm. For the DPP4T-CH2SAc oligomer instead, we record a remarkable trend in the formation of J-aggregates, with a bathochromic shift of the absorption maximum of 1660 cm−1. The spectrum presents a main peak at 691 nm, and two vibronic peaks at 623 and 570 nm (ΔE ≈ 1550 cm−1) with no clear evidence of H-species. The lower tendency to form stacked aggregates of acetylated derivative is supported also from the DSC analysis reported in Fig. 3b. No liquid crystal phase transition was apparently observed for any of the molecules. The melting (Tm) and crystallization (Tc) temperatures measured for the two dyes are lower with respect to those shown by other DPP compounds with unbranched chain substitution on the lactam nitrogen. This has been attributed by other authors to the increase of intermolecular distance in aggregates produced by the 2-ethylhexyl substitution.27 The DPP4TS-CH2SAc thermogram displays sharp and narrow melting and crystallization transitions, centred at 120 and 60 °C respectively. The presence of a single and sharp peak is consistent with the formation of a single aggregate species, likely J-like as already observed in the absorption spectra. Conversely, phase transitions for DPP4T-CH3 look more complex, with a broad endothermic phase transition appearing as a bi-modal peak at 133 and 151 °C, and the exothermic phase transition centred at 98 °C. The broad and higher transition temperatures are consistent with the hypothesis of the oligomer DPP4T-CH3 forming different aggregates with the highest melting energy required. The capability of the SAc-functionalization to trigger the formation of DPP4T J-aggregates independently on the processing technique was also tested on suspensions of organic nanoparticles (ONPs) in water. The normalized absorption spectra of the DPP4T-CH3 and DPP4T-CH2SAc ONPs are reported in Fig. 2. For both molecules, the ONP absorption spectra resemble those of the films. Actually, the major differences can be seen at higher frequency due to the scattering of particles. DPP4T-CH3 ONPs spectrum is characterized by peaks related to H- and J-aggregates at 598 and 663 nm respectively, as in the case of film. The peak at 663 nm results less intense in ONPs, suggesting a lower tendency to form J-like species in these conditions. Even the spectrum of DPP4T-CH2SAc retraces the film spectrum, with the maximum of absorption at 682 nm (691 nm for the film), and the same vibronic distribution. Contrarily to non-emissive DPP4T-CH3 ONPs, DPP4T-CH2SAc ONPs gives photoluminescence in the NIR. As can be seen from Fig. 4a, the maximum of emission shifts from 650 to 711 nm (ΔE ≈ 1300 cm−1) moving from the THF solution to the ONPs suspension, with a shoulder at 768 nm (ΔE ≈ 1050 cm−1). The coexistence of different kinds of aggregates in DPP4T-CH3 ONPs is confirmed from the analysis of the excitation spectra in Fig. 4b. In fact, the excitation spectra acquired close to the two maxima of emission (720 and 760 nm) have a different band-shape and they both differ from the absorption spectrum.9 In particular, the maximum of the excitation spectrum coincides with the maximum of absorption associated to the J-species at 663 nm. Instead, the excitation spectra measured at different wavelengths of DPP4T-CH2SAc ONPs are quite similar, indicating that the luminescence at the two maxima is originated by the same species. Moreover, the excitation band-shape perfectly resembles the absorption spectrum, with a maximum at 683 nm and the same vibronic distribution. Combining the information derived from DSC and the excitation data it is clear that the peripherical SAc-groups inhibit the H-organization and strongly promote the formation of J-aggregates, which exhibit a dipole allowed transition in the NIR.13 This aspect, together with the low absorbance of biological tissues in this region, makes this system extremely interesting for the development of fluorescent biomarkers for in vitro and in vivo experiments. Finally, we studied the colloidal stability of the two ONP suspensions. In Fig. 5 are reported the absorption spectra on DPP4T-CH3 and DPP4T-CH2SAc ONPs acquired during four weeks. Both systems are characterized by a remarkable stability. All the spectra maintain unaltered the band-shape without showing any increasement of the scattering, that implies that no further agglomeration
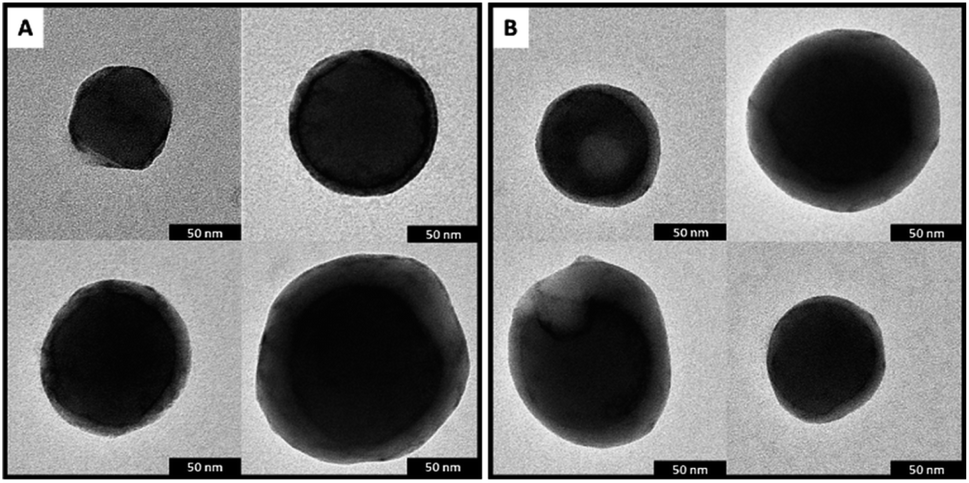
or precipitation occurred during this lapse of time. Results were confirmed by the dynamic light scattering (DLS) analysis (Fig. 5c). The DPP4T-CH3 ONPs show a monomodal distribution (Fig. S3†) with an average diameter of 150 nm and a width of about 70 nm (corresponding to a polydispersity index PDI = 0.22). The introduction of the thioacetyl group generates smaller nanoparticles. In fact, for the DPP4T-CH2SAc ONPs, the average size is 110 nm with a width of 70 nm (PDI = 0.40). The intensity distribution in this case exhibits a bi-modal distribution with two peaks at 15 and 110 mm (Fig. S4†). TEM images of samples, one day after their preparation, highlight the presence of nearly spherical objects in both DPP4T-CH3 and DPP4T-CH2SAc samples (Fig. 6). The statistical analysis of the nanoparticle size is in good agreement with DLS data. Indeed, the average diameter of sample DPP4T-CH3 is 114 nm (σ = 19%), while the mean diameter of sample DPP4T-CH2SAc is 106 nm (σ = 46%). During 20 days, storing the samples at 4 °C, no variations in the size distribution of the two species occurred. In order to better characterize the two systems, we studied the effects of the temperature and ionic strength on their colloidal stability. DPP4T-CH3 e DPP4T-CH2SAc ONP suspensions in water were heated in the temperature range 20–80 °C and the Z-average diameter was measured with a step of 5 °C. Finally, suspensions were cooled to room temperature and their size was measured again. As can be seen in Fig. 7, after this temperature scan, no variations in the average size of DPP4T-CH2SAc ONPs was observed, while the unsubstituted system exhibited a size reduction of 17%.
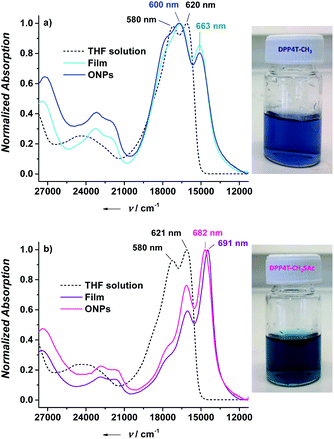 |
| | Fig. 2 Normalized absorption spectra for DPP4T-CH3 (a) and DPP4T-CH2SAc (b) in THF solution, as neat film and as ONP suspension in water. On the right are reported the picture of ONP suspensions of both species. | |
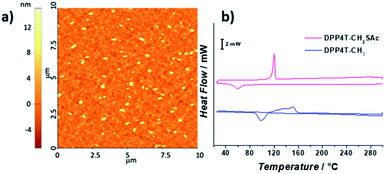 |
| | Fig. 3 (a) AFM 10 × 10 μm2 topography of DPP4TS-CH2SAc thin-film processed from a chloroform solution; (b) DSC thermograms of DPP4T-CH3 (blue line) and DPP4T-CH2SAc (pink line) recorded between 30 and 300 °C at a scan rate of 10 °C min−1. Scale bar 2 mW. | |
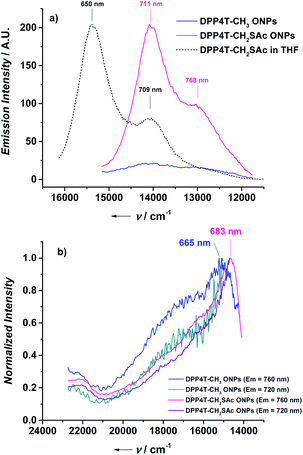 |
| | Fig. 4 (a) Emission spectra of DPP4T-CH3 ONPs (excited at 600 nm, absorbance = 0.295), DPP4T-CH2SAc (excited at 620 nm, absorbance = 0.536), and the normalized emission spectrum of DPP4T-CH2SAc in THF (dashed black line); (b) normalized excitation spectra of DPP4T-CH3 and DPP4T-CH2SAc ONPs recorded at different emission wavelength. | |
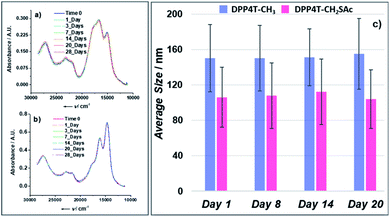 |
| | Fig. 5 (a) Absorption spectra of a DPP4T-CH3 ONP suspension acquired during 28 days; (b) absorption spectra of a DPP4T-CH2SAc ONP suspension acquired during 28 days; (c) evolution of the average size distribution of DPP4T-CH3 and DPP4T-CH2SAc ONPs during 20 days. The error bar represents the polydispersity index expressed in nm. | |
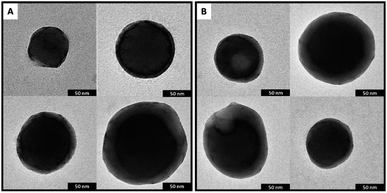 |
| | Fig. 6 (A) Representative TEM images of samples DPP4T-CH3 and, (B) DPP4T-CH2SAc 1 day after the preparation. | |
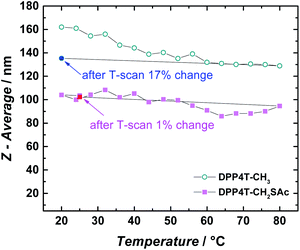 |
| | Fig. 7 Temperature dependence of the Z-average diameter of DPP4T-CH3 and DPP4T-CH2SAc ONP suspensions in water, varying the temperature in the range 20–80 °C. After the heating, suspensions were cooled at room temperature and the Z-average was measured again. | |
Interestingly, these temperature changes did not affect the supramolecular aggregation of the two oligomers in ONPs, since negligible variations in their absorption spectra were detected (Fig. S5†). Indeed, in the case of DPP4T-CH2SAc ONPs, varying the temperature from 25 to 65 °C, just an increment of about 7% was observe for the absorption peak at 660 nm (Fig. S5†).
Regarding the impact of ionic strength, in both cases, the addition of 1 ml of phosphate buffered saline to 1 ml of ONP suspension (final salt concentration: phosphate buffer 0.01 M, KCl 0.0027 M, NaCl 0.137 M) did not induce any precipitation at least for one week. Contrarily with what we observed in our previous studies on triazole–thiophene–DPP ONPs,14,15 where the J-aggregation was associated with a poor colloidal stability and with a remarkable tendency to aggregation, DPP4T-CH2SAc ONPs possess a high colloidal stability (observed also for the other derivative). Moreover, the presence of -SAc terminal group induces the formation of NIR emitting J-aggregates, opening the way to a promising system for the in vitro and in vivo bioimaging. In conclusion we demonstrated that the simple introduction of a bulky and polarizable terminal thioacetyl group on alkyl chains attached to a thiophene–DPP core, is able to improve the processability of the molecule and to strongly condition the supramolecular aggregation with the formation of stable NIR emitting J-aggregates, independently on the processing technique and conditions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by PRIN 2017 “CHIRALAB” (Prot. 20172M3K5N) and by University of Pisa through the project PRA_2020_21 “SUNRISE: Concentratori solari luminescenti NIR riflettenti”. The authors thank prof. Natalie Stingelin (Georgia Tech) for assistance in DSC measurements.
Notes and references
- D. Blasi, F. Viola, F. Modena, A. Luukkonen, E. MacChia, R. A. Picca, Z. Gounani, A. Tewari, R. Österbacka, M. Caironi, Z. M. Kovacs Vajna, G. Scamarcio, F. Torricelli and L. Torsi, J. Mater. Chem. C, 2020, 8, 15312–15321 RSC.
- L. Xu, D. Gao, J. Song, L. Shen, W. Chen, Y. Chen and S. Zhang, New J. Chem., 2015, 39, 5553–5560 RSC.
- Y. Gao, G. Feng, T. Jiang, C. Goh, L. Ng, B. Liu, B. Li, L. Yang, J. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857–2866 CrossRef CAS.
- A. Punzi, M. A. M. Capozzi, V. Fino, C. Carlucci, M. Suriano, E. Mesto, E. Schingaro, E. Orgiu, S. Bonacchi, T. Leydecker, P. Samorì, R. Musio and G. M. Farinola, J. Mater. Chem. C, 2016, 4, 3138–3142 RSC.
- M. A. M. Capozzi, A. Punzi, F. Babudri, R. Musio and G. M. Farinola, J. Org. Chem., 2018, 83, 14396–14405 CrossRef CAS PubMed.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- Y. Patil and R. Misra, J. Mater. Chem. C, 2019, 7, 13020–13031 RSC.
- N. Genevaz, P. Chávez, V. Untilova, A. Boeglin, C. Bailly, L. Karmazin and L. Biniek, J. Mater. Chem. C, 2018, 6, 9140–9151 RSC.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS PubMed.
- T. G. Hwang, J. Y. Kim, J. W. Namgoong, J. M. Lee, S. B. Yuk, S. H. Kim and J. P. Kim, Photochem. Photobiol. Sci., 2019, 18, 1064–1074 CrossRef CAS PubMed.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- X. A. Jeanbourquin, A. Rahmanudin, A. Gasperini, E. Ripaud, X. Yu, M. Johnson, N. Guijarro and K. Sivula, J. Mater. Chem. A, 2017, 5, 10526–10536 RSC.
- V. S. Gevaerts, E. M. Herzig, M. Kirkus, K. H. Hendriks, M. M. Wienk, J. Perlich, P. Müller-Buschbaum and R. A. J. Janssen, Chem. Mater., 2014, 26, 916–926 CrossRef CAS.
- A. Punzi, E. Maiorano, F. Nicoletta, D. Blasi, A. Ardizzone, N. Ventosa, I. Ratera, J. Veciana and G. M. Farinola, Eur. J. Org. Chem., 2016, 2016, 2617–2627 CrossRef CAS.
- A. Ardizzone, D. Blasi, D. Vona, A. Rosspeintner, A. Punzi, E. Altamura, N. Grimaldi, S. Sala, E. Vauthey, G. M. Farinola, I. Ratera, N. Ventosa and J. Veciana, Chem. –Eur. J., 2018, 24, 11386–11392 CrossRef CAS PubMed.
- M. Shaker, B. Park, J.-H. Lee, W. Kim, C. K. Trinh, H.-J. Lee, J. woo Choi, H. Kim, K. Lee and J.-S. Lee, RSC Adv., 2017, 7, 16302–16310 RSC.
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef.
- J. Li, Y. Liu, Y. Xu, L. Li, Y. Sun and W. Huang, Coord. Chem. Rev., 2020, 415, 213318 CrossRef CAS.
- D. Blasi, D. M. Nikolaidou, F. Terenziani, I. Ratera and J. Veciana, Phys. Chem. Chem. Phys., 2017, 19, 9313–9319 RSC.
- A. Barbieri, E. Bandini, F. Monti, V. K. Praveen and N. Armaroli, Top. Curr. Chem., 2016, 374, 47 CrossRef PubMed.
- Y.-F. Huang, S.-T. Chang, K.-Y. Wu, S.-L. Wu, G.-T. Ciou, C.-Y. Chen, C.-L. Liu and C.-L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 8869–8876 CrossRef CAS PubMed.
- G. Wiosna-Salyga, M. Gora, M. Zagorska, P. Toman, B. Luszczynska, J. Pfleger, I. Glowacki, J. Ulanski, J. Mieczkowski and A. Pron, RSC Adv., 2015, 5, 59616–59629 RSC.
- A. Punzi, A. Operamolla, O. Hassan Omar, F. Brunetti, A. D. Scaccabarozzi, G. M. Farinola and N. Stingelin, Chem. Mater., 2018, 30, 2213–2217 CrossRef CAS.
- A. Punzi, N. Zappimbulso and G. M. Farinola, European J. Org. Chem., 2020, 2020, 3229–3234 CrossRef CAS.
- A. Operamolla, S. Colella, R. Musio, A. Loiudice, O. Hassan Omar, G. Melcarne, M. Mazzeo, G. Gigli, G. M. Farinola and F. Babudri, Sol. Energy Mater. Sol. Cells, 2011, 95, 3490–3503 CrossRef CAS.
- M. Gora, S. Pluczyk, P. Zassowski, W. Krzywiec, M. Zagorska, J. Mieczkowski, M. Lapkowski and A. Pron, Synth. Met., 2016, 216, 75–82 CrossRef CAS.
- C. Kim, J. Liu, J. Lin, A. B. Tamayo, B. Walker, G. Wu and T.-Q. Nguyen, Chem. Mater., 2012, 24, 1699–1709 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, cyclic voltammetry, UV-vis and fluorescence characterization, film preparation and AFM characterization, ONPs preparation and DLS characterization. See DOI: 10.1039/d1ra01253c |
| ‡ Authors equally contributing. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Roberto Comparellic,
Gerardo Palazzo
b,
Roberto Comparellic,
Gerardo Palazzo