DOI:
10.1039/D1RA01173A
(Paper)
RSC Adv., 2021,
11, 18938-18944
Structural insights into targeting of the colchicine binding site by ELR510444 and parbendazole to achieve rational drug design†
Received
13th February 2021
, Accepted 27th April 2021
First published on 25th May 2021
Abstract
Microtubules consisting of α- and β-tubulin heterodimers have proven to be an efficient drug target for cancer therapy. A broad range of agents, including ELR510444 and parbendazole, can bind to tubulin and interfere with microtubule assembly. ELR510444 and parbendazole are colchicine binding site inhibitors with antiproliferative activities. However, the lack of structural information on the tubulin–ELR510444/parbendazole complex has hindered the design and development of more potent drugs with similar scaffolds. Therefore, we report the crystal structures of tubulin complexed with ELR510444 at a resolution of 3.1 Å and with parbendazole at 2.4 Å. The structure of these complexes revealed the intermolecular interactions between the two colchicine binding site inhibitors and tubulin, thus providing a rationale for the development of novel benzsulfamide and benzimidazole derivatives targeting the colchicine binding site.
Introduction
Microtubles, a key component of the eukaryotic cytoskeleton, are cylindrical molecules consisting of two subunits: α-tubulin and β-tubulin. It is known that α/β-tubulin heterodimers can be assembled into protofilaments with a head-to-tail formation, characterized by dynamic polymerization and depolymerization. Microtubules are involved in cell division, in which mitosis, morphogenesis, motility, and intracellular transport occur.1 In addition, microtubules have been reported to participate in the proliferation, invasion, and metastasis of tumor cells. In recent years, microtubules have been regarded as an important target for oncotherapy. At present, microtubule inhibitors, which disrupt microtubule dynamics, are used widely in cancer chemotherapy.2 Most of these molecules act by binding to the protein tubulin.3–8 There are several binding sites on the tubulin heterodimer, including for vinca alkaloids, taxanes, colchicine, and laulimalide,8 which are usually divided into two categories: microtubule stabilizers, including taxanes and laulimalide, which enhance tubulin polymerization; and microtubule depolymerizers, including vinca alkaloids and colchicine binding site agents, which inhibit tubulin polymerization. The microtubule destabilizers currently in clinical use include vinca alkaloids, vinblastine, vincristine, and vinorelbine, all of which bind to what has been termed the “vinca site” on tubulin. Another recognized binding site for microtubule-destabilizing agents is the colchicine binding site (CBS). Antitubulin agents are currently employed in cancer treatment. However, drug resistance arising from MDR1 expression in cancer cells leads to treatment failure. One drawback of microtubule-targeting agents is drug resistance, which can be innate or acquired. The most common form of clinical resistance to microtubule-targeting agents is overexpression of the MDR1 gene, which encodes the P-glycoprotein (Pgp) drug efflux pump.9 The other clinically relevant form of resistance to microtubule-targeting agents is overexpression of the type III isotype of tubulin.10 Extensive research has indicated that tubulin inhibitors targeting the CBS are candidate compounds to overcome the limitations of drug resistance and improve the associated clinical benefit.11,12
ELR510444, a sulfonamide derivative, was reported to inhibit tubulin polymerization, causing a loss of cellular microtubules (EC50 = 21 nM) and the formation of aberrant mitotic spindles, which led to mitotic arrest and apoptosis in cancer cells; in addition, significant antitumor activity was observed in the MDA-MB-231 xenograft model.13 Carew et al. reported that ELR510444 inhibits tumor growth and angiogenesis by abrogating HIF activity and disrupting microtubules in renal cell carcinoma.14 Extensive studies have indicated that ELR510444 is not a substrate for the Pgp drug transporter and retains activity in β III-tubulin-overexpressing cell lines, suggesting that it avoids both the clinically relevant mechanisms of resistance to this class of agents.13 However, the binding mode between ELR510444 and tubulin remains unclear. In addition, another chemotype of CBS inhibitors (CBSIs), benzimidazole carbamates, has been shown to inhibit the assembly of mammalian brain microtubules in vitro. Parbendazole was reported to be a potent inhibitor of microtubule assembly in vitro and in vivo, and was more effective than nocodazole.15,16 In a panel of four tested benzimidazoles, parbendazole exhibited the strongest antiproliferative activities against AsPC-1 (IC50 = 0.19 μM) and Capan-2 cells (IC50 = 0.36 μM), indicating its antitumor potential.17 Parbendazole binds to the tubulin dimer with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, and microtubules were reported to withdraw from the cell periphery within 30 min after treatment with parbendazole.15 We previously reported the crystal structure of the nocodazole–tubulin complex and revealed that nocodazole was bound deep within the CBS.18 However, the reason for the greater potency of parbendazole remains unknown.
:1 stoichiometry, and microtubules were reported to withdraw from the cell periphery within 30 min after treatment with parbendazole.15 We previously reported the crystal structure of the nocodazole–tubulin complex and revealed that nocodazole was bound deep within the CBS.18 However, the reason for the greater potency of parbendazole remains unknown.
To increase the efficacy of inhibition, it is necessary to explore the binding mode between ELR510444 and tubulin, which will facilitate subsequent structural optimization. To better understand the structure–activity relationship (SAR), and provide a solid structural basis for the development of novel benzimidazole derivatives, we also need to solve the structure of the parbendazole–tubulin complex.
Experimental methods
Special reagents
Porcine brain tubulin (catalog#: T-238P) was purchased from Cytoskeleton. The tubulin protein was supplied at 10 mg mL−1 in G-PEM (general tubulin buffer: 80 mM PIPES pH 6.9, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP) as a frozen liquid and stored at −80 °C until use. ELR510444 and parbendazole were purchased from MedChem Express. All reagents for crystallization (MES, tyrosine, DTT, and AMPPCP) were purchased from Sigma.
Protein expression and purification
The stathmin-like domain of RB3 (RB3-SLD) and tubulin tyrosine ligase (TTL) were overexpressed in E. coli strain BL21 (DE3). RB3-SLD was purified by anion-exchange chromatography (loading buffer: 20 mM Tris–HCl pH 8.0, 1 mM EGTA, 2 mM DTT; elution buffer: 20 mM Tris–HCl pH 8.0, 1 mM EGTA, 2 mM DTT, 2 M NaCl) and gel filtration chromatography (buffer: 10 mM HEPES pH 7.2, 150 mM NaCl, 2 mM DTT). The peak fractions from the gel filtration column were concentrated to 10 mg mL−1. TTL was purified through Ni-NTA affinity chromatography and gel filtration chromatography (buffer: bis–tris propane pH 6.5, 200 mM NaCl, 2.5 mM MgCl2, 5 mM DTT). The peak fractions of TTL were concentrated to 20 mg mL−1. All proteins were stored at −80 °C.
In vitro tubulin binding
Binding affinity with tubulin was analyzed using surface plasmon resonance (SPR) technology in a Reichert 4SPR system (Reichert Technologies, Depew, NY) equipped with a SPR sensor chip HC 1500 M. First, 50 μg mL−1 tubulin (Cytoskeleton, T-238P) was immobilized to the sensor chip surface to attain 10, 000 RU. One of the four flow channel on the chip was left free as a blank control. ELR510444, parbendazole or colchicine at different concentrations was injected over the sensor chip surface for association analysis, followed by dissociation analysis. The experiment data were obtained at 25 °C with running buffer PBST (8 mM Na2HPO4, 136 mM NaCl, 2 mM KH2PO4, 2.6 mM KCl, and 0.05% (v/v) Tween 20, pH 7.4). The equilibrium dissociation constant (KD) was calculated by a steady-state fitting mode with TraceDrawer software.
Crystallization and crystal soaking
Crystals of T2R-TTL complex (α/β tubulin, RB3-SLD, TTL) were cultivated by the sitting drop vapor diffusion method at 20 °C; details of the process have been reported previously.19 Briefly, the protein complex (α/β tubulin, RB3, and TTL at a 2:1.3:1.2 molar ratio) was incubated on ice with 1 mM AMPPCP, 5 mM tyrosine, and 10 mM DTT for 30 min, and the complex was then concentrated to 20 mg mL−1 at 4 °C. A drop of solution, consisting of 1 μL of the protein complex and 1 μL of crystallization buffer (6% PEG 4000, 5% glycerol, 100 mM MES pH 6.7, 30 mM CaCl2, 30 mM MgCl2), was placed in each well. Seeding was performed to improve the quality of crystals. The first crystals appeared after 2 days, and reached their final size within 3–5 days. Different concentrations of ELR510444 and parbendazole were dissolved in 10 mM DMSO, and the crystals were soaked with each compound (0.1 μL) at 20 °C for 24 h. Crystals with a good shape were picked and snap-frozen in liquid nitrogen with a cryoprotectant (100 mM MES pH 6.7, 30 mM CaCl2, 30 mM MgCl2, 20% glycerine).
Data collection and structure determination
The crystals of the T2R-TTL-ELR510444/parbendazole complexes were mounted in nylon loops and flash-cooled in a cold nitrogen stream at 100 K. Diffraction data were collected at the Shanghai Synchrotron Light Source (Shanghai, China); the data were indexed, integrated, and scaled using HKL2000.20 Structures were solved by molecular replacement with Phaser21 using the T2R-TTL structure (PDB ID: 4I55) as the template. The initial models were refined by alternating cycles of automatic refinement with Refmac5 (ref. 22) in the CCP4 program suite and manual model building with Coot.23 The quality of the model was checked with the PROCHECK program, and showed good stereochemistry according to the Ramachandran plot.24 PYMOL (http://www.pymol.org) was used to generate the figures.
Molecular docking
The receptor was extracted from the solved structure of the ELR510444–tubulin complex. The macromolecule and 24 inhibitors were then prepared using AutoDockTools.25 Each inhibitor was docked into the CBS by AutoDock Vina.26 The docking search space was a rectangular parallelepiped, 16 × 16 × 20 points in size, with a spacing of 1 Å. The exhaustiveness parameter was set to 12.
Results and discussion
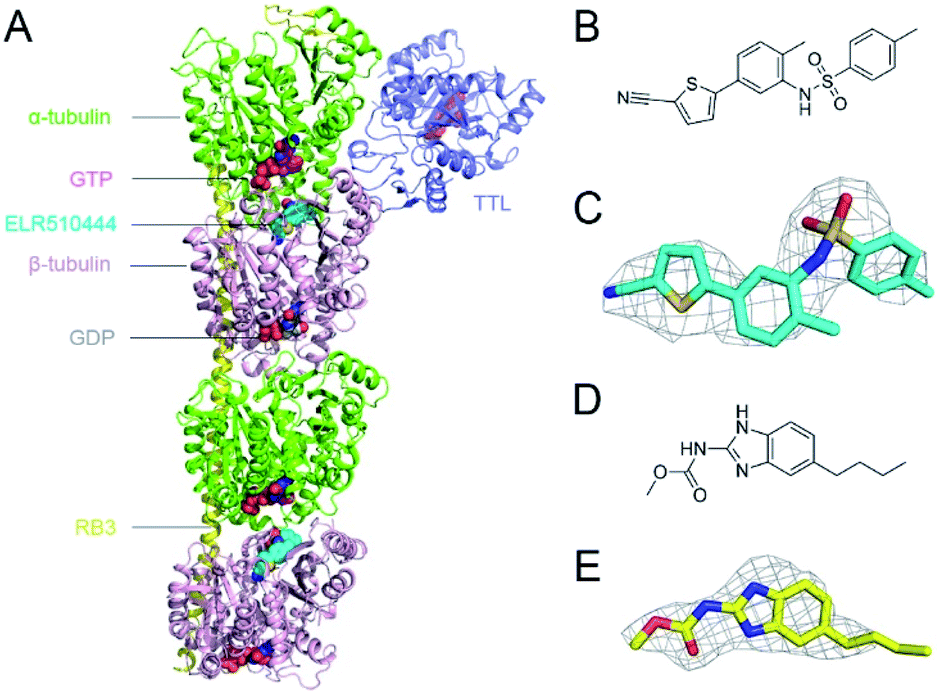
In this study, we obtained X-ray crystal structures of ELR510444 or parbendazole binding to tubulin. ELR510444 or parbendazole was soaked in a protein complex composed of α/β-tubulin, RB3-SLD, and TTL. We determined the complex structure at a resolution of 3.1 Å for ELR510444 (Fig. 1A) and 2.4 Å for parbendazole. Details of the data collection and refinement statistics are summarized in Table 1. The structures of ELR510444 and parbendazole are shown in Fig. 1B and D, respectively. The two inhibitors were well defined by electron density, allowing us to analyze the molecular interactions unambiguously (Fig. 1C and E).
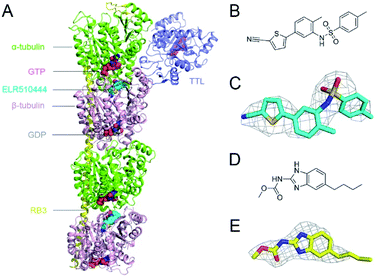 |
| | Fig. 1 Overview of the T2R-TTL-ligand complex structure and the chemical structures of the bound ligands. (A) Overall structure of the tubulin–ELR510444 complex. RB3-SLD is shown in yellow, TTL in blue, α-tubulin in green, β-tubulin in pink, GTP in red, GDP in grey, and ELR510444 in cyan. GTP, GDP, and ELR510444 are shown as spheres. (B) Chemical structure of ELR510444. (C) Experimental electron density map (Fo–Fc) contoured at 2.0 σ around ELR510444. (D) Chemical structure of parbendazole. (E) The electron densities of parbendazole. The Fo–Fc omit map is shown in grey and contoured at 1.5 σ. | |
Table 1 Data collection and refinement statistics for X-ray structuresa
| Parameters |
Tubulin/ELR510444 |
Tubulin/parbendazole |
| Data for the highest resolution shell are shown in parentheses. |
| PDB ID |
7DBD |
7DBC |
| Data collection |
| Space group |
P212121 |
P212121 |
|
| Cell dimension |
| a, b, c (Å) |
104.6 |
105.4 |
| 158.3 |
158.3 |
| 182.3 |
180.8 |
| α, β, γ (°) |
90 |
90 |
| 90 |
90 |
| 90 |
90 |
| Resolution (Å) |
49.87–3.1 (3.2–3.1) |
19.91–2.4 (2.49–2.4) |
| Rmerge |
0.303 (1.0) |
0.104 (0.772) |
| I/sigma |
8.7 (1.8) |
10.3 (2.2) |
| Completeness (%) |
100 (100) |
99.8 (100) |
| Redundancy |
12.8 (12.5) |
6.8 (6.7) |
|
| Refinement |
| No. of unique |
54906 |
118342 |
| Reflections Rwork/Rfree (%) |
19.10/24.40 |
19.79/23.37 |
| No. of atoms |
17563 |
17950 |
| Protein |
17332 |
17327 |
| Ligand |
231 |
192 |
| Water |
— |
431 |
|
| B-Factors (Å) |
| Protein |
62.90 |
56.56 |
| Ligand |
63.42 |
54.64 |
| Water |
— |
49.23 |
| RMS bond length (Å) |
0.01 |
0.01 |
| RMS bond angle (°) |
1.30 |
1.06 |
|
| Ramachandran plot statistics |
| Most favorable (%) |
94.41 |
96.95 |
| Allowed (%) |
5.59 |
3.05 |
| Outliers (%) |
0 |
0 |
Structure of ELR510444–tubulin complex
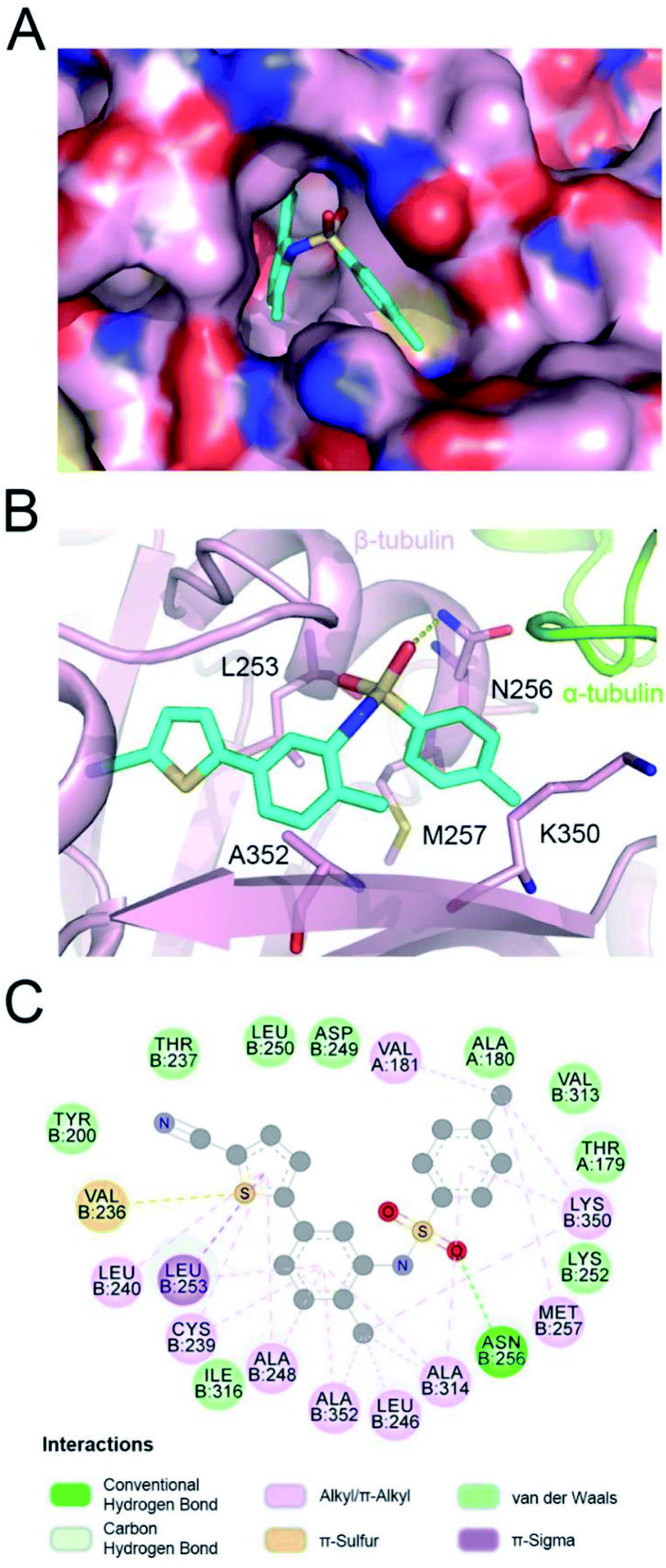
In the tubulin–ELR510444 crystal complex, ELR510444 occupied the CBS (Fig. 2A). As shown in Fig. 2A and B, the p-toluene sulfonamide group of ELR510444 was buried in a channel surrounded by the N256, M257, and K350 residues of β-tubulin. The thiophene-2-carbonitrile moiety was inserted deeply into the colchicine binding cavity. The sulfamide oxygen formed a hydrogen bond with the side chain of N256. As shown in Fig. 2C, there were extensive hydrophobic interactions between ELR510444 and β-tubulin, including at residues C239, L240, L246, A248, L253, A314, K350, and A352.
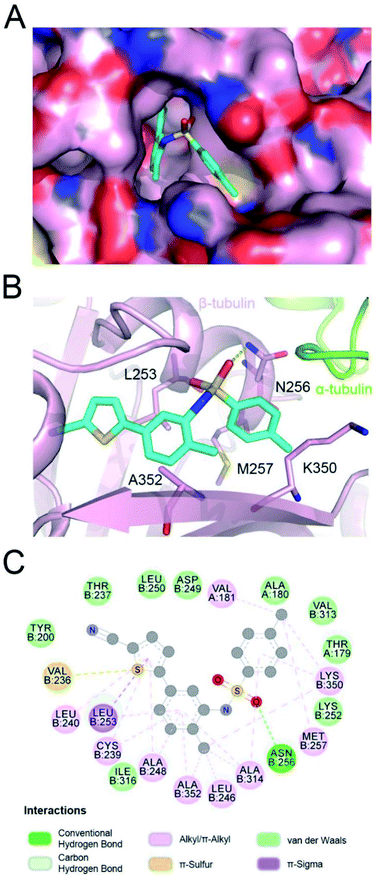 |
| | Fig. 2 Detailed interactions of ELR510444 with tubulin. Surface view of the ELR510444–tubulin complex (A) and intermolecular interactions of the binding (B). (C) Two-dimensional interaction map of the ELR510444 complex with β-tubulin. | |
Structure of parbendazole–tubulin complex
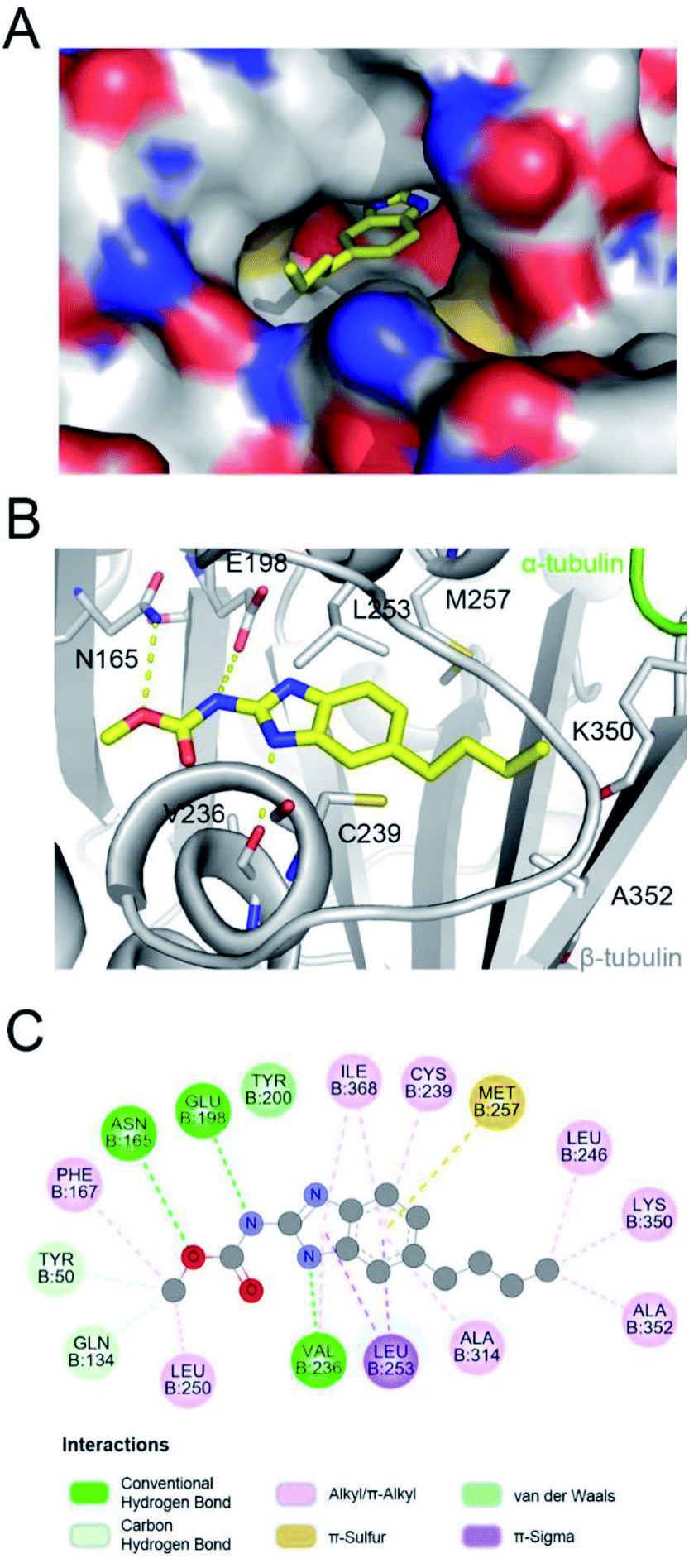
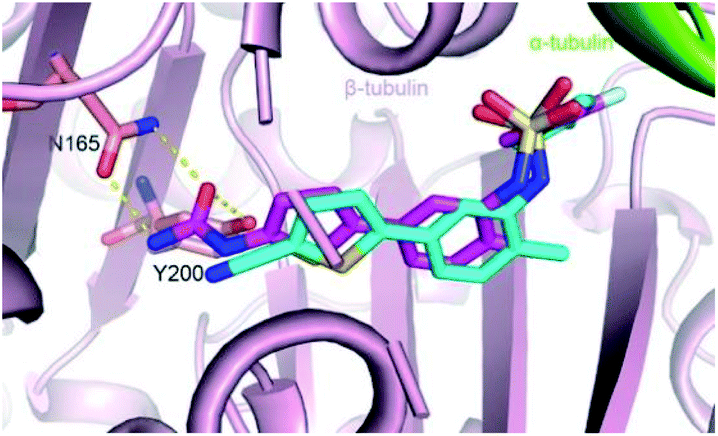
Parbendazole inserted straight and deeply into the CBS (Fig. 3A). The carbamate group was located at the bottom of the cavity and formed hydrogen bonds with the side chains of N165 and E198. The imidazole formed an additional hydrogen bond with the backbone carbonyl of the V236 residue. The butylbenzene moiety of parbendazole occupied a hydrophobic cavity formed by C239, L246, L253, M257, A314, K350, A352, and I368 (Fig. 3B and C). However, the hydrophobic channel surrounded by the N256, M257, and K350 residues remained unoccupied.
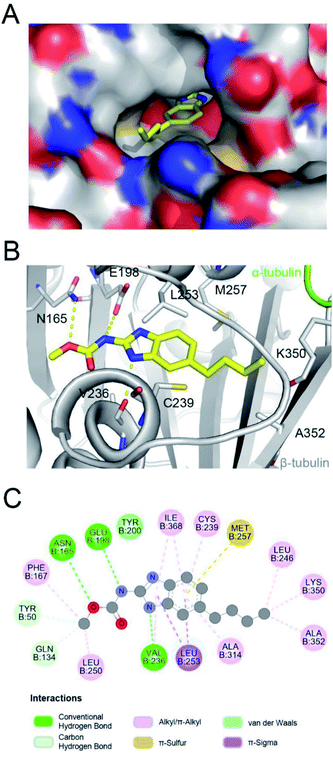 |
| | Fig. 3 Detailed interactions of parbendazole with tubulin. Surface view of the parbendazole–tubulin complex (A) and intermolecular interactions of the binding (B). (C) Two-dimensional interaction map of parbendazole complexes with β-tubulin. | |
Comparison of ELR510444, parbendazole, and colchicine binding
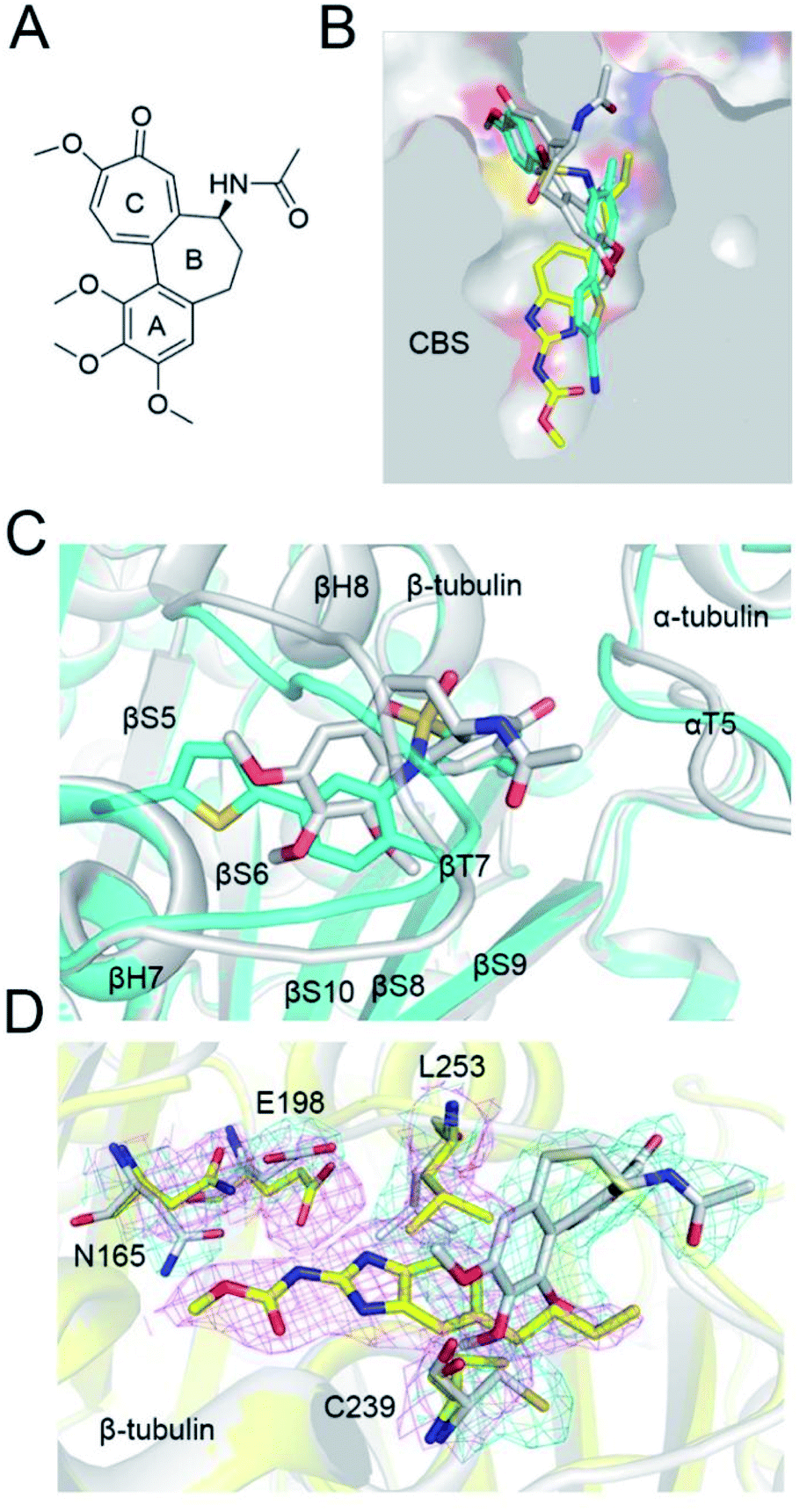
The colchicine site is a large pocket surrounded by two α-helices (H7 and H8) and strands of the two tubulin β-sheets (S1–S4–S5–S6 and S7–S10–S8–S9), which are from the β subunit and capped by two loops (βT7 and αT5). To compare the tubulin binding mode of ELR510444 and parbendazole with that of colchicine, we superimposed the two tubulin–CBSI structures onto the tubulin–colchicine structure (PDB ID: 4O2B), revealing that the toluene ring of ELR510444 superimposed well with the C-ring of colchicine. ELR510444 binds a little more deeply with β-tubulin than colchicine (Fig. 4B). The conformations of the two capped loops βT7 and αT5 differ significantly, indicating the flexibility required to accommodate the diverse structures of the CBSIs (Fig. 4C). Nevertheless, parbendazole is buried more deeply in β-tubulin (Fig. 5B). Unlike colchicine, parbendazole has no interaction with α-tubulin. Four residues (N165, E198, C239, L253) adopted very different conformations after parbendazole or colchicine binding (Fig. 4D). Interestingly, the binding affinity of the 3 CBSIs to β-tubulin seems to be correlated with the inserting depth in the CBS. The deeper inserted, the tighter binding to tubulin. Three hydrogen bonds and hydrophobic interactions also probably contribute to the higher binding affinity of parbendazole (KD = 3.39 μM) than that of ELR510444 (KD = 22.6 μM) and colchicine (KD = 28.1 μM) (ESI Fig. S1†).
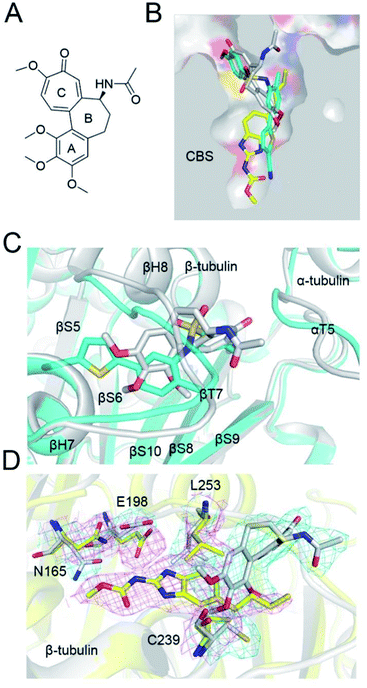 |
| | Fig. 4 Comparison of the binding modes of ELR510444, parbendazole, and colchicine. (A) Chemical structure of colchicine. (B) Superimposition of β-tubulin binding with ELR510444 (cyan sticks), parbendazole (yellow sticks), and colchicine (grey sticks) in CBS. (C) Conformation of the αT5 and βT7 loops differs significantly when ELR510444 or colchicine is bound. (D) Superimposition of parbendazole and colchicine. Residues with significant differences in conformation are highlighted with 2Fo–Fc map contours. | |
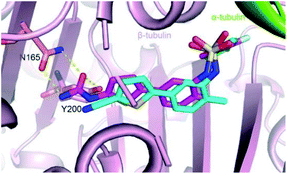 |
| | Fig. 5 Alignment of ELR510444 and C22. ELR510444 (cyan sticks) was copied from the crystal structure, whereas C22 (magenta sticks) was docked into the CBS by Autodock Vina. Interactions between C22 and β-tubulin differing from that of ELR510444 are highlighted by yellow dashed lines. | |
Insights into drug design and molecular docking
CBSIs are a class of structurally diverse agents. ELR510444 and parbendazole are CBSIs, and both showed inhibition of microtubule assembly. The binding modes identified in this work should allow subsequent structural optimization that will improve the potency of inhibition and reduce side effects. ELR510444 and parbendazole were located at the CBS. ELR510444 was bound a little deeper within the CBS than colchicine and made hydrophobic contacts with Val181 of α-tubulin. Parbendazole was located deep within the β subunit and did not interact with the α subunit. Consistent with our previous study,18 the two loops (T5 of the α subunit and T7 of the β subunit) capped the colchicine domain and adjusted their conformations to accommodate the diverse structures of the CBSIs (Fig. 4B).
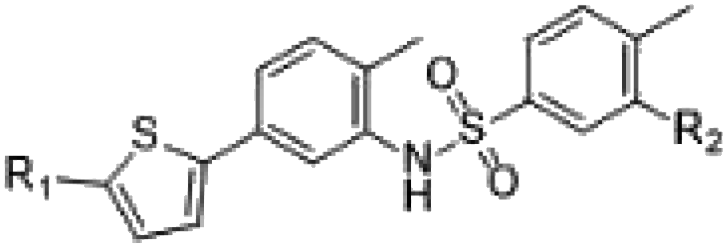
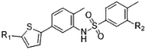
ELR510444 almost fully occupied the CBS, except the bottom of the pocket. A cyano group was located at the bottom of the binding pocket. Here, the cyano group could be extended to interact with Asn165. Since computer-aided drug design plays an important role in drug discovery,27 we also conducted molecular docking experiments on this tubulin complex. A series of ELR510444 derivatives with different substituents was designed and docked into the CBS by Autodock Vina.25 These compounds and their predicted binding affinities are listed in Table 2. ELR510444 had a strong binding affinity (−11.1 kcal mol−1). Compounds with substituents such as alkynyl (C7), formamido (C14), and urea (C15) groups at the R1 position had stronger binding affinities than ELR510444, whereas a hydrogen at the R2 position was preferentially displaced by fluorine (C19). With both the best predicted R1 and R2 substituents, compound C22 exhibited superior binding affinity (−11.7 kcal mol−1) compared with other ELR510444 derivatives. In addition, the predicted binding pose of C22 almost overlapped with ELR510444 (Fig. 5), indicating the reliability of the docking results.
Table 2 Predicted binding affinities of designed compounds
|
| Compound |
R1 |
R2 |
Affinity (kcal mol−1) |
| ELR510444 |
CN |
H |
−11.1 |
| C1 |
CF2 |
H |
−11.0 |
| C2 |
CF3 |
H |
−11.2 |
| C3 |
CH2CH3 |
H |
−10.5 |
| C4 |
CH2OH |
H |
−10.6 |
| C5 |
CH2NHCH3 |
H |
−10.1 |
| C6 |
CONHCH3 |
H |
−11.0 |
| C7 |
CCH |
H |
−11.3 |
| C8 |
CCCH3 |
H |
−11.2 |
| C9 |
CHCH2 |
H |
−10.6 |
| C10 |
CH(CH2)2 |
H |
−11.0 |
| C11 |
CH2CONH2 |
H |
−11.2 |
| C12 |
OCONH2 |
H |
−11.0 |
| C13 |
NHCOH |
H |
−10.6 |
| C14 |
NHCOCH3 |
H |
−11.3 |
| C15 |
NHCONH2 |
H |
−11.5 |
| C16 |
NHCOOCH3 |
H |
−10.9 |
| C17 |
NHSO2CH3 |
H |
−10.4 |
| C18 |
SO2NH2 |
H |
−10.5 |
| C19 |
CN |
F |
−11.2 |
| C20 |
CN |
Cl |
−10.5 |
| C21 |
CN |
CH3 |
−10.9 |
| C22 |
NHCONH2 |
F |
−11.7 |
Parbendazole was the most effective among the series of benzimidazole derivatives, including nocodazole, fenbendazole, mebendazole, and oxibendazole,17 indicating that the n-5-butyl group was preferred to the 2-acetylthiophene, phenyl, and propyl groups. Nocodazole and parbendazole have a same benzimidazole core with different hydrophobic substituents on the phenyl ring. It is a thiophene-2-carbonyl group for nocodazole while an n-butyl for parbendazole. Both two groups laid in a hydrophobic cavity surrounded by L246, L253, A314, I316, A352. n-Butyl is more hydrophobic than thiophene-2-carbonyl and be preferred in the cavity, thus explaining the reason of superior inhibitory effect of parbendazole.
Conclusions
We reported the X-ray structure of tubulin in complex with ELR510444 or parbendazole, and it showed that ELR510444 and parbendazole occupied the colchicine binding pocket. The structures represented the key interactions between tubulin and ligands, and the high-resolution structure of the tubulin–parbendazole complex facilitated the understanding of previous SARs of benzimidazoles. Structural alignment and molecular docking studies were used to achieve the theoretical structural optimization of ELR510444 and parbendazole. The docking results indicated that substitutions on the thienyl (R1) or phenyl (R2) groups of ELR510444 were compatible with binding to tubulin. Collectively, our results provide a solid foundation for the development of novel benzsulfamide and benzimidazole derivatives targeting the colchicine binding site, which will support further structural optimization.
Funding
This work was supported by National Natural Science Foundation of China (82073318, 81703553), National Major Scientific and Technological Special Project for ‘Significant New Drugs Development’ (2018ZX09201018-021), Sichuan Science and Technology Program (2019YFS0003), China Postdoctoral Science Foundation (2018T110984, 2017M610607), and Post-Doctoral Research Project, West China Hospital, Sichuan University (2018HXBH057).
Data availability
The atomic coordinates and structure factors for tubulin–CBSI complexes have been deposited in the Protein Data Bank under accession codes 7DBD (tubulin–ELR510444) and 7DBC (tubulin–parbendazole).
Author contributions
Conceptualization: Yu-Xi Wang; data curation: Jing-Hong Xian and Hai Chen; formal analysis: Yu-Yan Li; funding acquisition: Yu-Xi Wang; investigation: Ling-Ling Ma and Jia-Hong Lei; methodology: Jia-Hong Lei, Ling-Ling Ma and Jian-Jian Zhou; software: Qian Lei and Hai Chen; writing – original draft: Jia-Hong Lei; writing – review & editing: Hao Chen, Yan-Yan Wang, and Yu-Xi Wang. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the staff from the BL17U1, BL18U1, BL19U1 beamlines of the National Facility for Protein Science Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility for assistance with data collection.
Notes and references
- E. Nogales, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 397–420 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS PubMed.
- E. C. McLoughlin and N. M. O’Boyle, Pharmaceuticals, 2020, 13, 8 CrossRef CAS PubMed.
- M. O. Steinmetz and A. E. Prota, Trends Cell Biol., 2018, 28, 776–792 CrossRef CAS PubMed.
- W. Li, H. Sun, S. Xu, Z. Zhu and J. Xu, Future Med. Chem., 2017, 9, 1765–1794 CrossRef CAS PubMed.
- X. Wu, Q. Wang and W. Li, Anti-Cancer Agents Med. Chem., 2016, 16, 1325–1338 CrossRef CAS PubMed.
- M. J. Perez-Perez, E. M. Priego, O. Bueno, M. S. Martins, M. D. Canela and S. Liekens, J. Med. Chem., 2016, 59, 8685–8711 CrossRef CAS PubMed.
- Y. Lu, J. Chen, M. Xiao, W. Li and D. D. Miller, Pharm. Res., 2012, 29, 2943–2971 CrossRef CAS PubMed.
- T. Fojo and M. Menefee, Ann. Oncol., 2007, 18(suppl. 5), v3–8 CrossRef PubMed.
- P. Seve and C. Dumontet, Lancet Oncol., 2008, 9, 168–175 CrossRef CAS PubMed.
- K. E. Arnst, Y. Wang, D. J. Hwang, Y. Xue, T. Costello, D. Hamilton, Q. Chen, J. Yang, F. Park, J. T. Dalton, D. D. Miller and W. Li, Cancer Res., 2018, 78, 265–277 CrossRef CAS PubMed.
- C. Stengel, S. P. Newman, M. P. Leese, B. V. Potter, M. J. Reed and A. Purohit, Br. J. Cancer, 2010, 102, 316–324 CrossRef CAS PubMed.
- A. L. Risinger, C. D. Westbrook, A. Encinas, M. Mulbaier, C. M. Schultes, S. Wawro, J. D. Lewis, B. Janssen, F. J. Giles and S. L. Mooberry, J. Pharmacol. Exp. Ther., 2011, 336, 652–660 CrossRef CAS PubMed.
- J. S. Carew, J. A. Esquivel III, C. M. Espitia, C. M. Schultes, M. Mulbaier, J. D. Lewis, B. Janssen, F. J. Giles and S. T. Nawrocki, PLoS One, 2012, 7, e31120 CrossRef CAS PubMed.
- C. M. Ireland, K. Gull, W. E. Gutteridge and C. I. Pogson, Biochem. Pharmacol., 1979, 28, 2680–2682 CrossRef CAS PubMed.
- J. C. Havercroft, R. A. Quinlan and K. Gull, J. Cell Sci., 1981, 49, 195–204 CrossRef CAS PubMed.
- R. Florio, S. Veschi, V. di Giacomo, S. Pagotto, S. Carradori, F. Verginelli, R. Cirilli, A. Casulli, A. Grassadonia, N. Tinari, A. Cataldi, R. Amoroso, A. Cama and L. De Lellis, Cancers, 2019, 11, 2042 CrossRef CAS PubMed.
- Y. Wang, H. Zhang, B. Gigant, Y. Yu, Y. Wu, X. Chen, Q. Lai, Z. Yang, Q. Chen and J. Yang, FEBS J., 2016, 283, 102–111 CrossRef CAS PubMed.
- J. Yang, Y. Wang, T. Wang, J. Jiang, C. H. Botting, H. Liu, Q. Chen, J. Yang, J. H. Naismith, X. Zhu and L. Chen, Nat. Commun., 2016, 7, 12103 CrossRef CAS PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef PubMed.
- R. A. Laskowski, J. A. Rullmann, M. W. MacArthur, R. Kaptein and J. M. Thornton, J. Biomol. NMR, 1996, 8, 477–486 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- X. Chang, D. Sun, D. Shi, G. Wang, Y. Chen, K. Zhang, H. Tan, J. Liu, B. Liu and L. Ouyang, Acta Pharm. Sin. B, 2021, 11, 156–180 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01173a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ling-Ling Ma‡b,
Jing-Hong Xianc,
Hai Chenb,
Jian-Jian Zhoud,
Hao Chene,
Qian Leib,
Yu-Yan Liab,
Yan-Yan Wang*f and
Yu-Xi Wang
a,
Ling-Ling Ma‡b,
Jing-Hong Xianc,
Hai Chenb,
Jian-Jian Zhoud,
Hao Chene,
Qian Leib,
Yu-Yan Liab,
Yan-Yan Wang*f and
Yu-Xi Wang