
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical scale up synthesis of carboxylic acids and their bioisosteres 5-substituted-1H-tetrazoles catalyzed by a graphene oxide-based solid acid carbocatalyst†
Rupali Mittal a,
Amit Kumarb and
Satish Kumar Awasthi*a
a,
Amit Kumarb and
Satish Kumar Awasthi*a
aChemical Biology Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: satishpna@gmail.com
bDepartment of Chemistry, Jamia Millia Islamia, Jamia Nagar, New Delhi-110025, India
First published on 17th March 2021
Abstract
Herein, catalytic application of a metal-free sulfonic acid functionalized reduced graphene oxide (SA-rGO) material is reported for the synthesis of both carboxylic acids and their bioisosteres, 5-substituted-1H-tetrazoles. SA-rGO as a catalytic material incorporates the intriguing properties of graphene oxide material with additional benefits of highly acidic sites due to sulfonic acid groups. The oxidation of aldehydes to carboxylic acids could be efficiently achieved using H2O2 as a green oxidant with high TOF values (9.06–9.89 h−1). The 5-substituted-1H-tetrazoles could also be effectively synthesized with high TOF values (12.08–16.96 h−1). The synthesis of 5-substituted-1H-tetrazoles was corroborated by single crystal X-ray analysis and computational calculations of the proposed reaction mechanism which correlated well with experimental findings. Both of the reactions could be performed efficiently at gram scale (10 g) using the SA-rGO catalyst. SA-rGO displays eminent reusability up to eight runs without significant decrease in its productivity. Thus, these features make SA-rGO riveting from an industrial perspective.
1. Introduction
Oxidation reactions are one of the most fundamental areas of research in chemistry because of their persistent utilization in the chemical and pharmaceutical industries as well as in academic research.1 Among these, oxidation of aldehydes to carboxylic acids is highly desirable because of the common occurrence of the carboxylic acid moiety in the manufacturing of fine chemicals, drugs, soaps and detergents, cosmetics and protective coatings.2 In the previously available literature, the oxidation of aldehydes to carboxylic acids was either facilitated by using stoichiometric quantities of oxidants like potassium permanganate, chromium trioxide, periodate reagent, etc.3–5 or by a metal containing catalyst (Ag, Rh, Cu and Fe) in the presence of a ligand or a base.6–9 Thus, looking at the setbacks related to the utilization of hazardous oxidants, toxic metal catalysts, ligands, etc. in the previous reports, it is crucial to develop a sustainable, cost-effective and metal-free catalytic approach for the oxidation of aldehydes to carboxylic acids.Tetrazoles are a privileged class of five-membered synthetic heterocyclic rings composed of one carbon and four nitrogen atoms. The interesting biological characteristics of these nitrogen abundant heterocycles have triggered immense research, specifically 5-substituted-1H-tetrazoles in medicinal chemistry because they can be used as bioisosteric replacement of carboxylic acids.10 The presence of multiple nitrogen atoms in their structure renders them pharmacophore property. The drugs containing tetrazole ring belong to antibacterial,11 antifungal,12 anticancer,13 antitubercular and antimalarial category.14 Several commercial drugs like losartan, valsartan, cefazolin, azosemide, etc. contain tetrazole moiety.15 Over the years, several attempts have been made for the preparation of 5-substituted-1H-tetrazoles via [3 + 2] cycloaddition reaction between nitriles and sodium azide using diversified catalysts15 like Lewis acids,16,17 inorganic salts,18–20 etc. but regardless of the efficiency, their homogeneous nature posed difficulty in separation, recovery and reusability. Thereafter, to circumvent these issues, heterogeneous catalytic systems emerged but they have been mostly performed using metal-based catalysts which have drawbacks like metal toxicity and leaching of metal into the reaction medium.15,21–26 The reports of metal-free catalysis in this field are scarce and need attention for the development of sustainable and cost-effective methods.
Solid acid catalysts have emerged as viable alternatives to the conventional liquid acid catalysts that are susceptible to corrosion, toxicity, non-separability from the reaction mixture and the production of harmful wastes post reaction which require additional neutralization step.27 The development of solid acid catalysts by regulating the surface characteristics of a support material with acid functionalities results in improved catalytic performance due to the inherit attributes of the support material in addition to the grafted acidic groups. Graphene oxide is an attractive candidate as a support material because of its fascinating properties like hydrophilicity, large specific surface area, high thermal and mechanical stability, moisture insensitivity, easy and abundant availability and cost-effectiveness.28–30
In the current work, we report sulfonic acid functionalized reduced graphene oxide (SA-rGO) as a low-cost and efficient metal-free solid acid carbocatalyst for the oxidation of aldehydes to carboxylic acids using hydrogen peroxide as the green oxidant and for the synthesis of 5-substituted-1H-tetrazoles via [3 + 2] cycloaddition reaction between nitriles and inorganic sodium azide. The fascinating application of a common catalyst (SA-rGO) for the synthesis of carboxylic acid derivatives and their bioisosteres 5-substituted-1H-tetrazoles is unprecedented.
2. Experimental section
2.1. Preparation of SA-rGO
Firstly, graphene oxide (GO-H) was prepared using Hummers method.31 Thereafter, SA-rGO was prepared with modifications in the previously reported methods.30,32 Briefly, the GO-H powder was ultrasonicated in double distilled water for 30 min to achieve a dispersion of 0.5 mg mL−1. To 500 mL of this dispersion, 2.5 g of L-ascorbic acid was added under vigorous stirring. The reaction mixture was stirred at room temperature (∼35 °C) for 12 h. The reduced product was separated through centrifugation and washed thrice with double distilled water. Thereafter, 0.5 mg mL−1 dispersion of this reduced graphene oxide was prepared in double distilled water through ultrasonication for 30 min. A separately prepared diazonium salt solution of sulfanilic acid (ESI†) was directly added to this dispersion at 0 °C and stirred for 1 h. The solution was then stirred at room temperature for 18 h. The resulting black solution was centrifuged at 7000 rpm for 10 min, washed three times with double distilled water and finally with absolute ethanol. The precipitate was dried in an air oven at 80 °C for 4 h and coded as SA-rGO.2.2. General procedure for the synthesis of carboxylic acids
In a 10 mL RB flask, SA-rGO (10 mg) was added into the reaction mixture containing aldehyde (1 mmol), 30% H2O2 (1 equiv.) and H2O (2 mL). The reaction mixture was stirred for 6 h at 100 °C. After the completion of the reaction as indicated by TLC, the reaction mixture was allowed to cool to RT and vacuum filtered to separate out SA-rGO. Then, the obtained filtrate was washed with Na2CO3 solution and extracted with dichloromethane. The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo to yield the product.2.3. General procedure for the synthesis of 5-substituted-1H-tetrazoles
In a 10 mL RB flask, SA-rGO (10 mg) was added into the reaction mixture containing nitrile (1 mmol) in DMSO (3 mL) and NaN3 (97.5 mg, 1.5 mmol). The reaction mixture was then stirred for the required time (Table 2) at 120 °C. After the completion of the reaction as indicated by TLC, the reaction mixture was allowed to cool to RT and vacuum filtered to separate out SA-rGO. Then, the pH of the obtained filtrate was adjusted to 2 by addition of 5 N HCl and extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4 and concentrated in vacuo to yield the product.3. Results and discussion
In this report, we have prepared sulfonic acid functionalized reduced graphene oxide (SA-rGO) as a low-cost and efficient metal-free solid acid carbocatalyst. Initially, graphene oxide (GO-H) was prepared from graphite using Hummers method. Then, GO-H was chemically reduced using non-toxic L-ascorbic acid as a mild reducing agent to obtain reduced graphene oxide which was subsequently grafted with sulfonic acid containing aryl radicals prepared from the diazonium salt of sulfanilic acid, to attain SA-rGO. A systematic representation of the preparation of SA-rGO from GO-H is provided in Scheme 1. The graphene oxide acts as an ideal support material to anchor large number of acidic functionalities onto its surface with easy to access active sites.33 In SA-rGO, the presence of highly acidic –SO3H groups on the surface render it with Brønsted acid sites that are capable of furnishing organic transformations.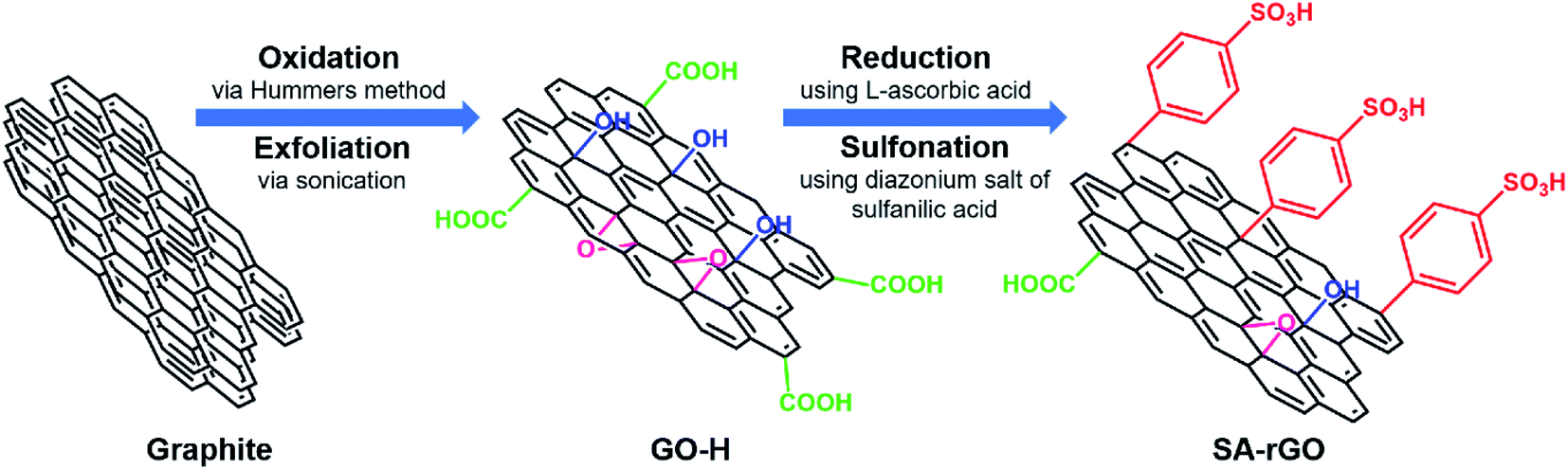 | ||
| Scheme 1 Schematic representation for the preparation of sulfonic acid functionalized reduced graphene oxide (SA-rGO). | ||
The functional group modifications from GO-H to SA-rGO were understood through FTIR spectroscopy (Fig. 1a). The FTIR spectrum of GO-H shows peaks at 3432, 1738, 1642 and 1090 cm−1. These peaks can be ascribed to the existence of O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for carbonyl and carboxylic acid, CC and C–O (epoxy) groups, respectively.31 In GO-H, peak at 1403 cm−1 can be attributed to the O–H bending mode of carboxylic acid. Further, in the SA-rGO spectrum, the severe attenuation of peaks at 1738 cm−1 and 1642 cm−1 after the sulfonation process suggest the partial reduction of GO-H and the partial restoration of the aromatic network of GO-H, respectively. Moreover, the appearance of peaks at 1383 and 1172 cm−1 attributed to SO stretching modes, respectively corroborate the grafting of sulfonic acid groups onto the surface of SA-rGO.30
O for carbonyl and carboxylic acid, CC and C–O (epoxy) groups, respectively.31 In GO-H, peak at 1403 cm−1 can be attributed to the O–H bending mode of carboxylic acid. Further, in the SA-rGO spectrum, the severe attenuation of peaks at 1738 cm−1 and 1642 cm−1 after the sulfonation process suggest the partial reduction of GO-H and the partial restoration of the aromatic network of GO-H, respectively. Moreover, the appearance of peaks at 1383 and 1172 cm−1 attributed to SO stretching modes, respectively corroborate the grafting of sulfonic acid groups onto the surface of SA-rGO.30
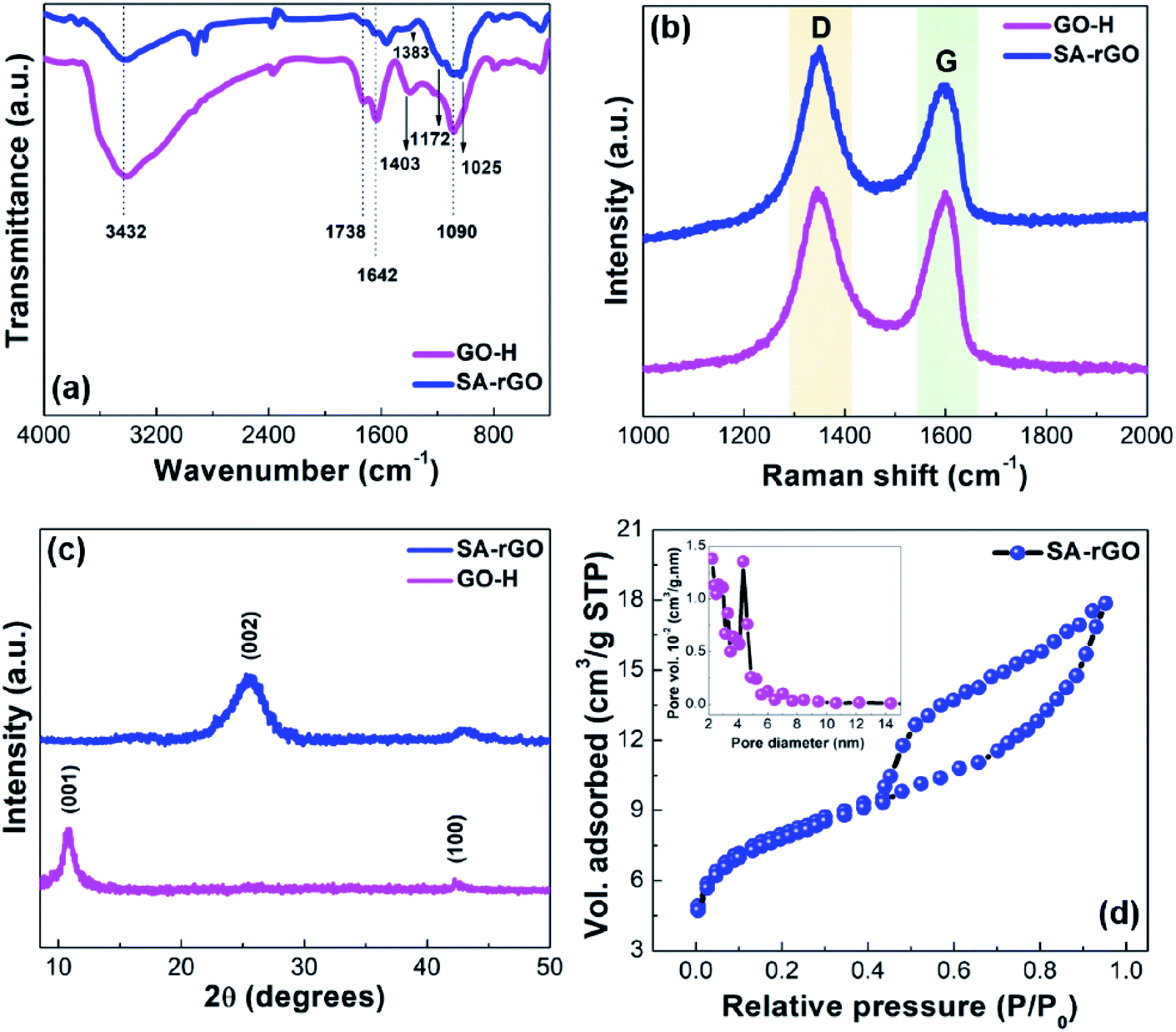 | ||
| Fig. 1 (a) FTIR spectra of GO-H and SA-rGO. (b) Raman spectra of GO-H and SA-rGO. (c) XRD patterns of GO-H and SA-rGO. (d) Nitrogen adsorption–desorption isotherm of SA-rGO. Inset: pore size distribution curve. | ||
The Raman spectra of GO-H and SA-rGO were recorded to understand their lattice structure and distortions (Fig. 1b). The two characteristic peaks around 1345 and 1597 cm−1 for GO-H and 1346 and 1602 cm−1 for SA-rGO corresponding to defects instigated D band and vibration of sp2 carbon atoms in graphitic hexagonal lattices (G band), respectively were observed. The modifications in the structure after the sulfonation process were assessed via the comparison of intensity ratio (ID/IG) values for GO-H and SA-rGO. The ID/IG ratio for SA-rGO (1.17) > GO-H (1.01) due to the introduction of acidic –SO3H groups into the aromatic network. The blue shift of ca. 5 cm−1 in the G band after sulfonation can be imputed to (a) doping which exposed the graphene oxide structure to large number of aryl radicals and (b) alterations in the conjugated double bond system of graphene oxide structure after sulfonation.33
The aqueous dispersions of GO-H and SA-rGO were subjected to UV-visible spectroscopy (Fig. S1†). UV-vis spectrum of GO-H depicts an absorption peak at 232 nm with a shoulder at 304 nm, which can be assigned to π–π* transitions due to CC bonds of the conjugated aromatic network and n–π* transitions due to CO bonds, respectively.31 Further, on partial reduction of GO-H with subsequent grafting of sulfonic acid groups onto its surface as indicated by FTIR and Raman spectroscopy, the absorption peak of GO-H at 232 nm red shifts to 260 nm while the shoulder at 304 nm completely disappears in the UV-vis spectrum of SA-rGO.34
To understand the crystallographic fine structure of GO-H and SA-rGO, their PXRD patterns were recorded (Fig. 1c). For GO-H, the diffraction peak at 2θ = 10.8° (d = 0.81 nm) corresponding to (001) plane of the hexagonal system was observed. This indicates the successful formation of graphene oxide due to the introduction of oxygen-containing functionalities and intercalation of water molecules between stacked sheets. The water molecules trapped within the interlayer spacing are responsible for the exfoliation of graphite oxide into individual graphene oxide sheets upon sonication.35 Moreover, a very low intensity peak at 42.2° (d = 0.21 nm) belongs to the (100) plane consistent with the hexagonal structure of carbon. For SA-rGO, the diffraction peak at 2θ = 10.8° disappeared and a broad peak at 2θ = 25.6° (d = 0.35 nm) indexed to (002) plane of hexagonal structure of carbon was observed as a result of partial reduction of graphene oxide using L-ascorbic acid followed by introduction of sulfonic acid containing aryl radicals, insinuating restacking of sheets through π–π interactions.36
To elucidate the textural and surface properties of SA-rGO, Brunauer–Emmett–Teller (BET) surface analysis was performed (Fig. 1d). The nitrogen adsorption–desorption isotherm of SA-rGO exhibits type IV isotherm with H3 type of hysteresis loop. The BET surface area of SA-rGO was found to be 26 m2 g−1. The inset of Fig. 3b displays the pore size distribution curve ascertained by BJH method, with maximum mesoporous distribution at 4.32 nm pore diameter and 0.014 cm3 g−1 pore volume.
The morphological attributes of GO-H and SA-rGO were studied using electron microscopy. The two-dimensional morphology of GO-H and SA-rGO was studied using TEM analysis. Fig. 2a depicts the TEM image of GO-H which manifests transparent crumpled sheet-like morphology. The TEM image of SA-rGO shows a similar corrugated sheet-like structure but with reduced transparency, suggesting restacking of sheets as also indicated by XRD results (Fig. 2b). Moreover, the SAED pattern of SA-rGO suggests semi-crystalline nature and random stacking of sheets (inset, Fig. 2b).37
 | ||
| Fig. 2 (a and b) TEM images of GO-H and SA-rGO, respectively. Inset of (b) SAED pattern of SA-rGO. (c and d) SEM images of GO-H and SA-rGO, respectively. | ||
Thereafter, to apprehend three-dimensional morphology, SEM analysis of GO-H and SA-rGO was performed (Fig. 2c and d). The SEM images of both GO-H and SA-rGO depict wrinkled sheet-like morphology. Thus, the TEM and SEM analyses of GO-H and SA-rGO depict similar microstructures, implying restoration of morphology after the sulfonation process. Furthermore, SEM-EDS analysis of SA-rGO was carried out to determine the composition of elements in SA-rGO which depicts the stoichiometric ratios of C, O and S elements. The sulfur content was found to be 5.11 wt% in SA-rGO (Fig. S2†).
To determine the elemental composition and concentration of –SO3H groups in SA-rGO, CHNS elemental analysis was performed (Table S1†). The sulfur content (%) was found to be 5.16, using which the concentration of –SO3H groups in SA-rGO was calculated to be 1.60 mmol g−1 (Table S2†). Further, to calculate the total acidic sites present in SA-rGO, back acid–base titration was performed (Section S1†). The total acidic sites in SA-rGO were found to be 2.40 mmol g−1 (Table S2†).
After successful synthesis and characterization of SA-rGO, the catalytic potential of the as-synthesized SA-rGO was assessed for the oxidation of aldehydes to carboxylic acids and for the synthesis of 5-substituted-1H-tetrazoles. Moreover, the catalytic efficacy of SA-rGO was further evaluated in the light of turnover frequency (TOF) values. Benzaldehyde and benzonitrile were selected as the model substrates for the oxidation reaction and synthesis of 5-substituted-1H-tetrazoles, respectively.
As shown in Table 1, the oxidation of benzaldehyde carried out at 1 mmol scale using 30% H2O2 (1.1 equiv.) as the oxidant and H2O (2 mL) as the solvent at 100 °C in the absence of any catalyst did not proceed towards product formation even after 24 h of reaction (entry 1, Table 1). The completion of the reaction was discerned by thin-layer chromatography indicated by the complete consumption of benzaldehyde. Subsequently, the importance of SA-rGO catalyst was explored for the oxidation of benzaldehyde to benzoic acid. The reaction was performed using O2 balloon and 30% H2O2 (1.1 equiv.) as the oxidant in the presence of SA-rGO (5 mg) as the catalyst and H2O (2 mL) as the solvent at 100 °C which produced the desired product in 72% and 88% yield in 16 h and 6 h, respectively (entries 2 and 3, Table 1). Clearly, H2O2 was better oxidant for the present protocol, therefore, the amount of H2O2 oxidant was optimized. The highest yield of the desired product was achieved using 1.1 equiv. of H2O2 (entries 3–5, Table 1). The best solvent for the present protocol was selected by performing the reaction in 1,4-dioxane, ethanol (EtOH) and neat conditions which resulted in 71%, 73% and 79% yield of the desired product, respectively after 8 h of reaction (entries 6–8, Table 1). This displayed that H2O is the optimum solvent for the current protocol. Next, the reaction temperature was optimized by performing the reaction at room temperature and 80 °C which resulted in trace amount and 75% yield of the product, respectively (entries 9 and 10, Table 1). The amount of SA-rGO catalyst required for the reaction was optimized which showed that 10 mg of SA-rGO produces the highest yield (entries 11–13, Table 1). Finally, under the optimized reaction conditions, GO-H was scrutinized as catalyst which could furnish the desired product in only 63% yield after 16 h of the reaction (entry 14, Table 1).
| Entry | Catalyst amount (mg) | Oxidant (equiv.) | Solvent | Temp. (°C) | Timeb (h) | Yieldc (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: benzaldehyde (1 mmol), oxidant (equiv.), solvent (2 mL) and catalyst (mg).b Complete consumption of benzaldehyde.c Isolated yields. N.R. = no reaction. RT = room temperature. | ||||||
| 1 | — | H2O2 (1.1) | H2O | 100 | 24 | N.R. |
| 2 | SA-rGO (5 mg) | O2 balloon | H2O | 100 | 16 | 72 |
| 3 | SA-rGO (5 mg) | H2O2 (1.1) | H2O | 100 | 6 | 88 |
| 4 | SA-rGO (5 mg) | H2O2 (1.0) | H2O | 100 | 7 | 84 |
| 5 | SA-rGO (5 mg) | H2O2 (1.5) | H2O | 100 | 6 | 89 |
| 6 | SA-rGO (5 mg) | H2O2 (1.1) | 1,4-Dioxane | 100 | 8 | 71 |
| 7 | SA-rGO (5 mg) | H2O2 (1.1) | CH3CN | 100 | 8 | 73 |
| 8 | SA-rGO (5 mg) | H2O2 (1.1) | Neat | 100 | 8 | 79 |
| 9 | SA-rGO (5 mg) | H2O2 (1.1) | H2O | RT | 24 | Trace |
| 10 | SA-rGO (5 mg) | H2O2 (1.1) | H2O | 80 | 6 | 75 |
| 11 | SA-rGO (2.5 mg) | H2O2 (1.1) | H2O | 100 | 8 | 80 |
| 12 | SA-rGO (10 mg) | H2O2 (1.1) | H2O | 100 | 6 | 94 |
| 13 | SA-rGO (15 mg) | H2O2 (1.1) | H2O | 100 | 6 | 94 |
| 14 | GO-H (10 mg) | H2O2 (1.1) | H2O | 100 | 16 | 63 |
After achieving the optimal reaction conditions, substrate scope for SA-rGO catalyzed oxidation reaction was evaluated. Table 2 summarizes the substrate scope results displaying good to excellent yields (87–95%) of carboxylic acids. Aromatic aldehydes substituted with electron-withdrawing groups produced higher yields of the corresponding carboxylic acids as compared to those substituted with electron-donating groups. Terephthalaldehyde could also be oxidized to disubstituted terephthalic acid using 2.2 equiv. H2O2 (entry 2 g, Table 2). The aliphatic long chain aldehyde also produced the corresponding carboxylic acid in good yield (entry 2 h, Table 2). Further, the catalytic activity of SA-rGO was assessed by determining the TOF values for the synthesized carboxylic acid derivatives which were found to be between 9.06–9.89 h−1. The details regarding the calculation of TOF values of SA-rGO is given in Section S2.†

Further, the green chemistry metrics for the oxidation reaction of benzaldehyde to benzoic acid 2a were calculated. The calculations tabulated in Table S3† and the results depicted as radar plot in Fig. 3 show the closeness of the calculated values for the current protocol to the ideal values of the green chemistry parameters. This manifests the sustainability of the present method.
 | ||
| Fig. 3 Radar plot depicting Green chemistry metrics for the synthesis of benzoic acid 2a. | ||
To decipher the reaction pathway, oxidation reaction was carried out in the presence of a free radical scavenger, hydroquinone. The addition of hydroquinone to the reaction mixture did not affect the oxidation reaction suggesting that a non-radical pathway is followed. Therefore, in Fig. 4, a plausible mechanism demonstrating the role of SA-rGO for the oxidation of aldehydes to carboxylic acids is depicted using benzaldehyde as the representative substrate. The interaction between SA-rGO and H2O2 leads to the formation of peroxysulfuric acid intermediate (I) which on further reaction with the aldehyde results in the peroxyacetal intermediate (II). Finally, the decomposition of peroxyacetal intermediate (II) generates the carboxylic acid and SA-rGO.
 | ||
| Fig. 4 Plausible mechanism for the oxidation of aldehydes to carboxylic acids using SA-rGO. | ||
As depicted in Table 3, the [3 + 2] cycloaddition reaction between benzonitrile and sodium azide for the preparation of 5-phenyl-1H-tetrazole was performed at 1 mmol scale without any catalyst using DMF and DMSO as solvents which resulted in very low yield (20–22%) of the desired product after 24 h of reaction (entries 1 and 2, Table 3). The completion of the reaction was discerned by thin-layer chromatography indicated by the complete consumption of benzonitrile. Subsequently, the importance of SA-rGO catalyst was explored for the synthesis of 5-phenyl-1H-tetrazole. The efficacy of SA-rGO (5 mg) as a catalyst was evaluated in the presence of various solvents (DMF, DMSO, CH3CN, EtOH and H2O) at varying temperatures (entries 3–7, Table 3). Among these, the highest yield of 5-phenyl-1H-tetrazole (89%) was achieved when DMSO was used as solvent at 120 °C (entry 4, Table 3). Further, the amount of SA-rGO required for the catalysis was optimized. The amount of SA-rGO was varied from 2.5–15 mg in the presence of DMSO as solvent at 120 °C (entries 8–10, Table 3). Thus, the optimized reaction conditions were established to be SA-rGO (10 mg) as catalyst in the presence of DMSO (3 mL) at 120 °C (entry 9, Table 3). Under these conditions, the product 5-phenyl-1H-tetrazole was isolated in 94% yield after 4 h of the reaction. Finally, under the optimized reaction conditions, GO-H was scrutinized as catalyst which could furnish the desired product in only 42% yield after 12 h of the reaction (entry 11, Table 3).
| Entry | Catalyst amount (mg) | Solvent | Temp. (°C) | Timeb (h) | Yieldc (%) |
|---|---|---|---|---|---|
| a Reaction conditions: benzonitrile (1 mmol), NaN3 (1.5 mmol), solvent (3 mL) and catalyst (mg).b Complete consumption of benzonitrile.c Isolated yields. | |||||
| 1 | — | DMF | 140 | 24 | 20 |
| 2 | — | DMSO | 120 | 24 | 22 |
| 3 | SA-rGO (5 mg) | DMF | 140 | 8 | 88 |
| 4 | SA-rGO (5 mg) | DMSO | 120 | 8 | 89 |
| 5 | SA-rGO (5 mg) | CH3CN | 80 | 12 | 38 |
| 6 | SA-rGO (5 mg) | EtOH | 78 | 12 | 51 |
| 7 | SA-rGO (5 mg) | H2O | 90 | 12 | 42 |
| 8 | SA-rGO (2.5 mg) | DMSO | 120 | 12 | 76 |
| 9 | SA-rGO (10 mg) | DMSO | 120 | 4 | 94 |
| 10 | SA-rGO (15 mg) | DMSO | 120 | 4 | 95 |
| 11 | GO-H (10 mg) | DMSO | 120 | 12 | 42 |
After achieving the optimal reaction conditions, substrate scope for SA-rGO catalyzed 5-substituted-1H-tetrazole synthesis was extended to aromatic, heteroaromatic and aliphatic nitriles. Table 4 summarizes the substrate scope results displaying good to excellent yields (82–95%) of 5-substituted-1H-tetrazoles. Aromatic nitriles substituted with electron-withdrawing groups produced higher yields of the corresponding 5-substituted-1H-tetrazoles as compared to those substituted with electron-donating groups. The heteroaromatic and aliphatic nitriles also produced the corresponding 5-substituted-1H-tetrazoles in good yields (entry 5f and 5k, Table 4, respectively). The benzyl carbamate substituted nitrile produced the corresponding 5-substituted-1H-tetrazole in moderate yield (entry 5m, Table 4). Further, the catalytic activity of SA-rGO was assessed by determining the TOF values for the synthesized 5-substituted-1H-tetrazole derivatives which were found to be between 12.08–16.96 h−1. The details regarding the calculation of TOF values of SA-rGO is given in Section S2.†


Further, the current approach for the synthesis of 5-substituted-1H-tetrazoles was authenticated using single crystal X-ray analysis of compound 5k. The X-ray quality crystals were grown in 50% ethyl acetate in hexane using slow evaporation of solution growth method. Fig. 5 depicts the ORTEP diagram of compound 5k which was crystallized in monoclinic cell and P21/n space group having following lattice parameters: a = 4.8058 (5) Å, b = 26.6823 (19) Å, c = 7.1899 (7) Å, α = 90°, β = 106.575° (14), γ = 90°, V = 883.65 (15) Å3 and ρ (calculated) = 1.463 g cm−3. The other single crystal X-ray crystallographic details of compound 5k are given in Table S4.†
 | ||
| Fig. 5 ORTEP diagram of compound 5k. | ||
In Fig. 6, the plausible mechanism demonstrating the role of SA-rGO for the synthesis of 5-substituted-1H-tetrazoles is depicted using benzonitrile as the representative substrate. Initially, the coordination of nitrogen atom of benzonitrile to the sulfonic acid group (acidic sites) of SA-rGO results in step I. The activation of nitrogen atom of benzonitrile by the acidic sites of SA-rGO facilitates the [3 + 2] cycloaddition reaction between the nitrile group (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and the azide ion smoothly, resulting in the step II. Finally, SA-rGO is separated out through vacuum filtration and the pH of the solution is adjusted to 2 to yield 5-phenyl-1H-tetrazole.
N) and the azide ion smoothly, resulting in the step II. Finally, SA-rGO is separated out through vacuum filtration and the pH of the solution is adjusted to 2 to yield 5-phenyl-1H-tetrazole.
 | ||
| Fig. 6 Plausible mechanism for the synthesis of 5-substituted-1H-tetrazoles using SA-rGO. | ||
Further, the computational calculations were performed to get a better insight into the proposed reaction mechanism and to comprehend the importance of SA-rGO catalyst for the synthesis of 5-substituted-1H-tetrazoles. Fig. 7 depicts the energy profile diagram for the reaction between benzonitrile and azide ion via an uncatalyzed (blue line) and SA-rGO catalyzed (pink line) reaction pathway. The computational calculations suggest that both uncatalyzed (blue line) and SA-rGO catalyzed (pink line) reaction pathway involve the addition of azide ion to nitrile group followed by cyclization step which finally results into 5-substituted-1H-tetrazole product. It is also predicted that the formation of 5-substituted-1H-tetrazole occurs through two transition states, viz. TS-1A and TS-2A for uncatalyzed reaction while TS-1B and TS-2B for SA-rGO catalyzed reaction. The energy difference between TS-1A and TS-1B was found to be 7.5 kJ mol−1 while that between TS-2A and TS-2B was found to be 11.7 kJ mol−1. Thus, the computational calculation outlined in Fig. 7 clearly indicates that the formation of 5-substituted-1H-tetrazole is faster for SA-rGO catalyzed reaction pathway compared to that of the uncatalyzed reaction pathway, which is in correlation with our experimental findings. This data is in agreement with the proposed reaction mechanism within the thermodynamic limitations.
 | ||
| Fig. 7 The free energy profiles for the synthesis of 5-substituted-1H-tetrazoles via an uncatalyzed (blue line) and SA-rGO catalyzed (pink line) reaction pathway. | ||
The practicality of the oxidation reaction and 5-substituted-1H-tetrazoles synthesis for the potential industrial application were studied. Keeping the reaction stoichiometry unaltered, gram scale synthesis of benzoic acid and 5-phenyl-1H-tetrazole was carried out at 100 mmol scale (10 g). The results are summarized in Tables S5 and S6† for benzoic acid and 5-phenyl-1H-tetrazole, respectively. The reaction occurred steadily and encouraging yields were obtained. A plot showing the effect of scale (mmol) on the isolated yield (%) of benzoic acid and 5-phenyl-1H-tetrazole and reaction time (h) is represented in Fig. 8a and b, respectively.
 | ||
| Fig. 8 (a) Plot representing the effect of substrate scale (mmol) on isolated yield (%) of benzoic acid 2a and 5-phenyl-1H-tetrazole 5a. (b) Plot representing the effect of substrate scale (mmol) on reaction time (h) for the synthesis of benzoic acid 2a and 5-phenyl-1H-tetrazole 5a. (c) Plot representing the reusability of SA-rGO for the synthesis of benzoic acid 2a and 5-phenyl-1H-tetrazole 5a over eight catalytic cycles. | ||
The prowess of a heterogeneous catalyst to show easy recoverability and effective reusability are necessary for their worthwhile industrial application. In the current study, the recoverability and reusability of SA-rGO was investigated for the synthesis of benzoic acid and 5-phenyl-1H-tetrazole under the optimized reaction conditions. After the completion of each cycle, SA-rGO could be easily recovered by vacuum filtration, washed with ethanol and subsequently dried in an air oven at 70 °C for 3 h which was then reused for the next cycle. SA-rGO was reused for eight consecutive catalytic cycles without significant depreciation in its activity. The isolated yield (%) for the eight catalytic cycles for both the reactions is plotted in Fig. 8c and tabulated in Tables S7 and S8,† respectively.
The stability of SA-rGO reused over eight consecutive catalytic cycles was examined through FTIR, PXRD and Raman studies (Fig. S3a–c†). The peaks in FTIR and Raman spectra match well with that of fresh SA-rGO sample insinuating structural integrity even after eight consecutive catalytic cycles. The resemblance of PXRD pattern with fresh SA-rGO sample implies phase purity. Further, SEM image of the SA-rGO reused over eight cycles was collected which did not display any noteworthy changes indicating morphology preservation (Fig. S3d†).
To rationalize the use of SA-rGO for the synthesis of 5-substituted-1H-tetrazoles, the present work was compared with the available literature. The comprehensive literature survey suggested that the use of SA-rGO as a catalyst for the oxidation reaction and the synthesis of 5-substituted-1H-tetrazoles has several advantages over earlier work (Tables S9 and S10†). The present catalyst was synthesized keeping in mind the low-cost, easy preparation of graphene oxide-based materials and the usefulness of solid acid catalysts. It is evident from Tables S9 and S10,† that SA-rGO produces carboxylic acid derivatives and 5-substituted-1H-tetrazoles in high yields with short reaction time using low amount of catalyst. Specifically, the comparison with previously reported sulfonated carbon analogues also reveal better catalytic output of the current SA-rGO catalyst in terms of better reaction conditions with higher yields in shorter reaction times (entry 3, Table S9 and entry 10, Table S10†). Also, the previous sulfonated carbon analogues were prepared using harmful and highly corrosive concentrated sulfuric acid while SA-rGO was prepared via diazonium salt solution of sulfanilic acid. Moreover, both the reactions in the current work could be performed at gram scale with high efficiency, which makes the current methodology using SA-rGO catalyst interesting from industrial viewpoint.
4. Conclusions
In conclusion, we have successfully developed a methodology using metal-free sulfonic acid functionalized reduced graphene oxide (SA-rGO). SA-rGO was implemented as solid acid carbocatalyst for the oxidation of aldehydes to carboxylic acids and for the synthesis of 5-substituted-1H-tetrazoles. The amalgamation of fascinating properties, easy and low-cost preparation of graphene oxide-based materials with additional benefits of highly acidic sites due to the grafting of sulfonic acid groups was found quite valuable. The oxidation of substituted aromatic and aliphatic aldehydes to their corresponding carboxylic acids could be efficiently achieved using H2O2 as green oxidant and H2O as green solvent with high TOF values (9.06–9.89 h−1). The calculation of their green chemistry metrics displayed an E-factor value of 0.24 which is very close to ideal value. Further, the 5-substituted-1H-tetrazoles could be effectively synthesized from their corresponding substituted aromatic, heteroaromatic and aliphatic nitrile substrates with high TOF values (12.08–16.96 h−1). The current approach for the synthesis of 5-substituted-1H-tetrazoles was corroborated by computational calculations of the proposed reaction mechanism which was found to be in agreement with the experimental findings. Moreover, both the reactions could be performed at gram scale with high efficiency. Thus, low-cost, easy recovery, eminent reusability and ability to perform gram scale reactions makes this metal-free solid acid carbocatalyst riveting for industrial applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
RM is grateful to UGC, New Delhi, India for providing senior research fellowship. SKA acknowledges the financial support from Institution of Eminence, University of Delhi (IoE/FRP/PCMS/2020/27) and University of Delhi, Delhi, India. Authors are sincerely thankful to University Science Instrumentation Centre (USIC) and Department of Chemistry, University of Delhi, Delhi, India for providing instrumentation facilities. We are also grateful to Sophisticated Analytical Instrument Facility (SAIF)-AIIMS, New Delhi, India for providing TEM facility.References
- Y. Zhang, Y. Cheng, H. Cai, S. He, Q. Shan, H. Zhao, Y. Chen and B. Wang, Green Chem., 2017, 19, 5708 RSC.
- L. Sancineto, C. Tidei, L. Bagnoli, F. Marini, E. J. Lenardão and C. Santi, Molecules, 2015, 20, 10496 CrossRef CAS PubMed.
- A. Mahmood, G. E. Robinson and L. Powell, Org. Proc. Res. Dev., 1999, 3, 363 CrossRef CAS.
- J. K. Thottathil, J. L. Moniot, R. H. Mueller, M. K. Y. Wong and T. P. Kissick, J. Org. Chem., 1986, 51, 3140 CrossRef CAS.
- M. Hunsen, Synthesis, 2005, 15, 2487 CrossRef.
- M. Liu, H. Wang, H. Zeng and C.-J. Li, Sci. Adv., 2015, 1, e1500020 CrossRef PubMed.
- X. Wang, C. Wang, Y. Liu and J. Xiao, Green Chem., 2016, 18, 4605 RSC.
- M. Liu and C.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 10806 CrossRef CAS PubMed.
- H. Yu, S. Ru, G. Dai, Y. Zhai, H. Lin, S. Han and Y. Wei, Angew. Chem., Int. Ed., 2017, 56, 3867 CrossRef CAS PubMed.
- H. Singh, A. S. Chawla, V. K. Kapoor, D. Paul and R. K. Malhotra, Prog. Med. Chem., 1980, 17, 151 CrossRef CAS PubMed.
- Y. Méndez, G. D. Armas, I. Pérez, T. Rojas, M. E. Valdés-Tresanco, M. Izquierdo, M. A. Rivero, Y. M. Álvarez-Ginarte, P. A. Valiente, C. Soto, L. León, A. V. Vasco, W. L. Scott, B. Westermann, J. González-Bacerio and D. G. Rivera, Eur. J. Med. Chem., 2019, 163, 481 CrossRef PubMed.
- S.-Q. Wang, Y.-F. Wang and Z. Xu, Eur. J. Med. Chem., 2019, 170, 225 CrossRef CAS PubMed.
- Z. Yan, S. Chong, H. Lin, Q. Yang, X. Wang, W. Zhang, X. Zhang, Z. Zeng and Y. Su, Eur. J. Med. Chem., 2019, 164, 562 CrossRef CAS PubMed.
- C. Gao, L. Chang, Z. Xu, X.-F. Yan, C. Ding, F. Zhao, X. Wu and L.-S. Feng, Eur. J. Med. Chem., 2019, 163, 404 CrossRef CAS PubMed.
- R. Mittal and S. K. Awasthi, Synthesis, 2019, 51, 3765 CrossRef CAS.
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS PubMed.
- S. D. Guggilapu, S. K. Prajapti, A. Nagarsenkar, K. K. Gupta and B. N. Babu, Synlett, 2016, 27, 1241 CrossRef CAS.
- L. Bosch and J. Vilarrasa, Angew. Chem., Int. Ed., 2007, 46, 3926 CrossRef CAS PubMed.
- F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983 CrossRef CAS PubMed.
- P. Mani, A. K. Singh and S. K. Awasthi, Tetrahedron Lett., 2014, 55, 1879 CrossRef CAS.
- P. Mani, C. Sharma, S. Kumar and S. K. Awasthi, J. Mol. Catal. A Chem., 2014, 392, 150 CrossRef CAS.
- S. Kumar, A. Kumar, A. Agarwal and S. K. Awasthi, RSC Adv., 2015, 5, 21651 RSC.
- M. L. Kantam, K. B. S. Kumar and C. Sridhar, Adv. Synth. Catal., 2005, 347, 1212 CrossRef CAS.
- T. Tamoradi, A. Ghorbani-Choghamarani and M. Ghadermazi, New J. Chem., 2017, 41, 11714 RSC.
- D. Khalili and M. Rezaee, Appl. Organomet. Chem., 2019, 33, e5219 CrossRef CAS.
- H. Sharghi, S. Ebrahimpourmoghaddam and M. M. Doroodmand, J. Organomet. Chem., 2013, 738, 41 CrossRef CAS.
- B. Majumdar, S. Mandani, T. Bhattacharya, D. Sarma and T. K. Sarma, J. Org. Chem., 2017, 82, 2097 CrossRef CAS PubMed.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023 RSC.
- A. Bahuguna, A. Kumar and V. Krishnan, Asian J. Org. Chem., 2019, 8, 1263 CrossRef CAS.
- R. Mittal, A. Mishra and S. K. Awasthi, Synthesis, 2020, 52, 591 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed.
- J. Zhang, H. Yang, G. Shen, P. Cheng, J. Zhang and S. Guo, Chem. Commun., 2010, 46, 1112 RSC.
- J. Ji, G. Zhang, H. Chen, S. Wang, G. Zhang, F. Zhang and X. Fan, Chem. Sci., 2011, 2, 484 RSC.
- L. Q. Xu, Y. B. Liao, N. N. Li, Y. J. Li, J. Y. Zhang, Y. B. Wang, X. F. Hu and C. M. Li, J. Colloid Interf. Sci., 2018, 514, 733 CrossRef CAS PubMed.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711 CrossRef CAS PubMed.
- M. Brahmayya, S. A. Dai and S.-Y. Suen, Sci. Rep., 2017, 7, 4675 CrossRef PubMed.
- W. Zhang, W. He and X. Jing, J. Phys. Chem. B, 2010, 114, 10368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1953617. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01053k |
| This journal is © The Royal Society of Chemistry 2021 |