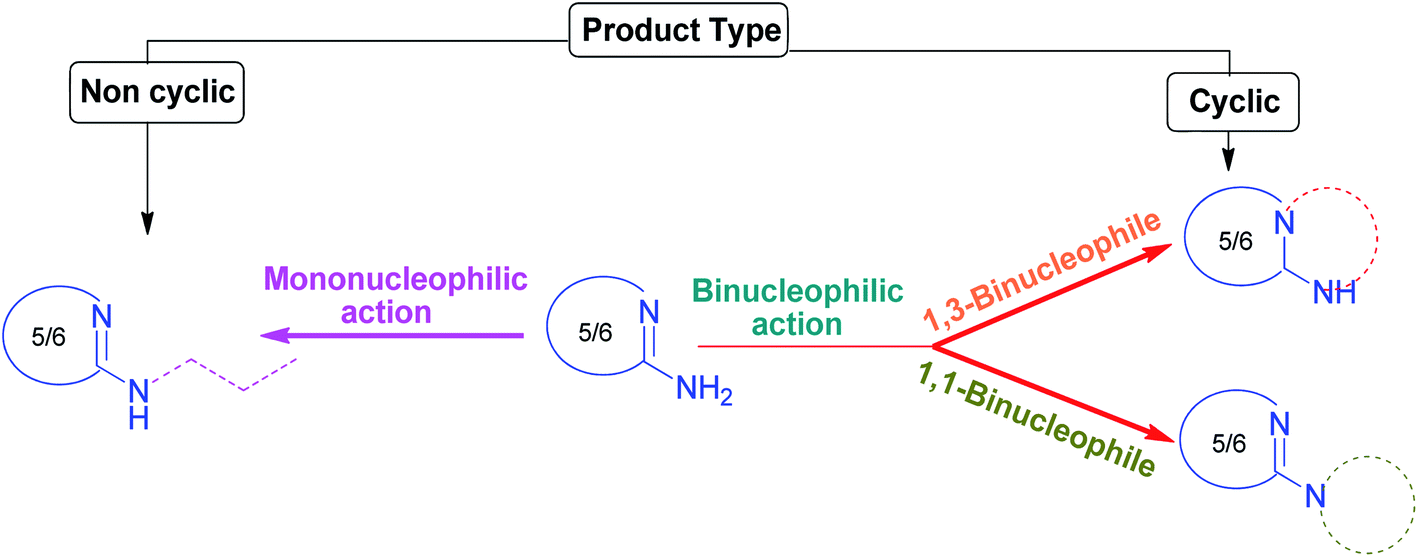
Structurally, these amino heterocycles can be considered as the dinitrogen analogs of carboxylic acids and esters in which the carbonyl, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and hydroxyl, OH, groups are replaced by azomethine, CNR, and amino, NH2, groups, respectively. The cyclic amidine functionality of amino heterocycles is obtained when the substituents of both the carbon and nitrogen atoms of the azomethine group are replaced by the same and only one five- or six-membered cycle in which both atoms are embedded and the primary amino group is flanked outside the ring known as an exocyclic amino group. Therefore, they display rich chemistry by combining the effect of an azomethine-like C–N double bond with an amide-like C–N single bond having some partial double bond character.
O, and hydroxyl, OH, groups are replaced by azomethine, CNR, and amino, NH2, groups, respectively. The cyclic amidine functionality of amino heterocycles is obtained when the substituents of both the carbon and nitrogen atoms of the azomethine group are replaced by the same and only one five- or six-membered cycle in which both atoms are embedded and the primary amino group is flanked outside the ring known as an exocyclic amino group. Therefore, they display rich chemistry by combining the effect of an azomethine-like C–N double bond with an amide-like C–N single bond having some partial double bond character.
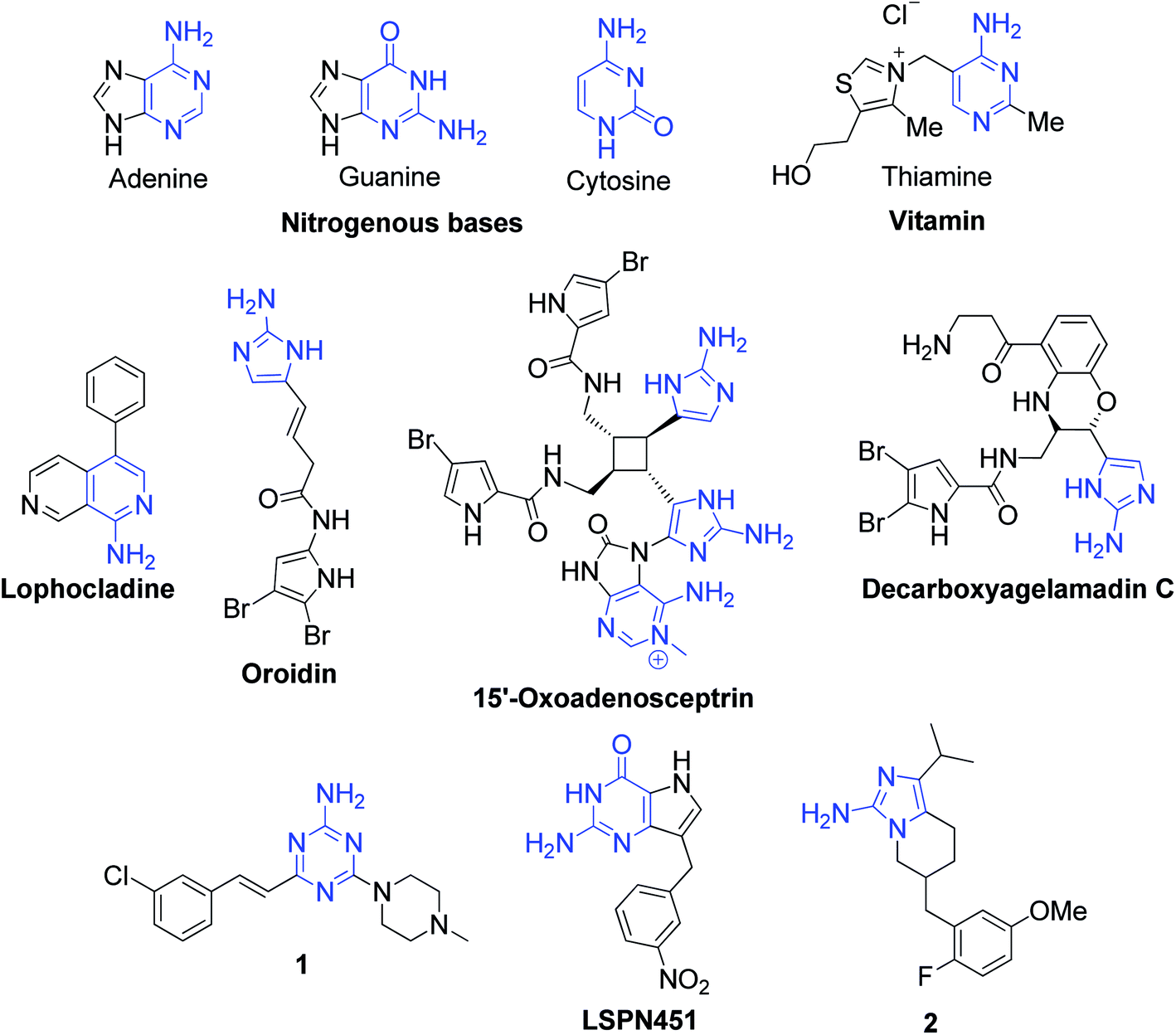
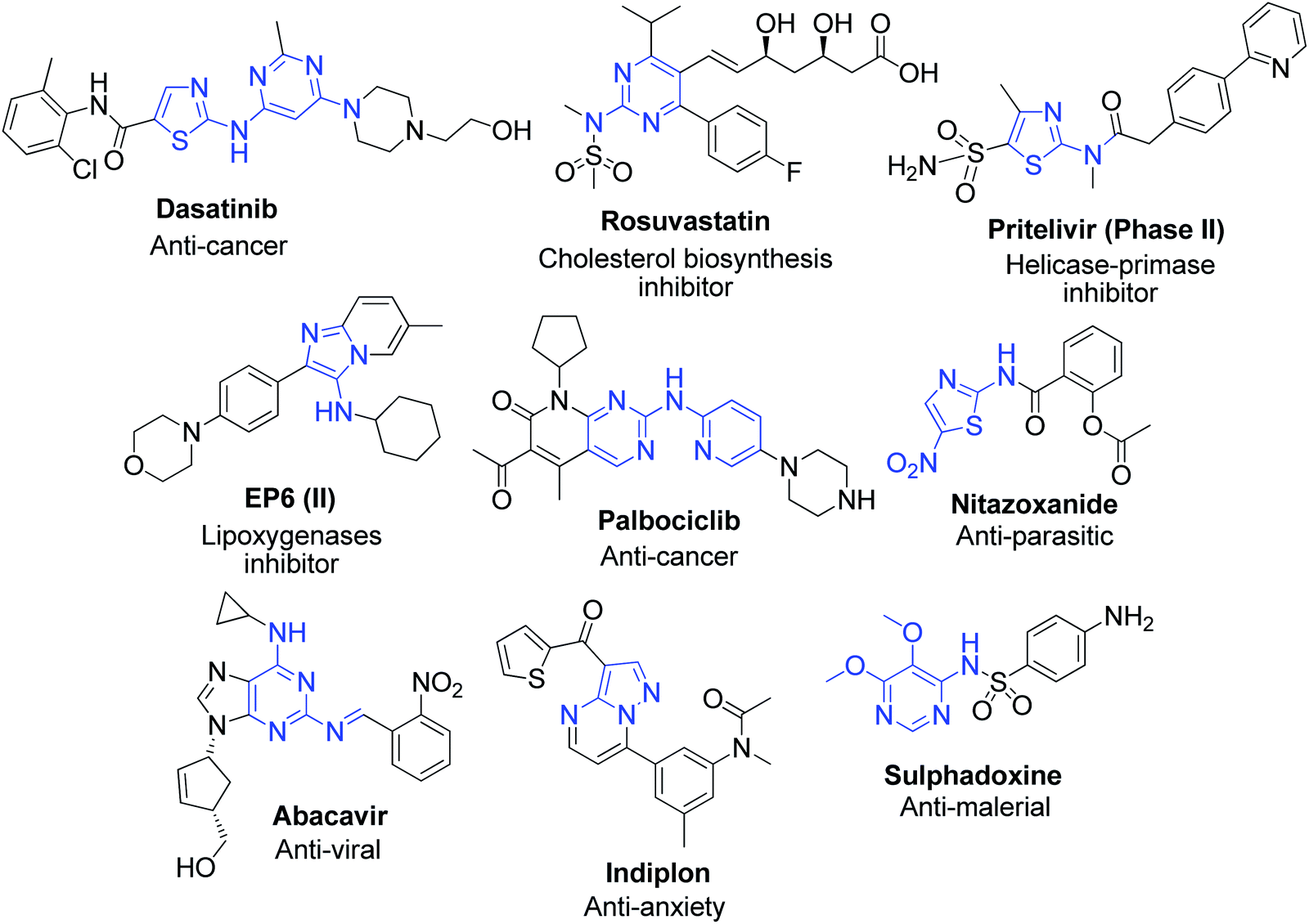
In relation to their utility in the synthesis of various derivatives, aromatic α-aminoazaheterocycles represent important structural motifs for the creation of ring junction nitrogen heterocyclic compounds, and therefore have become promising building blocks for the discovery of novel molecules in the drug and agrochemical industries.7,8 They are also the source of important precursors for the production of valuable heteroaromatics such as benzimidazoles,9 quinazolines and derivatives of substituted imidazoles, pyrimidines, dihydropyridines,10 etc., which are known to possess robust biological profiles. The methods exploring the reaction of the amidine functionality in α-aminoazaheterocycles have gained immense popularity for obtaining biological leads and exploring drug discovery programs in the past few decades. In most of reactions, they act as 1,3-dinucleophiles and deliver the triad of N–C–N atoms into the ring of the target heterocycle, and hence are widely used in the synthesis of fused pyrimidines and imidazoles constituting the core ring junction structure of several currently marketed drugs. Considering that several natural and biologically active molecules possess nitrogen-fused heterocyclic fragments, they occupy a central place in pharmaceutical and biomedical research. Fig. 2 shows some representative examples of well-known drug and medicinally active compounds prepared as derivatives of α-aminoazaheterocycles.
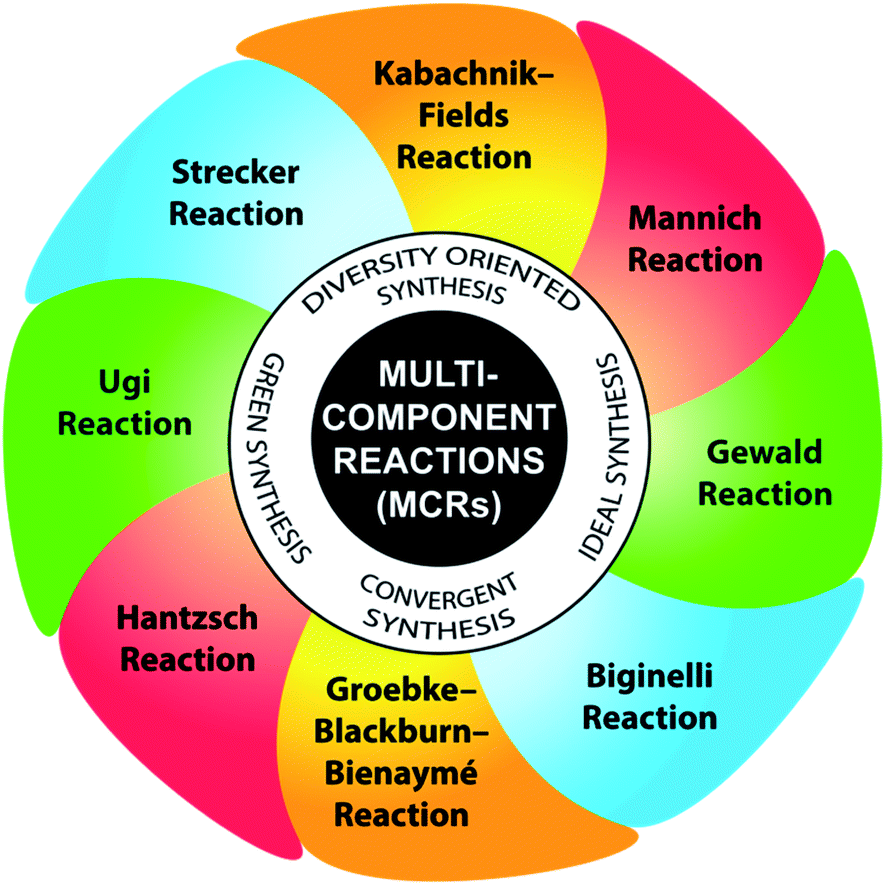
In the past few decades, the development of processes that allow the formation of several bonds in one operation has greatly overcome the burden to synthetic organic chemists by creating molecular diversity and complexity via the rapid the assembly of simple and readily available substrates. The beginning of the 21st century marked an increase in the interest in multicomponent reactions (MCRs) as a highly impressive tool to access elaborate molecular scaffolds, while combining structural diversity with eco-compatibility to achieve the diversity-orientated synthesis of a variety of molecules, especially bioactive heterocycles.11 MCRs are now well-established convergent chemical approaches, in which a single operation is sufficient to build one product from the well-defined condensation of three or more reactant molecules with high atom economy and multiple-bond-forming efficiency.12 With a reduced number of reaction steps and starting from simple, inexpensive starting materials, MCRs render the library production of substantially diverse range of small molecules less costly compared to conventional approaches. Further, the time-consuming isolation and purification of synthetic intermediates are not required, and hence, both the production of waste and expenditure of human labor are significantly reduced, complying with the principles of green chemistry. Their experimental simplicity lies in the fast and elegant access of compounds that allow systematic variations, and consequently they allow the possibility of automatization.13 Therefore, their extensive application in the synthesis of densely functionalized molecules with potential bioactivity has undergone explosive growth in recent years. Moreover, their high atom economy, high efficiency, mild conditions, high convergence and concomitant step economy together with their general compatibility with green solvents justify their central place in the toolbox of sustainable synthetic methodologies.14 Some promising and well-established MCRs include the Ugi-4-component condensation, Strecker amino acid synthesis, Hantzsch dihydropyridine synthesis, Biginelli reaction, Mannich reaction, Kabachnik–Fields reaction, Groebke–Blackburn–Bienaymé (GBB) reaction, and Gewald reaction (Fig. 3).15
The present review is devoted to gathering and generalizing the reported data obtained from a literature survey from 2010 to 2019 and some recently available references from 2020 concerning the application of α-aminoazaheterocycles in MCRs and attempt to fulfil the conception of these reactions. Among the aromatic α-aminoazaheterocycles undergoing MCRs, it has been observed that five-membered (fused or non-fused) azoles are the most studied and frequently employed azaheterocyclic compounds, and V. A. Chebanov's group covered most of the publications up to 2010 regarding the utilization of azoles in MCRs.16 Therefore, we incorporate the references starting from 2010.
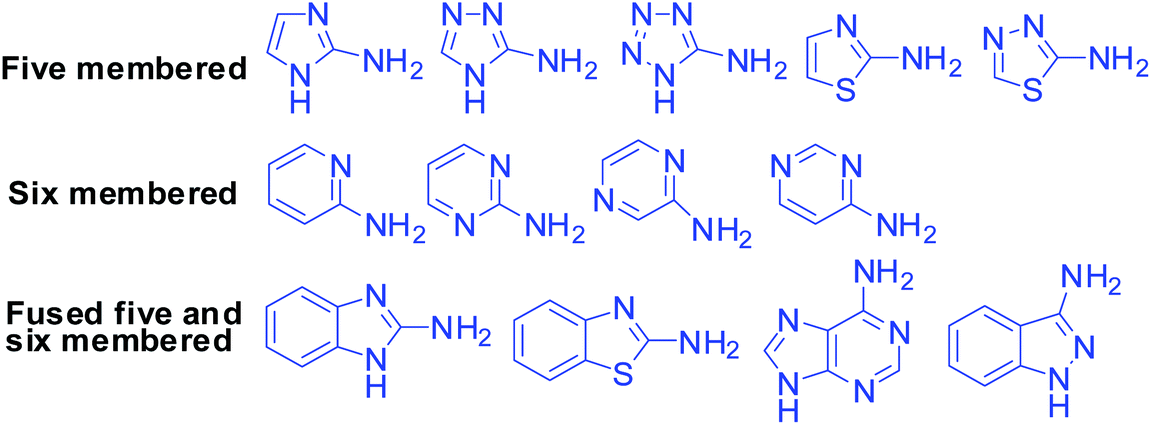
Although there is a huge variety of five- and six-membered α-aminoazaheterocycles with fused and non-fused structures, fortunately only the simplest of them are reported with their applicability in MCRs as they are readily available and inexpensive. Fig. 4 shows these heterocyclic compounds, which will be encountered in this review frequently. However, only the α-aminoazaheterocycles that are part of a heteroaromatic ring system are covered in this review.
Further, the multicomponent treatments of AAH as binucleophile are subdivided into two categories:
(ii) AAH as 1,1-binucleophiles (less diverse).
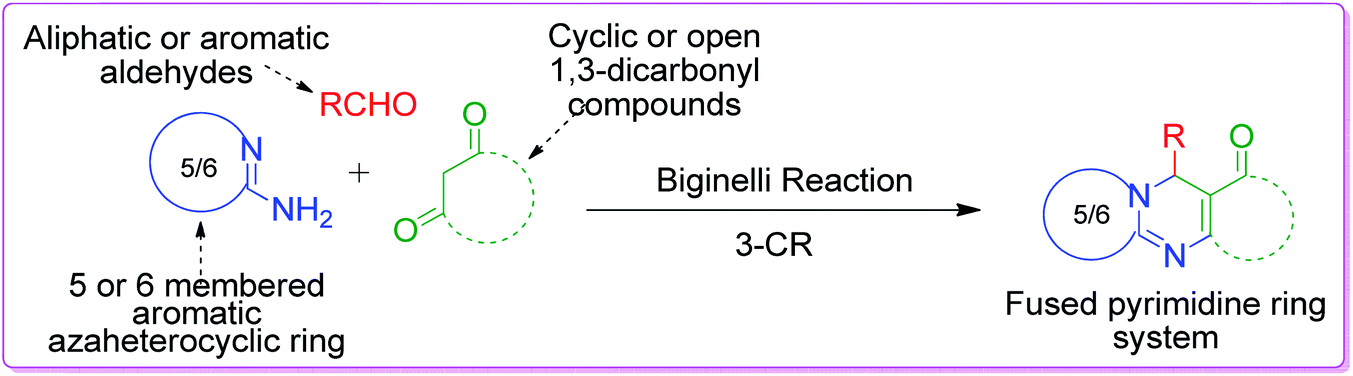
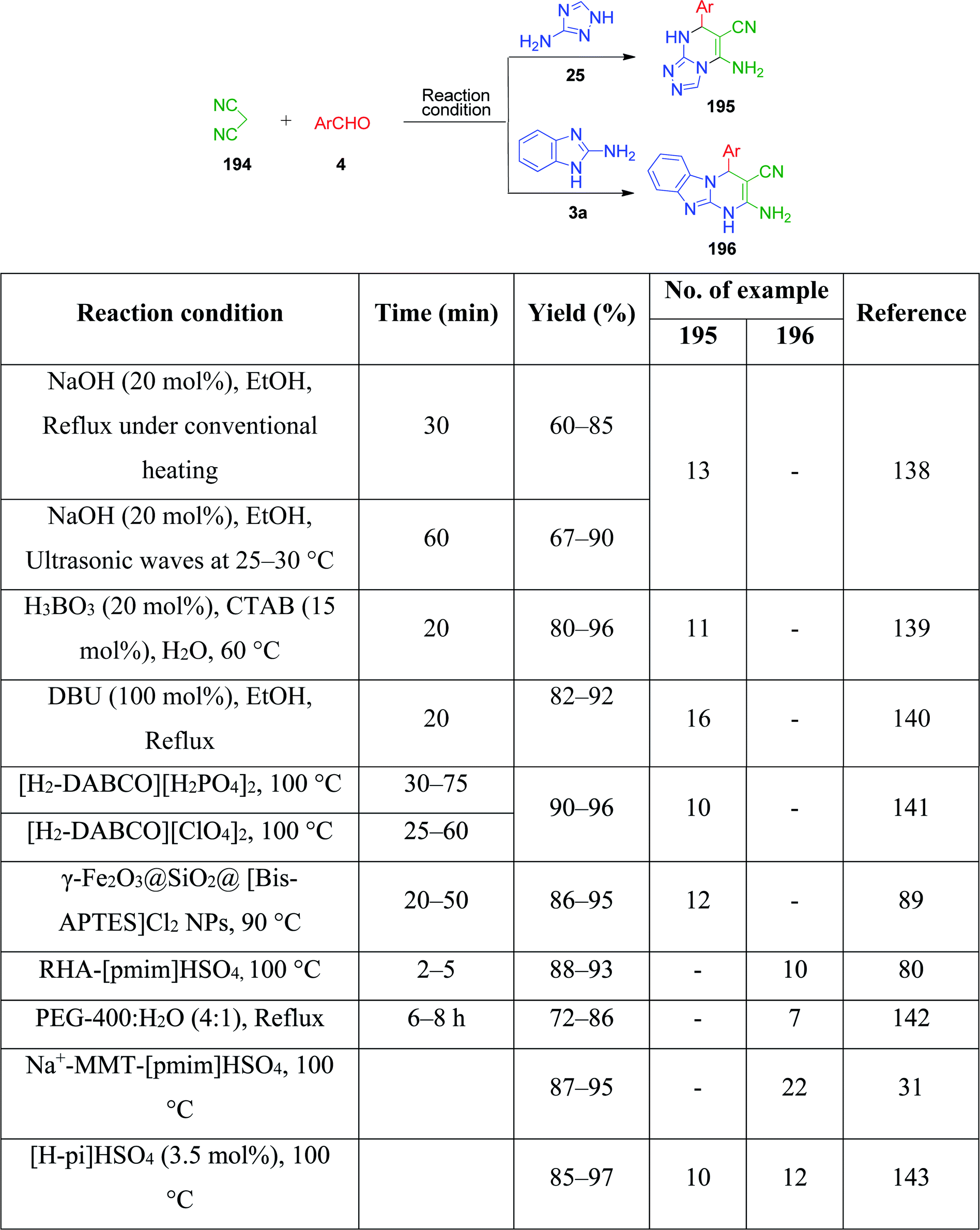
2.1.1 As 1,3-binucleophile. A vast number of MCRs have been reported in the literature in the past decade, where both nitrogens of the amidine group of AAH function as nucleophiles. Pyrimidine and imidazole derivatives are the usual structures resulting from these binucleophilic actions and the heterocycles are classed according to their known biological profiles. Therefore, the study of these MCRs is the central principle for the synthetic application of aminoazaheterocycles. In the formation of compounds containing these core nuclei, AAHs are combined with components that can act simultaneously as an electrophile and nucleophile together with an electrophilic component such as aldehyde and ketone. Based on the type of components that react with AAHs other than aldehydes and ketones, this subgroup is classified as follows.By definition, active methylene compounds are compounds that possess a methylene group between two strong electron withdrawing groups such as carbonyl, nitrile, and nitro. Because of the strong deactivating nature of these group, the hydrogens of the methylene bridge become acidic in nature, and consequently the methylene carbon acquires nucleophilic character, while the group adjacent to it behaves as an electrophile.
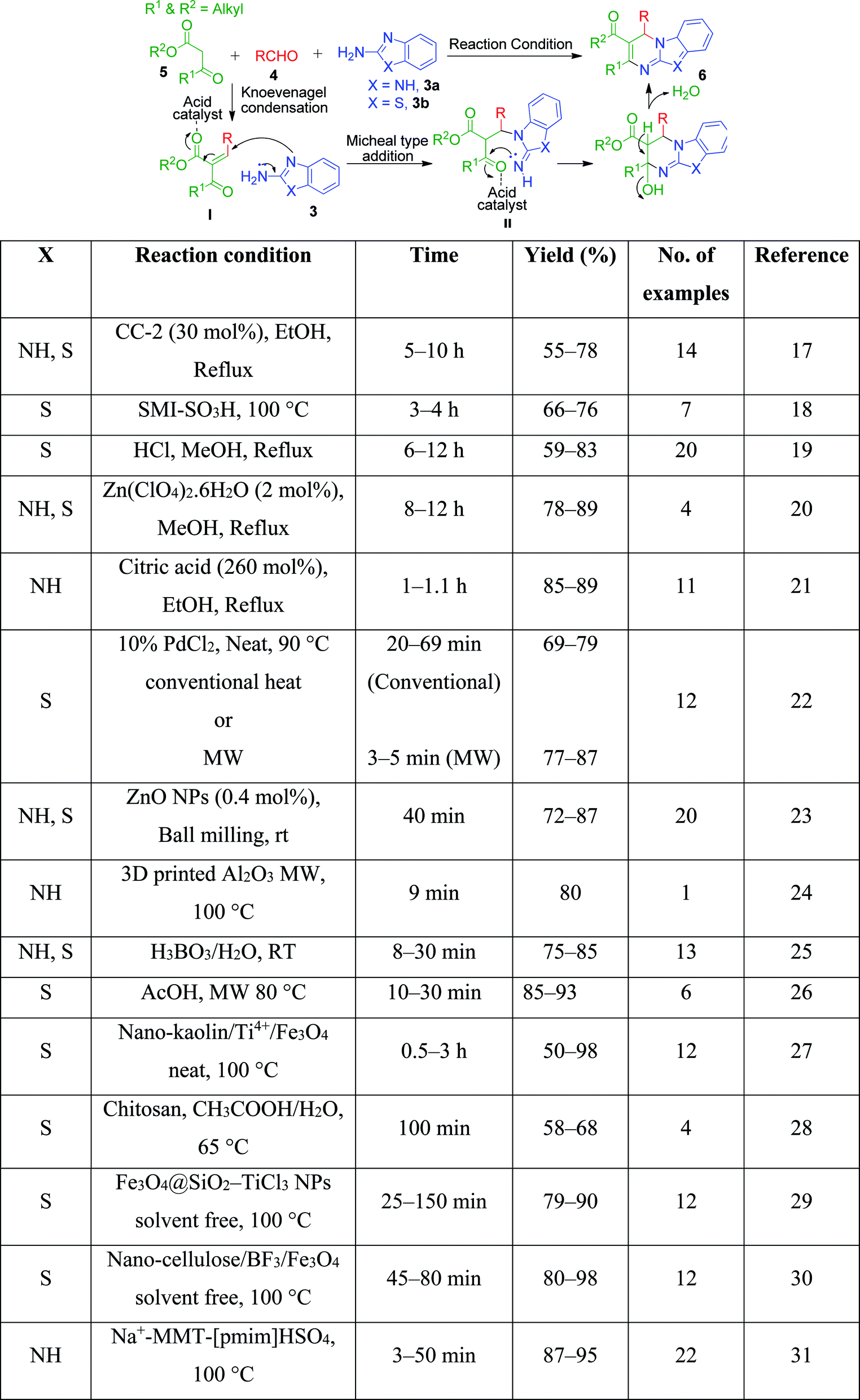
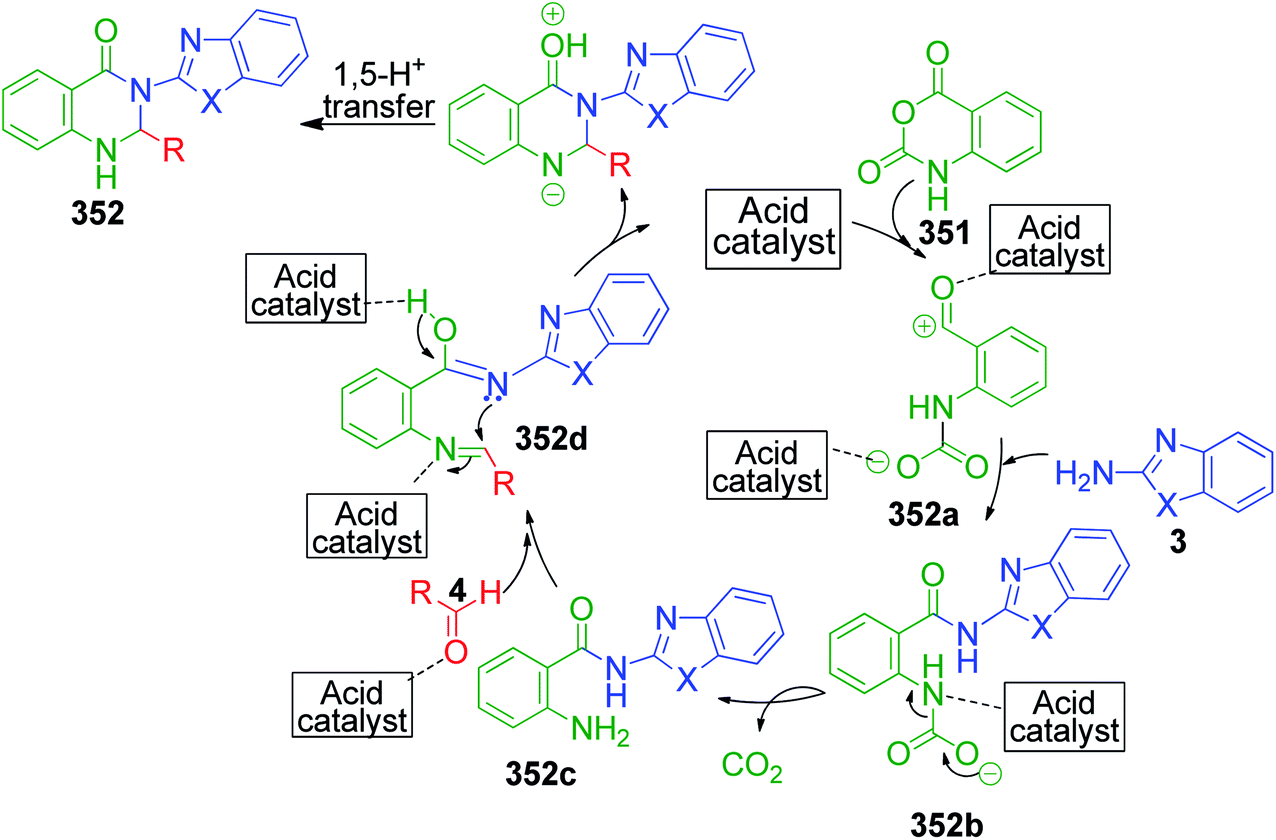
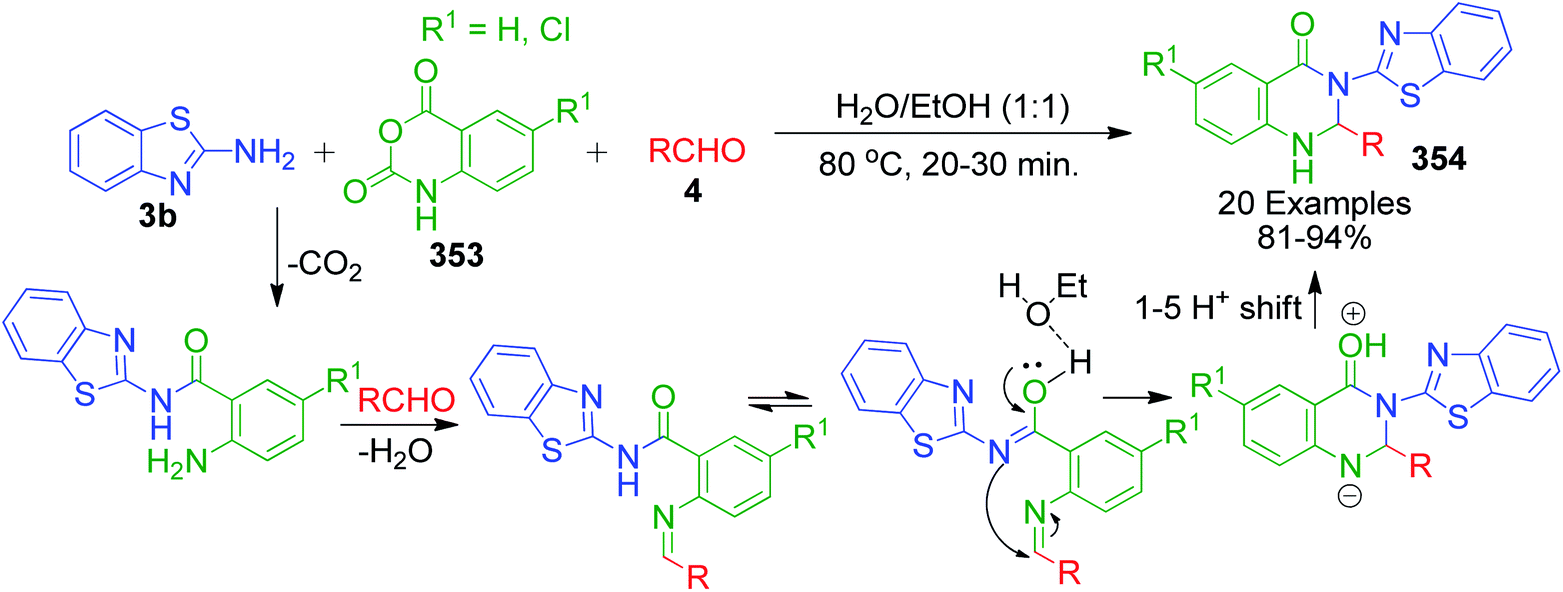
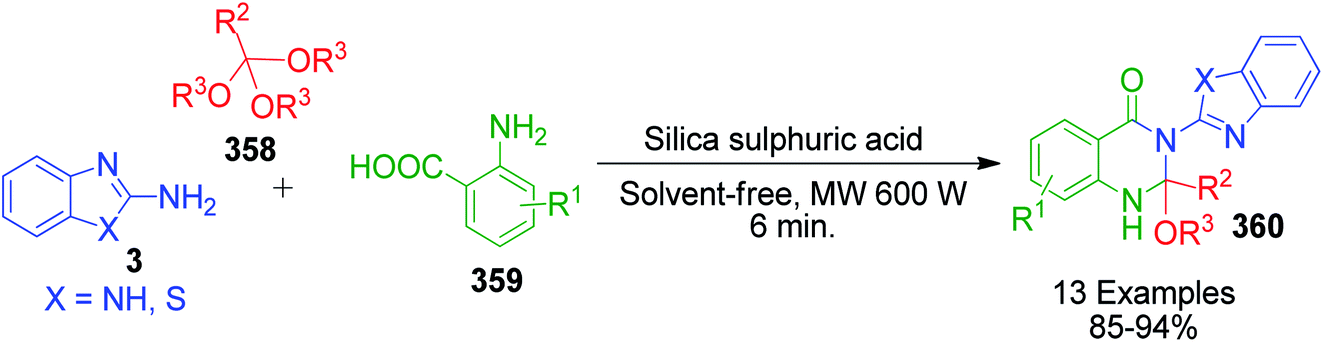
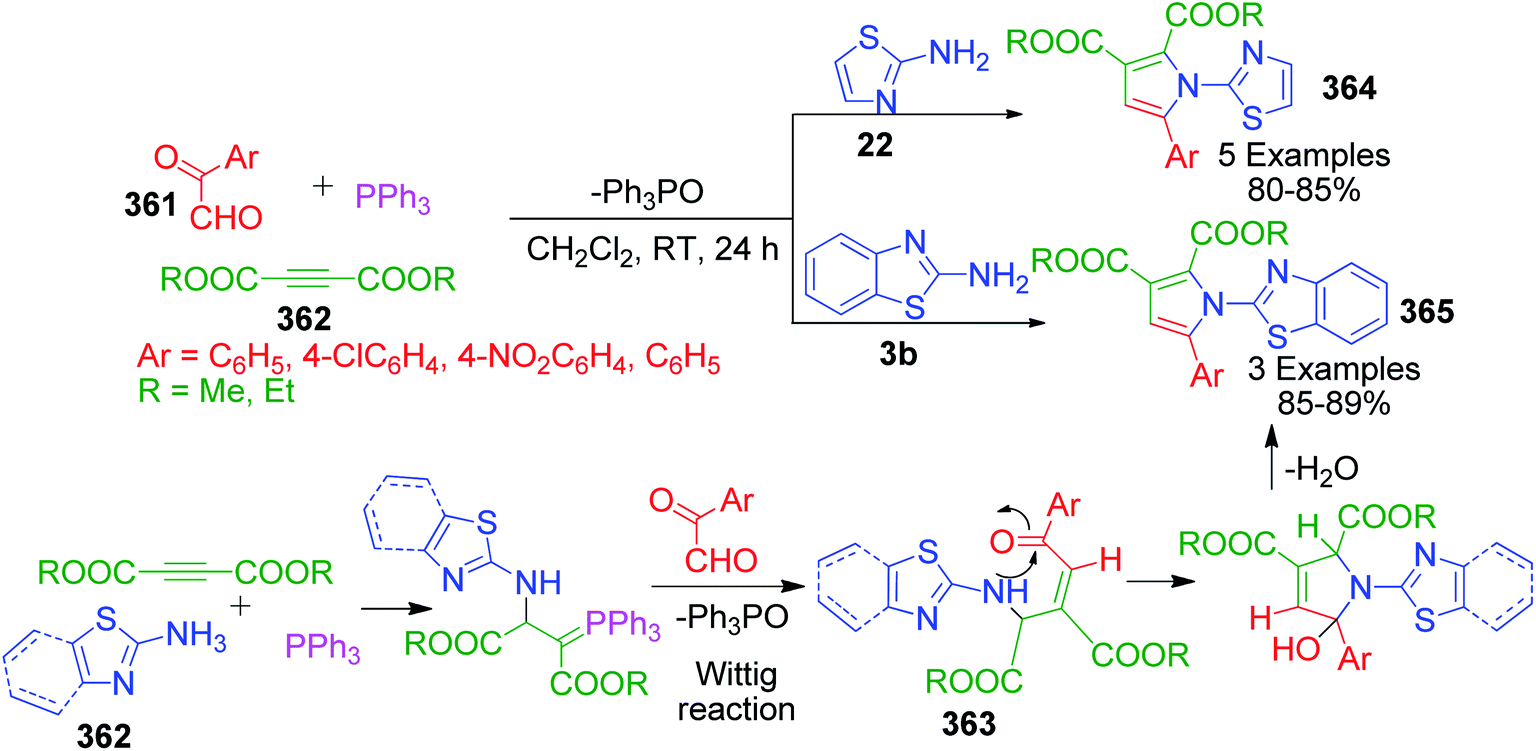
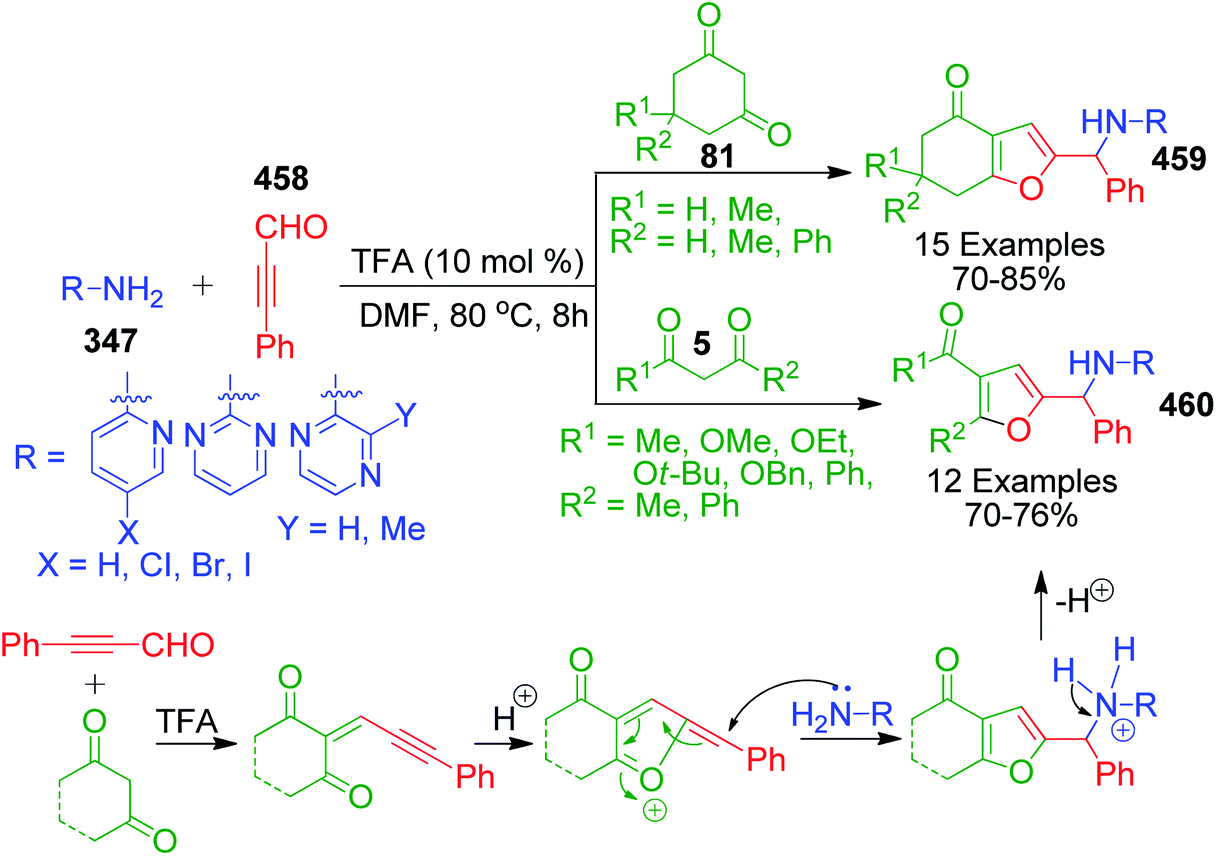
β-Keto/β-dicarbonyl compounds, both non-cyclic and cyclic, are the most investigated components used to explore the 1,3-binucleophilic nature of the amidine functionality of 2-aminoazaheterocycles through MCR chemistry. According to our literature survey, the majority of publications in this review comprise MCRs related to non-cyclic β-keto compounds such as β-ketoesters and β-ketoamides and cyclic β-keto compounds such as cyclohexanedione and analogues, Meldrum's acid, and 4-hydroxycoumarin. Accordingly, the Biginelli reaction, a three-component reaction (3-CR) that employs β-ketoesters, amines and aldehydes has emerged as a prototype for the construction of dihydropyrimidine scaffolds containing carboxylic ester or carboxamide groups. The versatility of this reaction allows the incorporation of a vast range of substrate molecules including amines, and therefore remains one of the most common reactions studied among chemists utilizing α-aminoazaheterocycles as amine components. A general view of the Biginelli reaction is depicted in Scheme 1.
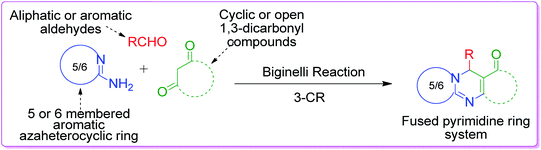 |
| | Scheme 1 General form of the Biginelli reaction. | |
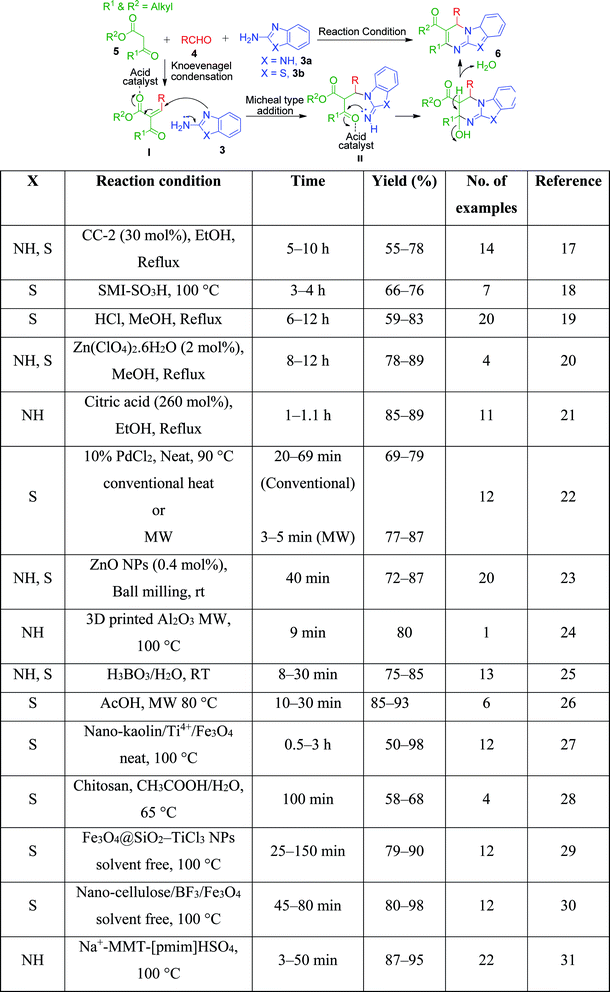 |
| | Scheme 2 Three-component synthesis of benzoazole-fused pyrimidine derivatives through the reported acid catalysts. | |
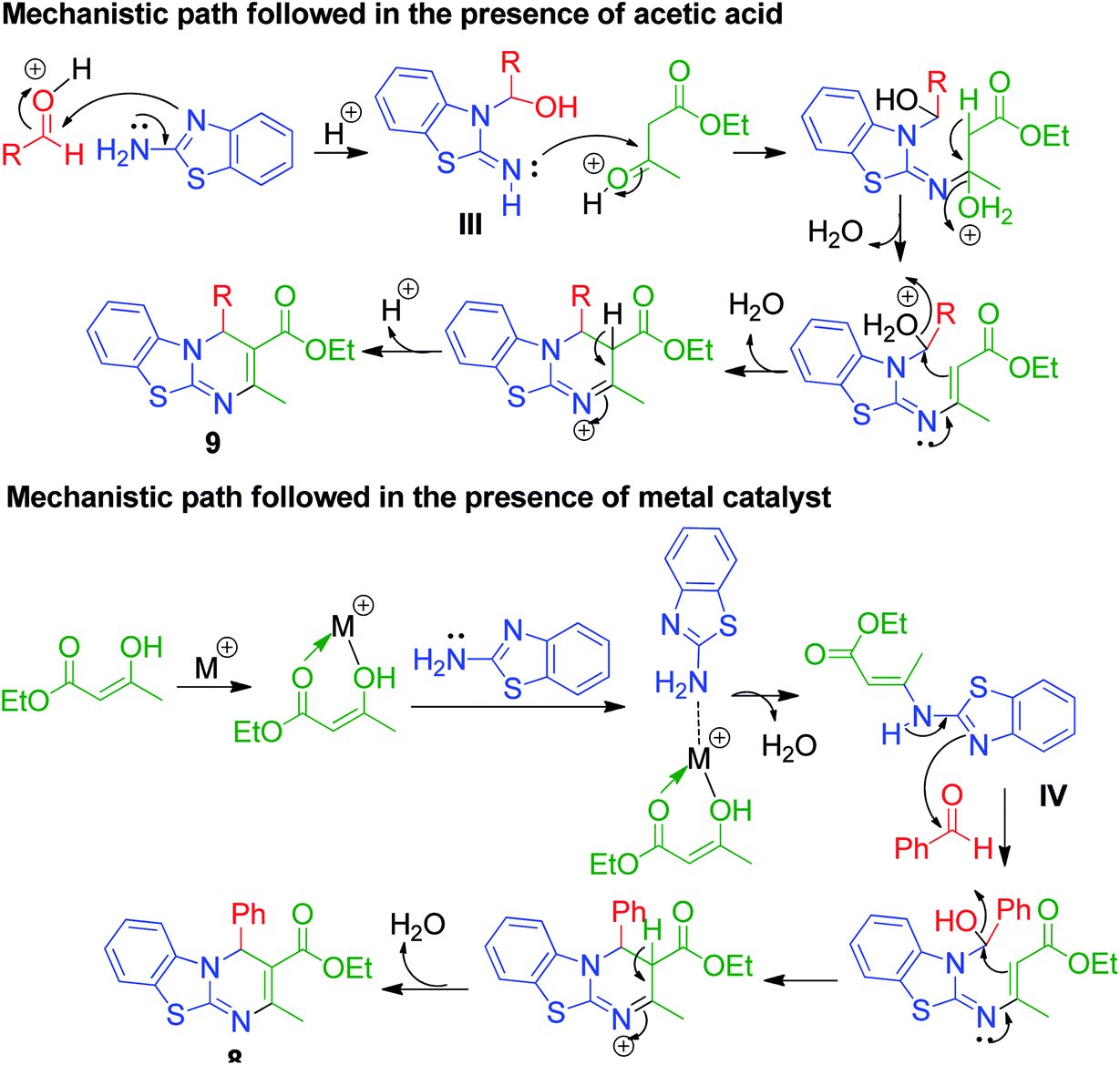
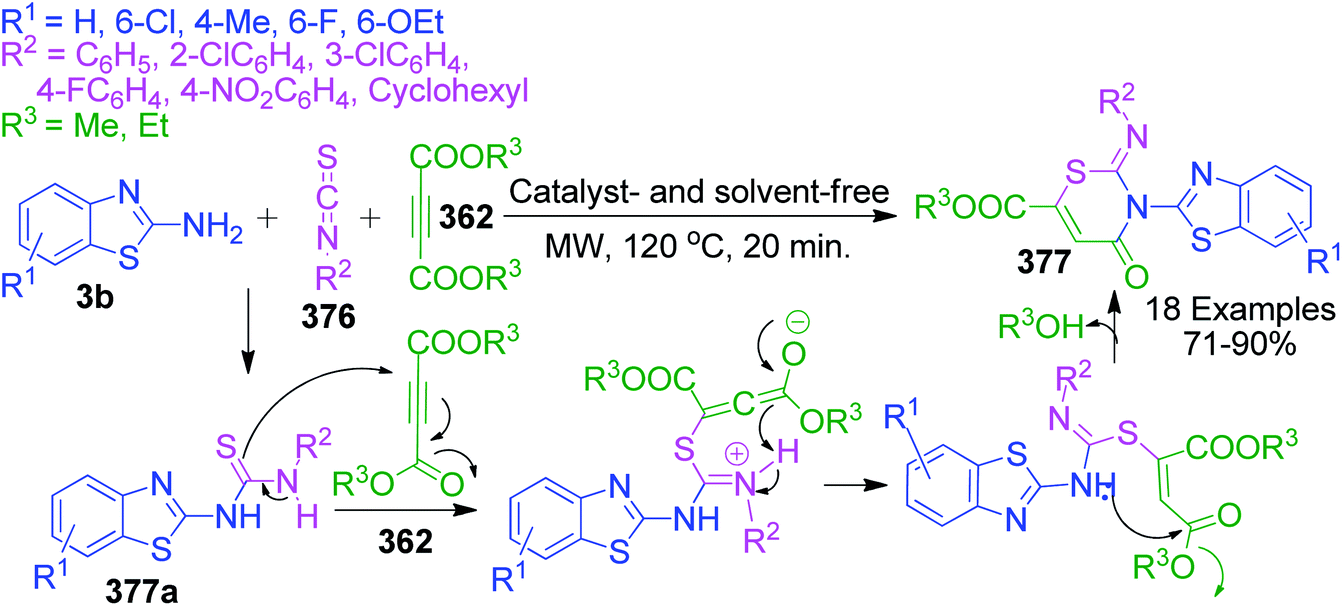
An interesting case was studied by P. K. Sahu and coworkers,32 where the synthesis of these pyrimidine derivatives was achieved in the presence of a solvent and under solvent-free conditions using various catalysts. When the condensation among 2-amino benzothiazole 3b, aldehydes 4 and β-ketoester 7 was carried out in the presence of acetic acid as the catalyst and methanol as the solvent at 65 °C, the corresponding products were obtained after 12–20 h. Conversely, the same reaction was completed in 70–120 min when carried out with various metal catalysts under solvent-free conditions. The highest yield was obtained when CuCl2 and LiCl were used as catalysts, but the use of LiCl ensured minimum time for completion of the reaction (Scheme 3). The experimental results suggested that the reaction follows different mechanistic routes depending on the type of catalyst. In the presence of acetic acid, 2-amino benzothiazole 3b first reacts with aldehyde 4 to give imine as intermediate III, which then reacts with ethyl acetoacetate 7 to give the final product 9, whereas 2-amino benzothiazole 3b and ethyl acetoacetate 4 first combine in the presence of metal catalysts and resultant intermediate IV adds to the benzaldehyde to give pyrimidobenzothiazole 8 (Scheme 4).
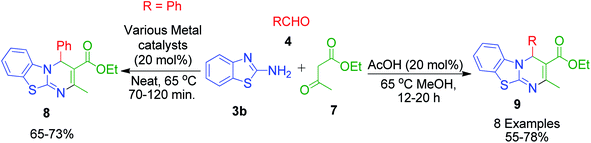 |
| | Scheme 3 Synthesis of 4H-pyrimido[2,1-b]benzothiazole derivatives under two different conditions. | |
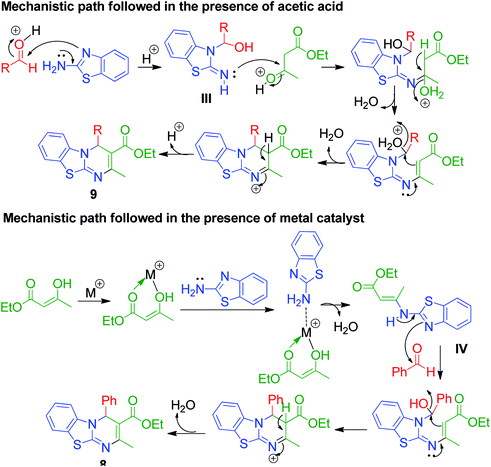 |
| | Scheme 4 Mechanistic routes explaining the formation of 4H-pyrimido[2,1-b]benzothiazoles in the presence of acetic acid and metal catalyst. | |
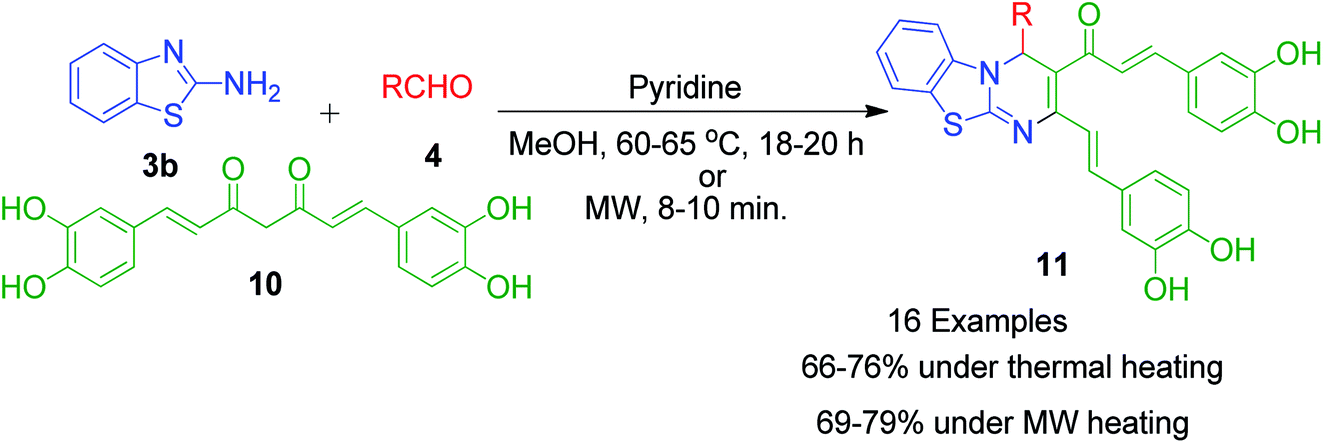
Besides these acid-catalyzed methods, P. K. Sahu et al. adapted the synthesis of benzothiazolopyrimidine 11 starting from a completely different β-dicarbonyl, known as curcumin 10, 2-aminobenzothiazole 3b and aromatic aldehydes 4 using pyridine as a catalyst under thermal heating involving methanol as the solvent as well as microwave irradiation solvent-free conditions (Scheme 5).33 The synthesis of curcumin derivatives 11 was found to be completed within 18–20 h when carried out under conventional heating. Conversely, the reaction time was significantly reduced to 8–10 min when the same reaction mixture was irradiated with microwaves under solvent-free conditions. Thus, synthesis under microwaves provides a very simple and efficient way to obtain curcumin derivatives in a shorter time with the advantage of reducing environmental pollution by eliminating the use of volatile organic solvents. The detailed mechanistic study shows that the reaction proceeds through a Knoevenagel-type intermediate and follows the path of the usual Biginelli reaction.
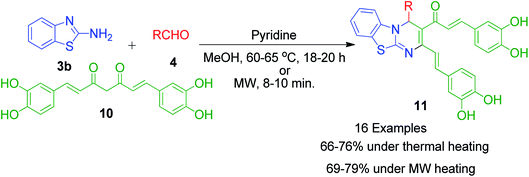 |
| | Scheme 5 Synthesis of benzothiazolopyrimidine using pyridine under thermal and microwave heating. | |
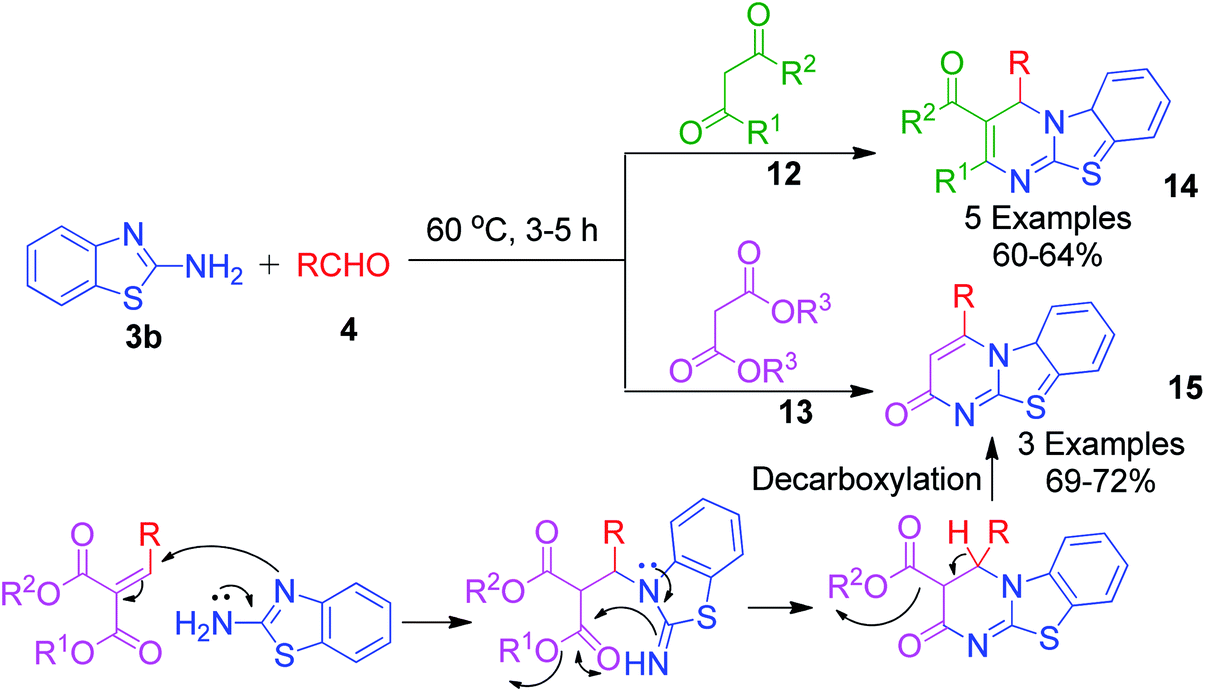
A catalyst-free and solvent-free approach based on the Biginelli reaction was developed by F. Chadegani et al., wherein the one-pot three-component reaction between 2-aminobenzothiazole 3b, benzaldehyde derivatives 4 and β-ketoester or β-diketone derivatives (12 or 13) was performed at 60 °C, which afforded the corresponding pyrimido[2,1-b]benzothiazole derivatives 14 in 3–5 h.34 The same reaction was also studied with malonic acid derivatives as the β-diketo component, and subsequent annulation reactions provided 2-oxo-pyrimido[2,1-b]benzothiazoles 15 with no substitutions at the 2 and 3 positions. The mechanism for the formation of 4H-pyrimido[2,1-b]benzothiazole and 2-oxo-pyrimido[2,1-b]benzothiazole derivatives 15 was the same, except in the latter case, malonates with two properly placed leaving groups (two alkoxy groups) at sufficiently high temperature underwent decarboxylation to afford the final product (Scheme 6).
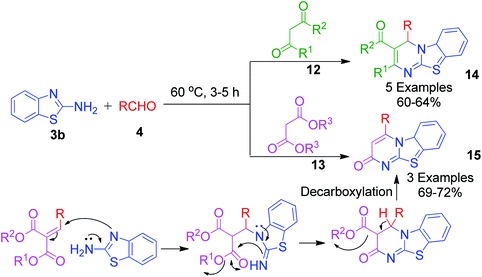 |
| | Scheme 6 Catalyst-free and solvent-free synthesis of 4H-pyrimido[2,1-b]benzothiazole and 2-oxo-pyrimido[2,1-b]benzothiazole derivatives. | |
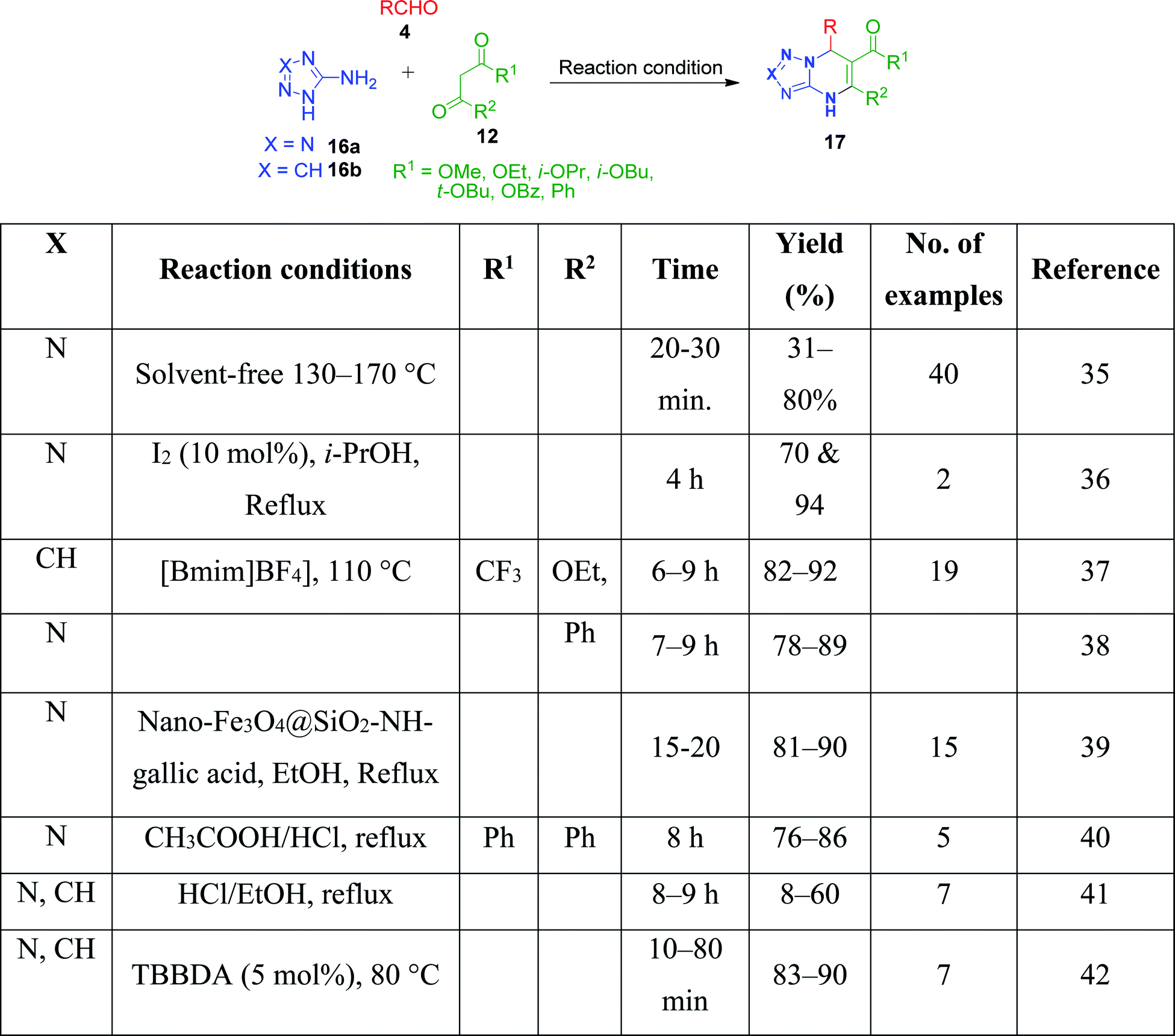
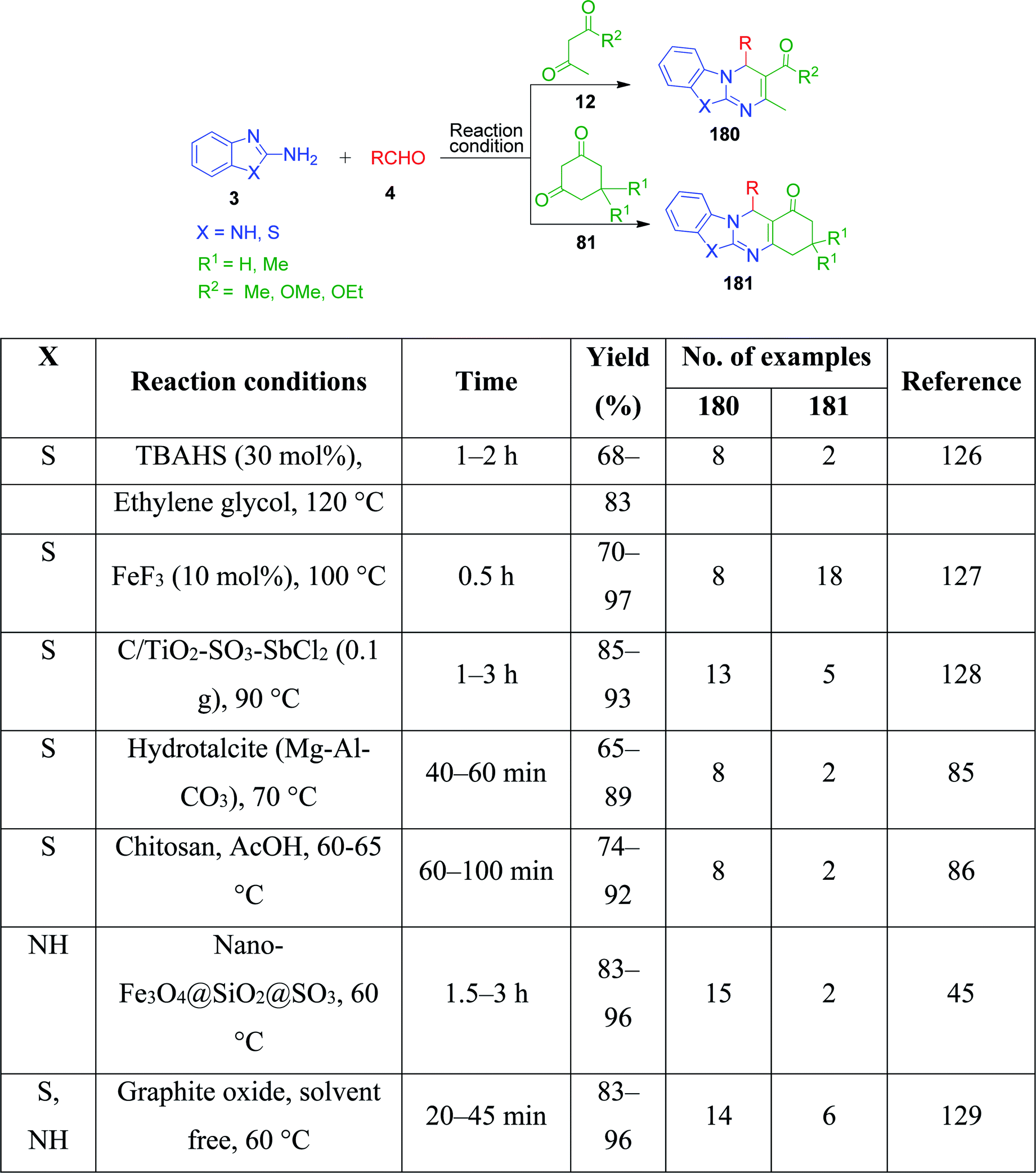
Simple aminoazoles parallel to benzo-fused aminoazoles have also been found to take part in the Biginelli reaction to generate azole-fused pyrimidine derivatives. However, although the obtained product is slightly less fused, it possesses equivalent biological features. Further among the non-fused aminoazoles, 5-aminotetrazole, 3-aminotriazole, and 2-aminothiazole are widely employed in MCRs for the synthesis of various heterocyclic derivatives upon reaction with 1,3-diketocompounds and aldehydes under different conditions. In this context, a catalyst- and solvent-free method was employed by V. L. Gein's research group for the synthesis of tetrazolo-fused pyrimidines by heating a mixture of acetoacetic esters, 5-aminotetrazole and aromatic aldehydes at 130–170 °C until the evolution of bubbles of gaseous side product ceased. The yield of the products was found to be low in the case of electron-donor substituents in the aldehyde. Conversely, the character of substituents in the acetoacetic esters did not notably affect the yields.35 Haleel and coworkers developed a protocol for the synthesis of analogues of tetrazolopyrimidine derivatives 17 in the presence of iodine under refluxing conditions.36 The synthesis was achieved by reacting ethyl acetoacetic esters 12, 5-aminotetrazole 16 and heteroaryl aldehydes (2-pyridinecarboxaldehyde and 4-pyridinecarboxaldehyde) 4 in isopropanol medium at 82–85 °C in the presence of 10 mol% of catalyst. The same three-component reaction was also established using catalysts such as [bmim]+[BF]− as a green ionic liquid,37,38 nano-Fe3O4@SiO2–NH-gallic acid39 and CH3COOH/HCl,40 conc. HCl,41 and N,N,N′,N′-tetrabromobenzene-1,3-disulfonamide (TBBDA)42 (Scheme 7).
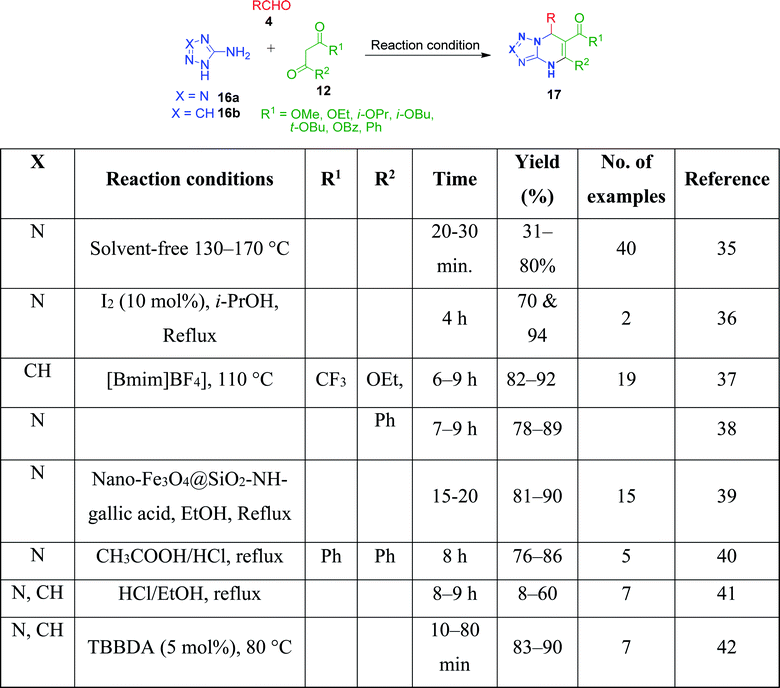 |
| | Scheme 7 Three-component synthesis of triazolo/tetrazolo-fused pyrimidines using various catalysts. | |
E. S. Filatova et al. utilized 3-oxobutanoate-containing polyethers 19 as 1,3-diketocompounds in the reaction with benzaldehyde 18 and 5-aminotetrazole 16a involving polyphosphoric acid as the catalyst to carry out the synthesis of podands bearing two terminal tetrazolo[1,5-a]dihydropyrimidine groups 20 along with byproducts such as podands containing one 4,7-dihydrotetrazolo[1,5-a]pyrimidine ring system 21 and a free hydroxy group at the other terminus of the polyether chain (Scheme 8).43
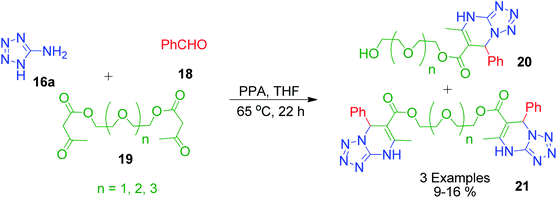 |
| | Scheme 8 Synthesis of podands containing two terminal tetrazolo[1,5-a]dihydropyrimidine groups. | |
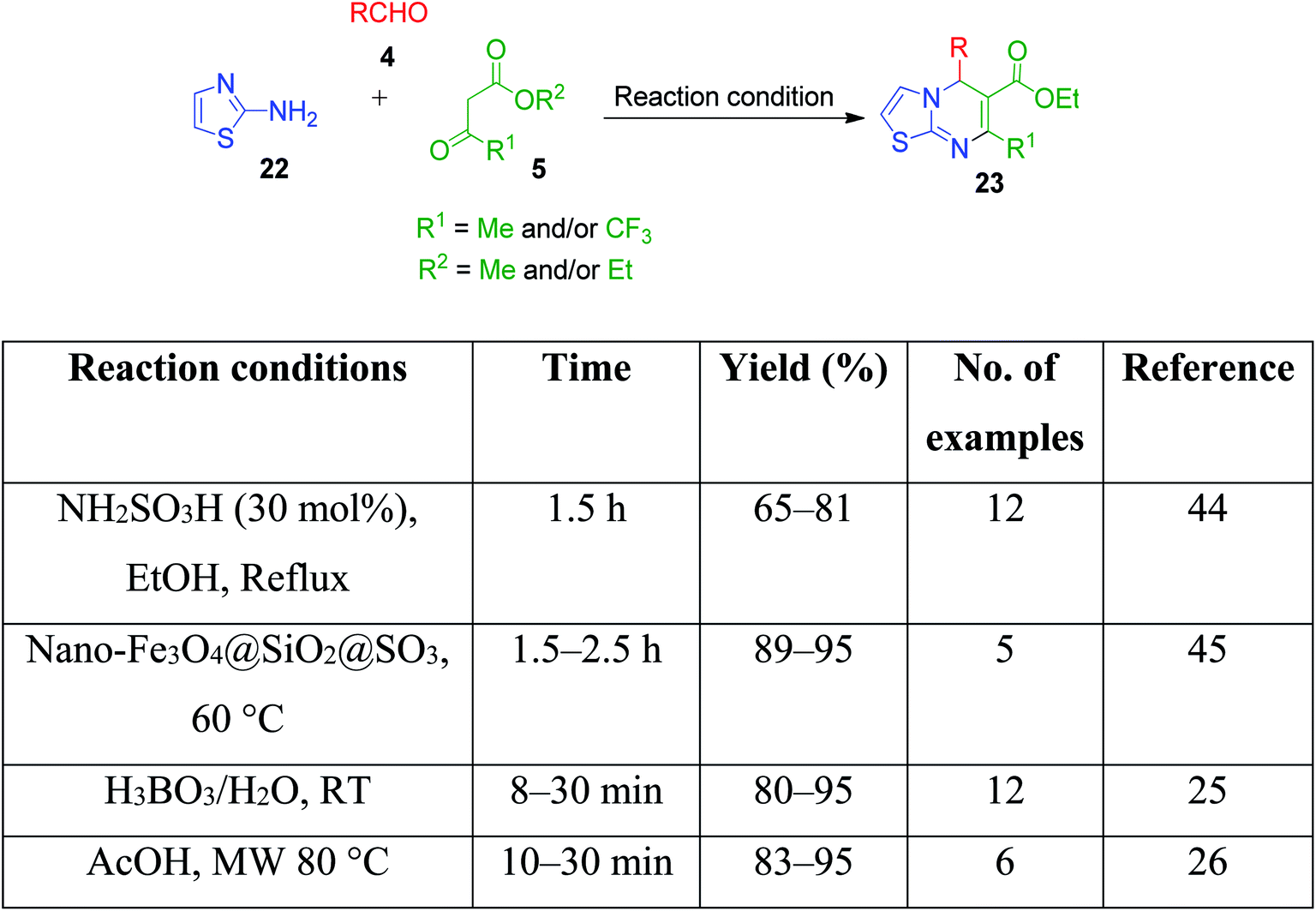
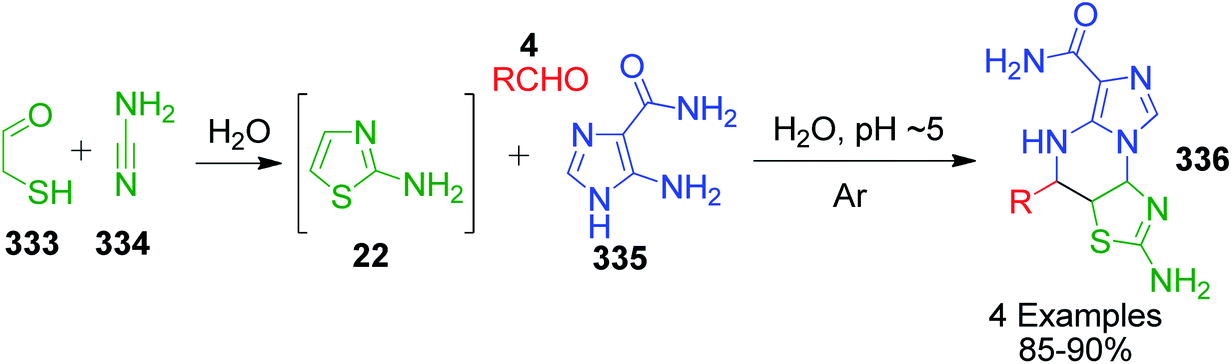
I. Batool and coworkers disclosed a novel multicomponent strategy that affords the synthesis of thiazolopyrimidines 23 through the condensation reaction among 2-aminothiazole 22, ethylacetoacetate 5 and differently substituted aromatic aldehydes 4. The reaction, which was catalyzed by sulfamic acid in ethanol under reflux conditions, was established to find potent antidiabetic and antibacterial drugs (Scheme 9).44 This reaction was also performed using nano-Fe3O4@SiO2@SO3,45 H3BO3,25 acetic acid,26 etc.
 |
| | Scheme 9 Protic acid-catalysed synthesis of thiazolopyrimidines from 2-aminothiazole, ethylacetoacetate and aromatic aldehydes. | |
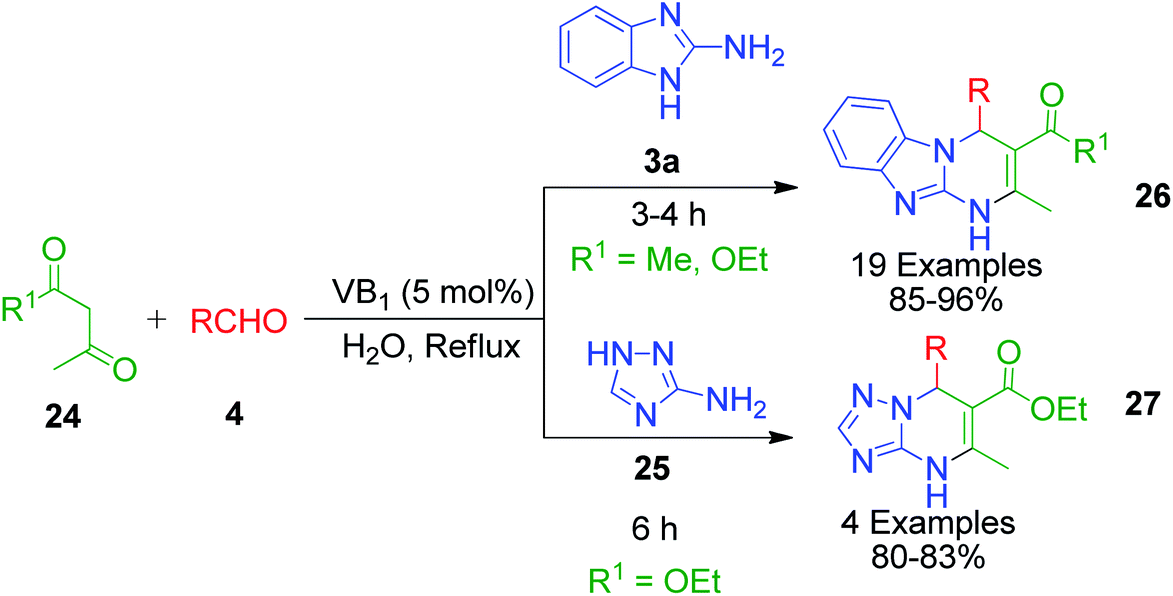
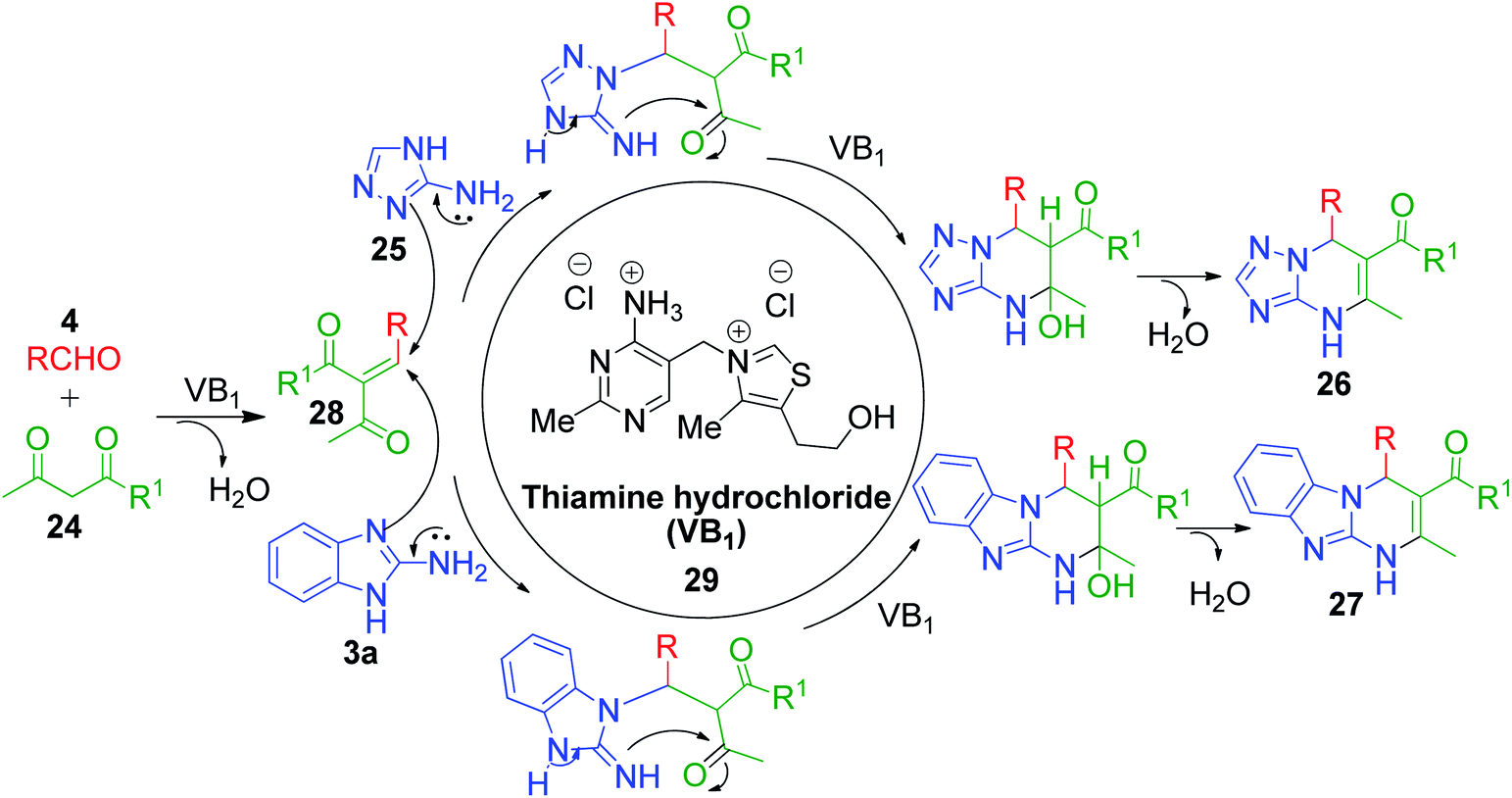
Moreover, some reported methods demonstrated their versatility by using both amino azole and its benzo analogues as AAH for the synthesis of the corresponding pyrimidine scaffolds under the same reaction conditions. For example, the thiamine hydrochloride (VB1)-catalyzed synthesis of benzo[4,5]imidazo[1,2-a]pyrimidine 26 and [1,2,4]triazolo[1,5-a]pyrimidine 27 derivatives was developed by heating a reaction mixture containing 2-aminobenzimidazole/3-amino-1,2,4-triazole 3a and 25 with various aldehydes 4 and ethyl acetoacetate/acetyl acetone 24 in water under refluxing conditions (Scheme 10).46 VB1 is a nonflammable, inexpensive and non-toxic reagent, which is comprised of a pyrimidine ring and a thiazole ring linked by a methylene bridge. Target compounds 26 and 27 were obtained in high yields (80–96%) in 3–6 min with the employment of merely 5 mol% of the catalyst. A plausible mechanism illustrating the reaction pathway is summarized in Scheme 11. After the initial formation of benzylidene-type intermediate 28 from the VB1 29-catalyzed Knoevenagel condensation of dicarbonyl compound 24 and aldehyde 4, a nucleophilic attack from amino azoles 3a and 25 occurs in Michael-type addition manner, followed by intramolecular cyclisation, yielding the desired pyrimidines 26 and 27 after dehydration.
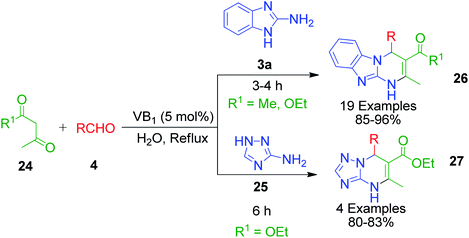 |
| | Scheme 10 Synthesis of benzoimidazolo pyrimidine and triazolopyrimidine derivatives based on thiamine hydrochloride (VB1)-catalyzed three-component reaction. | |
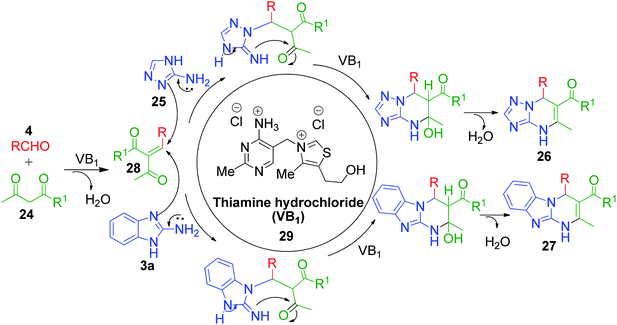 |
| | Scheme 11 Mechanism proposed for VB1-catalyzed synthesis of fused pyrimidines. | |
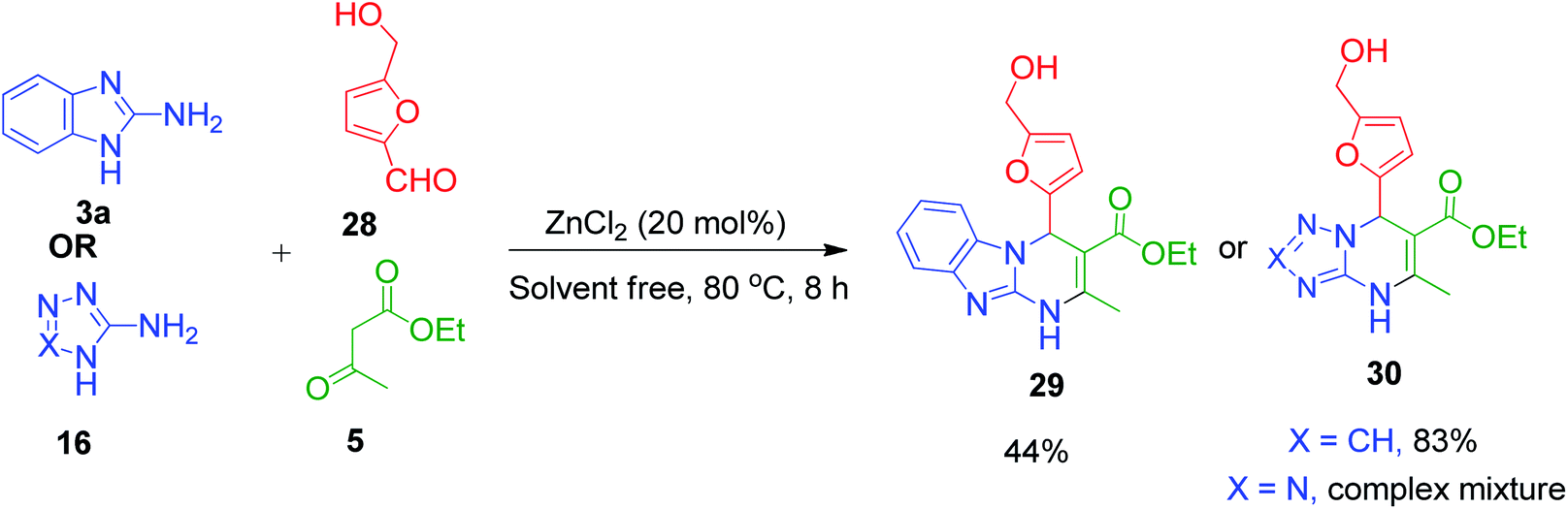
W. Fan et al. utilized 5-hydroxymethylfurfural (HMF) 28 in the multi-component Biginelli reaction with 2-aminobenzimidazole/aminotriazole 3a and 16 and ethyl acetoacetate 5 together with the catalyst ZnCl2 at 80 °C, which led to the generation of dihydropyrimidinones 29 and 30 in moderate to good yields (Scheme 12). The advantage of using HMF lies in the CH2OH motif, which can be functionalized further to the desired target molecules.47
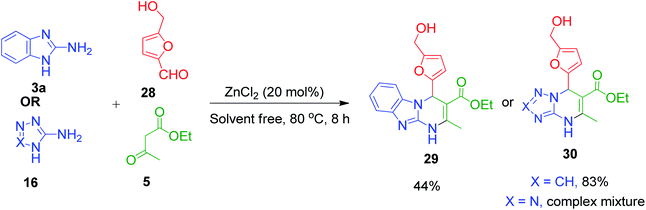 |
| | Scheme 12 ZnCl2-catalysed synthesis of benzimidazole/triazole-fused dihydropyrimidinones. | |
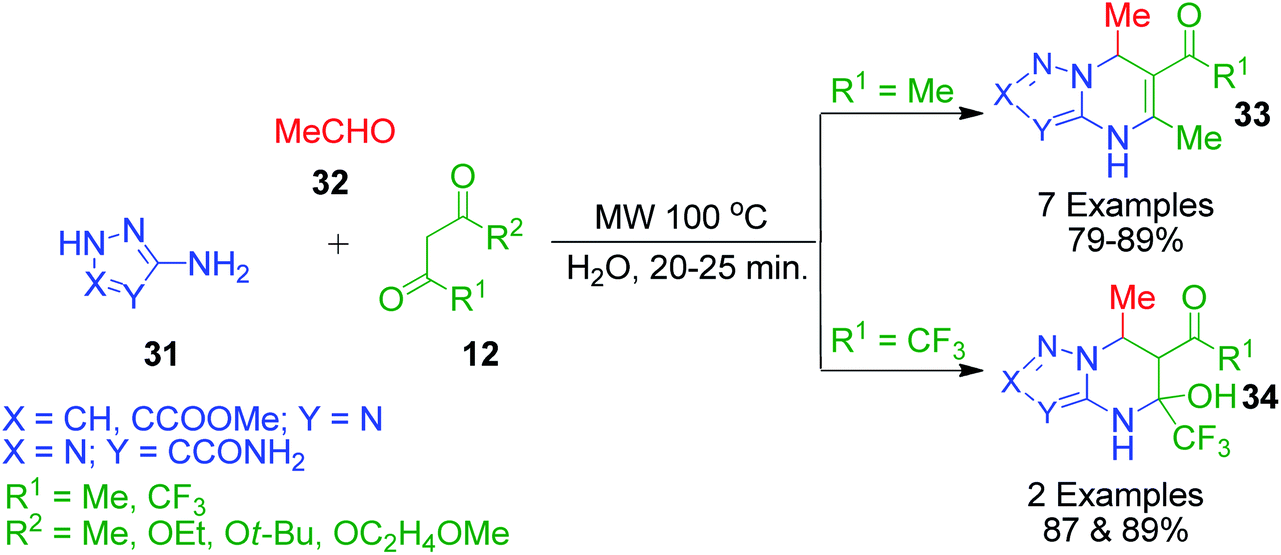
I. G. Tkachenko et al. reported a three-component synthesis for the regioselective formation of 4,7-dihydro[1,2,4]-triazolo- and 4,7-dihydro[1,2,3]triazolo[1,5-a]pyrimidines 33 involving a reaction between 3-amino-1H-1,2,4-triazole/5-amino-2H-1,2,3-triazole derivatives 31, acetaldehyde 32 and acetylacetone/β-keto esters 12 in water under microwave irradiation. Using ethyl 4,4,4-trifluoro-3-oxobutanoate as the 1,3-dicarbonyl compound under the same reaction conditions, the corresponding 5-hydroxy-4,5,6,7-tetrahydro derivatives 34 were obtained, which failed to dehydrate at the reaction temperature (Scheme 13).48
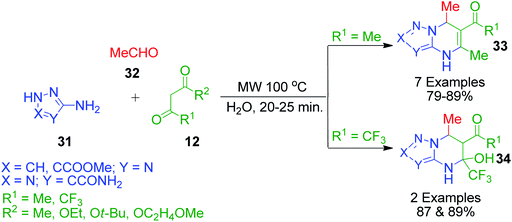 |
| | Scheme 13 Microwave heating-assisted synthesis of azolopyrimidine derivatives. | |
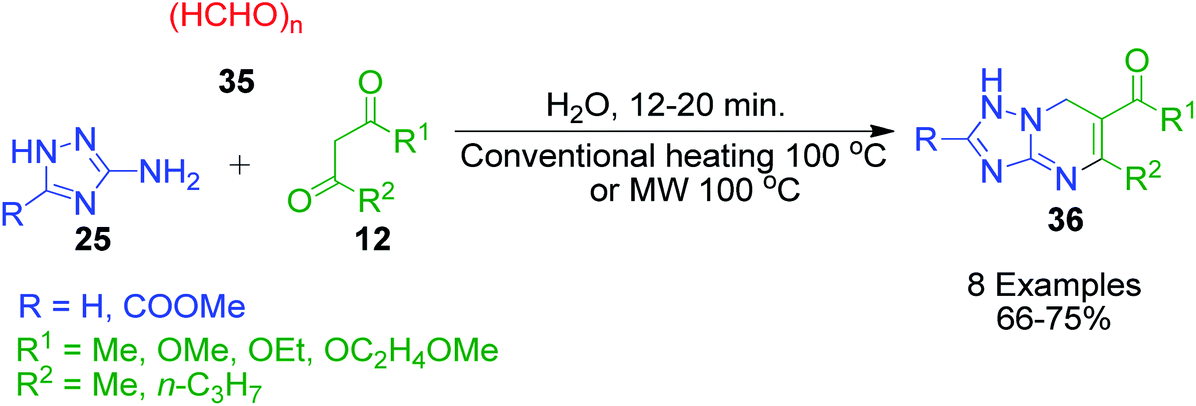
The simplest aldehyde, formaldehyde (in the form of paraformaldehyde) was also effectively incorporated into the structure of azole-fused pyrimidine derivatives. Specifically, S. A. Komykhov et al. reported a method for the preparation of 8 unsubstituted 4,7-dihydro-1,2,4-triazolo[1,5-a]pyrimidines 36, which presumed the three-component reaction of 3-amino-1,2,4-triazole 25 with paraformaldehyde 35 and different 1,3-dicarbonyl compounds 12 (Scheme 14).49 This method is highly environment-friendly as no catalyst and organic solvent are required. Comparable yields of the target compounds were isolated by refluxing the starting components in hot water under either conventional heating or microwave irradiation.
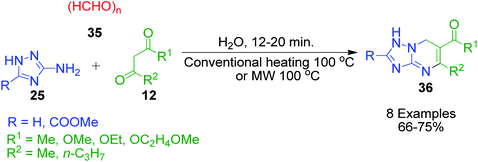 |
| | Scheme 14 Conventional heating or microwave heating-assisted synthesis of 7-unsubstituted 4,7-dihydro-1,2,4-triazolo[1,5-a]pyrimidines 36. | |
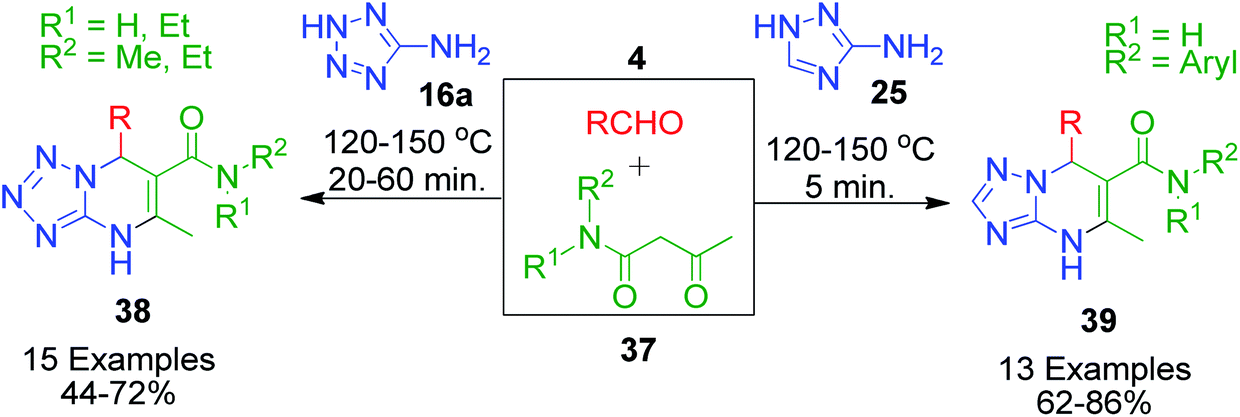
In the synthesis of triazole/tetrazole-fused pyrimidine derivatives through the involvement of N-aryl/alkyl acetoacetamides as one component acting as a β-keto compound, Gein et al. made a significant contribution. In this context, a three-component reaction mixture containing N-alkylamides of acetoacetic acid such as N-methyl- or N,N-diethyl-3-oxobutanamide 37, aromatic aldehydes 4 and 5-aminotetrazole 16a was condensed to afford the corresponding N-substituted 7-aryl-5-methyl-4,7-dihydrotetrazolo-[1,5-a]pyrimidine-6-carboxamides 38.50 Based on a similar method, the same group also reported the condensation of 3-amino-1,2,4-triazole 25 with N-arylamides of acetoacetic acid 37 and aromatic aldehyde 4, which resulted in N,7-diaryl-5-methyl-4,7-dihydro-1,2,4-triazolo[1,5-a]pyrimidine-6-carboxamides 39.51 In both cases, a solventless mixture of starting materials was heated at high temperature (120–150 °C) for a few minutes, resulting in the synthesis of tetrazolo and triazolopyrimidine rings in high yields, and the formation of product was indicated by the solidification of the mixture together with no further evolution of water vapor (Scheme 15).
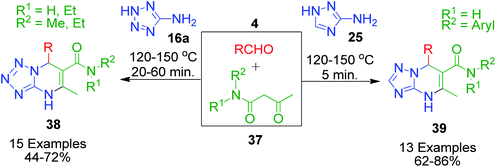 |
| | Scheme 15 Construction of triazolo 39 and tetrazolopyrimidines 38 under solvent-free and catalyst-free conditions. | |
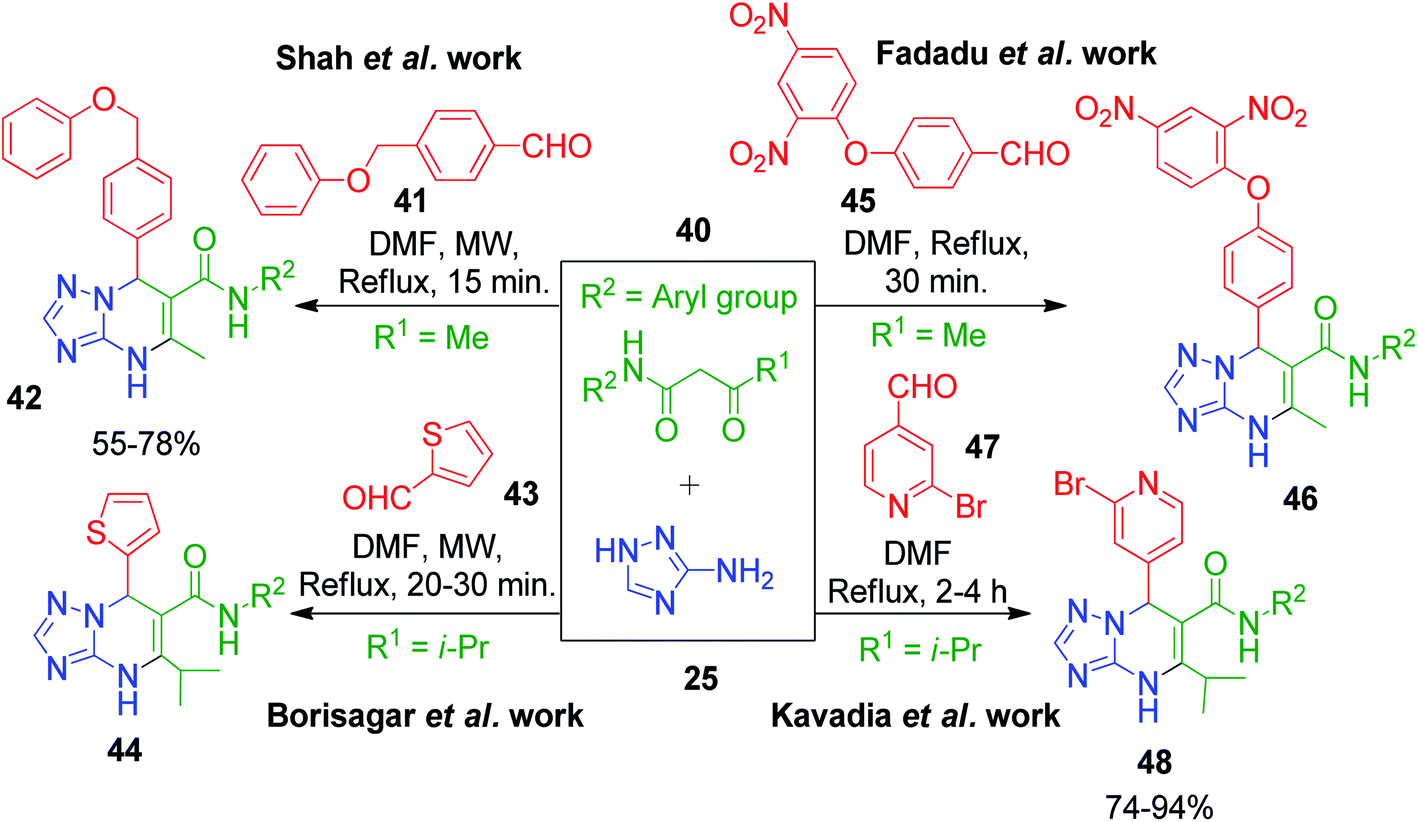
Several other synthetic strategies have also been reported, where triazolopyrimidine derivatives were prepared using N-arylamides of β-keto acid as the active methylene compound. For example, R. Pada et al. reported a simple method involving heating a DMF solution containing 5-amino-1,2,4-triazole 25, N-aryl acetoacetamides 40 and 4-(phenoxymethyl)benzaldehyde 41 for 15 min under conventional and microwave heating conditions, which afforded the corresponding triazole-fused pyrimidines 42 in 55–78% yield.52 The same approach in the same year was developed by M. Borisagar's group with thiophene-2-carbaldehyde 43 as the aldehydic component, and the reaction of the starting materials was carried out under microwave heating.53 Another similar method under conventional heating of the starting components involving 4-(2,4-dinitrophenoxy)benzaldehyde 45 as the specific aldehyde, 5-amino-1,2,4-triazole 25 and N-aryl substituted acetoacetamides 40 was demonstrated by P. D. Fadadu et al. in DMF solvent.54 Following these results, Kavadia and coworkers explored 2-bromopyridine-4-carbaldehyde 47 as an aldehydic component in an analogous methodology. The approach utilized conventional heating conditions for a DMF solution of the three starting reagents to construct the corresponding pyrimidine ring 48. A comparatively longer reaction time (2–4 h) was needed in this strategy to obtain yields in the range of 74–94% (Scheme 16).55
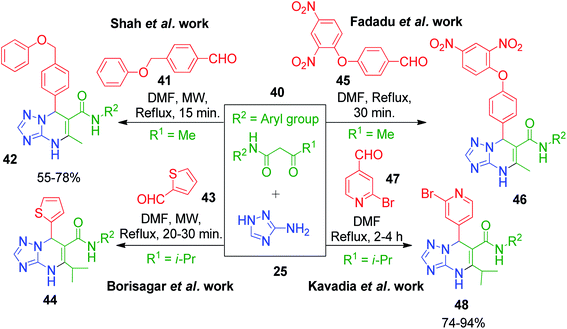 |
| | Scheme 16 Catalyst-free synthesis of various triazolopyrimidine derivatives. | |
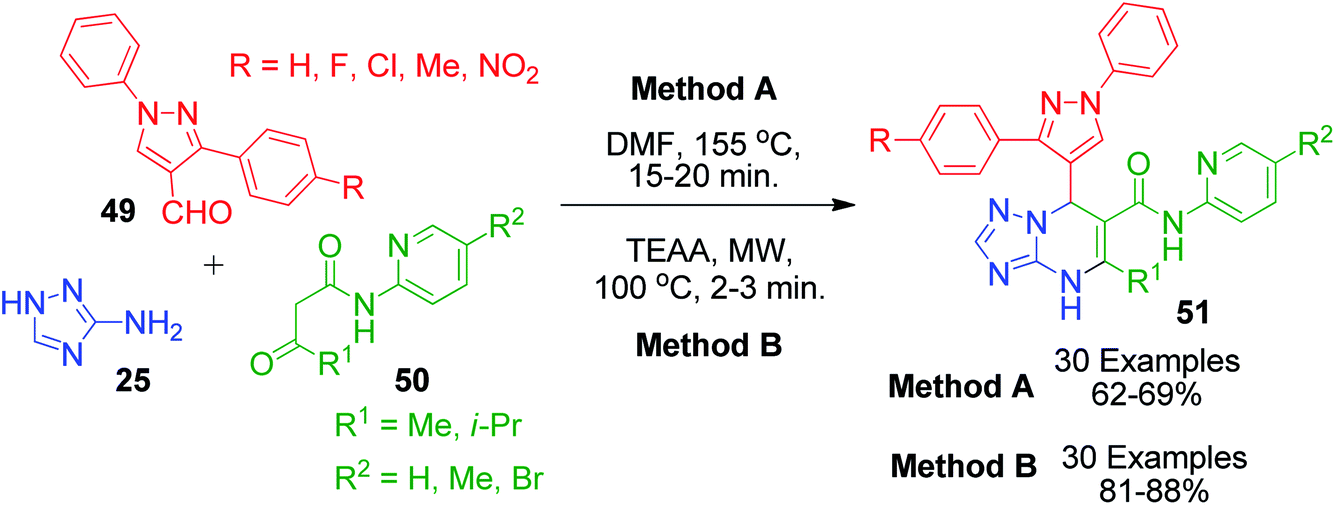
J. D. Bhatt and coworkers also contributed to this field by synthesizing pyrazole-linked triazolopyrimidines 51 using two different methods. In the catalyst-free method, the Biginelli-type reaction involving starting materials 25, 49 and 50 was heated conventionally at high temperature (155 °C) in DMF solvent (Scheme 17, Method A).56 In the other method, the same reaction was performed by heating the starting materials in the presence of triethylammonium acetate (TEAA) under microwave irradiation at 350 W (Scheme 17, Method B).57 The catalytic efficiency of TEAA was also studied by recovering the catalyst from previous runs up to five successive reaction cycles, which showed that there was a notable decrease in the efficiency of the catalyst only after four successive cycles.
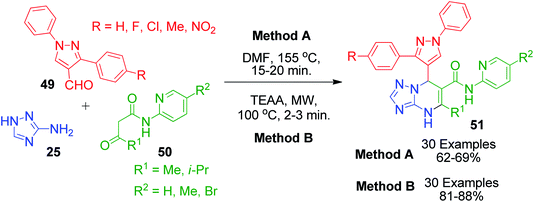 |
| | Scheme 17 Pyrazole-linked triazolopyrimidines 51 synthesised under conventional and microwave heating. | |
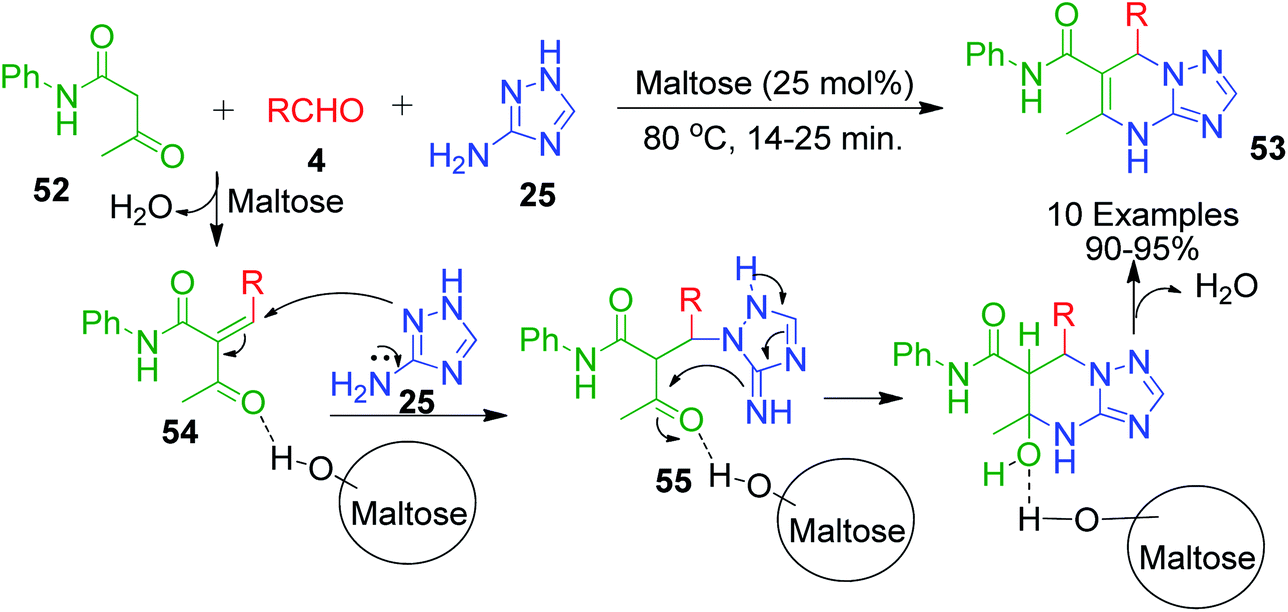
The sugar-catalysed solvent-free one-pot synthesis of [1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide derivatives 53 was accomplished by B. Adrom and co-workers. In this method, a mixture of 3-amino-1,2,4-triazole 25, aryl aldehyde 4 and acetoacetanilide 52 was heated with maltose as the acidic catalyst at 80 °C under solvent-free conditions (Scheme 18).58 The reaction was proposed to proceed through the usual pathway, in which the in situ formation of intermediate 54 from acetoacetamide 52 and activated aldehyde 4 occurred followed by nucleophilic attack of 3-amino-1,2,4-triazole 25 in the presence of the catalyst. The resulting species 55 upon cyclisation afforded the desired product. Maltose, as a carbohydrate, was supposed to promote the reaction through hydrogen bonding.
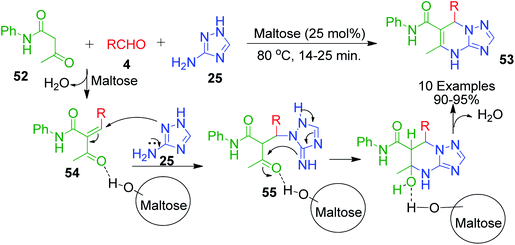 |
| | Scheme 18 Maltose-catalysed solvent-free synthesis of triazolopyrimidine derivatives 53 and its proposed mechanistic route. | |
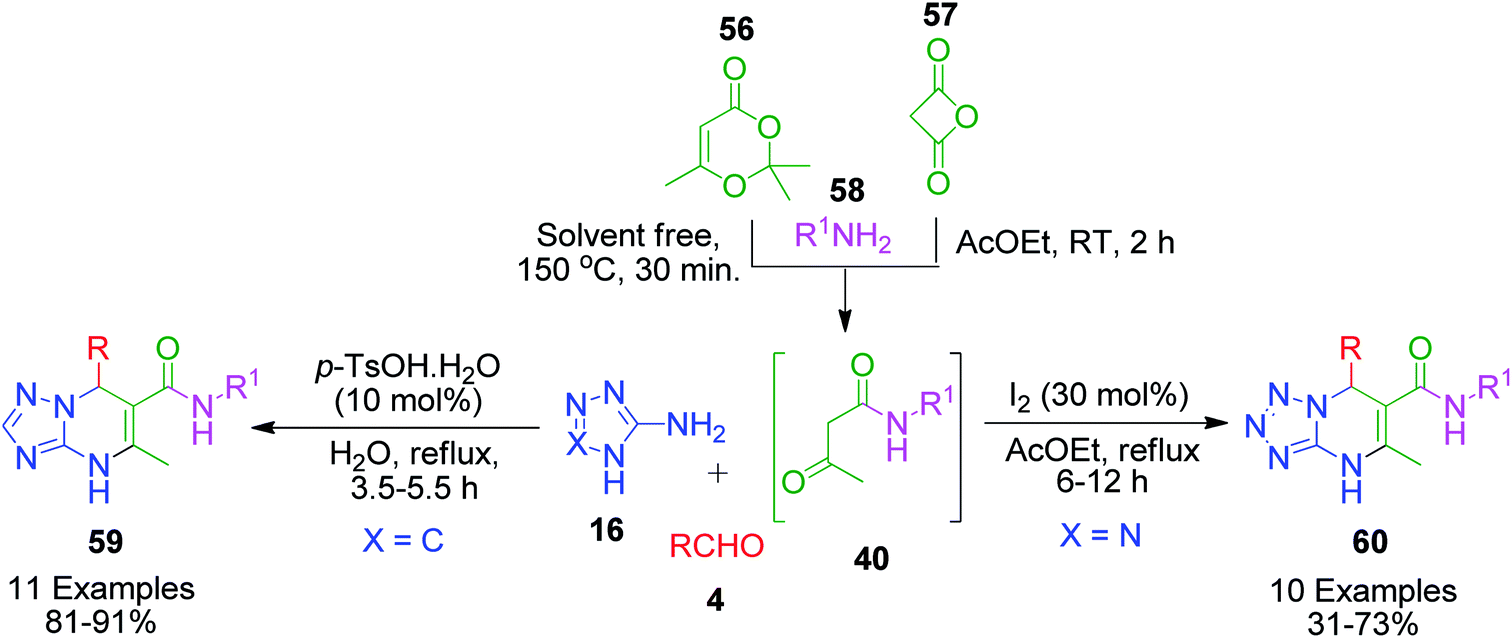
Owing to the pharmacological properties associated with the numerous derivatives of azole-fused pyrimidines, an extended Biginelli reaction was reported, wherein N-alkyl acetoamide 40 was generated in situ by the reaction of amines 58 with compounds such as diketene 56, and 2,2,6-trimethyl-4H-1,3-dioxin-4-one 57. For instance, Zeng et al. presented a four-component tandem procedure to prepare a series of dihydrotetrazolopyrimidinyl carbamides 60.59 N-alkyl acetoamide 40 was generated by stirring a solution containing amines 58 and diketen 56 in ethyl acetate at room temperature for 2 h. Subsequently, the temperature was increased to 78 °C and 5-aminotetrazole 16 and aldehyde 4 were added together with iodine (30 mol%). It was observed that aryl amine did not work given that combining aniline with diketen did not afford the corresponding product even after 24 h. A similar and greener approach was introduced by A. Shaabani et al., in which 2,2,6-trimethyl-4H-1,3-dioxin-4-one 56 was combined with different alkyl amines 58 at 150 °C for the in situ generation of N-alkyl acetoamide 40. The intermediate was then allowed to participate in the MCR with various aldehyde 4 and 3-amino-1,2,4-triazole 16 in the presence of a catalytic amount of p-toluenesulfonic acid (PTSA) in water for the synthesis of [1,2,4]triazolo[1,5-a]pyrimidine-6-carboxamide derivatives 59 (Scheme 19).60
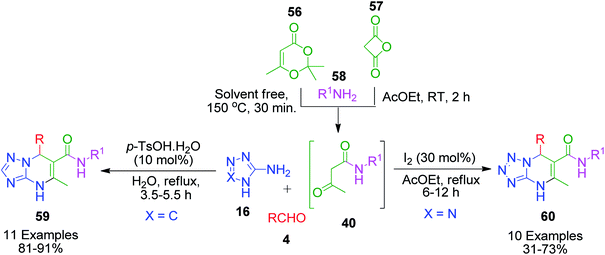 |
| | Scheme 19 In situ generation of N-alkyl acetoamide 60 and its use in the construction of triazole 61 and tetrazole fused pyrimidines 62. | |
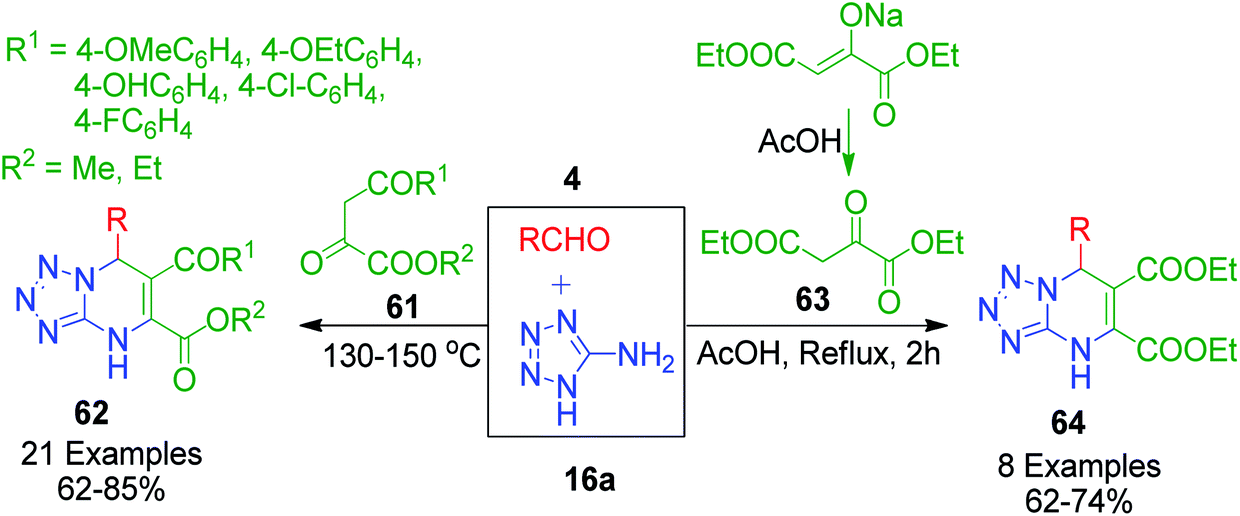
The synthesis of tetrazolo-fused pyrimidines utilizing pyruvic acid derivatives as the 1,3-dicarbonyl compound was accomplished by V. L. Gein et al. through multicomponent heterocyclisation reactions. In a catalyst-free and solvent-free approach, they performed a three-component condensation reaction between various pyruvic acid derivatives 61 and 63 with 5-aminotetrazole 16a and aromatic aldehydes 4 at 130–150 °C. The corresponding products were obtained in 62–85% yield after 30 min when the evolution of gas ceased and mixture was solidified.61 The same authors also described a condensation protocol in which diethyl 2-oxobutanedioate sodium salt was in situ converted into diethyl 2-oxobutanedioate 61, which was reacted with aromatic aldehydes 4 and tetrazol-5-amine 16a in acetic acid under reflux conditions for 2 h. Subsequently, the mixture was then kept at room temperature for 24 h and the corresponding diethyl 6-aryl-3,6-dihydrotetrazolo[1,5-a]pyrimidine-4,5-dicarboxylates 62 and 64s were filtered as solid precipitates (Scheme 20).62 These condensations were assumed to proceed through the initial Knoevenagel condensation reaction of aldehydes and pyruvic acid derivatives, and then the as-formed unsaturated intermediate experiences nucleophilic attack from the tetrazolo amine followed by intramolecular cyclisation and dehydration. Moreover, the data before 2015 concerning the application of pyruvic acid in multicomponent heterocyclisation with aminoazoles was compiled and presented in the form of a review titled “Heterocyclization reactions of pyruvic acids and aminoazoles with controlled chemoselectivity” by Y. I. Sakhno et al.63
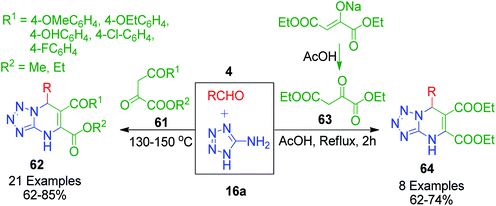 |
| | Scheme 20 Synthesis of tetrazolopyrimidines using pyruvic acid and its sodium salt as 1,3-dicarbonyl compounds. | |
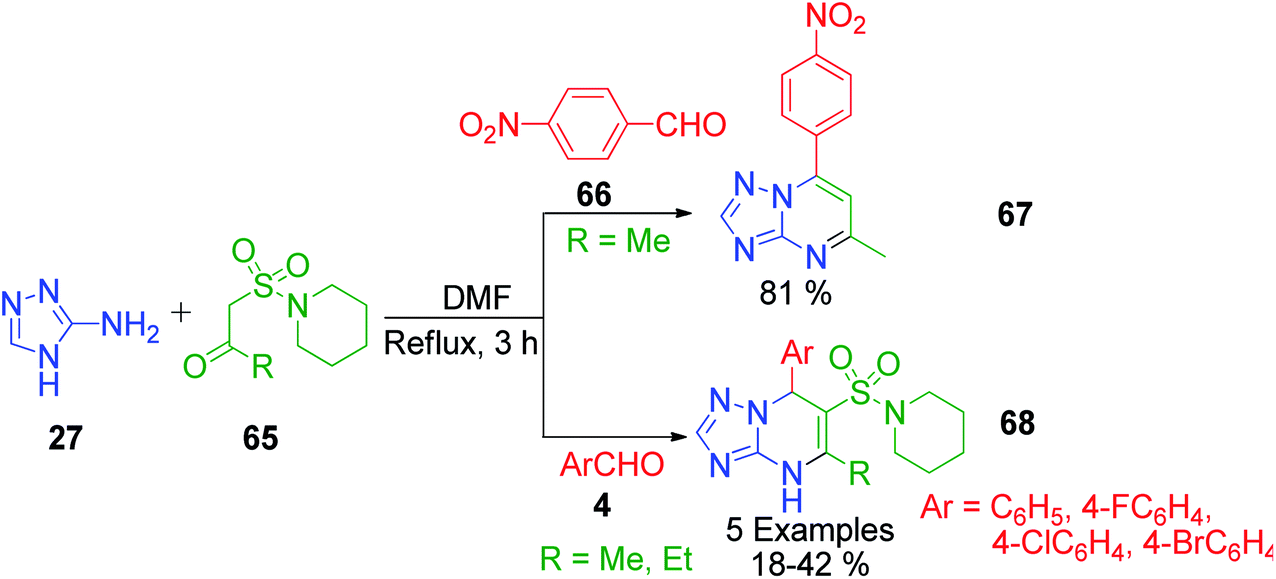
New pyrimidine nuclei containing scaffolds with increased biological application are the basic demands for the development of new strategies. In this context, sulfonamide-linked pyrimidine derivatives possessing exciting biological properties were prepared by M. A. Kolosov et al. through a three-component condensation protocol. 5-Alkyl-6-(1-piperidinylsulfonyl)-4,7-dihydro[1,2,4]triazolo[1,5-a]pyrimidine derivatives 68 were obtained when a mixture containing β-sulfonamide ketone, aromatic aldehydes 4, and 3-amino-1,2,4-triazole 27 was refluxed in DMF for 3 h (Scheme 21).64 The starting material, 1-(1-piperidinylsulfonyl)acetones 65, was synthesized sequentially from 1-(methylsulfonyl)piperidine undergoing metalation with n-butyllithium, reaction with aliphatic aldehydes, and oxidation of the obtained sulfoalcohols. The interesting feature of the present method is the formation of a different type of heteroaromatic derivative, i.e., 7-(4-nitrophenyl)[1,2,4]triazolo[1,5-a]pyrimidine 68, if 4-nitrobenzaldehyde 66 is employed as the aromatic aldehyde. This heteroaromatization is generally characteristic to 4-nitro derivatives of dihydroazoloazines, and in this case, 4-nitrobenzaldehyde 66 triggered the elimination of the sulfonamide moiety.
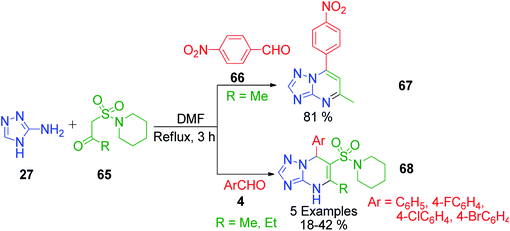 |
| | Scheme 21 Synthesis of 7-(4-nitrophenyl)[1,2,4]triazolo[1,5-a]pyrimidine 69 and 5-alkyl-6-(1-piperidinylsulfonyl)-4,7-dihydro[1,2,4]triazolo[1,5-a]pyrimidine 70 derivatives. | |
Next, the utility of β-ketosulfones as molecular scaffolds for the synthesis of pyrazolo[1,5-a]pyrimidine was described by M. R. Shaabana et al. with the development of one-pot three-component procedures, wherein the reaction between 1-aryl-2-(phenylsulfonyl)ethanone derivatives 69, 3-amino-1,2,4-triazole/2-aminobenzimidazole (3a, 28 and 71) and triethyl orthoformate 70 was carried out at refluxing temperature for 4 h in the presence of piperidine (Scheme 22). Here, triethyl orthoformate seems to act as a methyne-furnishing agent to form the ring structure of the products. The corresponding products 7-aryl-6-(phenylsulfonyl)[1,2,4]triazolo[1,5-a]pyrimidines 72 and 2-aryl-3-(phenylsulfonyl)pyrimido[1,2-a]benzimidazoles 73 were obtained in 80–83% yield and evaluated for Aurora-A kinase inhibitor activity.65
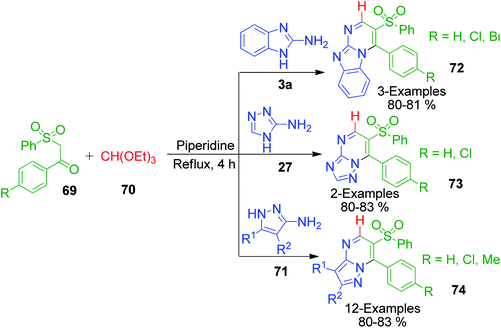 |
| | Scheme 22 Synthesis of pyrazolo[1,5-a]pyrimidines 72, 73 and 74 tethered with a sulfone moiety. | |
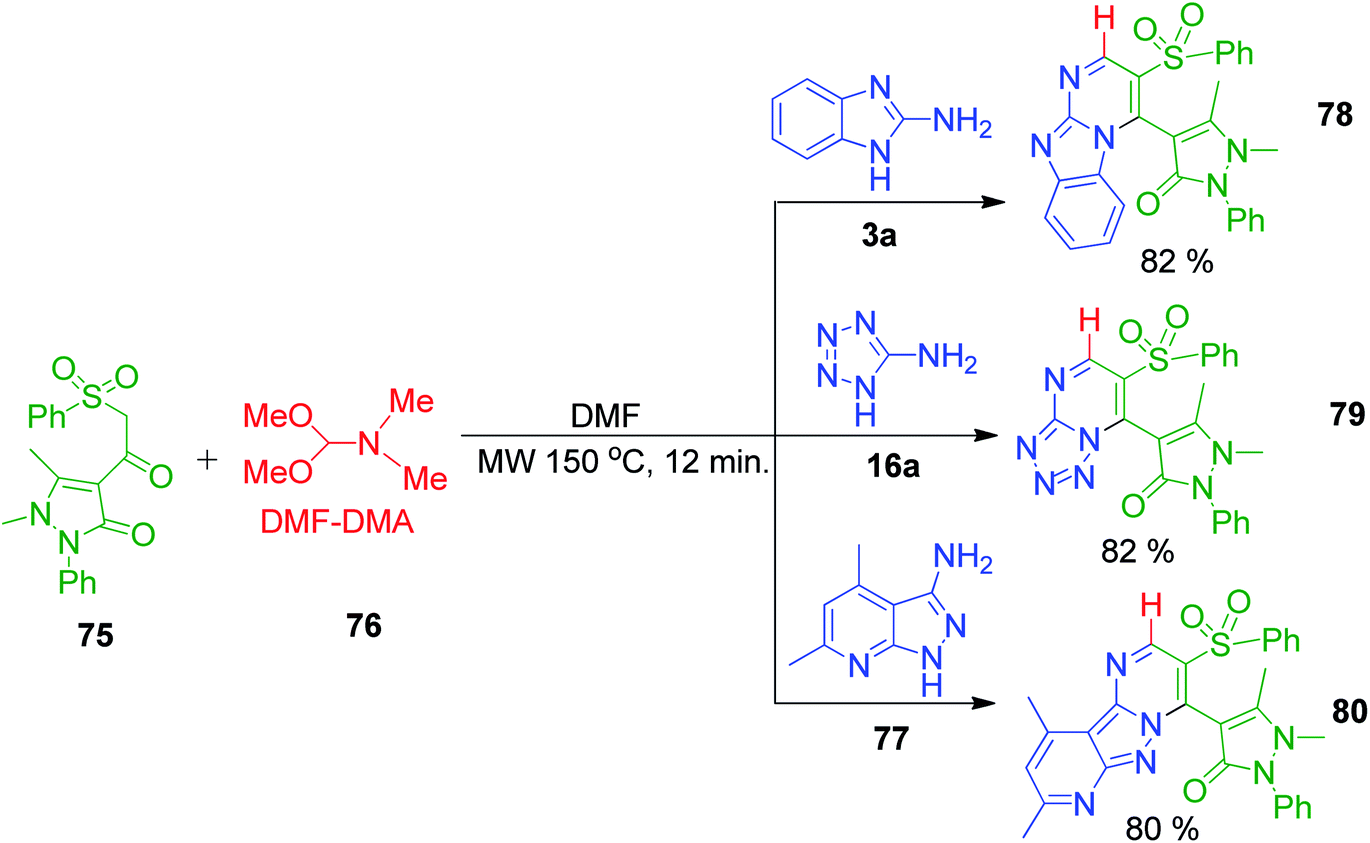
Furthermore, S. M. Gomha and coworkers also demonstrated the similar utility and greater versatility of the complex β-ketosulfones, 1,5-dimethyl-2-phenyl-4-(2-(phenylsulfonyl)acetyl)-1H-pyrazol-3(2H)-one 75. They performed a reaction between dimethylformamide dimethylacetal (DMF-DMA) 76, a methyne-furnishing reagent, various amino azoles (3a, 16a and 77) and β-ketosulfones under microwave irradiation at 150 °C for 12 min to synthesize several azole-fused pyrimidine derivatives 78, 79 and 80 (Scheme 23).66
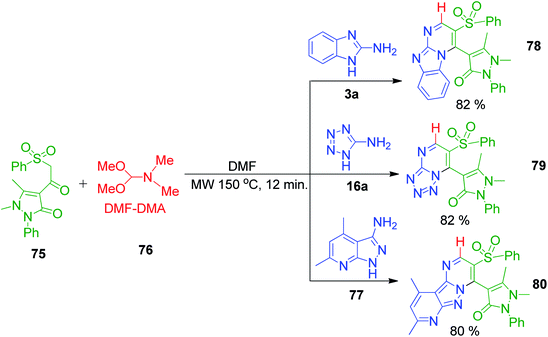 |
| | Scheme 23 Microwave-assisted synthesis of azole-fused pyrimidine derivatives 78, 79 and 80. | |
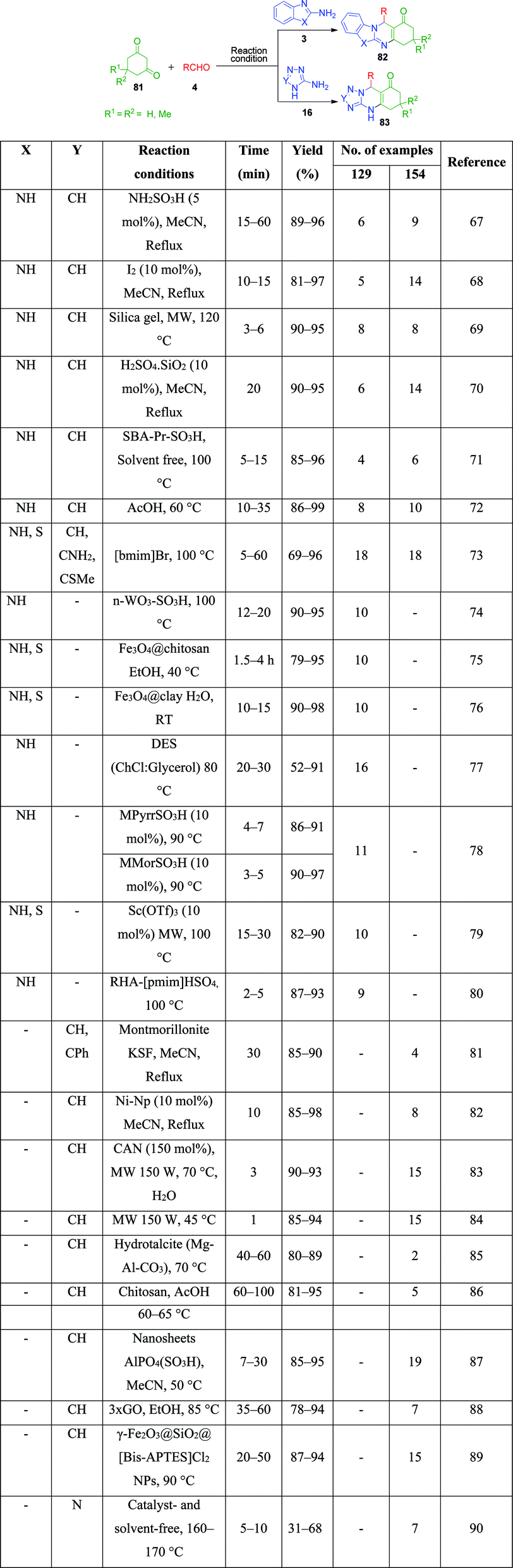
Furthermore, a number of methods have been reported where triazolo and benzimidazolo quinazolinone rings are constructed with the same catalytic system, exhibiting their versatility. In 2010, the use of sulfamic acid as a green and reusable catalyst-based approach was developed by M. M. Heravi et al. for the efficient and convenient one-pot three-component condensation of 2-amino benzimidazole 3/3-amino-1,2,4-triazole 16 as amine sources with dimedone 81 and different aldehydes 4 to afford corresponding triazolo/benzimidazoloquinazolinones 82 and 83.67 They also examined the same reaction using indandione instead of dimedone, but the reaction did not proceed beyond the Knoevenagel condensation of indandione and aldehyde. Other strategies to obtain these quinazolinone systems with catalytic systems include the use of iodine,68 silica gel,69 silica-supported sulfuric acid,70 sulfonic acid-functionalized SBA-15 as a heterogeneous nanoporous solid acid catalyst,71 acetic acid (performing dual role of acid and solvent),72 and 1-butyl-3-methylimidazolium bromide ([bmim][Br]).73
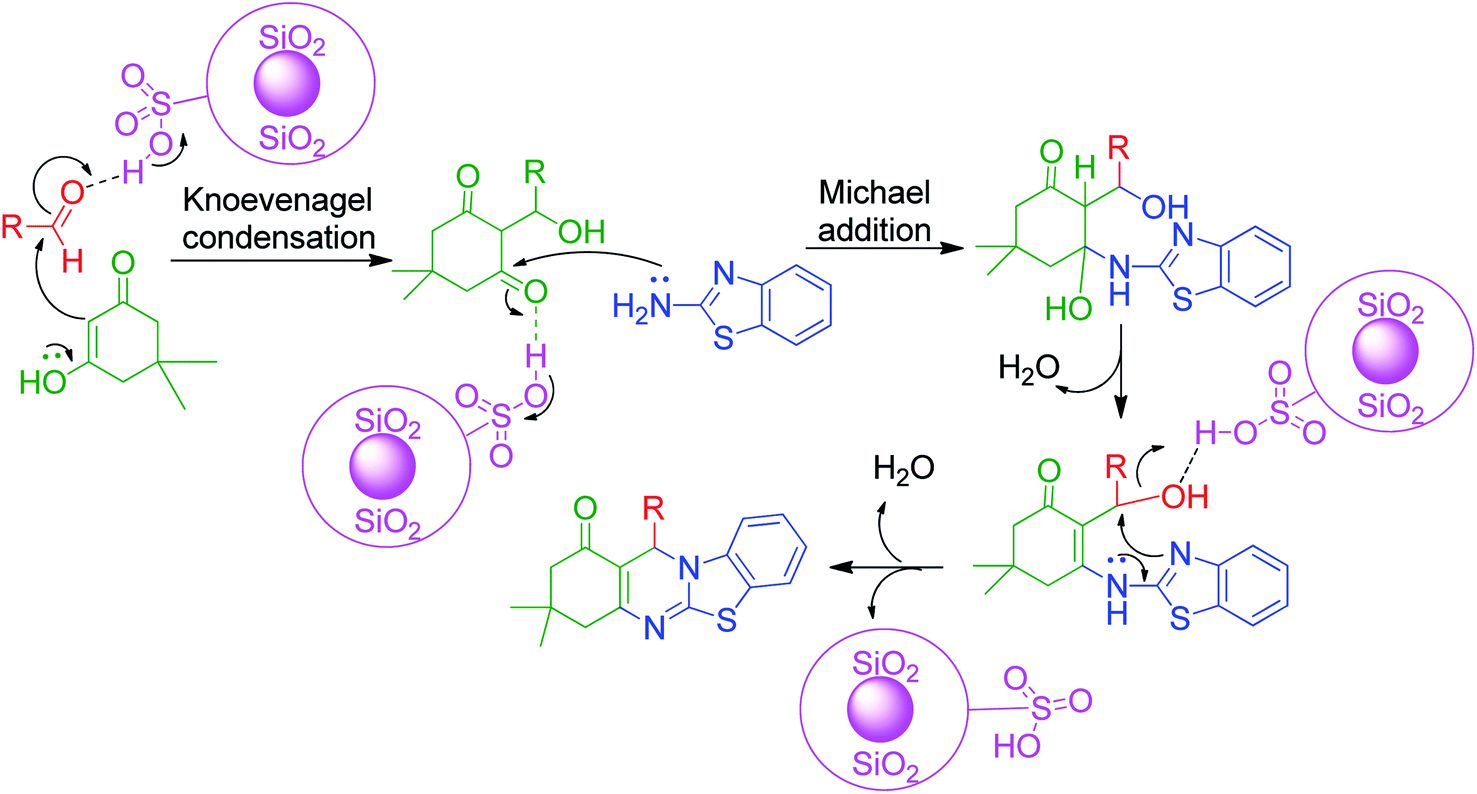
Nano-WO3-supported sulfonic acid was employed by A. Amoozadeh and S. Rahmani as an efficient and recyclable catalyst for the synthesis of benzimidazolo quinazolinone derivatives based on the three-component reaction between 2-aminobenzimidazole, 1,3-cyclohexandione derivatives and various aldehydes under solvent-free conditions. The products were isolated after 12–20 min in excellent yields.74 To date, various practical approaches have been developed, wherein the same starting materials are being reacted in the presence of catalysts such as Fe3O4@chitosan,75 Fe3O4@clay nanocomposite,76 choline chloride:glycerol as a deep eutectic solvent,77 1-methyl-1-sulfonic acid pyrrolidinium chloride [MPyrrSO3H]Cl/4-methyl-4-sulfonic acid morpholinium chloride [MMorSO3H]Cl,78 Sc(OTf)3,79 Brönsted acidic ionic liquid supported on rice husk ash (RHA-[pmim]HSO4),80 montmorillonite KSF as a heterogeneous catalyst,81 nickel nanoparticles,82 CAN,83 microwave irradiation at 150 W,84 hydrotalcite (Mg–Al–CO3),85 chitosan,86 nanosheets AlPO4(SO3H),87 graphene oxide (GO),88 γ-Fe2O3@SiO2@[bis-APTES]Cl2 nanoparticles,89 and catalyst- and solvent-free conditions at 160–170 °C90 by various research groups. All these methods were proven to be highly efficient, giving the products in excellent yields in a shorter time (Scheme 24).
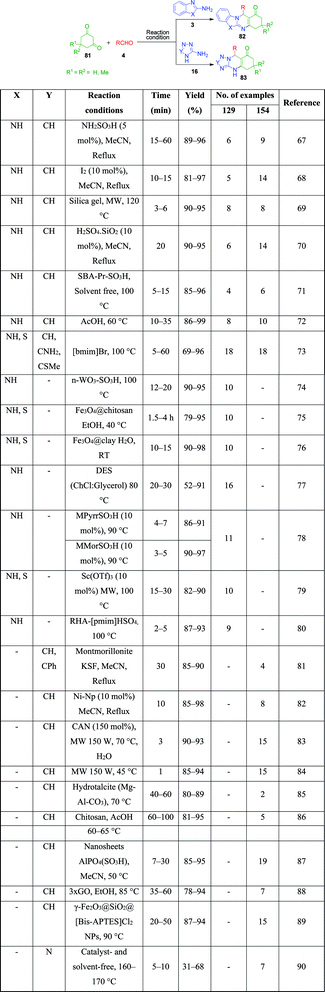 |
| | Scheme 24 Variety of acid catalysts used for the construction of triazolo 82 and benzimidazoloquinazolinone ring 83. | |
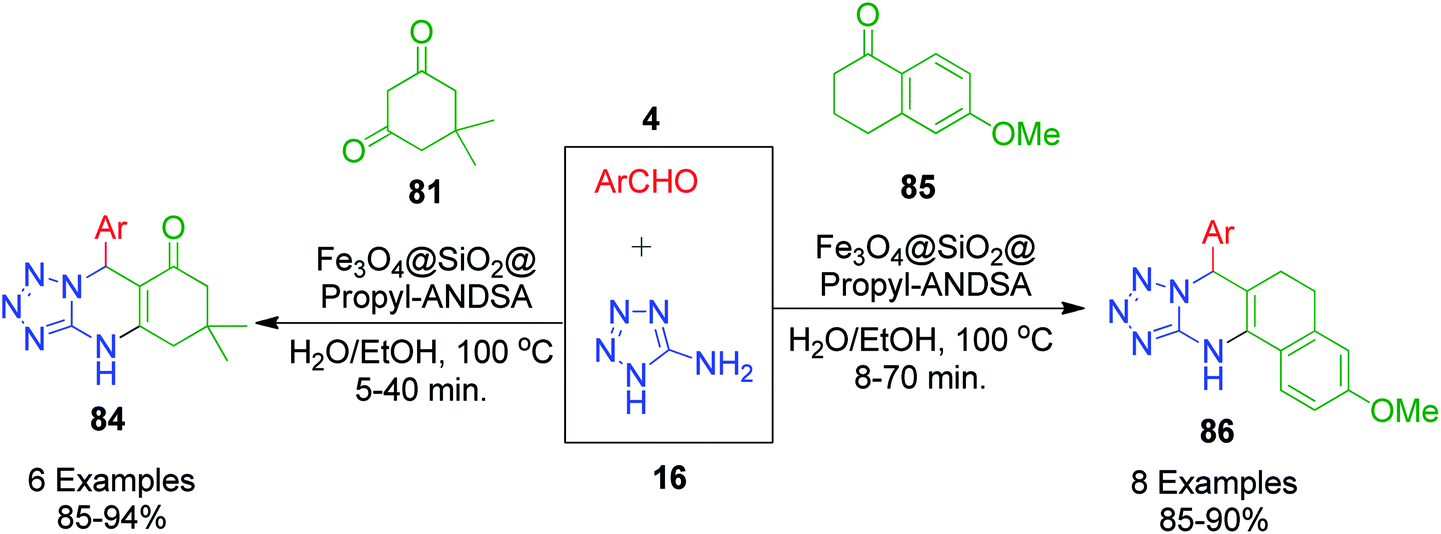
R. Ghorbani-Vagheia et al. reported the 7-aminonaphthalene-1,3-disulfonic acid-functionalized magnetic Fe3O4 nanoparticles (Fe3O4@SiO2@Propyl-ANDSA)-catalyzed one-pot synthesis of tetrahydrotetrazolo[1,5-a]quinazolines/tetrahydrobenzo[h]tetrazolo[5,1-b]quinazolines 84 and 86 by the reaction of 5-aminotetrazole 16 aromatic aldehydes 4, and dimedone/6-methoxy-3,4-dihyronaphtalen-1(2H)-one (81 and 85 at 100 °C in H2O/EtOH medium (Scheme 25).91
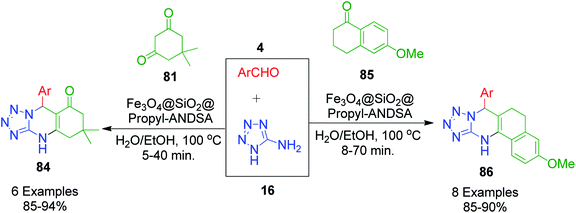 |
| | Scheme 25 Magnetic Fe3O4 nanoparticle-catalyzed synthesis of quinazolines derivatives 84 and 86. | |
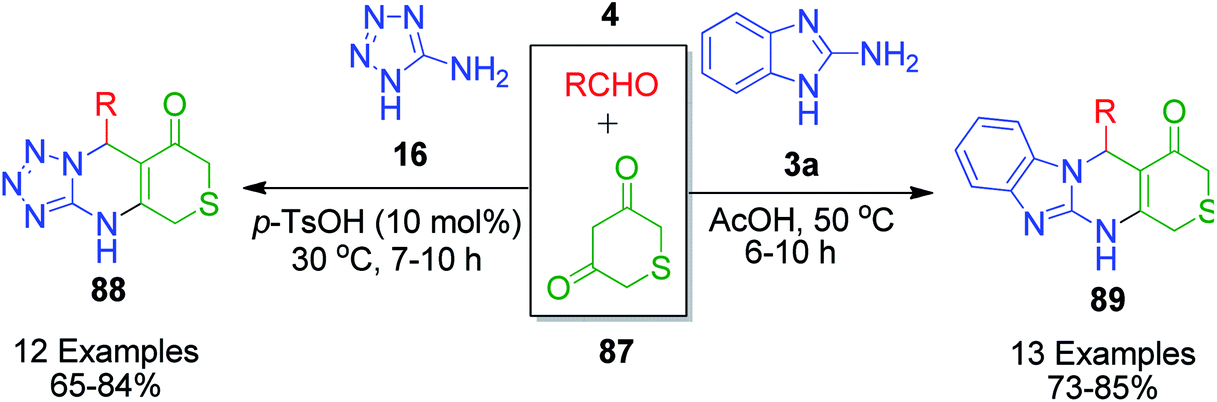
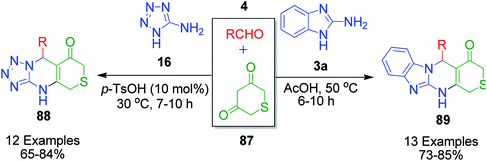
A closely related structural analogue of dimedone, 2H-thiopyran-3,5(4H,6H)-dione 87, was utilized by S. Shen's group as 1,3-cyclicdicarbonyl compounds in their two successive publications to synthesize a series of novel thiopyran-fused pyrimidine derivatives 88 and 89. In 2011, benzoimidazole and thiopyran-fused novel pyrimidine frameworks were constructed through the MCR between 2-aminobenzothiazole 3a, aryl aldehyde and 2H-thiopyran-3,5(4H,6H)-dione in glacial acetic acid at moderate temperature (50 °C).92 The reaction proceeded smoothly with aromatic aldehydes, bearing both electron-donating and electron-withdrawing groups, whereas the same reaction with aliphatic and heteroaromatic aldehydes did not afford the expected products. After the successful synthesis of these fused pyrimidine derivatives, they further explored 2H-thiopyran-3,5(4H,6H)-dione 87 in the synthesis of a novel series of tetrazolo[1,5-a]thiopyrano[3,4-d]pyrimidine 88 derivatives by the reaction of 5-aminotetrazole 16, aryl aldehyde 4 and 2H-thiopyran-3,5(4H,6H)-dione 87 in the presence of PTSA at 30 °C (Scheme 26).93 Besides PTSA, other Brønsted acidic catalysts such as sulfamic acid, sulfuric acid, hydrochloric acid, and nitric acid and Lewis acidic catalysts such as ferric chloride, ferrous chloride and aluminium chloride were also utilized to optimize the reaction conditions.
 |
| | Scheme 26 Synthesis of thiopyran-fused pyrimidine derivatives. | |
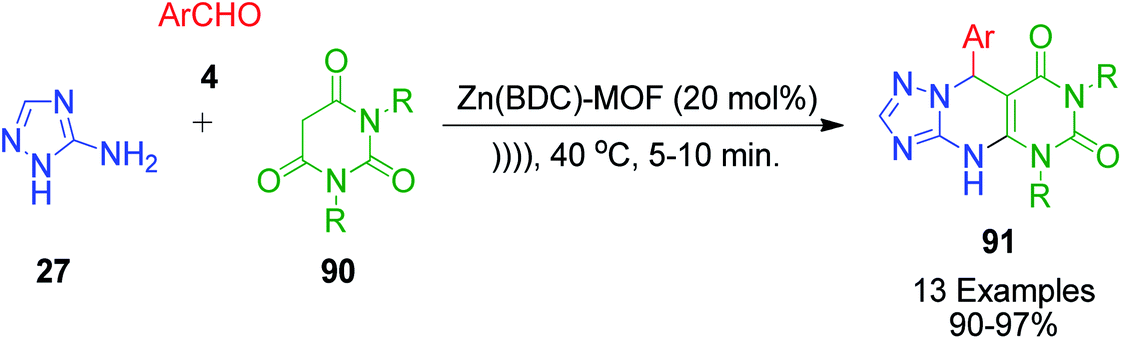
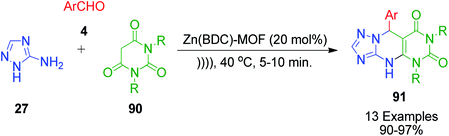
Barbituric acid derivatives 90 were explored as cyclic 1,3-dicarbonyl compounds in the reaction with 3-amino-1H-1,2,4-triazoles 27 and aromatic aldehydes 4 by M. H. Abdollahi-Basir and coworkers. This zinc terephthalate metal–organic framework catalyzed one-pot, three-component reaction was conducted under ultrasonic irradiation and solvent-free conditions, affording pyrimido[4,5-d][1,2,4]triazolo[1,5-a]pyrimidinediones 91 with good to excellent yields (Scheme 27).94 The advantage of using the zinc terephthalate metal–organic framework catalyst is that it can be easily recovered after the reaction is over by simple filtration. The recovered catalyst was used in consecutive runs with no significant decrease in the product yield.
 |
| | Scheme 27 Synthesis of pyrimido[4,5-d][1,2,4]triazolo[1,5-a]pyrimidinediones. | |
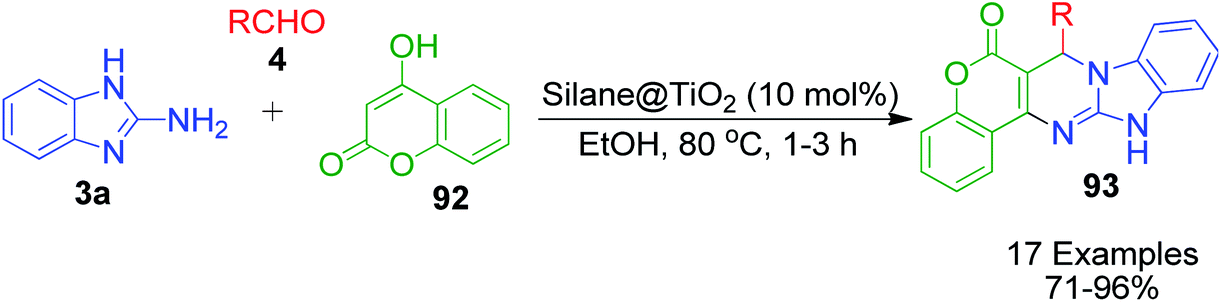
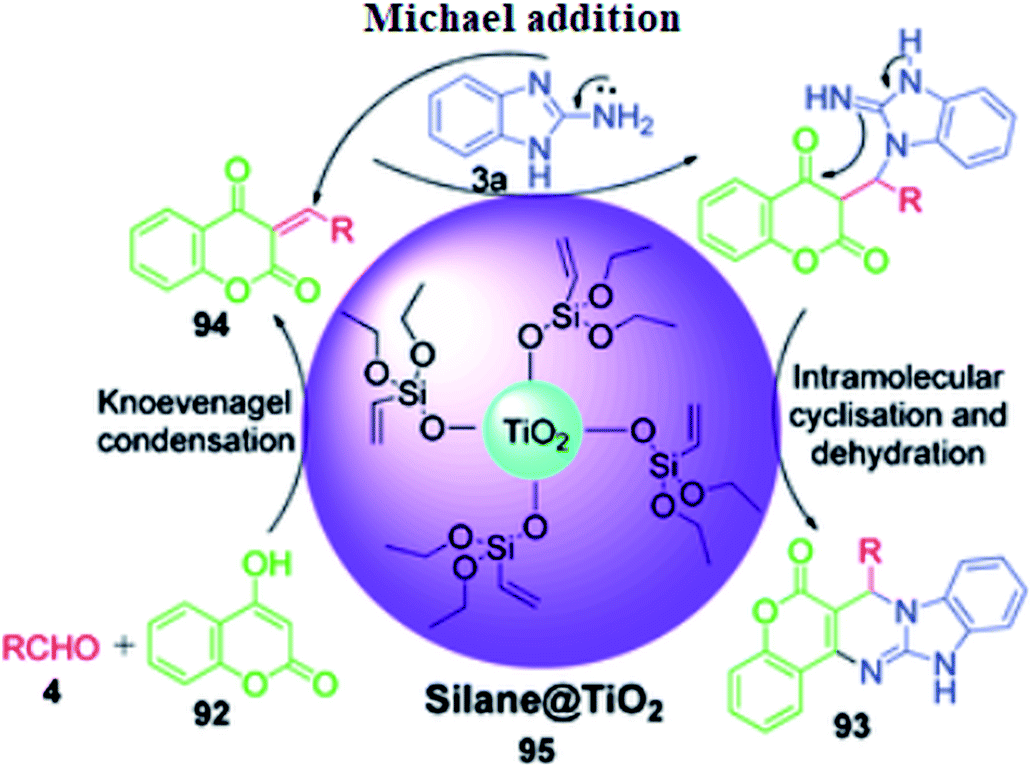
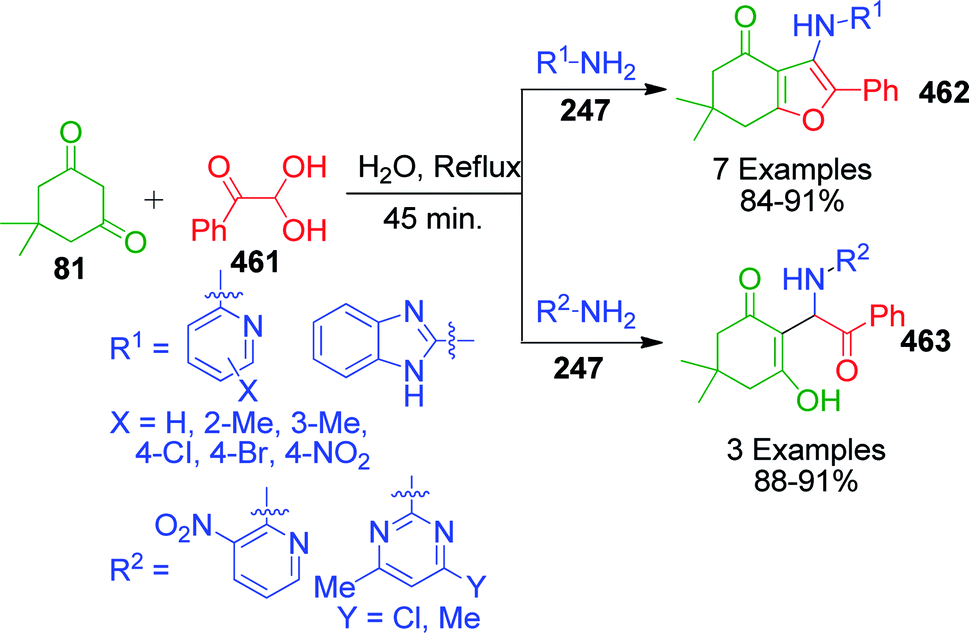
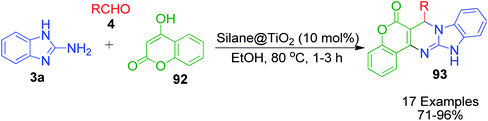
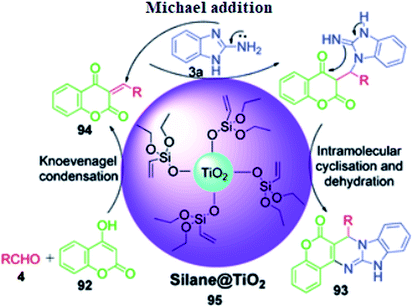
4-Hydroxycoumarine 92 and analogous compounds are interesting cases of 1,3-dicarbonyl-type compounds with which the scope of the Biginelli reaction has greatly been studied for the synthesis of extensively fused pyrimidine structures. Developments in this regard include V. N. Mahire and coworker's environmentally benign protocol, which afforded chromene fused benzoimidazopyrimidinones 93 via the reaction of 4-hydroxycoumarin 92, aldehydes 4 and 2-aminobenzimidazole 3a using silane@TiO2 nanoparticles 95 as a heterogeneous catalyst under reflux conditions in ethanol (Scheme 28).95 The mechanism proposed to explain this conversion starts with the usual Knoevenagel condensation between 4-hydroxycoumarin 92 and benzaldehyde 4 to form a Schiff base as intermediate 94 and it is assumed that silane@TiO2 nanoparticles 95 activate the carbonyl carbon of the aldehyde through hydrogen bonding. Next, 2-aminobenzimidazole 3a reacts with the intermediate 94 through Michael addition followed by cyclization and dehydration to give the desired products (Scheme 29).
 |
| | Scheme 28 Synthesis of thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one derivatives. | |
 |
| | Scheme 29 Proposed mechanism for the synthesis of thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one derivatives 93. | |
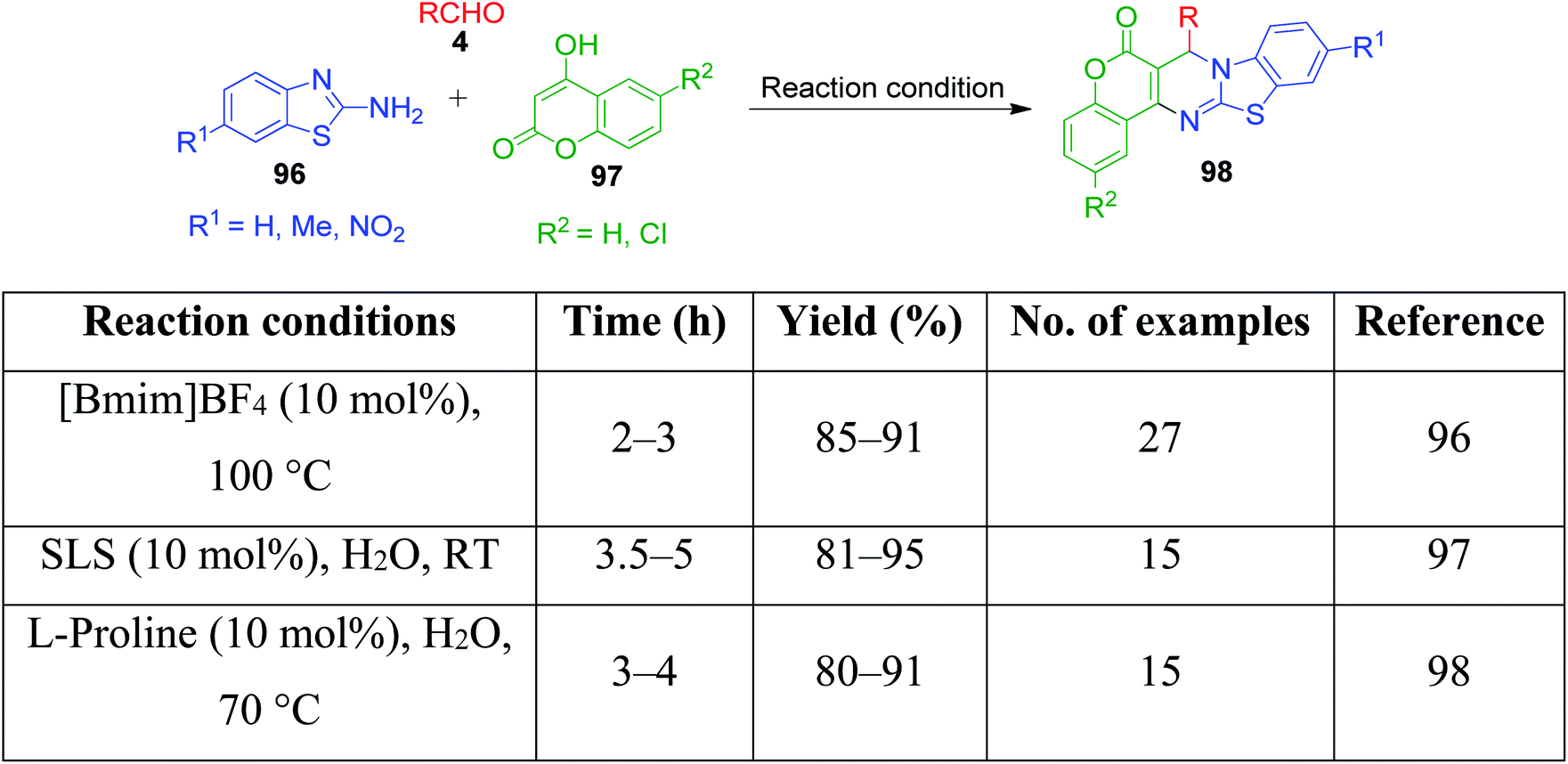
A combination of substituted 2-benzothiazole 96 and 4-hydroxycoumarine 97 with different aldehydes 4 was employed by A. V. S. Reddy and Y. T. Jeong to present a versatile one-pot multicomponent approach for the preparation of substituted fused chromenopyrimidine rings 98. In this approach, a mixture of the above-mentioned reactants was condensed in the presence of an ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate. The optimization of the reaction conditions over the reaction mixture of 4-hydroxycoumarin, 4-ethoxybenzaldehyde and 2-aminobenzothiazole under solvent-free conditions revealed that the best result in terms of reaction time and product yield was achieved with 10 mol% of ionic liquid at 100 °C. The authors also showed that the ionic liquid can be reused and recycled for at least five consecutive runs without the loss in activity.96 Conversely, P. K. Sahu catalyzed a one-pot reaction containing an aqueous mixture of the same three components using a surfactant, sodium lauryl sulphate (SLS), at room temperature. The study of the influence of the sodium lauryl sulphate (SLS) micelles and their different concentrations on reactivity showed that the best yield was obtained with 10 mol% catalyst loading in the least time compared to the other surfactants and catalysts.97 P. K. Sahu et al. developed a mild and efficient variant of the above-mentioned protocol, wherein the reaction involving the same three-components was catalyzed by the L-proline in water.98 A brief summary regarding the reaction conditions, reaction time, range of the product yields, etc. is given in Scheme 30.
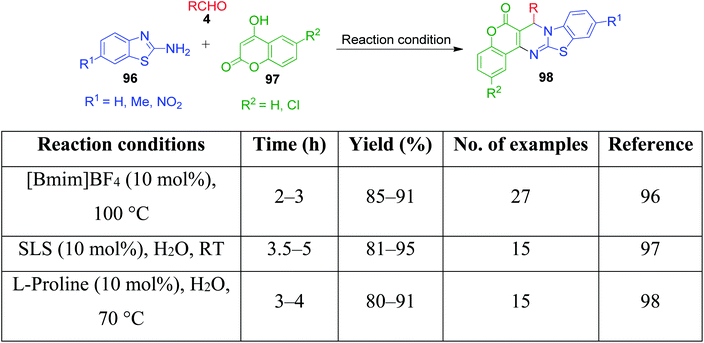 |
| | Scheme 30 Different methods to synthesize thiazolo[3,2-a]chromeno[4,3-d]pyrimidin-6(7H)-one derivatives 98. | |
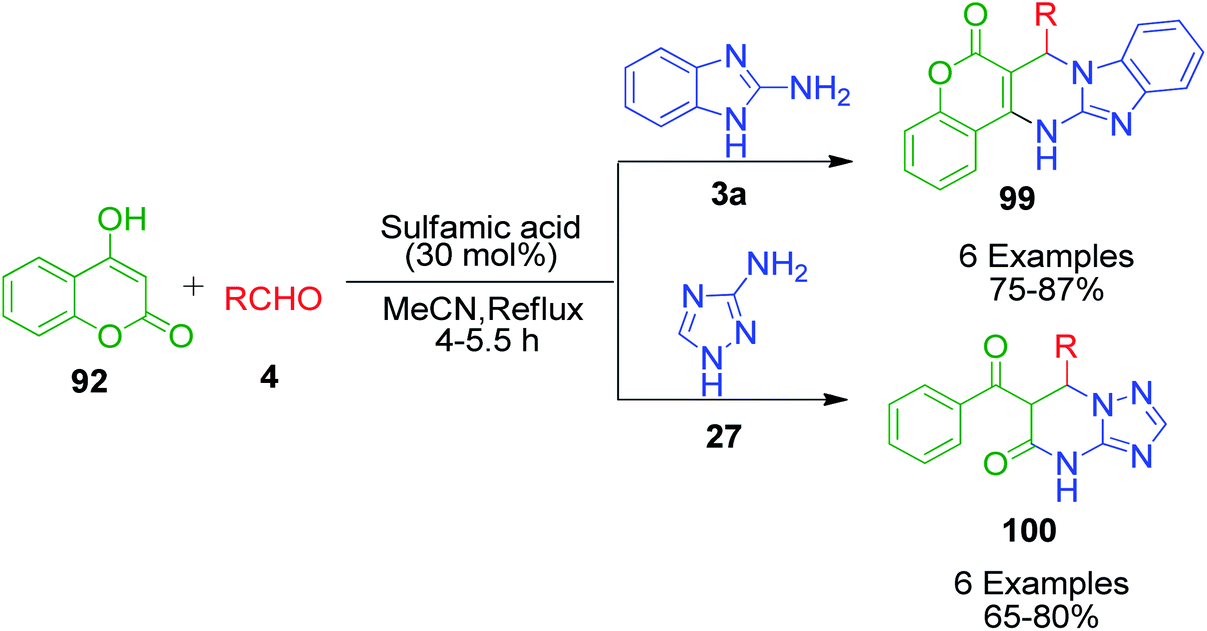
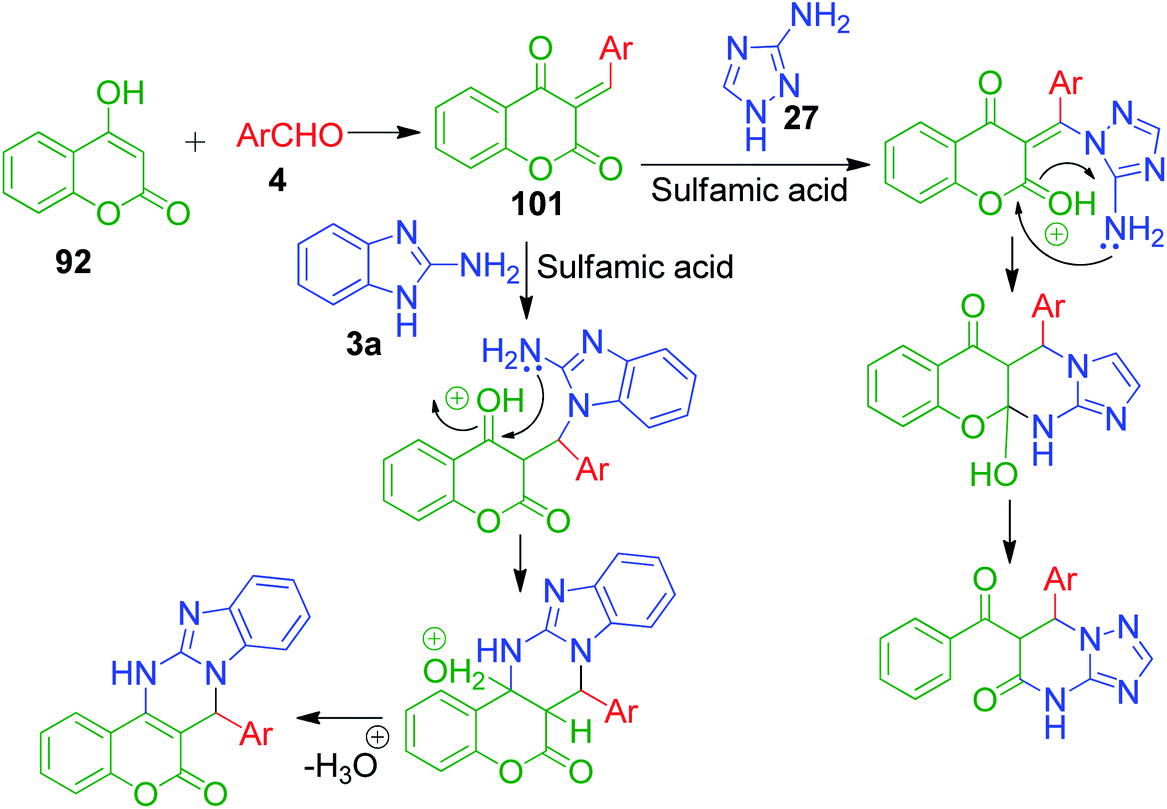
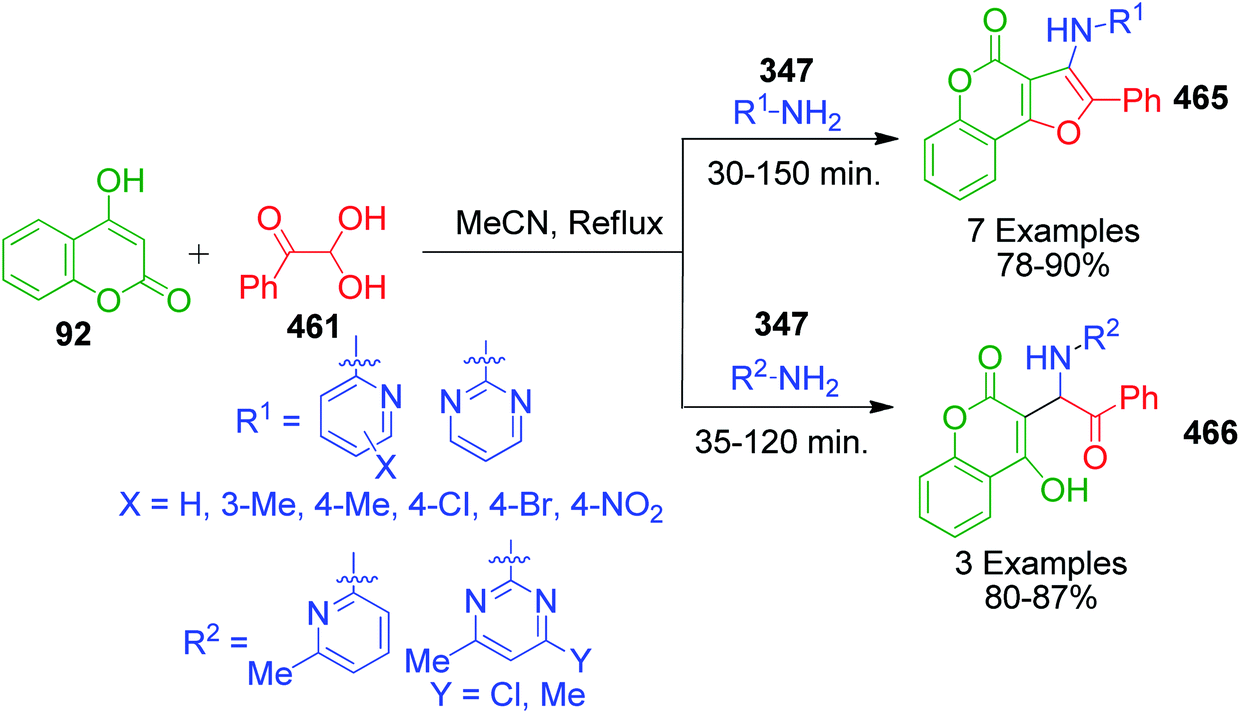
An interesting strategy in which the ester linkage of 4-hydroxycoumarine 92 is opened upon the sulfamic acid-catalyzed reaction of 4-hydroxycoumarin 92, aromatic aldehydes 4 and 3-amino-1H-1,2,4-triazole 27 was reported by M. M. Heravi and coworkers. The resulting compound is the simple triazolopyrimidinone derivatives 100. When the authors performed this one-pot three-component reaction with 2-aminobenzimidazole 3a, an usual chromene-fused benzimidazolopyrimidinone 99 was obtained (Scheme 31).99 Based on the proposed mechanism, the lactone carbonyl group of intermediate 101, which is formed as a result form the combination of 4-hydroxycoumarin 92 and aldehyde 4, is prone to attack by the nucleophilic nitrogen of 3-amino-1H-1,2,4-triazole 27 or 2-aminobenzimidazole 3a (Scheme 32).
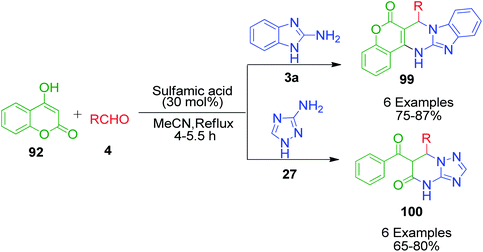 |
| | Scheme 31 Sulfamic acid-catalyzed synthesis of fused pyrimidine derivative. | |
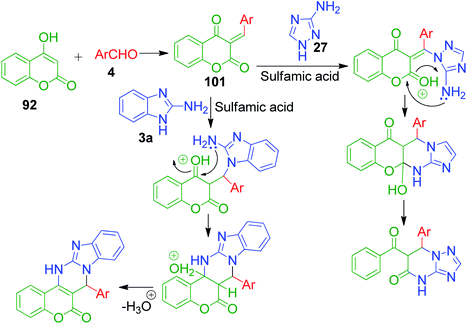 |
| | Scheme 32 Proposed pathways to fused pyrimidine derivative. | |
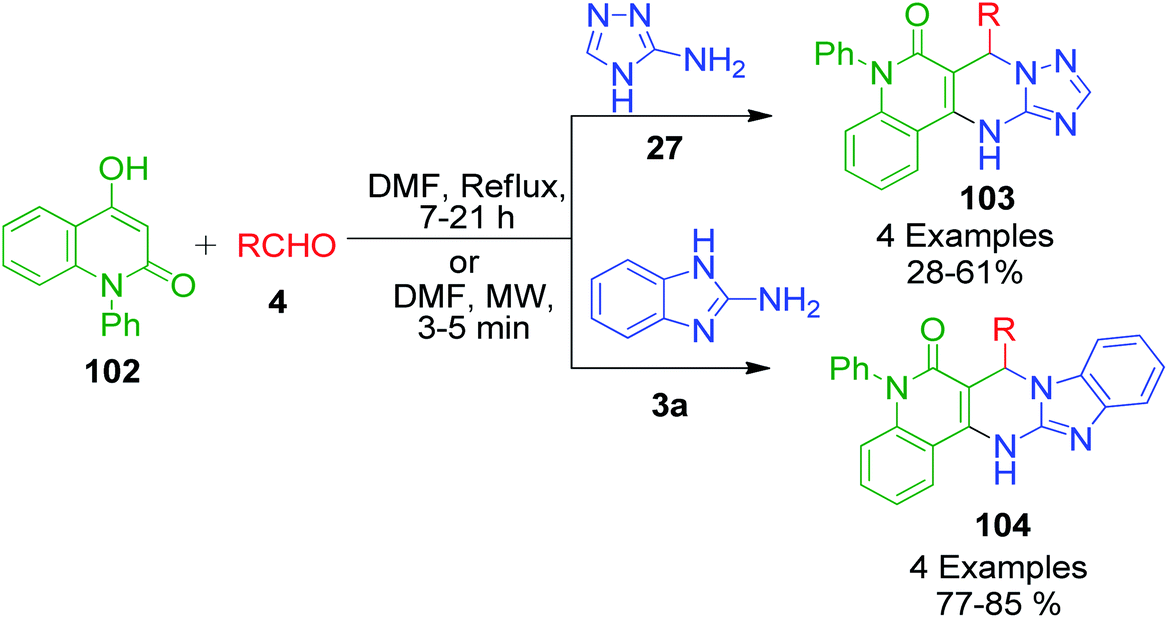
4-Hydroxy-1-phenylquinolin-2(1H)-one 102, a structural variant of 4-hydroxycoumarine, was used in the Biginelli reaction together with aryl aldehydes 4 and aminotriazole/aminobenzimidazole 27 and 3a by Mourad et al. under the classical and microwave heating.100 Around 7–21 h was required to complete the reaction when a DMF solution of all the starting materials was allowed to stir under classical heating, whereas microwave heating triggered the same reaction to completion within 5 min (Scheme 33). Besides the advantageous reduction in reaction time in the latter case, the yields of products 103 and 104 were also observed to be greatly enhanced to an excellent level.
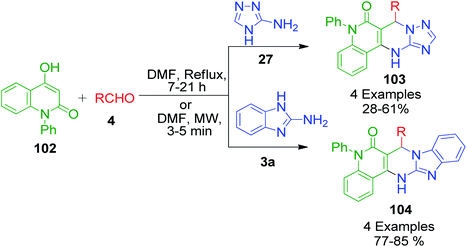 |
| | Scheme 33 Synthesis of triazole/benzimidazole-fused pyrimidine derivatives. | |
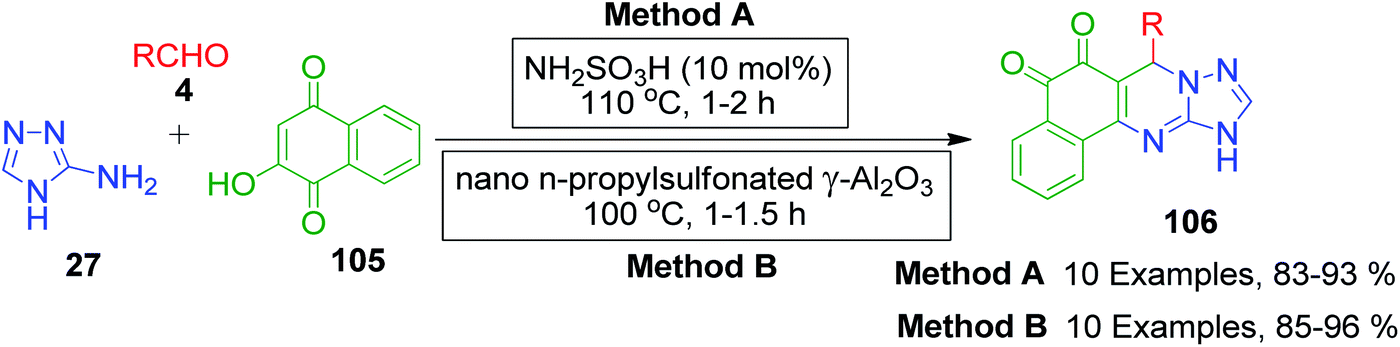
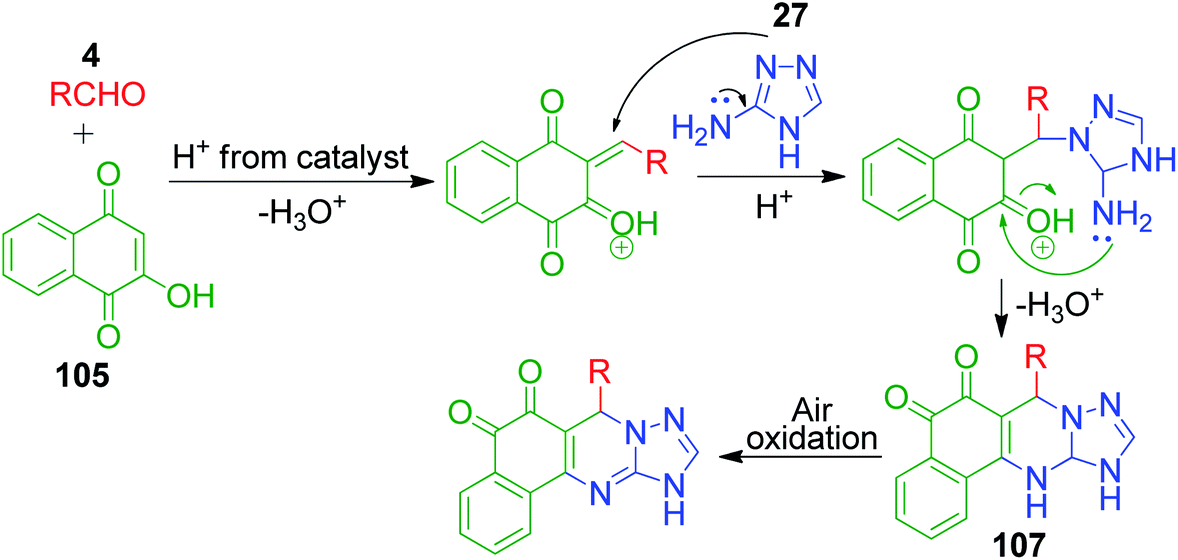
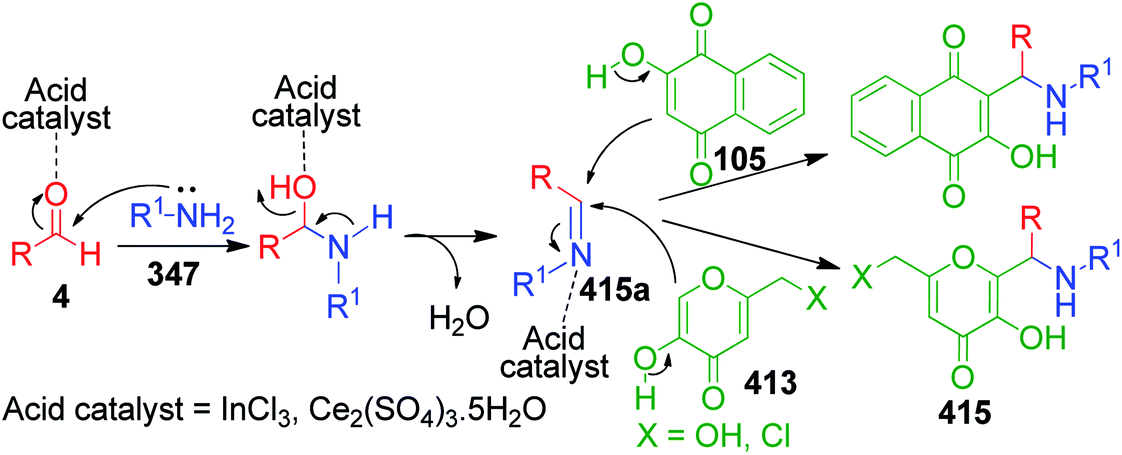
Naphthoquinones, which possess a cyclic β-keto structural unit, constitute a major class of naturally occurring compounds with potential medicinal properties. L. Wu et al. employed this precursor in the MCR synthesis of various structurally diverse quinazolinedione derivatives under different catalytic systems. The three-component coupling of aldehyde 4, 2-hydroxy-1,4-naphthoquinone 105 and 3-amino-1,2,4-triazole 27 was achieved in the presence of sulfamic acid under solvent-free conditions to produce a novel series of 6-aryl substituted triazoloquinazolindione derivatives 106 in good yields and high regioselectivity.101 In another paper published in the same year, they demonstrated nano n-propylsulfonated γ-Al2O3 as an efficient and reusable catalyst to promote the same conversion. Both solvent-free approaches provided excellent yields of the products within 1–2 h of heating (Scheme 34).102 The common mechanism based on the proposed synthetic route is shown in Scheme 35, which follows the sequence of Knoevenagel reaction, Michael addition reaction, intramolecular cyclisation and dehydration in the presence of catalyst to afford compound 107, which is prone to undergo aerial oxidation to give the target compound.
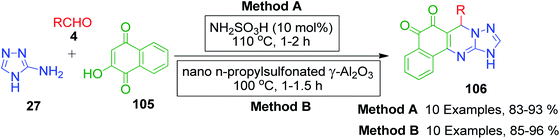 |
| | Scheme 34 Solvent-free approaches for the synthesis of 6-aryl-substituted triazoloquinazolindiones. | |
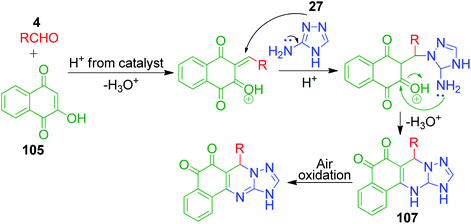 |
| | Scheme 35 Mechanism proposed by L. Wu et al. to explain the synthesis of substituted triazoloquinazolindiones. | |
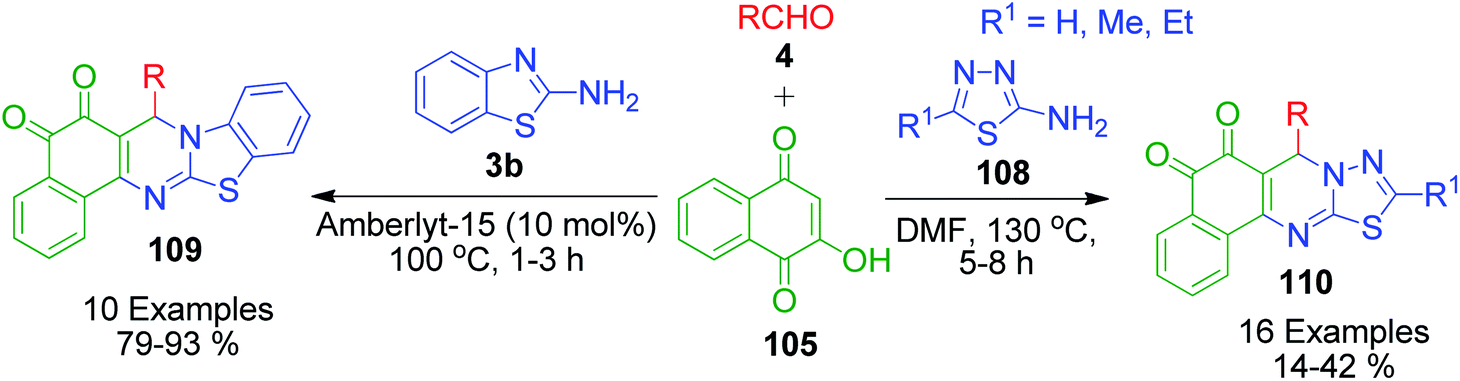
After getting successful results from previous reports, the same authors employed other amino azoles as precursors under modified reaction conditions. A one-pot three-component reaction involving 2-aminobenzothiazole 3b, aromatic aldehydes 4 and 2-hydroxy-1,4-naphthoquinone 105 in the presence of Amberlyst-15 was accomplished under solvent-free conditions to afford the synthesis of novel 13-aryl-13H-benzo[g]benzothiazolo[2,3-b]quinazoline-5,14-dione derivatives 109.103 They also synthesized a series of novel substituted 5H-benzo[i][1,3,4]thiadiazolo[3,2-a]quinazoline-6,7-diones 110 in very good yields through the one-pot condensation of 5-substituted-2-amino-1,3,4-thiadiazole 108 with 2-hydroxy-1,4-naphthoquinone 105 and aldehydes 4 in DMF at 130 °C (Scheme 36).104
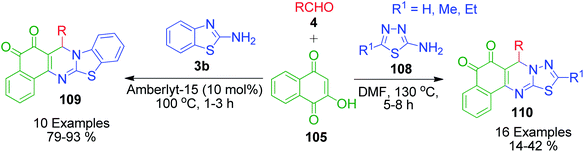 |
| | Scheme 36 Synthesis of novel benzothiazole/thiadiazole fused quinazoline-5,14-dione derivatives 109 and 110 under different reaction conditions. | |
A. Maleki et al. employed a Brönsted acid-functionalized magnetic polymeric nanocomposite, Ba0.5Sr0.5Fe12O19@PU-SO3H, for the synthesis of 7-aryl-benzo[h]tetrazolo[5,1-b]quinazoline-5,6-diones 111 in a deep eutectic solvent (DES) based on choline chloride and urea (Scheme 37).105 The catalyst was readily recovered from the reaction mixture with the help of an external magnet and could be reused 6 times without significant loss in activity.
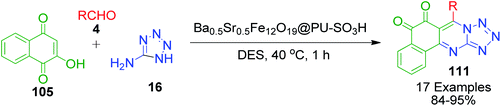 |
| | Scheme 37 Synthesis of 7-aryl-benzo[h]tetrazolo[5,1-b]quinazoline-5,6-diones 111 under heterogenous catalysis. | |
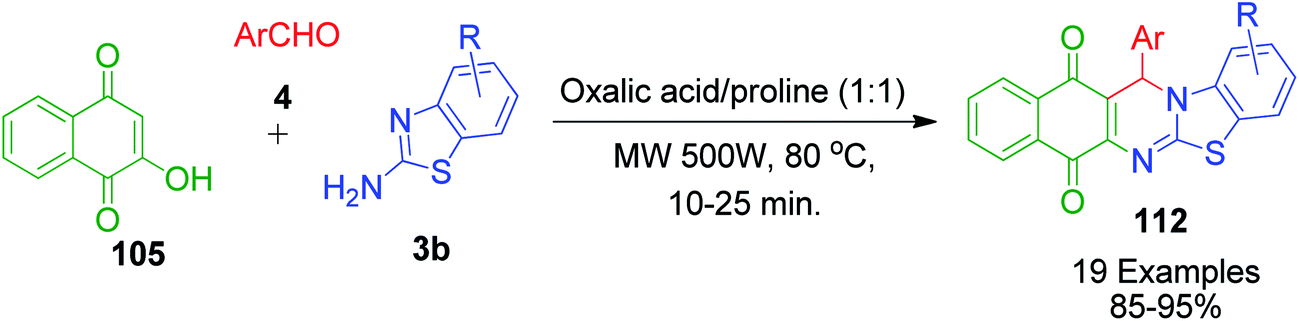
C.-T. Ma et al. identified an oxalic acid and proline-based deep eutectic solvent (DES) as an effective catalyst and environmentally benign reaction medium for the one-pot synthesis of 13-aryl-13H-benzo[g]benzothiazolo[2,3-b]quinazoline-5,14-diones 112 via the microwave-assisted three-component reaction between aromatic aldehydes 4, 2-aminobenzothiazole 3b and 2-hydroxy-1,4-naphthoquinone 105 (Scheme 38).106
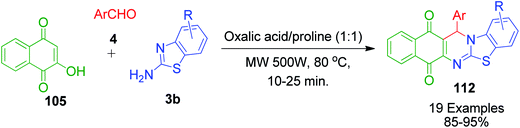 |
| | Scheme 38 Microwave-assisted synthesis of 13-aryl-13H-benzo[g]benzothiazolo [2,3-b]quinazoline-5,14-diones 112. | |
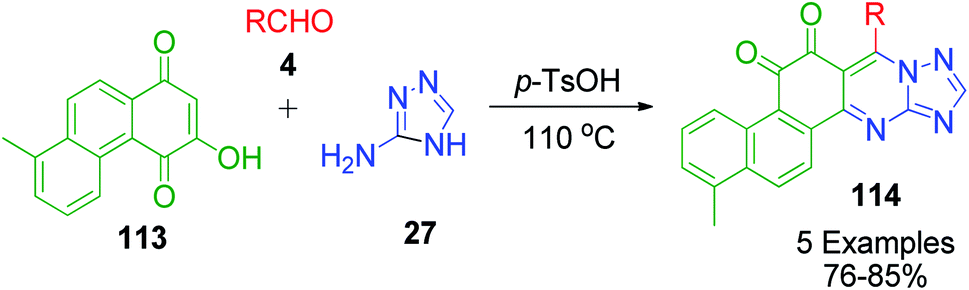
Furthermore, a slightly modified form of naphthoquinone, 3-hydroxy-8-methyl-1,4-phenanthrenequinone 113, was utilized by L. Wu and X. Yang in a three-component coupling reaction with 3-amino-1,2,4-triazole 27 and aldehydes 4. The reaction was carried out in the presence of a catalytic amount of p-TsOH under solvent-free conditions to produce a novel series of tanshinone I derivatives 114 having an azacyclo ring with good yields and high regioselectivity (Scheme 39).107
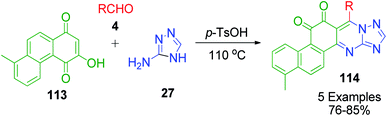 |
| | Scheme 39 Solvent-free synthesis of a novel series of tanshinone I derivatives 115 having an azacyclo ring. | |
P. T. Patil et al. reported the synthesis of novel pyrazolo[3,4-b][1,8]naphthyridine 118 and pyrazolo[3,4-d]pyrimido[1,2-a]pyrimidine 119 derivatives through an efficient one-pot method. In this method, the reactions between 2-aminopyrimidine/2-aminopyridine 116 and 117, aromatic aldehydes 4 and 3-methyl-1-phenyl-2-pyrazolin-5-one 115 were performed in the presence of 15 mol% phosphorus oxide under refluxing ethanol (Scheme 40).108 The authors also studied the structure–activity relationship, which showed that the anti-inflammatory activity of product 118 is much better than that of product 119.
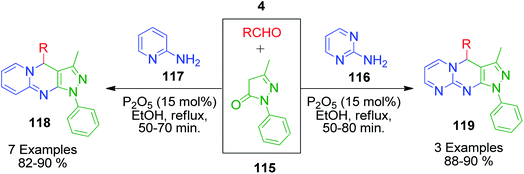 |
| | Scheme 40 Synthesis of pyrazolo[3,4-b][1,8]naphthyridine and pyrazolo[3,4-d]pyrimido[1,2-a]pyrimidine derivatives 118 and 119. | |
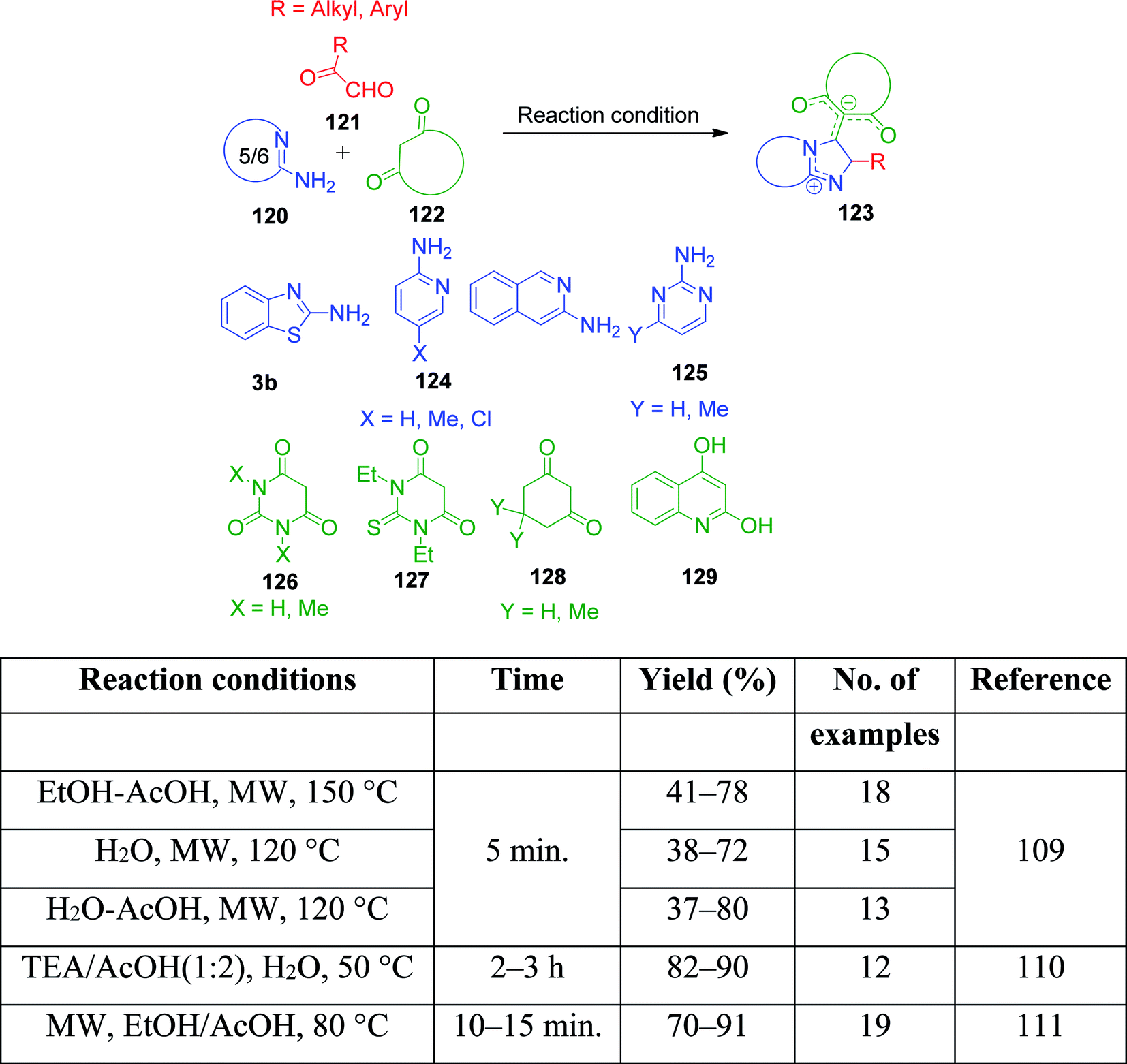
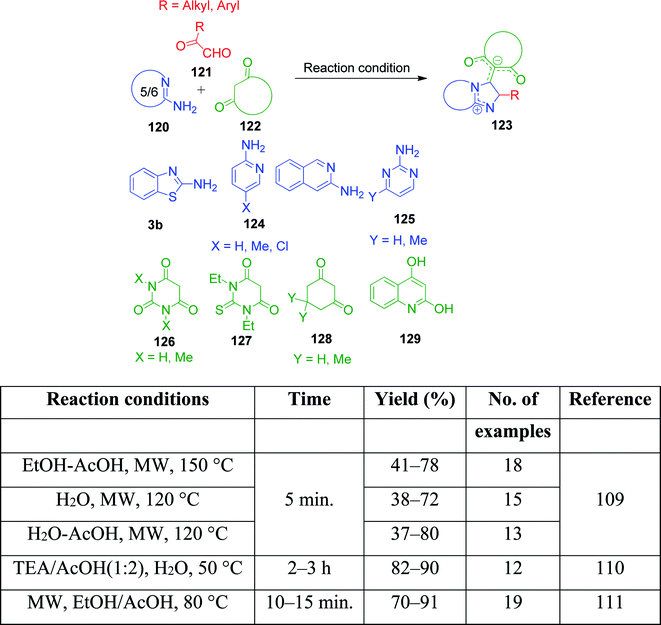
A variation to this reaction was investigated by V. A. Peshkov et al., wherein 2-ketoaldehyde is the main component to introduce the deviation in the synthesized product by the involvement of both carbonyl groups, thus providing a novel class of imidazoazine derivatives 3b, 124 and 125.109 Three different reaction conditions were used to study the scope of the process, where 2-aminoazines 120, 2-oxoaldehydes 121 and cyclic 1,3-dicarbonyl compounds 122 were taken in a combination of two solvents and heated at high temperature under microwave irradiation for 5 min, resulting in the generation of a small library of title compounds and highlighting the possibility of a case-specific approach. The same reactions were also performed in the presence of TEA/AcOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) by N. Etivand et al.110 and EtOH/AcOH under microwave irradiation by J. Wang and coworkers (Scheme 41).111
:2) by N. Etivand et al.110 and EtOH/AcOH under microwave irradiation by J. Wang and coworkers (Scheme 41).111
 |
| | Scheme 41 Different approaches for the synthesis of imidazo[1,2-a]azine derivatives. | |
K. Meena et al. developed the catalyst-free synthesis of benzo[d]imidazo[2,1-b]thiazoles 131, wherein 2-aminobenzothiazole derivatives 3a and dimedone 81 were combined with arylglyoxal 130 instead of aromatic aldehydes at 80 °C in glycerol. The same starting materials were also ground at room temperature in glycerol. All the reactions were completed in ∼30 min (Scheme 42).112
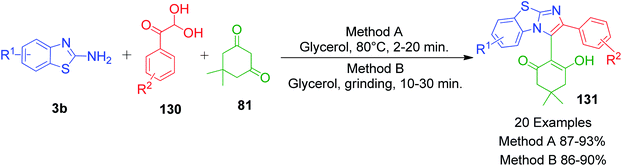 |
| | Scheme 42 Synthesis of benzo[d]imidazo[2,1-b]thiazole derivatives 131 via ecofriendly methods. | |
A lemon juice-mediated ecofriendly one-pot three-component approach for the synthesis of diverse pharmaceutically important tricyclic fused imidazoles 134 tethered with aryl and various cyclic 1,3-dicarbonyls was developed by A. Saha and coworkers. This metal-free strategy involved reactions between arylglyoxals 132 and N-alkyl-4-hydroxyquinolone/4-hydroxycoumarin 133 with 2-aminobenzothiazoles/2-aminobenzimidazoles 3 using lemon juice as a natural acid catalyst under refluxing temperature giving the products in good to excellent yields (Scheme 43).113
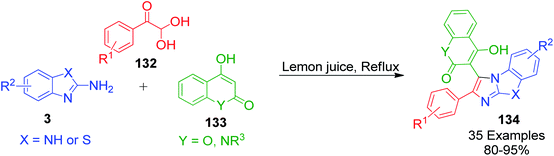 |
| | Scheme 43 Lemon juice-mediated synthesis of tricyclic fused imidazoles 134. | |
A. Nouri et al. developed a novel approach for the synthesis of benzo[g]thiazolo[2,3-b]quinazolin-4-ium hydroxide derivatives 135 using aryl glyoxal monohydrates 132 instead of aldehyde in a triethylamine and p-toluenesulfonic acid system-catalyzed one-pot, three-component reaction with 2-hydroxy-1,4-naphthoquinone 105 and 2-aminothiazole/2-aminobenzothiazole 3b under H2O/acetone (2:1) at room temperature (Scheme 44).114
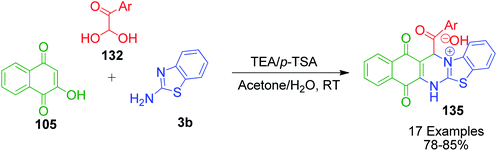 |
| | Scheme 44 Synthesis of novel benzo[g]thiazolo[2,3-b]quinazolin-4-ium hydroxide derivatives. | |
Instead of using arylglyoxals directly, A. Jana et al. utilized aryl acetylenes and aryl methyl ketones for the in situ generation of arylglyoxals and developed a metal-free I2/DMSO-based oxidation–cyclization protocol for the synthesis of medicinally important structures having 2-arylbenzo[d]imidazo[2,1-b]thiazoles linked with a barbituric acid moiety. These three-component reactions were performed between 2-aminobenzothiazole derivatives 3b, barbituric acid derivatives 136 and aryl acetylenes/aryl methyl ketones 137 and 138 at 110 °C under microwave heating. Mechanistically, aryl acetylenes 137 and aryl methyl ketones 138 under the reaction conditions generate arylglyoxals, which are then reacted with 2-aminobenzothiazole derivatives and barbituric acid derivatives. Consequently, a series of benzothiazole-fused imidazoles 139 and 140 was prepared in good yield (Scheme 45).115
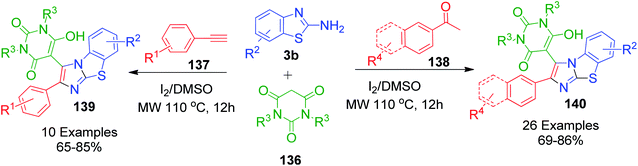 |
| | Scheme 45 Synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazoles 139 and 140 linked with a barbituric acid moiety. | |
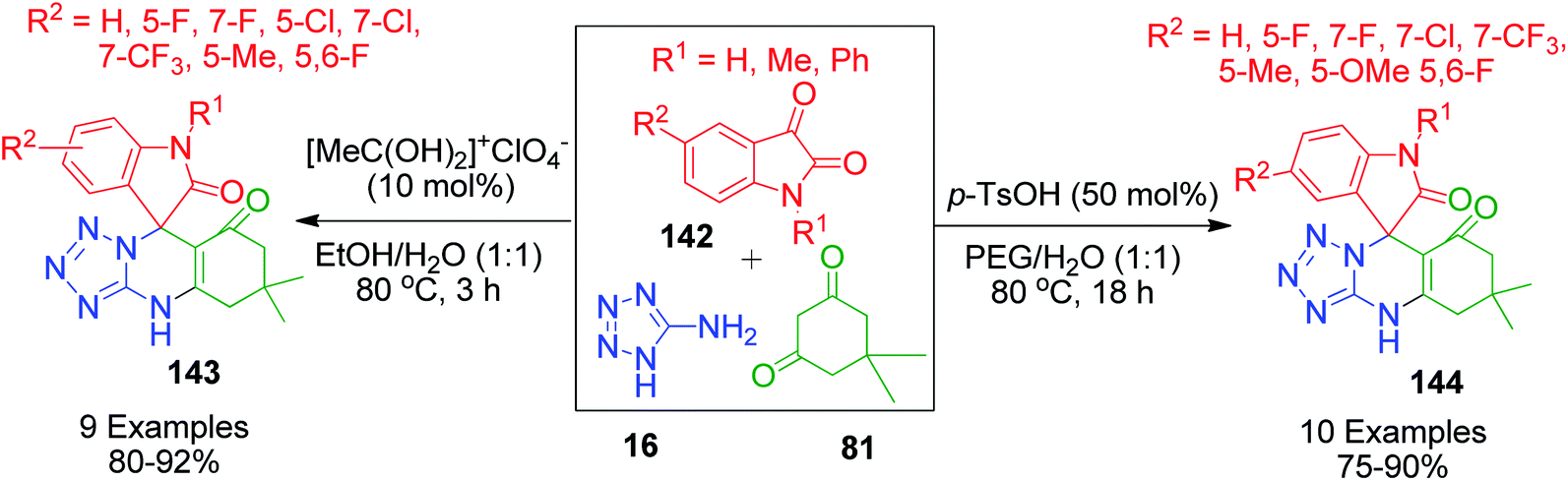
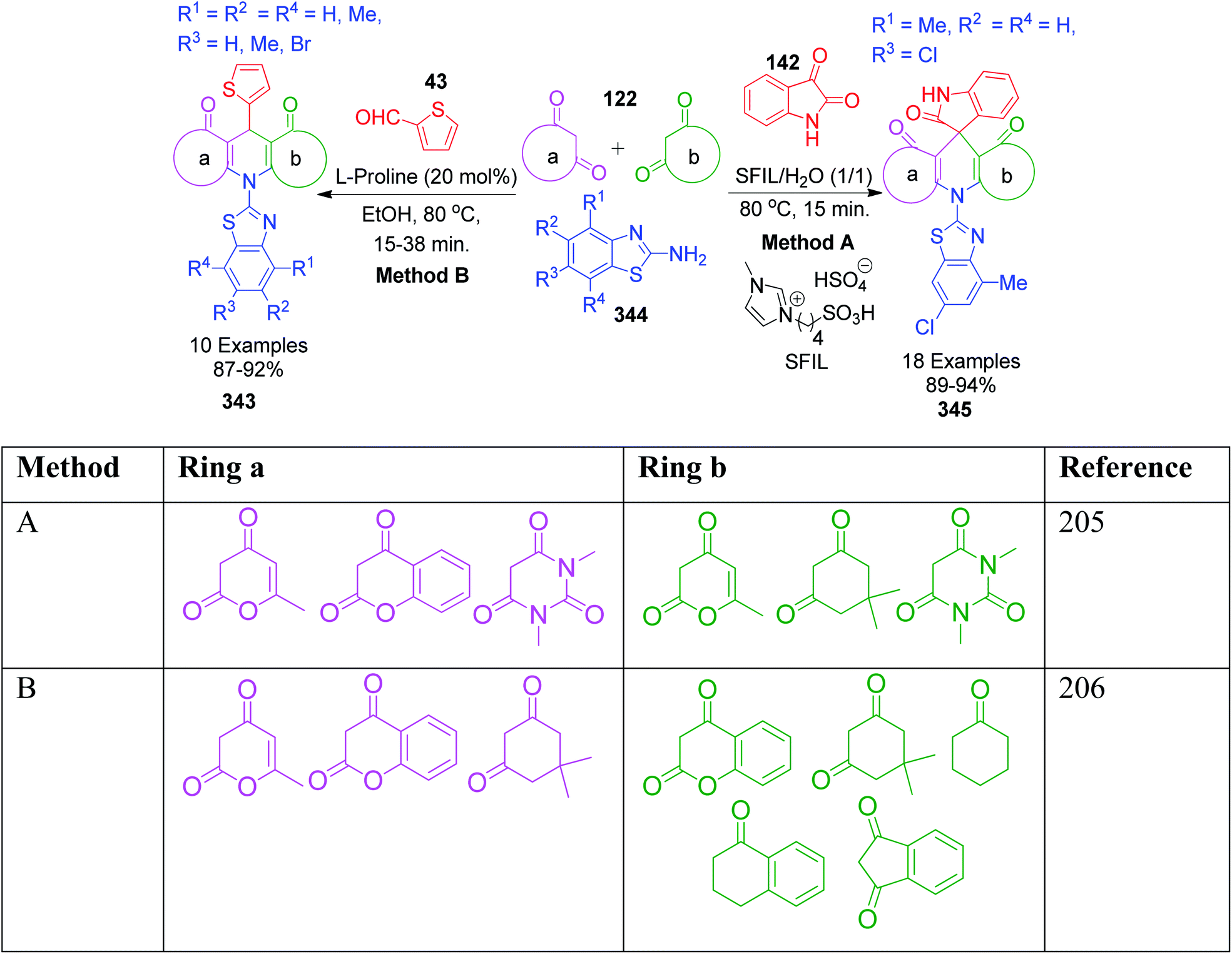
When aldehydes are replaced by ketones such as isatin, acenaphthoquinone and related compounds in three-component Biginelli-like condensation reactions, a pyrimidine scaffold with spiro linkage results in 143. This was exemplified by Y. Dai et al. through an efficient one-pot strategy, where isatin 142 was mixed with 5-aminotetrazole 16 and dimedone 81 in the presence of a super acid catalyst ([MeC(OH)2]+ClO4−) in an aqueous medium.116 The spiro-tetrazoloquinazolinones 144 were also synthesized by M. Shen's group, wherein the same combination of reactants was treated in PEG-H2O medium together with PTSA as the catalyst.117 Both procedures were conducted under refluxing temperature (80 °C) and exhibit a great level of tolerance for isatin (Scheme 46).
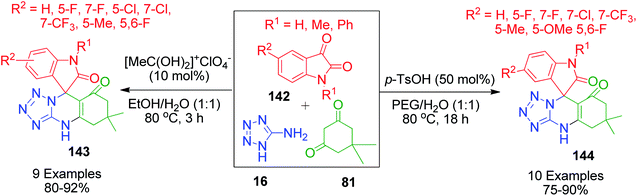 |
| | Scheme 46 Synthesis of spiro-imidazoquinazolinones 143 and 144 in the presence of acid catalysts. | |
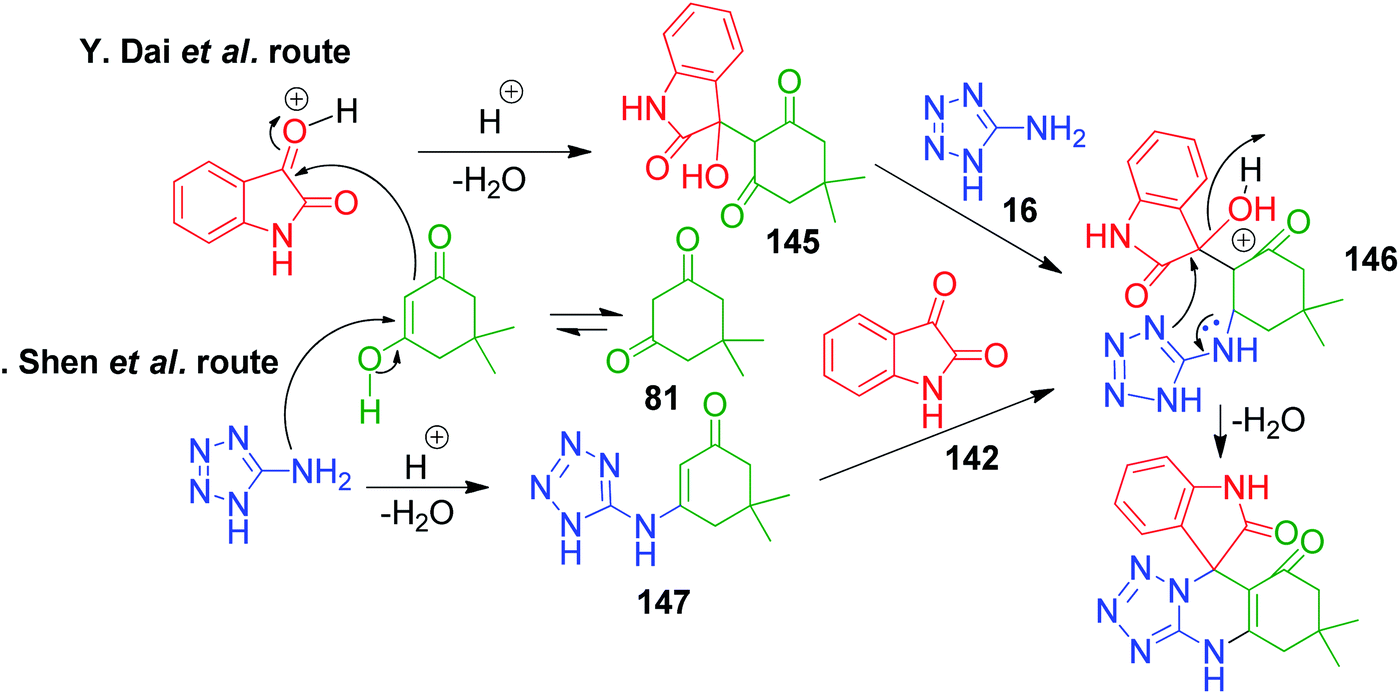
In the first step of the mechanistic route described by Y. Dai et al., the condensation of isatin and dimedone was proposed to give intermediate 145, which is then attacked by 5-amino-1H-tetrazole to provide intermediate 146. This intermediate 147 in the study by M. Shen et al. was achieved through addition of isatin to the condensed intermediate 147 formed from 5-aminotetrazole and dimedone. The final product is obtained from the intramolecular condensation and cyclisation of the intermediate 146 with the loss of a water molecule (Scheme 47).
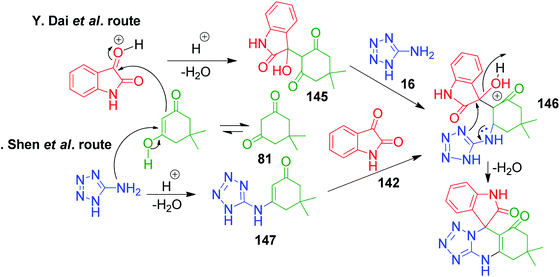 |
| | Scheme 47 Mechanistic routes illustrating the generation of a common intermediate 146 through two different pathways for the synthesis of spiro-quinazolinones. | |
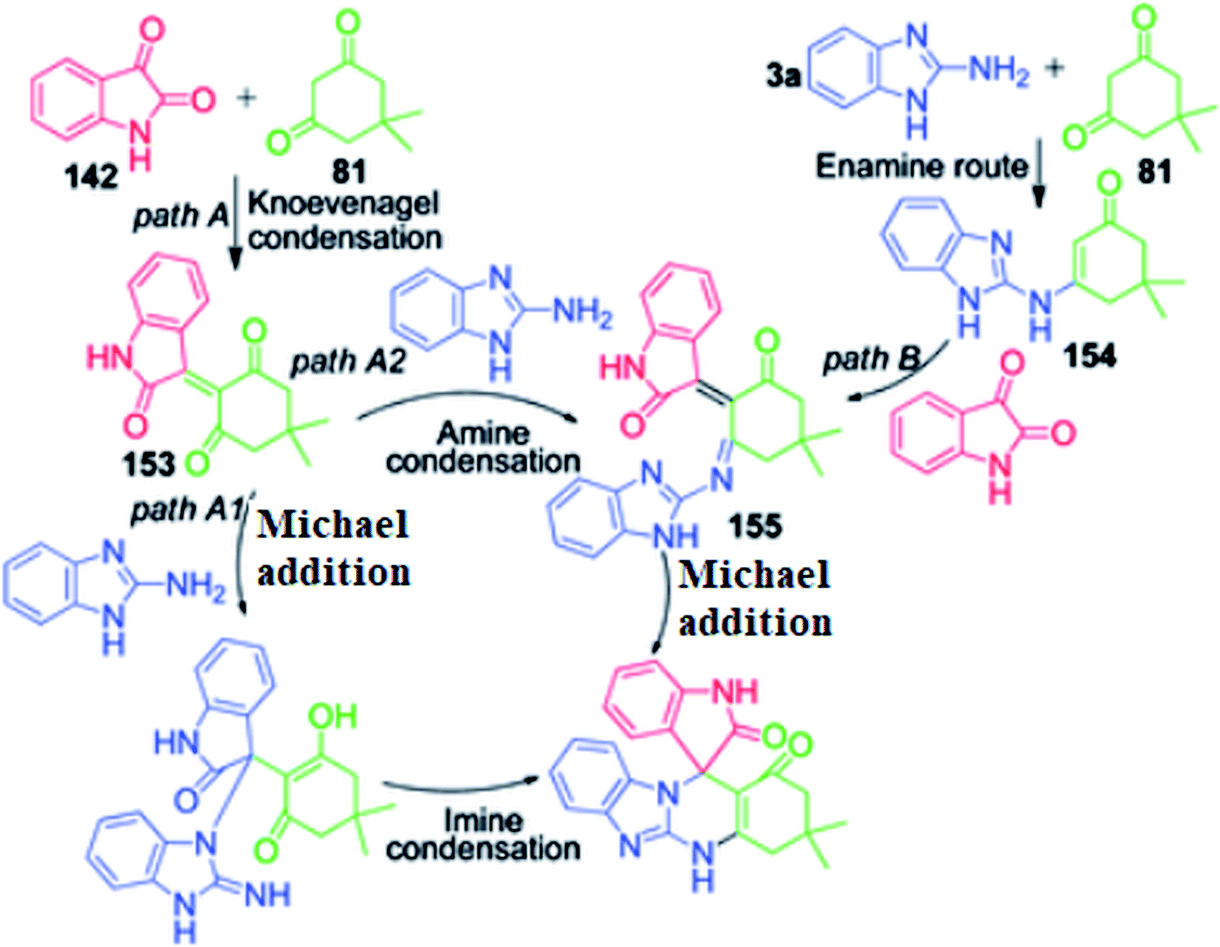
P. Maloo's group reported a straightforward novel multi-component route to access spiro-benzimidazoquinazolinones 150, 151 and 152.118 It involves a one-pot three-component reaction of acenaphthoquinone/isatin 148 and 142, 1,3-diketone 81 and 2-aminobenzimidazole 3a in ethanol at 160 °C using 180 W power microwave irradiation. An attractive feature of this method is the utilization of 1,3-indanedione 149 as an active methylene compound for the construction of the corresponding fused spiropyrimidine ring 151 in 72% yield under mild and operationally simple conditions (Scheme 48). The computational study provided insight into the mechanistic aspects of the reaction. The mechanism proposed by the authors to explain this reaction is based on two paths, A and B. In path A, the Knoevenagel condensation of dimedone and isatin generates intermediate 153, which undergoes reaction with 2-aminobenzimidazole 3a in two possible ways via the Knoevenagel-Michael-imine route (path A1) or Knoevenagel–imine-Michael route (path A2). In path B, enamine 154 is generated through dimedone 81 and 2-aminobenzimidazole, which upon condensation with isatin in the Knoevenagel fashion, generates intermediate 155, followed by intramolecular Michael addition to furnish the final spiro product (Scheme 49).
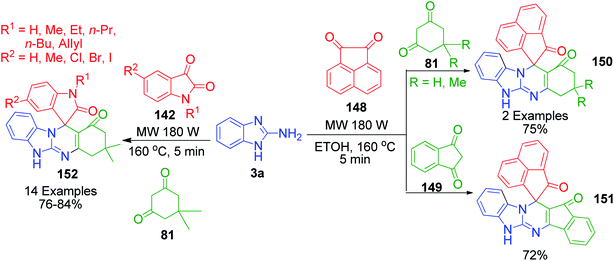 |
| | Scheme 48 Synthesis of spiro-benzimidazoquinazolinones under microwave irradiation. | |
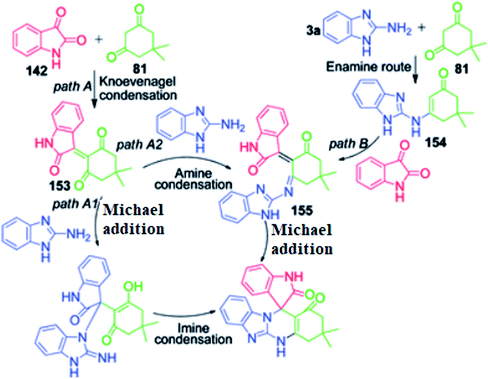 |
| | Scheme 49 Proposed mechanistic pathways for the synthesis of spiro-benzimidazoquinazolinones. | |
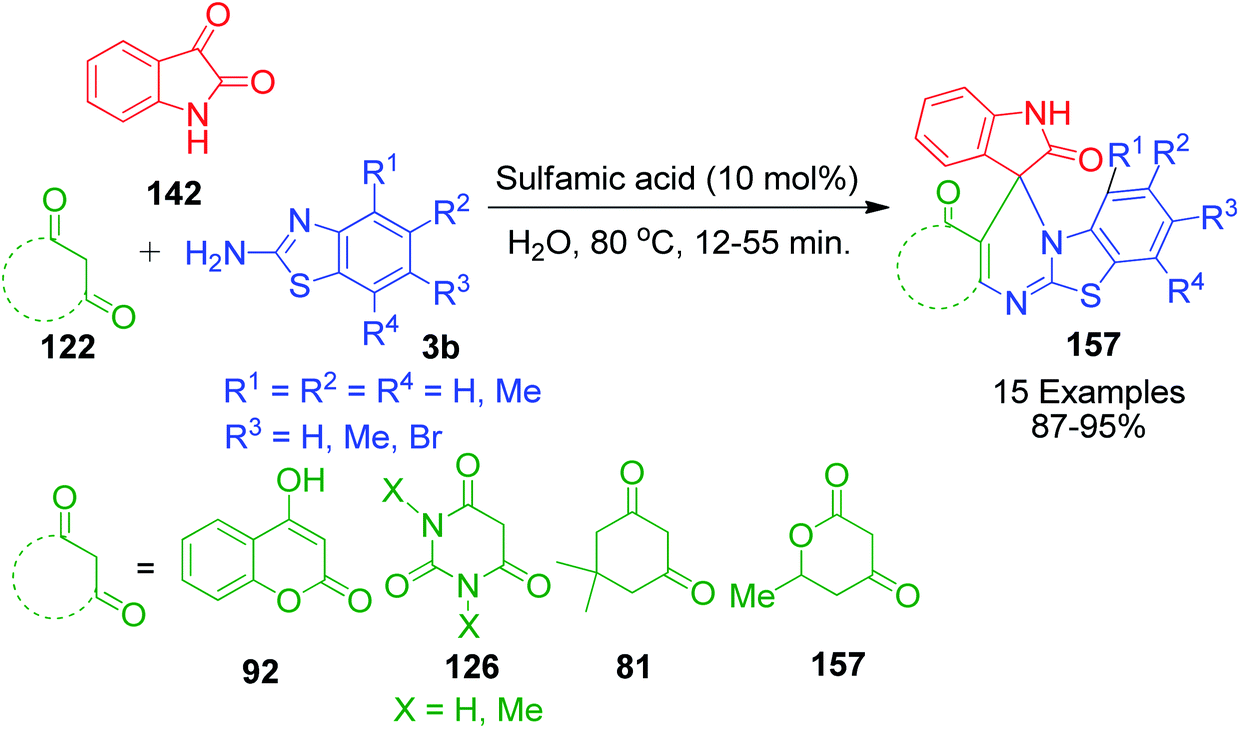
Next, sulfamic acid-catalysed spiro analogue derivatives with fused heterosystems were prepared by A. K. Arya and M. Kumar using isatin instead of aldehyde via a three-component domino reaction. This method involves heating a reaction mixture containing isatin 142, various cyclic 1,3-diketones 122 and 2-aminobenzothiazole 3b in aqueous medium in the presence of 10 mol% sulfamic acid at 80 °C for 12–55 min to afford the corresponding spiroheterocycles 157 (Scheme 50).119
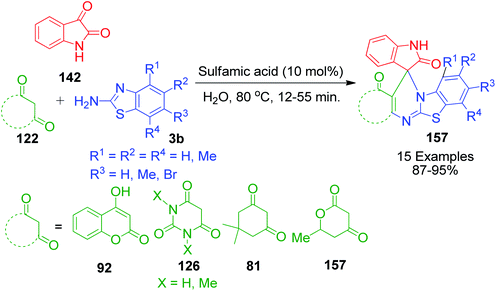 |
| | Scheme 50 Synthesis of structurally diverse spiroheterocycles with fused heterosystems. | |
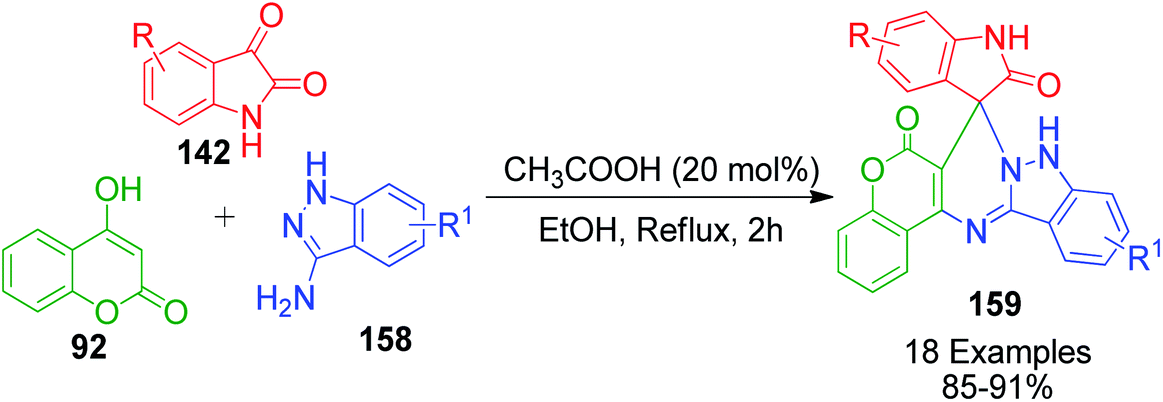
A similar strategy utilizing 1H-indazole-3-amine 158 instead of 2-aminobenzothiazole 3b in a one-pot three-component condensation with 4-hydroxy-2H-chromen-2-one 92 and isatin 142 was developed by A. M. Jadhav et al. in the presence of acetic acid in EtOH for the rapid synthesis of novel spiro[chromeno[40,30:4,5] pyrimido[1,2-b]indazole-7,30-indoline]-20,6(9 H)-dione derivatives 159 (Scheme 51).120
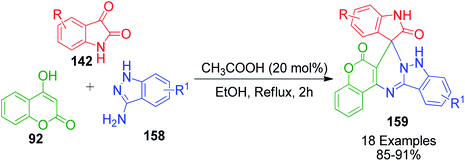 |
| | Scheme 51 Synthesis of indazole-fused spiroheterocycles 159 catalyzed by acetic acid. | |
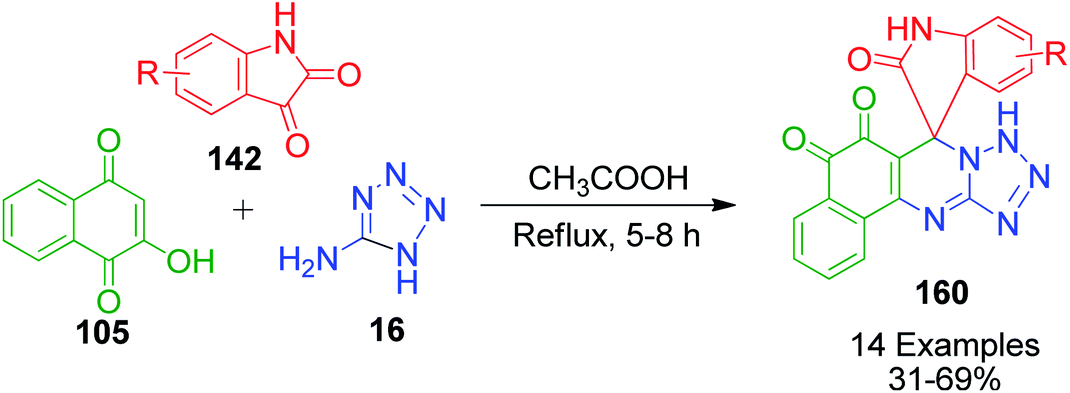
A series of novel spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine hybrids 160 was synthesized and evaluated as potent antitumor agents by L. Wu and coworkers. The synthesis of these ternary hybrid molecules was carried out through the condensation reaction between 2-hydroxy-1,4-naphthoquinone 105, isatin 142 and 5-aminotetrazole 16 in the presence of acetic acid under reflux conditions (Scheme 52).121
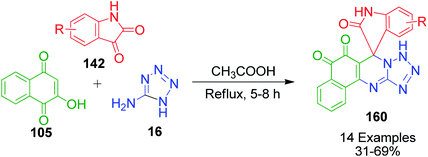 |
| | Scheme 52 Synthesis of spirooxindole-O-naphthoquinone-tetrazolo[1,5-a]pyrimidine. | |
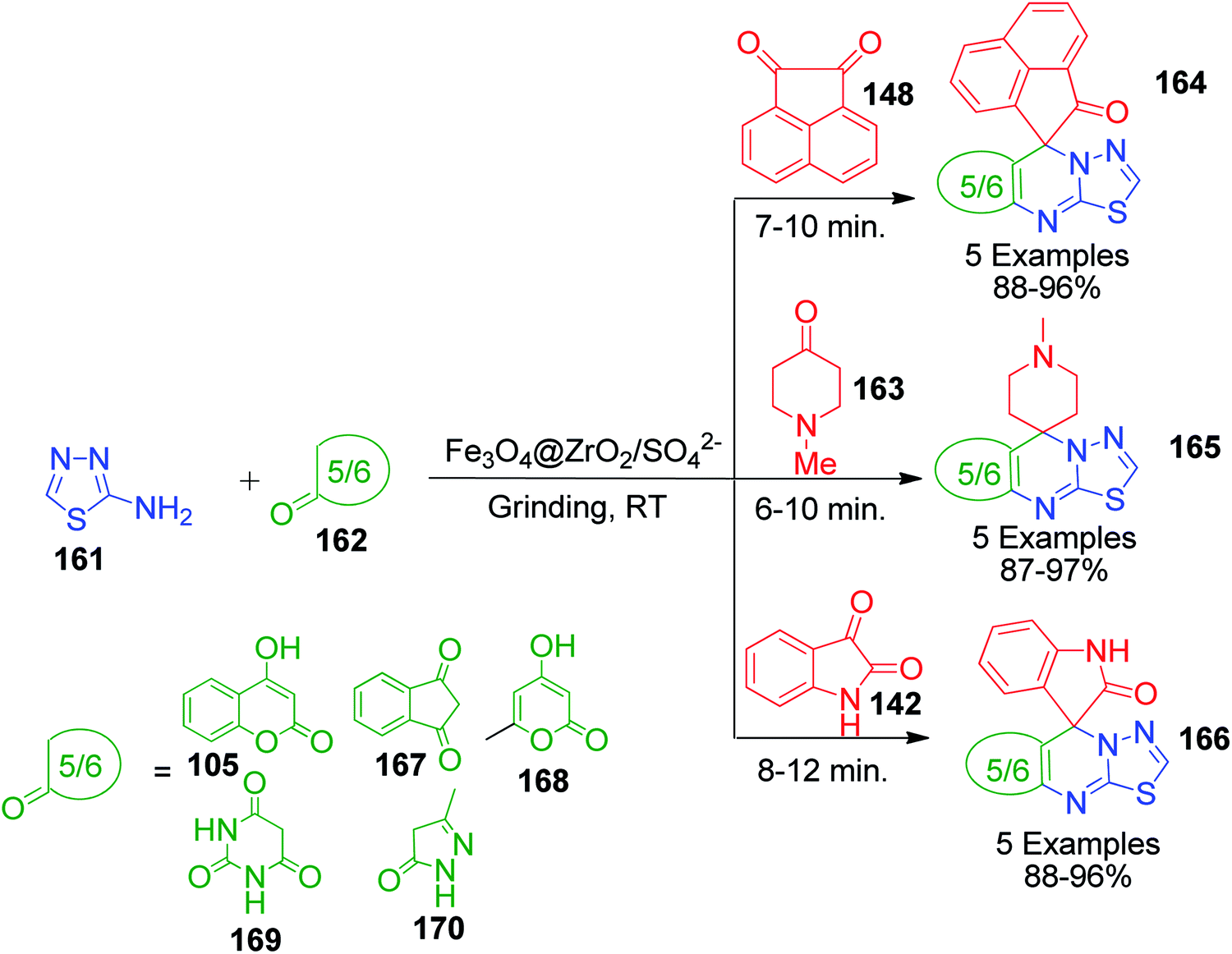
Y. K. Tailor et al. Presented an efficient and environmentally benign domino protocol for the synthesis of structurally diverse spiroheterocycles 164, 165 and 166 spiroannulated with 1,3,4-thiadiazolo[3,2-a]pyrimidine, involving the three-component reaction of 2-amino-1,3,4-thiadiazole 161, isatin 142/N-methyl-4-piperidone 163/1,2-acenaphthylenedione 148 and carbonyl compounds 162 catalyzed by magnetically recoverable and reusable nanocrystalline sulfated zirconia (Fe3O4@ZrO2/SO42−) under solvent-free conditions with grinding at room temperature (Scheme 53).122
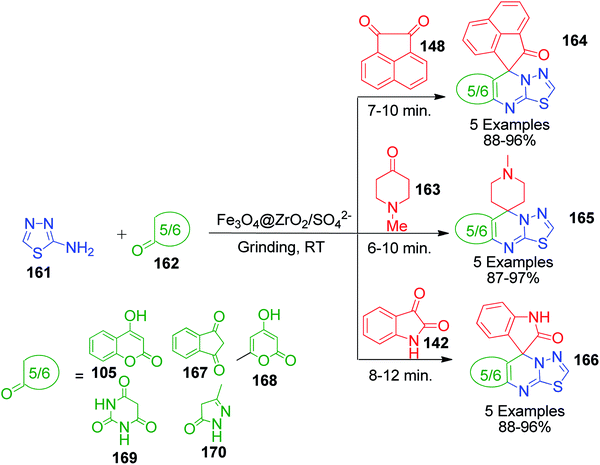 |
| | Scheme 53 Synthesis of spiroheterocycles spiroannulated with 1,3,4-thiadiazolo[3,2-a]pyrimidine. | |
K. Verma et al. developed a domino protocol to construct spiroannulated pyrimidophenazines 174, 175 and 176. The synthesis of these hybrid molecules with advantageous heterocyclic substructures involves the four-component reaction of 2-hydroxynaphthalene-1,4-dione 105, benzene-1,2-diamine 171, cyclic ketones (163, 173 and 148) and 2-aminoazines 172 in the presence of erbium-doped TiO2 NPs as a recyclable and reusable heterogeneous acid catalyst under refluxing ethanol (Scheme 54).123 The doping of TiO2 with erbium was found to enhance the catalytic efficiency of TiO2, facilitating the reaction to provide the products with comparatively better yields.
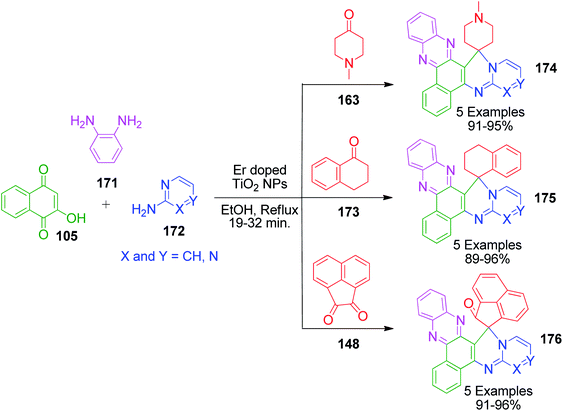 |
| | Scheme 54 Four-component synthesis of spiroannulated pyrimidophenazines. | |
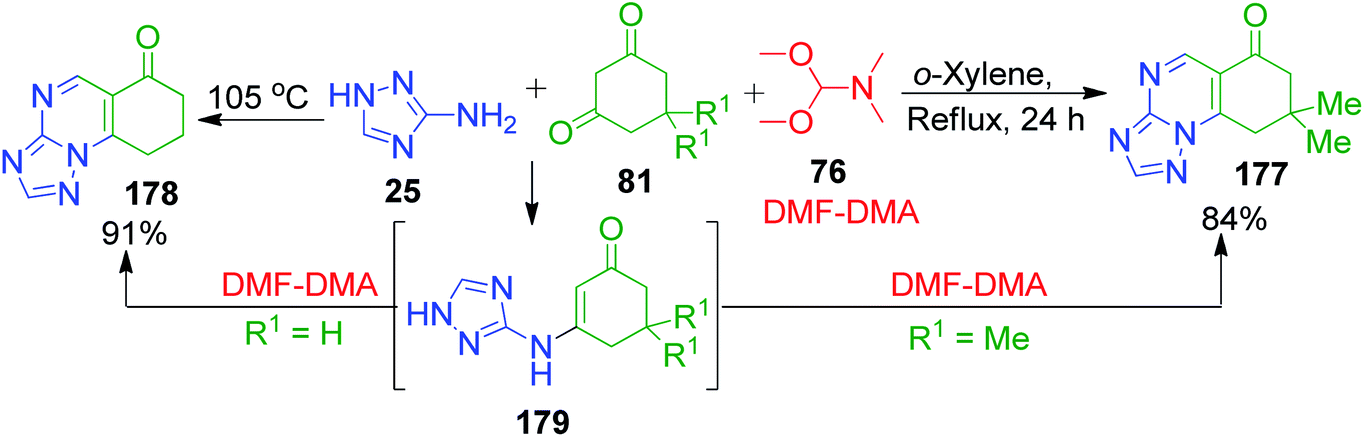
Formaldehyde equivalents, dimethoxy N,N-dimethylmethanamine (DMF-DMA) 76, provide less substituted fused quinazolinone rings, as shown in the approach developed by C.-B. Zhang and coworkers.124 In their reactions, cyclohexane-1,3-dione 81 and DMF-DMA 76 were mixed and heated at 60 °C. After the mixture was completely homogenized, 3-amino-1,2,4-triazole 25 was added and then the reaction temperature was increased to 105 °C. After a few minutes, the solidified mixture was cooled and diluted with propan-2-ol to obtain the final product 178 with 91% yield. The utilization of DMF-DMA as an aldehydic component in the three-component reaction with aminotriazole and dimedone was also illustrated by A. A. Petrov and A. N. Kasatochkin. The reaction, which was performed in xylene under reflux, was shown to proceed via the generation of enamino ketone 179 and the corresponding pyrimidine ring was formed regioselectively by examining the mixture with high-resolution mass spectrometry (Scheme 55).125
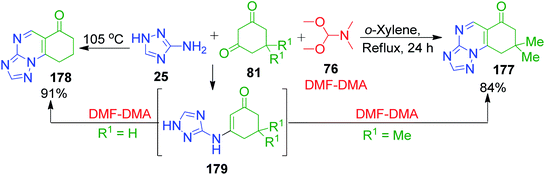 |
| | Scheme 55 Three-component synthesis of 7-unsubstituted 4,7-dihydro-1,2,4-triazolo[1,5-a]pyrimidines 177 and 178. | |
 |
| | Scheme 56 Some reported methods for the synthesis of substituted benzothiazole/thiazole fused pyrimidines. | |
 |
| | Scheme 57 Proposed reaction pathway to explain the synthesis of benzothiazolopyrimidines. | |
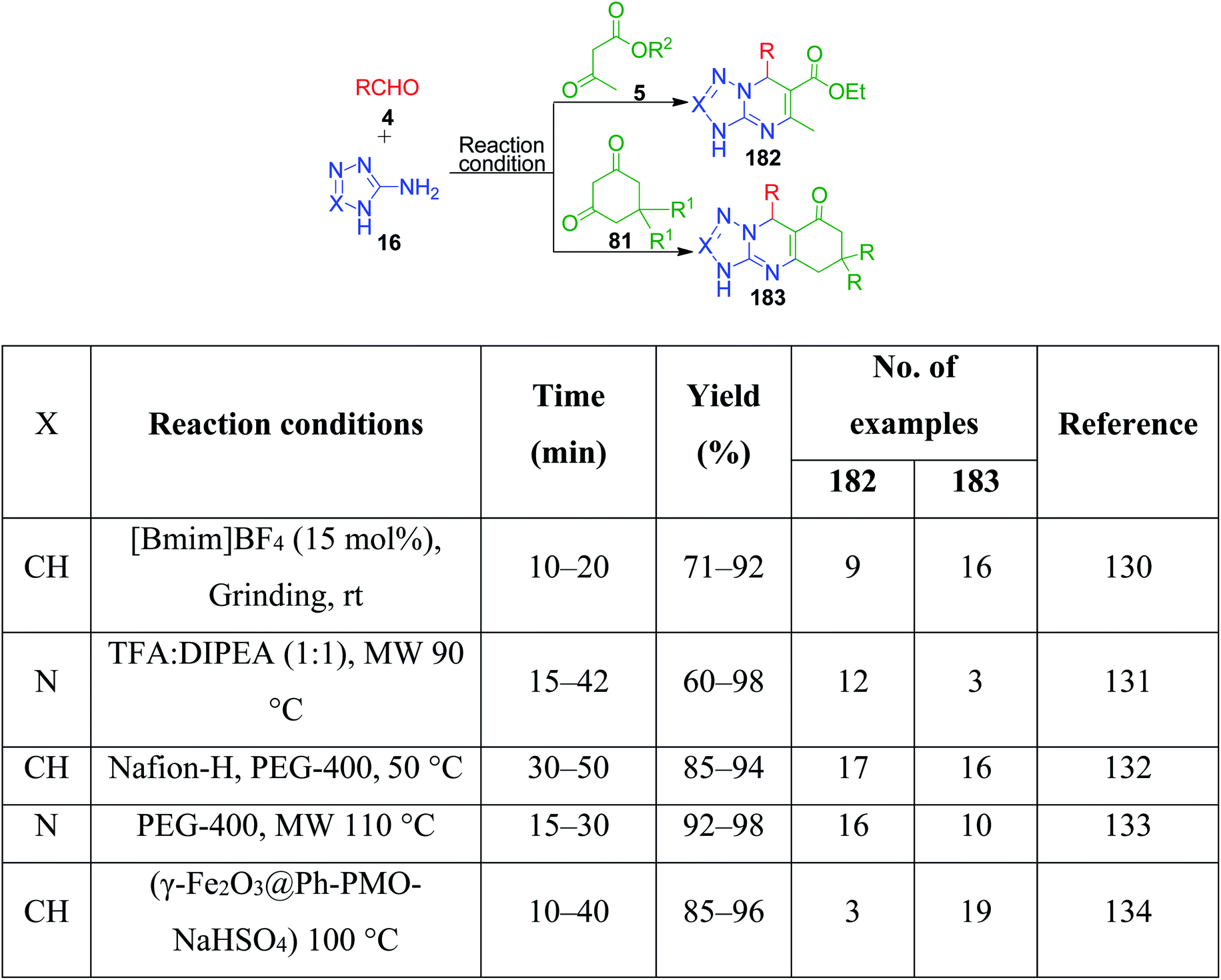
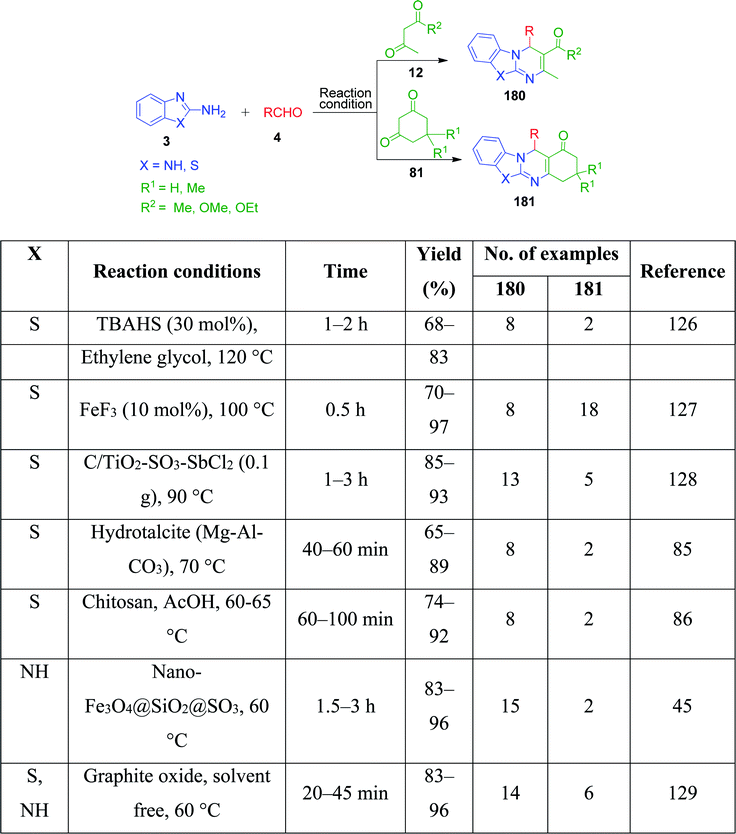
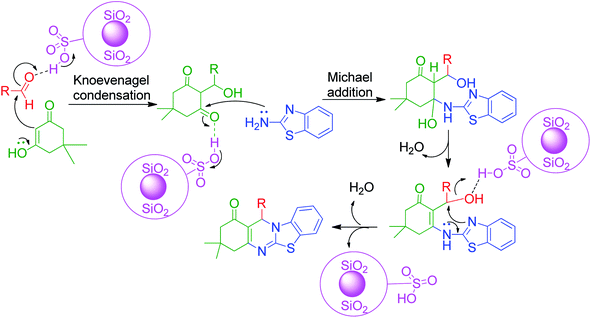
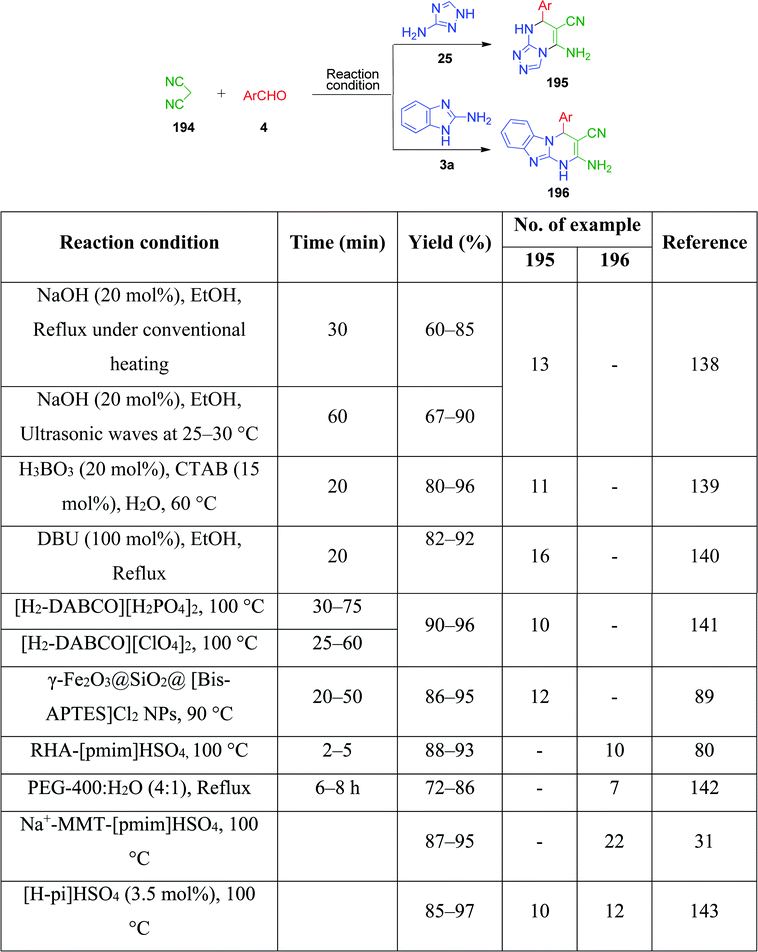
3-Amino-1,2,4-triazole/5-aminotetrazole 16 has also been utilized together with cyclic and non-cyclic β-ketocompounds for the synthesis of various tetrazolo/triazolo-fused pyrimidine moieties 182 and 183. K. Kumari et al. demonstrated a three-component reaction, wherein a mixture containing 3-amino-1,2,4-triazole 17, aromatic aldehyde 3, dimedone/ethyl acetoacetate 83 and 4 was grounded using an agate mortar in the presence of [Bmim]BF4.130 The TFA:DIPEA (1:1)-catalysed version of this protocol was developed by C. Raju et al. utilizing 5-aminotetrazole instead of 3-amino-1,2,4-triazole in a reaction with aromatic aldehyde and dimedone/ethyl acetoacetate, and the synthesized tetrazole-fused compounds were evaluated for their antimicrobial and antioxidant activity.131 Similarly, other catalysts such as Nafion-H,132 PEG-400,133 and NaHSO4 immobilized on core/shell phenylene-bridged periodic mesoporous organosilica magnetic nanoparticles (γ-Fe2O3@Ph-PMO-NaHSO4)134 were used (Scheme 58).
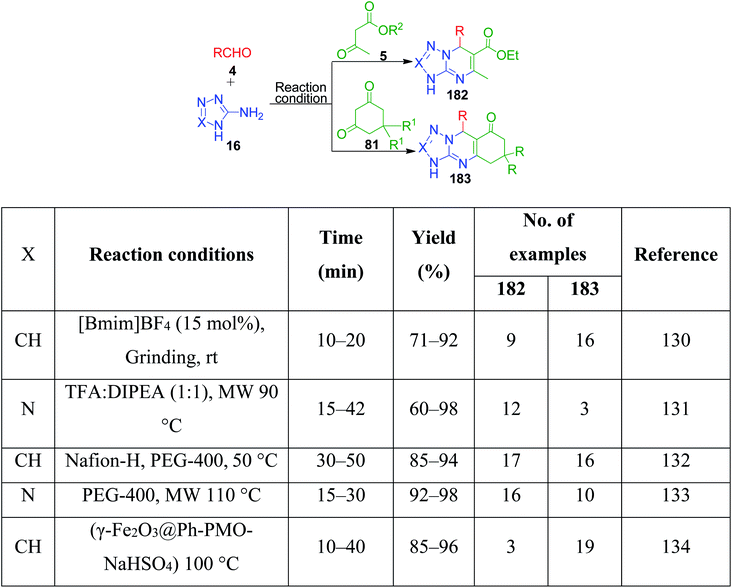 |
| | Scheme 58 Some reported methods for the synthesis of tetrazolo/triazolo fused pyrimidines. | |
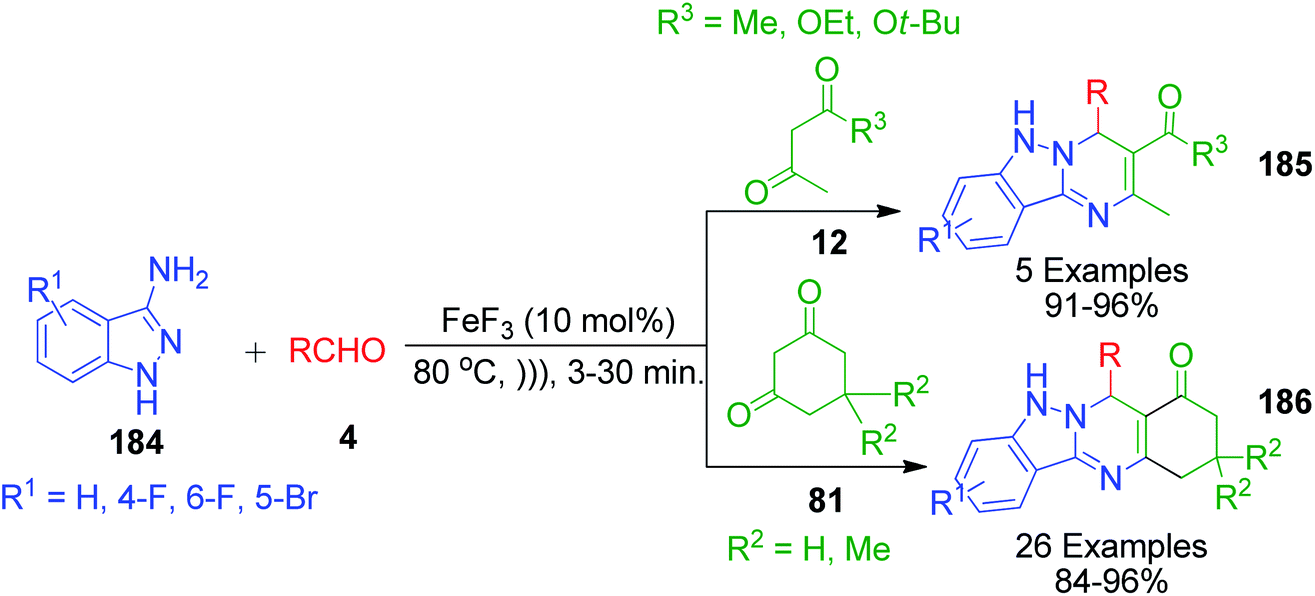
A synthetic approach for densely functionalized tetrahydroindazolo[3,2-b]quinazoline 185 and 186 catalyzed by iron fluoride under ultrasonication in solvent-free conditions was developed by V. V. Shinde and Y. T. Jeong by utilizing 1H-indazole-3-amines 184, cyclic 81 and acyclic 1,3-dicarbonyl compounds 12 and various aldehydes 4 as starting components (Scheme 59).135 This reaction was completed in a short time and it possesses advantages in comparison to the conventional heating such as good to excellent yields, easy work-up procedure, and avoiding the use of solvent and column chromatographic purification of products.
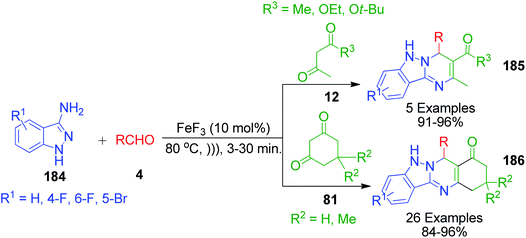 |
| | Scheme 59 Ultrasonic wave-assisted synthesis of tetrahydroindazolo[3,2-b]quinazoline. | |
A straightforward and green route to 4-aryl-3,4-dihydro-1H-pyrimido[1,2-a]benzimidazole derivatives 188, 189 and 190 was accomplished in excellent yields via the reaction of aryl aldehyde 3, 1,3-dicarbonyl compounds 187, 81 and 12 and 2-aminobenzimidzole 3a in ionic liquid, [bmim][BF4], by C. Yao and coworkers.136 At high temperature, the three-component reaction with 2,2-dimethyl-1,3-dioxane-4,6-dione 187 yielded pyrimidone ring as the final product through sequential Knoevenagel condensation, Michael addition, intramolecular cyclization and elimination reaction sequence (Scheme 60). During intramolecular cyclisation, the temperature plays a key role in rearranging the cyclized structure into a thermodynamically more stable product upon the elimination of acetone and carbon dioxide as gaseous side products.
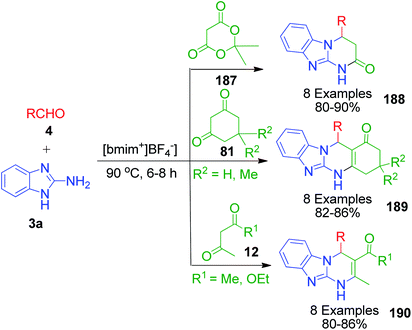 |
| | Scheme 60 Ionic liquid-mediated synthesis of various pyrimidine derivatives. | |
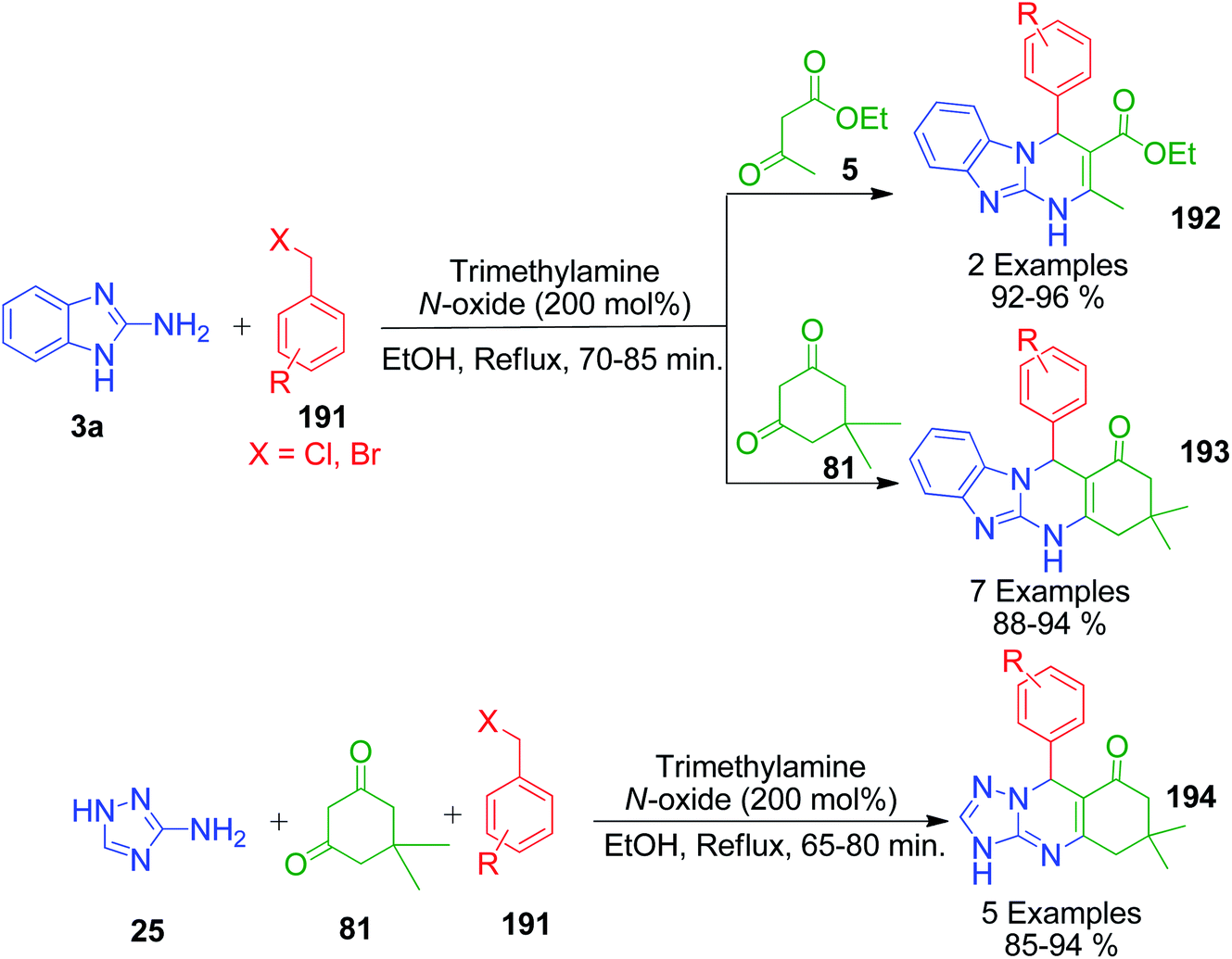
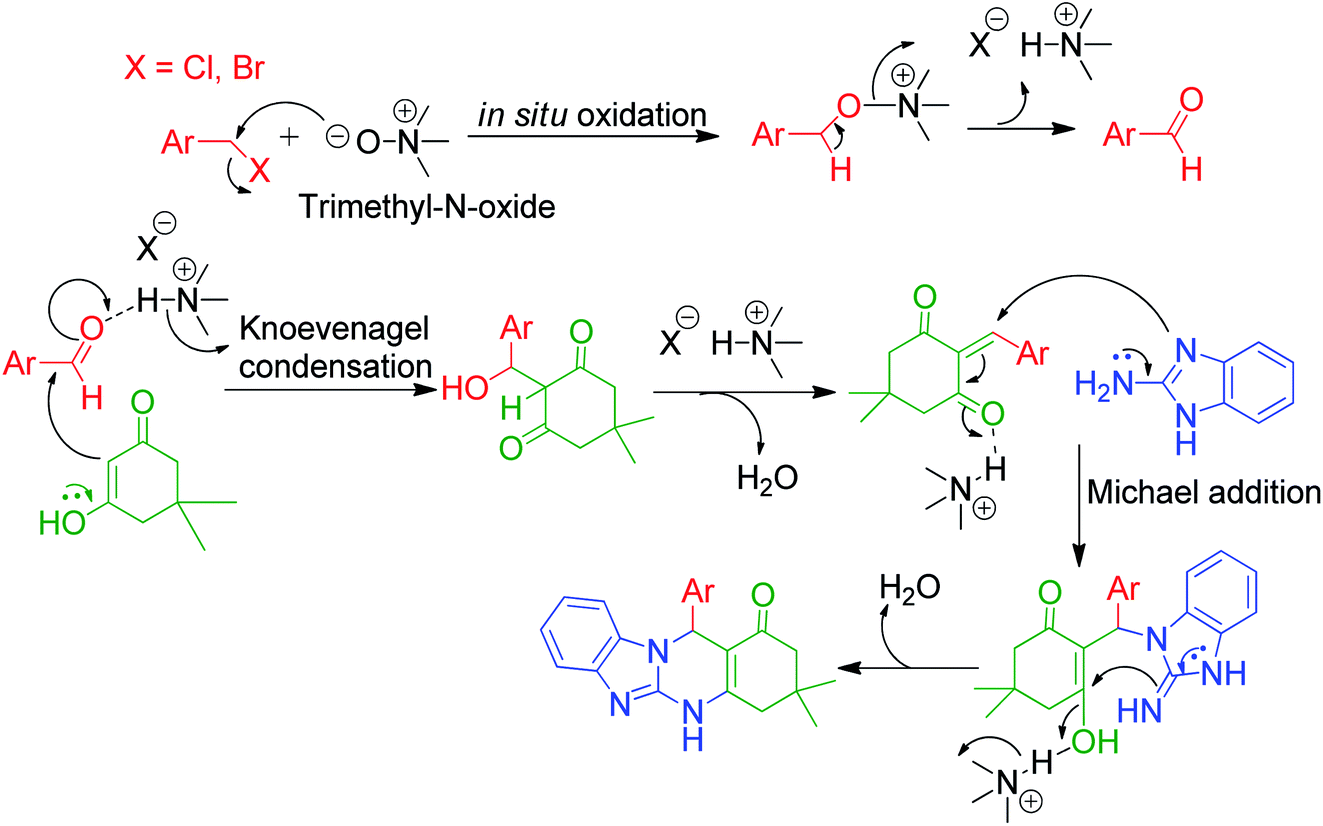
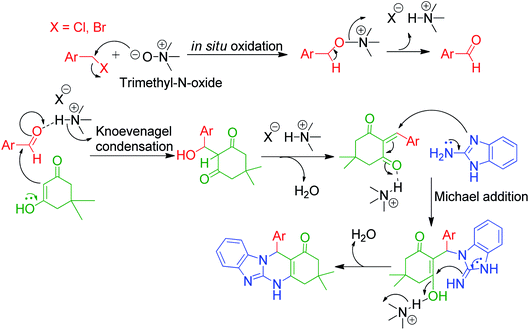
An interesting protocol developed by M. Beerappa and K. Shivashankar revealed that benzyl halide can be used as an aldehydic precursor to construct triazolo/benzimidazolo quinazolinones 192, 193 and 194 via MCRs. This method involves the one-pot three-component reaction between 2-amino benzimidazole/3-amino-1,2,4-triazole 3a and 25, dimedone/ethylacetoacetate 81 and 5 and various benzyl halides 191 in the presence of trimethyl amine N-oxide as a catalyst under refluxing ethanol (Scheme 61).137 Mechanistically, the reaction proceeds through the cascade of in situ oxidation of benzyl halides into benzaldehydes by trimethyl amine N-oxide, Knoevenagel condensation between aldehyde and β-dicarbonyl compound, and finally Michael addition followed by cyclization and dehydration (Scheme 62).
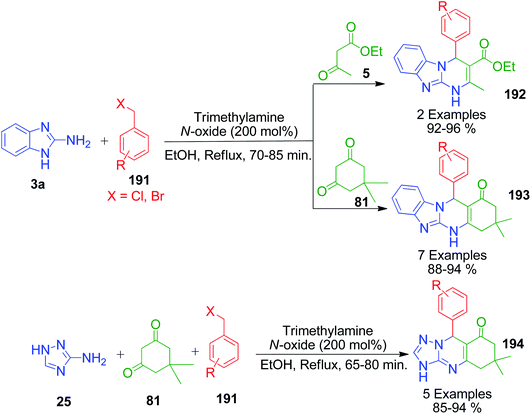 |
| | Scheme 61 An interesting protocol for the synthesis of triazolo/benzimidazolo quinazolinones. | |
 |
| | Scheme 62 Proposed mechanistic route to explain the synthesis of triazolo/benzimidazolo quinazolinones. | |
 |
| | Scheme 63 Synthesis of multi-substituted [1,2,4]-triazolo-[4,3-a-]pyrimidines. | |
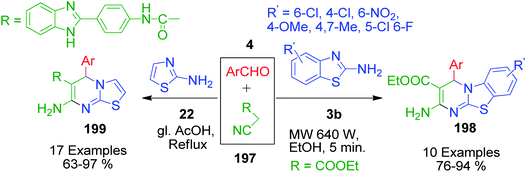
S. Nalawade and coworkers reported a method to achieve the regioselective synthesis of pyrimido[2,1-b][1,3]benzothiazole-3-carboxylate 198 by the reaction of ethylcyanoacetate with substituted benzaldehyde 4 and 2-aminobenzothiazol 3b in ethanol under microwave irradiation operating at 640 W.144 These one-pot three-component reactions were completed within a few minutes, affording the desired products in good to excellent yields. A newly modified form of the cyano group 197-bearing active methylene compound was prepared and utilized in the MCR synthesis of thiazolo[3,2-a]pyrimidine derivatives 199 by A. S. Abd El-All's group. N-(4-(1H-Benzo[d]imidazol-2-yl)phenyl)-2-cyanoacetamide 199 was prepared by mixing ethyl cyanoacetate with 4-(1H-benzo[d]imidazol-2-yl)benzenamine in refluxing ethanol. The newly synthesized cyanoacetamide was then reacted with 2-aminothiazole 22 and aromatic aldehyde 4 in the presence of glacial acetic acid under reflux conditions to afford the corresponding thiazole ring-fused pyrimidine derivatives (Scheme 64).145
 |
| | Scheme 64 Synthesis of thiazole/benzothiazole ring-fused pyrimidine derivatives. | |
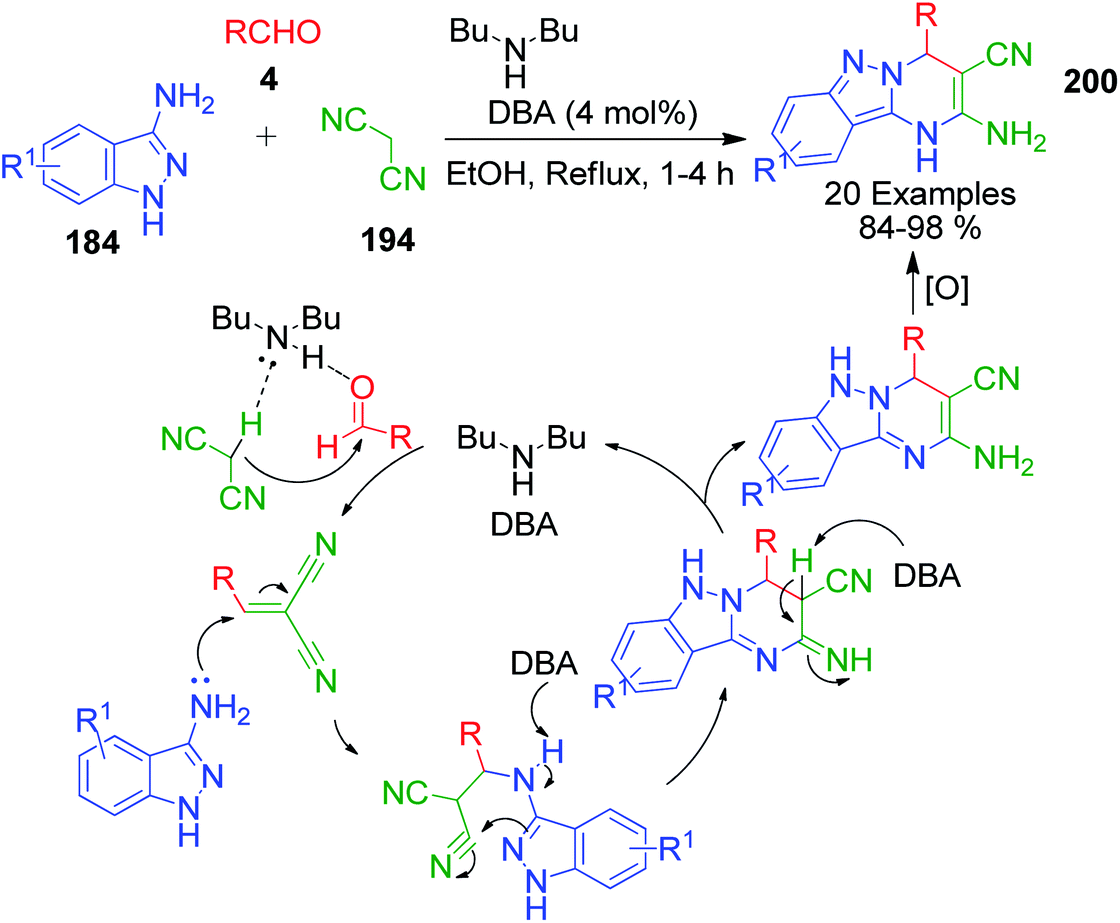
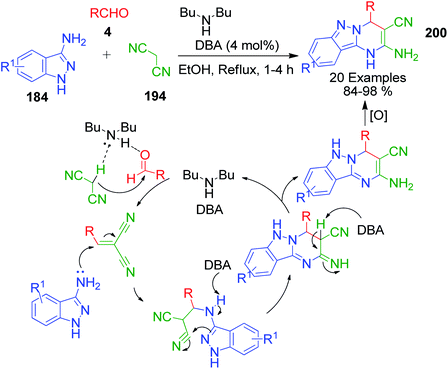
Novel tricyclic pyrimido[1,2-b]indazole-3-carbonitrile derivatives 200 containing both biologically active pyrimidine and indazole templates were synthesized via the three-component reaction of 1H-indazol-3-amine 184, aldehydes 4, and malononitrile 194 catalyzed by the base di-n-butylamine (DBA) in ethanol.146 This group-assisted-purification (GAP) chemistry process follows a proposed mechanism, as shown in Scheme 65.
 |
| | Scheme 65 Synthesis and proposed mechanism for tricyclic pyrimido[1,2-b]indazole-3-carbonitriles 200. | |
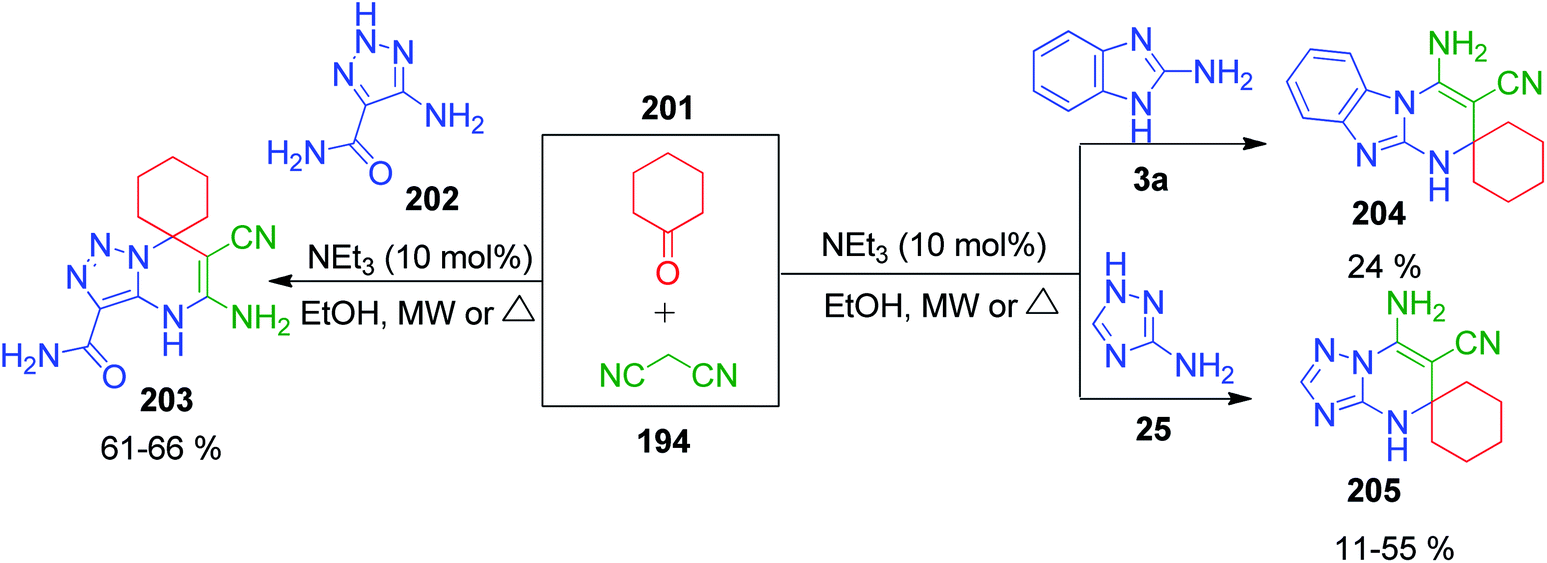
E. S. Gladkov et al. reported the synthesis of spiro derivatives of pyrimidine by methods involving a three-component reaction under microwave and conventional heating. The triazole and benzimidazole-fused spiro derivatives of pyrimidine were prepared by mixing cyclohexanone 201 with malononitrile 194 and 5-amino-1,2,3-triazole/2-aminobenzimidazole 25, 202 and 3a using 10 mol% trimethylamine in ethanol (Scheme 66).147
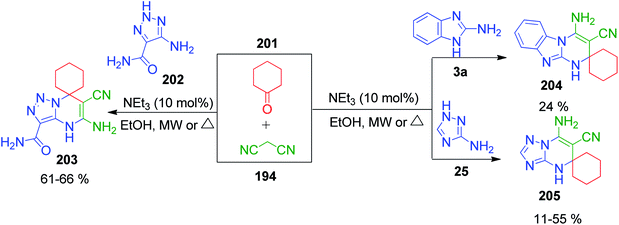 |
| | Scheme 66 Synthesis of triazole and benzimidazole-fused spiro derivatives under microwave and conventional heating. | |
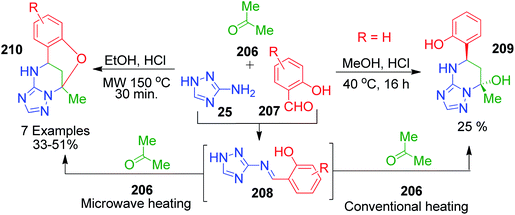 |
| | Scheme 67 Synthesis of two different products under microwave and conventional heating. | |
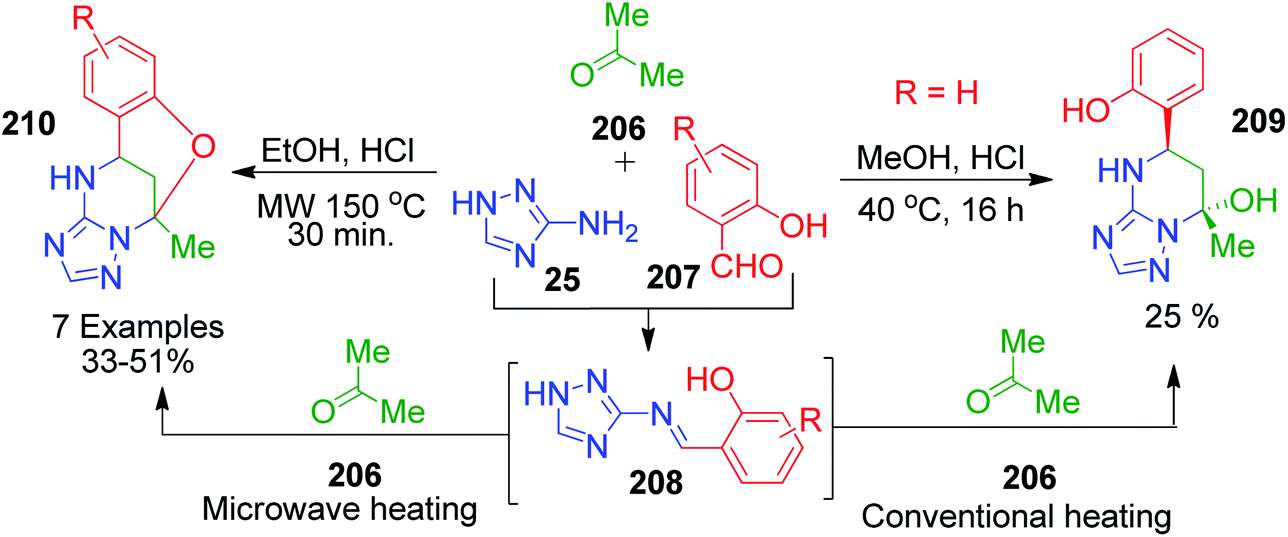
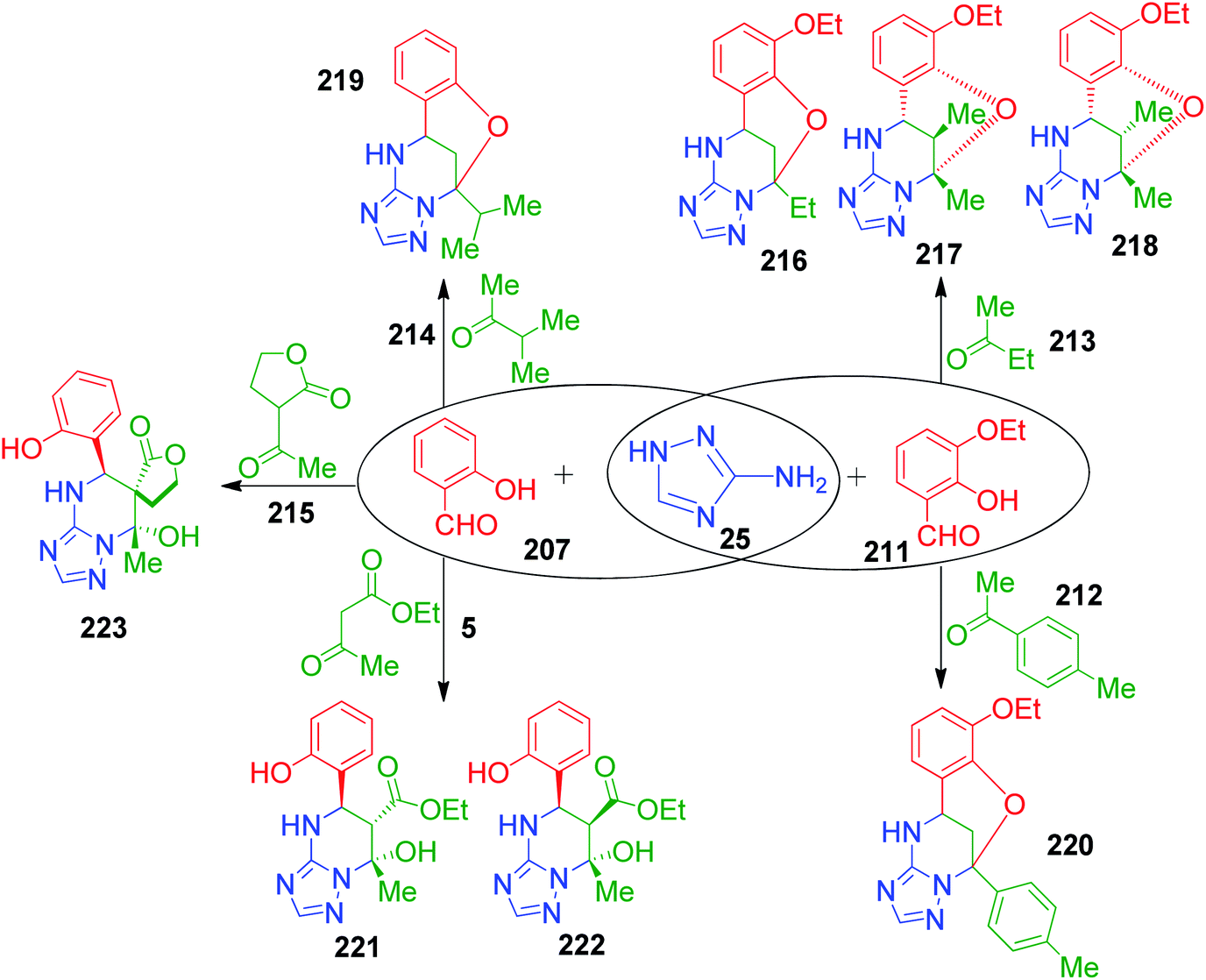
The versatility of the method was demonstrated by employing various ketones such as butan-2-one 213, 3-methylbutan-2-one 214, 4-methylacetophenone 212 and ethyl acetoacetate 4. Under similar reaction conditions, butan-2-one 213 afforded a mixture of three isomers 216, 217 and 218 in a ratio of 3:3:1 with total yield of 42%. The NOESY correlation technique was used to determine the structure of 217 and 218 diastereoisomers and correlations between the protons of the aryl ring and methyl group at position 13 were observed in of case 218 only. The corresponding bridged compounds constructed with 3-methylbutan-2-one 214 and 4-methylacetophenone 212 were isolated upon precipitation in poor yields of 25% and 21%, 219 and 220, respectively, due to the incomplete reaction. Next, an equimolar mixture of ethyl acetoacetate 5 with 3-amino-1,2,4-triazole 25 and salicylic aldehyde acetoacetate 211 in absolute ethanol (instead of methanol to avoid transesterification) and HCl solution in dioxane resulted in the formation of dihydroxy derivative 221. The relative configuration 5R,6S,7S at the stereocenters of 221 was assigned by NMR. During NMR measurement in DMSO-d6 solution, diastereoisomer 222 with the 5R,6R,7S configuration was detected due to the isomerization of 221. Further, the regio- and stereoselective formation of spiropyrimidine 223 was observed with 71% yield when 3-acetyl-dihydrofuran-2(3H)-one 215 was used in the present Biginelli-like multicomponent reaction (Scheme 68).
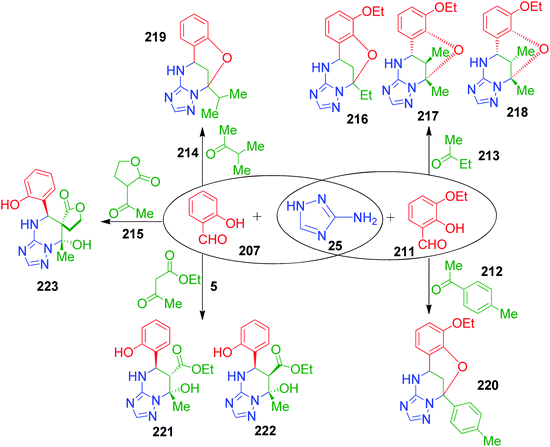 |
| | Scheme 68 Synthesis of various tetrahydropyrimidines depending upon the structure of acetones used. | |
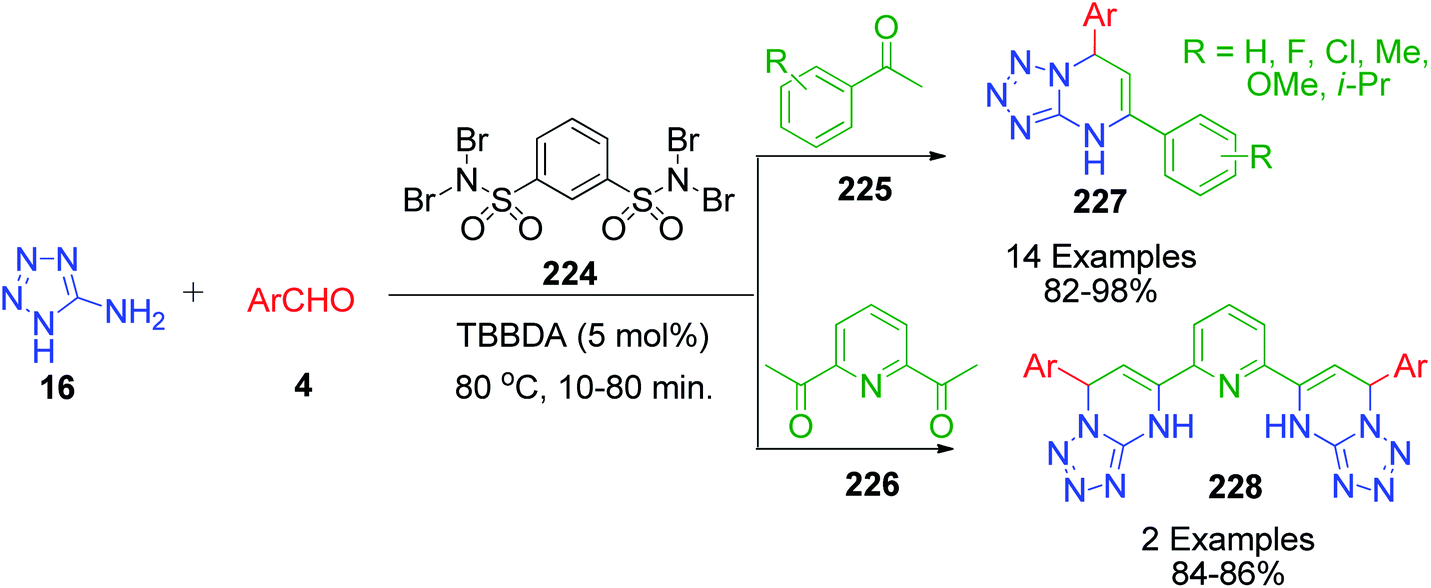
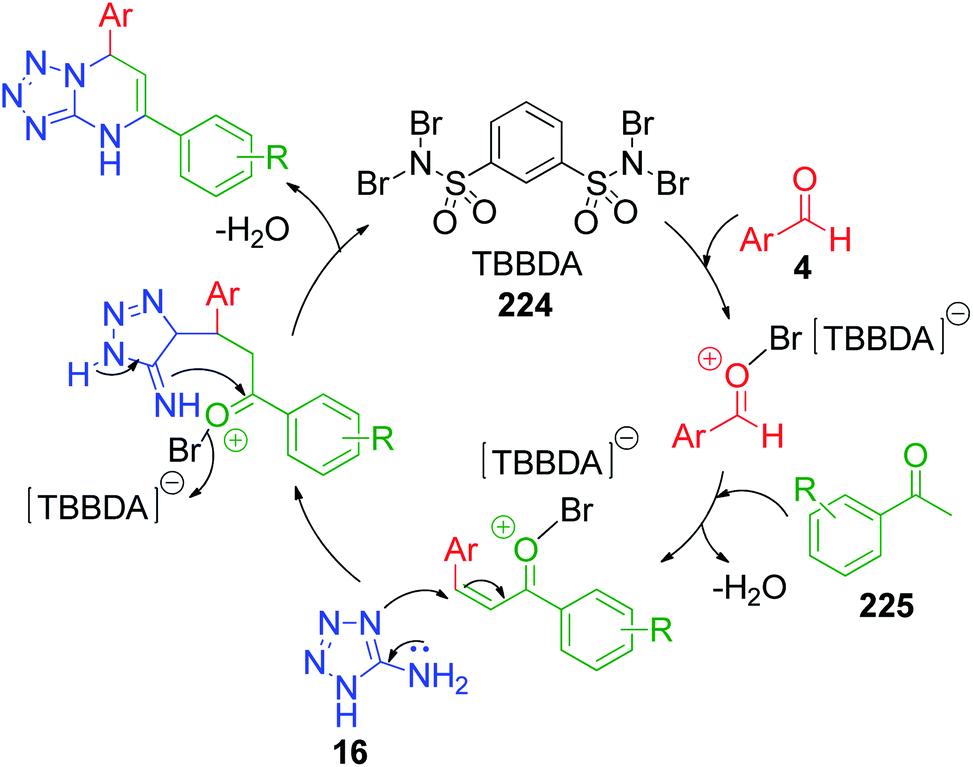
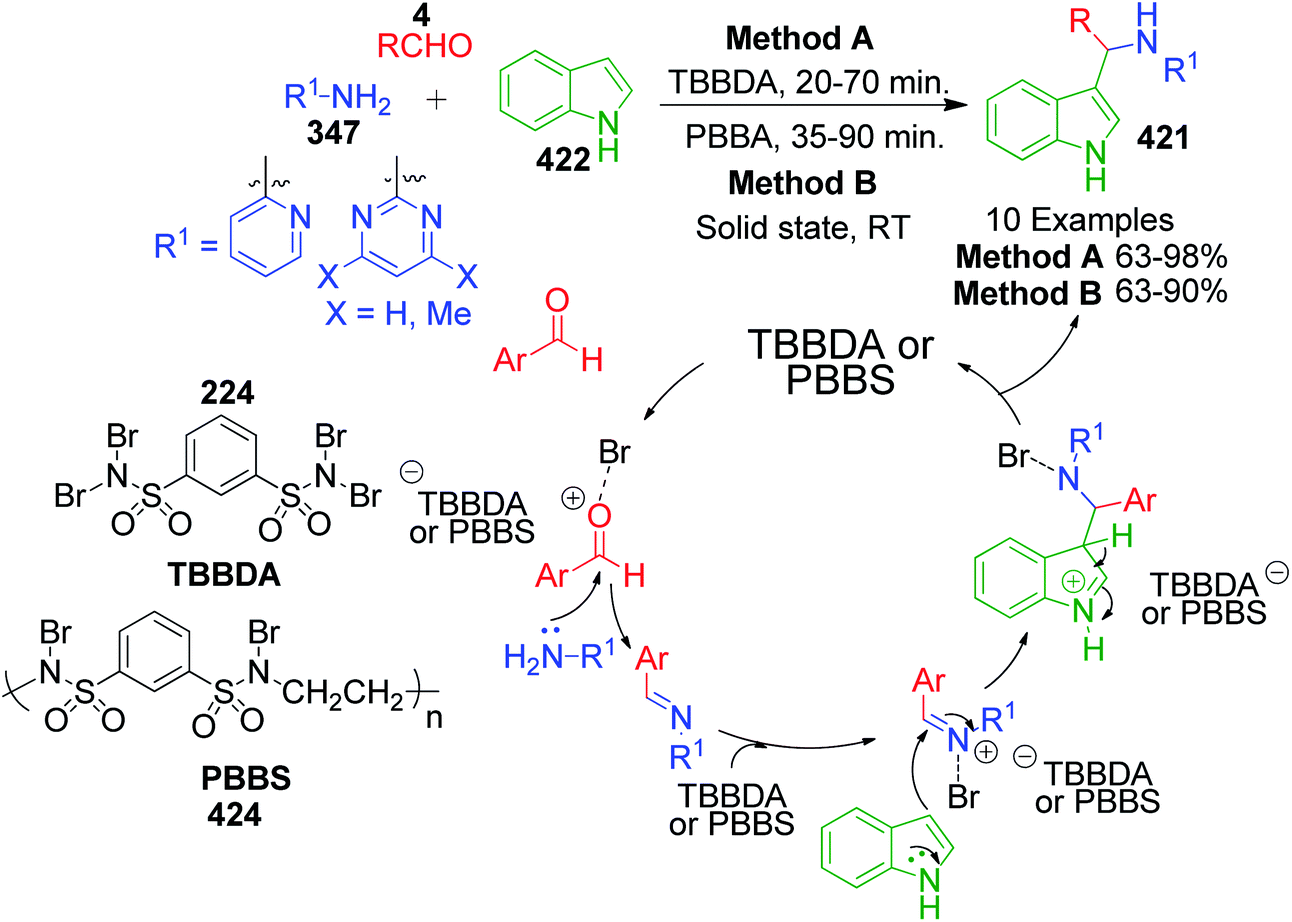
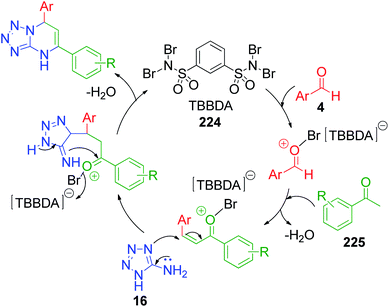
Methyl ketones work analogously to β-dicarbonyl compounds in the MCR but have less acidic protons compared to the latter due to the presence of only one electron-withdrawing keto group. R. Ghorbani-Vaghei et al. developed a series of tetrazole-fused pyrimidine derivatives utilizing acetophenone 225, 5-aminotetrazole 16 and various aromatic aldehydes 4.42 Condensation of these reactants at 80 °C under solvent-free conditions together with N,N,N′,N′-tetrabromobenzene-1,3-disulfonamide (TBBDA) 224 as a catalyst afforded 5,7-diaryl-4,7-dihydrotetrazolo[1,5-a]pyrimidines 227. They also reported the synthesis of novel bis-dihydrotetrazolo[1,5-a]pyrimidine 228 via the reaction of diacetylpyridine 226 with 2 equivalents of each aldehyde 4 and 5-aminotetrazole 16 under the optimized reaction conditions (Scheme 69). The plausible mechanism as suggested by the authors for this reaction is shown in Scheme 70, where bromine atoms are detached in situ from TBBDA 224 as bromonium ions and act as the oxidant in the reaction medium.
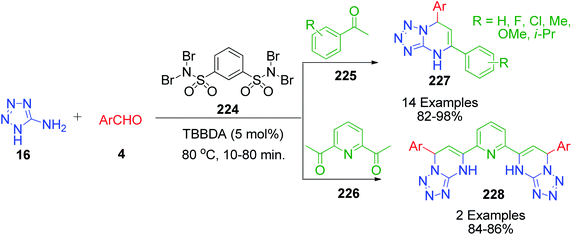 |
| | Scheme 69 Solvent-free synthesis of dihydrotetrazolo[1,5-a]pyrimidines and bis-dihydrotetrazolo[1,5-a]pyrimidine. | |
 |
| | Scheme 70 Proposed mechanistic pathway to dihydrotetrazolo[1,5-a]pyrimidines. | |
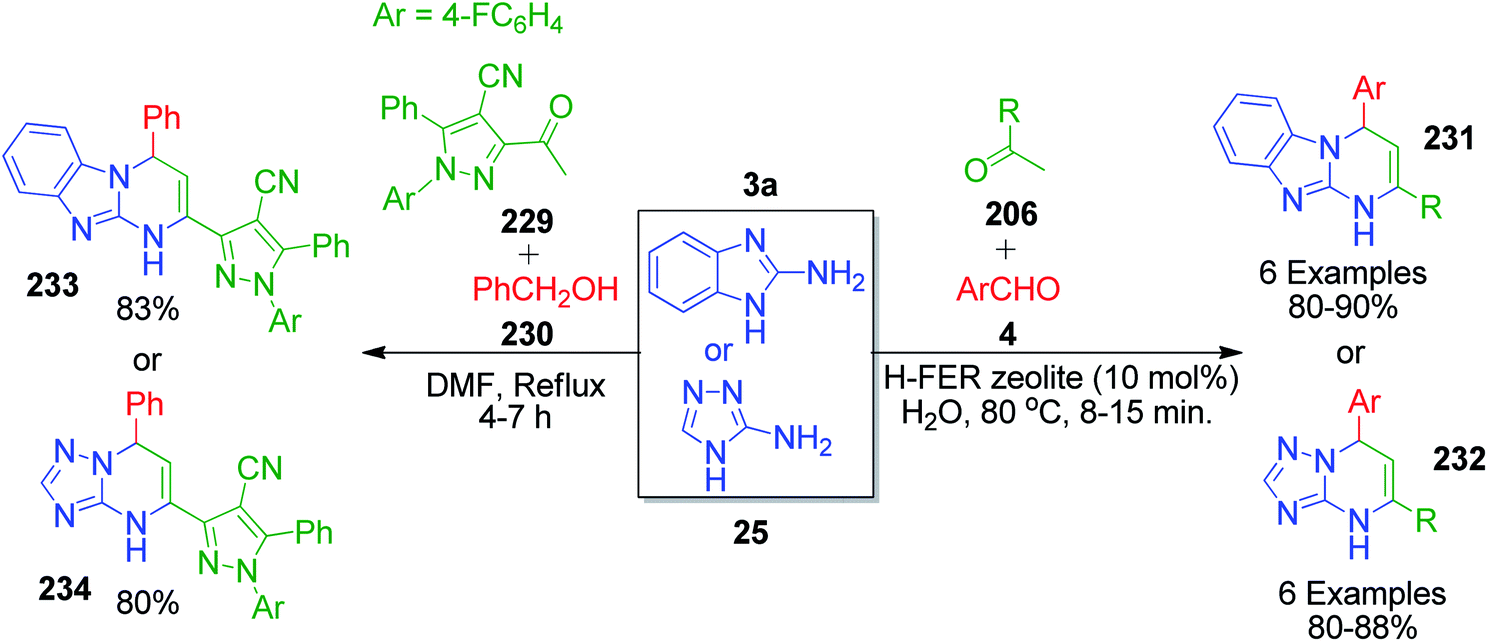
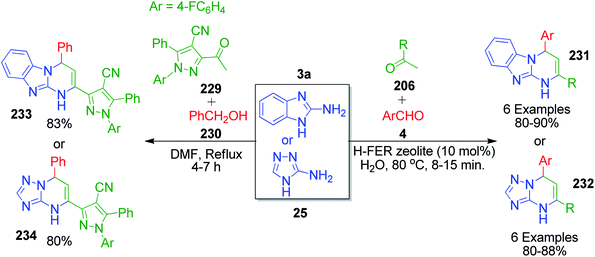
H. M. E. Hassaneen and T. A. Farghaly also explored acetophenone derivatives in a one-pot three-component reaction with 2-aminobenzimidazole/3-amino-[1,2,4]triazole 3a and 25 and aromatic aldehyde 22. The reaction mixture was taken in aqueous medium together with H-ferrierite zeolite as a catalyst and heated under reflux conditions for 8–15 min, affording a novel series of [1,2,4]triazolo[1,5-a]pyrimidines 232 and pyrimido[1,2-a]benzimidazole derivatives 231.149 A different methyl ketone-linked compound, 3-acetyl-1-(4-fluorophenyl)-5-phenyl-1H-pyrazole-4-carbonitrile, was synthesized and utilized by K. A. Ali et al. as one of the MCR components for the synthesis of novel substituted pyrimidine scaffolds. This component was mixed with benzaldehyde and 2-aminobenzimidazole/3-amino-[1,2,4]triazole in DMF solvent under reflux to afford 1-(4-fluorophenyl)-5-phenyl-3-(4-phenyl-1,4-dihydropyrimido[1.2-a]benzimidazol-2-yl)-1H-pyrazole-4-carbonitrile/1-(4-fluorophenyl)-3-(5,8-dihydro-5-phenyl-[1,2,4]triazolo[4,3-a]pyrimidin-7-yl)-5-phenyl-1H-pyrazole-4-carbonitriles 233 and 234 (Scheme 71).150
 |
| | Scheme 71 Synthesis of [1,2,4]triazolo[1,5-a]pyrimidines and pyrimido[1,2-a]benzimidazole derivatives. | |
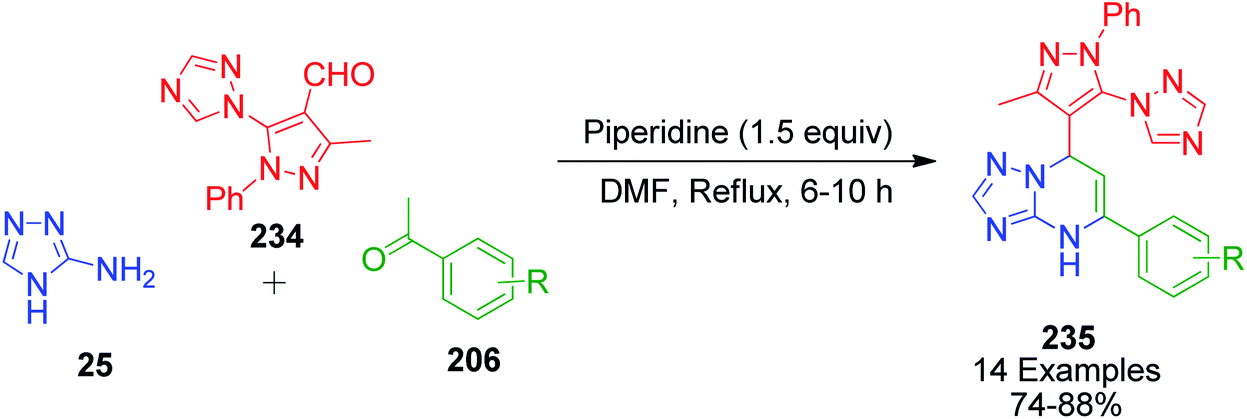
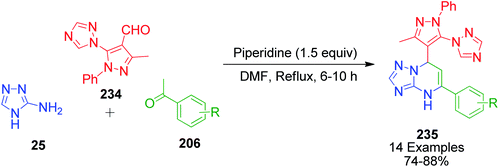
V. Pogaku et al. developed a one-pot three-component approach to synthesize novel pyrazole-linked triazolopyrimidines as a potent α-glucosidase inhibitor by performing a reaction using acetophenone derivatives 206, 3-methyl-1-phenyl-5-(1H-1,2,4-triazol-1-yl)-1H-pyrazole-4-carbaldehyde 234 and 4H-1,2,4-triazol-3-amine 25 as starting materials. This piperidine-catalyzed reaction was executed in DMF under reflux conditions to afford the target products 235 with 74–88% yield (Scheme 72).151
 |
| | Scheme 72 Piperidine-catalyzed synthesis of new pyrazole–triazolopyrimidine hybrids. | |
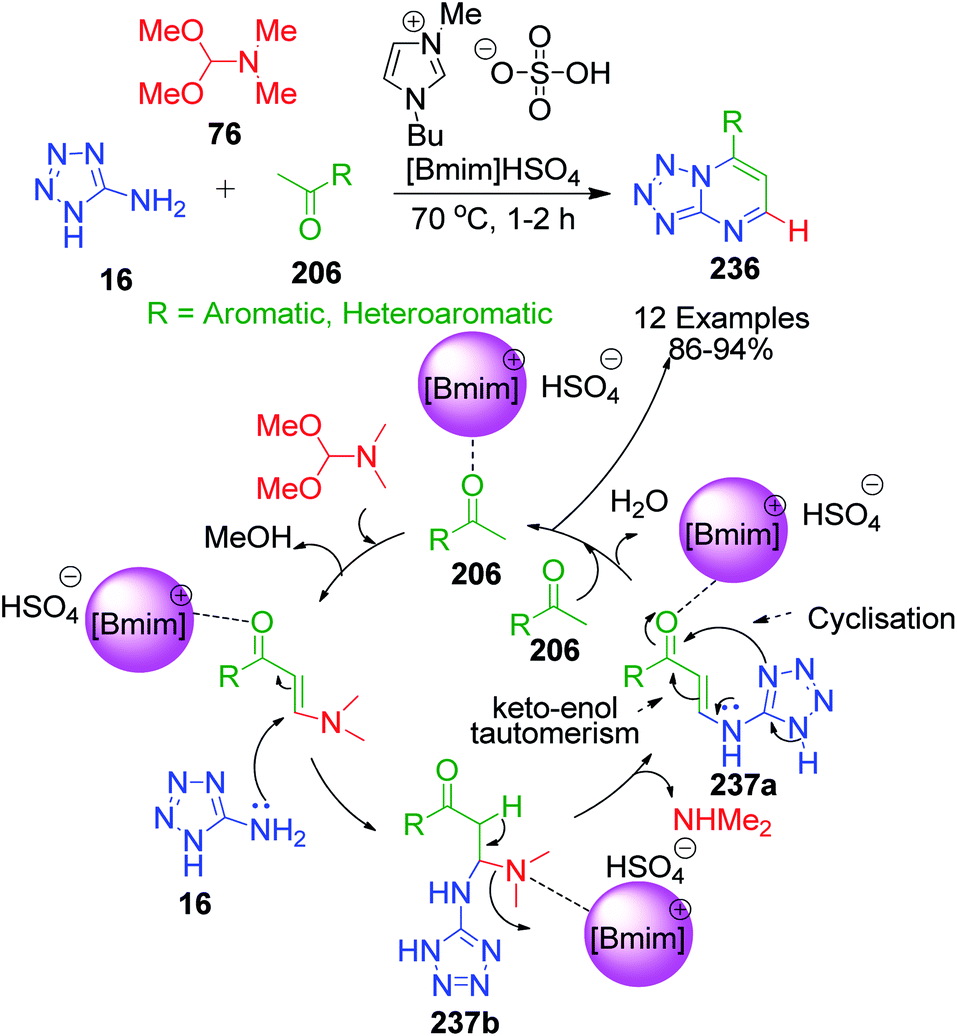
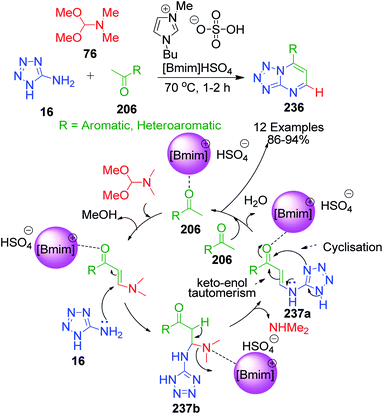
N,N-Dimethylformamidedimethylacetal 76, a formaldehyde analogue, was employed in place of substituted aromatic aldehyde by L. Suresh and coworkers to achieve fused tetrazolo[1,5-a]pyrimidine derivatives 236.152 The multicomponent condensation of 5-aminotetrazole 16, dimethylformamidedimethylacetal 76 and acetophenones 206 was carried out in the ionic liquid 1-butyl-3-methylimidazolium hydrogen sulfate [bmim]HSO4 at 70 °C. The formation of tetrazolo[1,5-a]pyrimidine 236 was proposed to proceed through α,β-unsaturated ketone intermediate 237a generated in situ from the ionic liquid-supported condensation of acetophenone 206 and N,N-dimethylformamidedimethylacetal 76. This intermediate is then attacked by 5-aminotetrazole 16 followed by the subsequent loss of dimethyl amine to form another α,β-unsaturated ketone intermediate 237b. The final step involves the keto–enol tautomerism of this intermediate, followed by cyclization via the elimination of a water molecule (Scheme 73).
 |
| | Scheme 73 Ionic liquid-mediated synthesis of fused tetrazolo[1,5-a]pyrimidines and proposed mechanistic pathway. | |
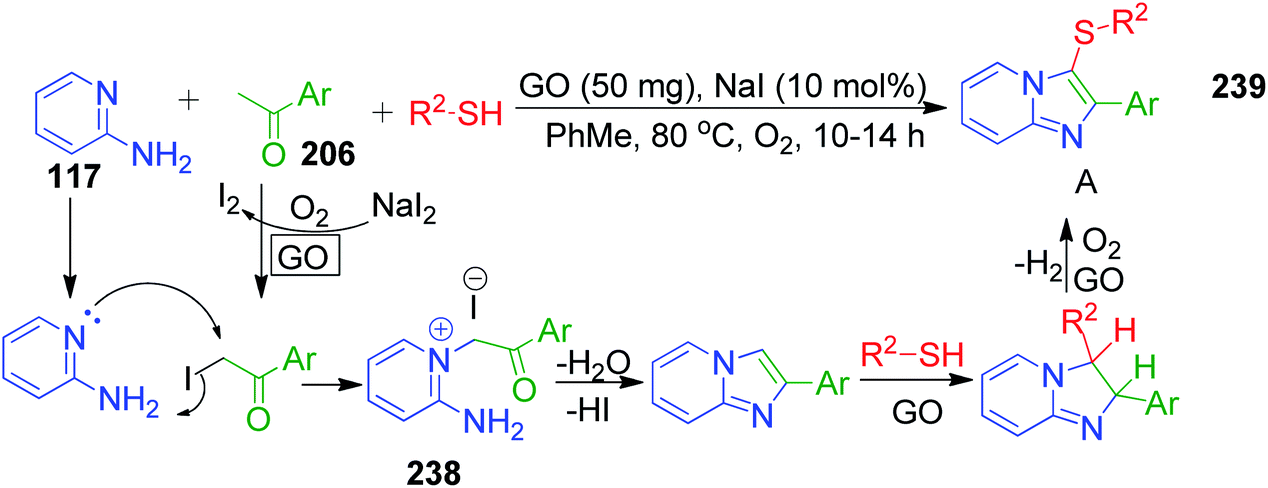
The green carbocatalyst graphene oxide (GO) was employed by S. Kundu and B. Basu in a one-pot multi-component approach for the synthesis of biologically important 3-sulfenylimidazo[1,2-a]pyridine scaffolds 239.153 The present method involves a reaction between 2-aminopyridine 117, acetophenones 206 and various thiols in the presence of GO and NaI (as the additive) at 80 °C under aerial conditions. The reactions are believed to proceed via a selective and tandem manner involving an Ortoleva–King-type intermediate 238 and the catalyst GO was found to be recyclable with appreciable conversions (Scheme 74).
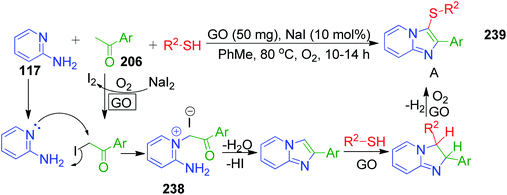 |
| | Scheme 74 Selective and tandem protocol to access 3-sulfenylimidazo[1,2-a]pyridines. | |
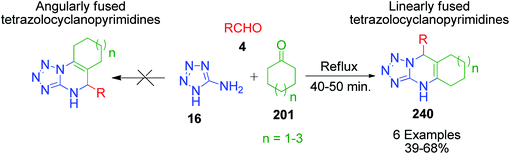 |
| | Scheme 75 Synthesis of tetrazolopyrimidines annulated to C6–C8 carbocycles. | |
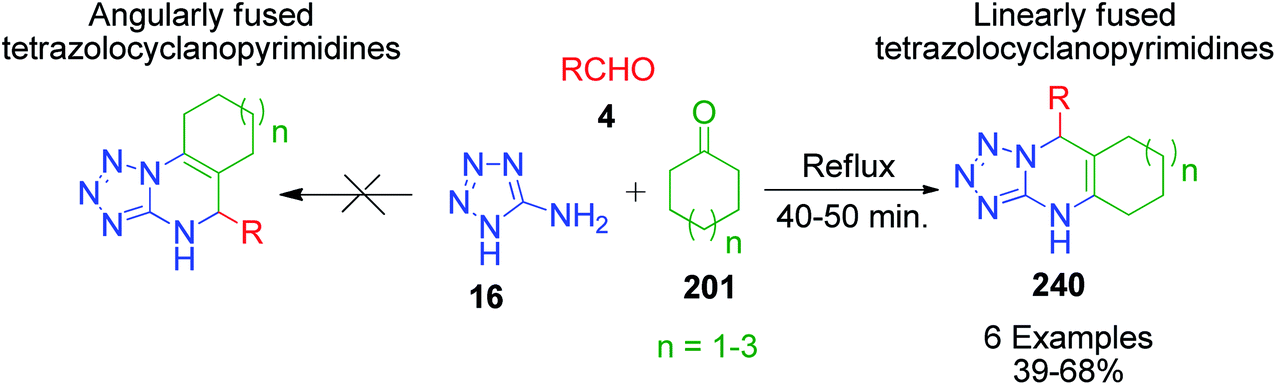
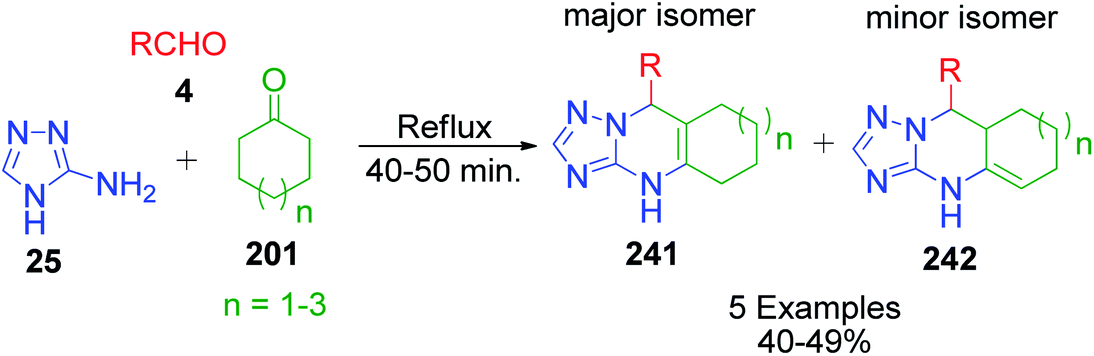
Next, the same researcher described the synthesis of two isomers of linearly fused pyrimidine derivatives with a C6–C8 cycloalkene ring fused at the C5–C6 bond, differing by the position of the double bond in the cycloalkene ring.156 In this method, a more nucleophilic aminoazole, 4H-1,2,4-triazol-5-amine 25, was heated with equimolar amounts of benzaldehyde/furfural 4 and cycloalkanone 201 (C6–C8) under refluxing conditions (Scheme 76). Phenyl(furyl)-substituted linearly fused cycloalkatriazolopyrimidines 241 and 242 containing a common double bond to the cycloalkene and tetrahydropyrimidine rings were obtained as the major isomers together with a minor isomer having a C4C5 double bond. The position of the CC double bond in the minor isomer was determined using 1H COSY and 1D NOESY data.
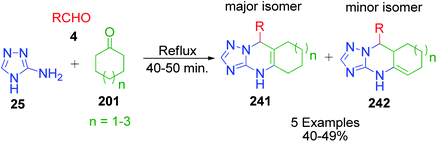 |
| | Scheme 76 Synthesis of two isomers of linearly fused pyrimidine derivatives. | |
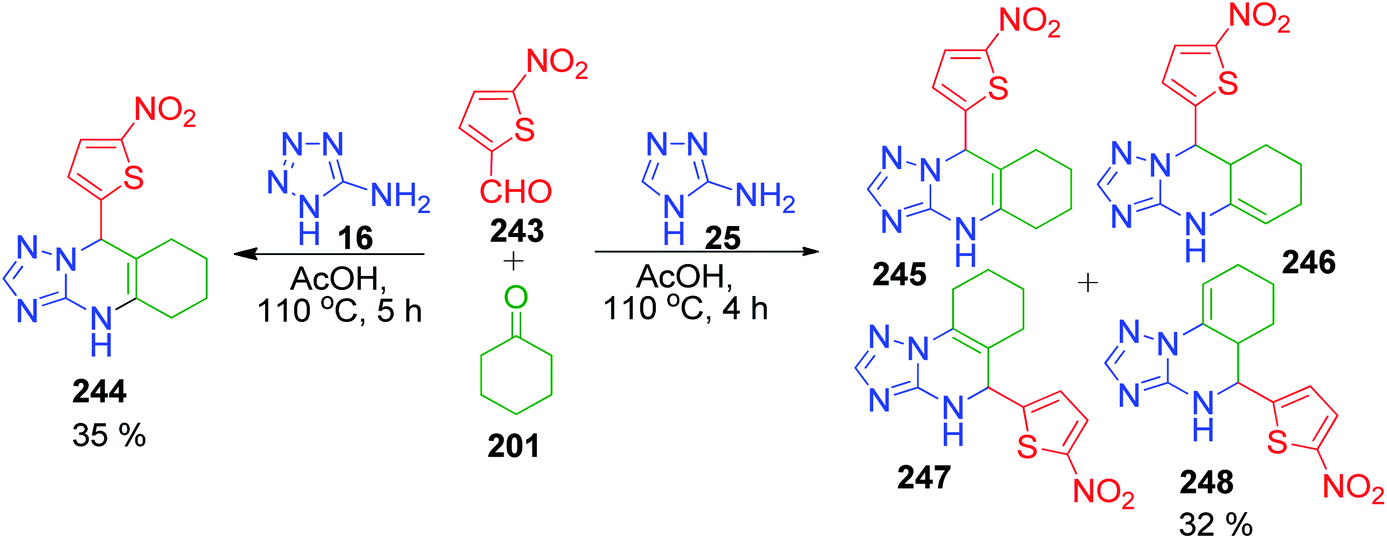
Following the above results, subsequently they performed a three-component condensation reaction between 5-aminotetrazole/3-amino-4H-1,2,4-triazole 16 and 25 with 5-nitrothiofene-2-carbaldehyde 243 and cyclohexanone 201 in acetic acid at 110 °C. The obtained product, 9-(5-nitrothiophen-2-yl)-4,5,6,7,8,9-hexahydrotetrazolo[5,1-b]-quinazoline 244 was found to be linearly fused and consistent with previous reports. An analogous reaction with 3-amino-4H-1,2,4-triazole afforded a mixture of linearly and angularly fused isomeric hexahydrotriazoloquinazolines 245, 246, 247 and 248 (Scheme 77).157
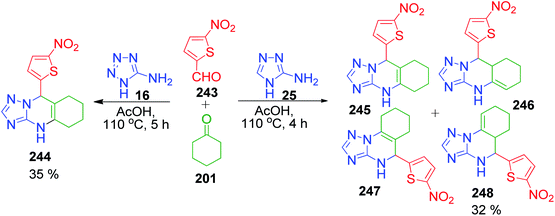 |
| | Scheme 77 Synthesis of linearly and angularly fused tetrazolo/triazoloquinazolines. | |
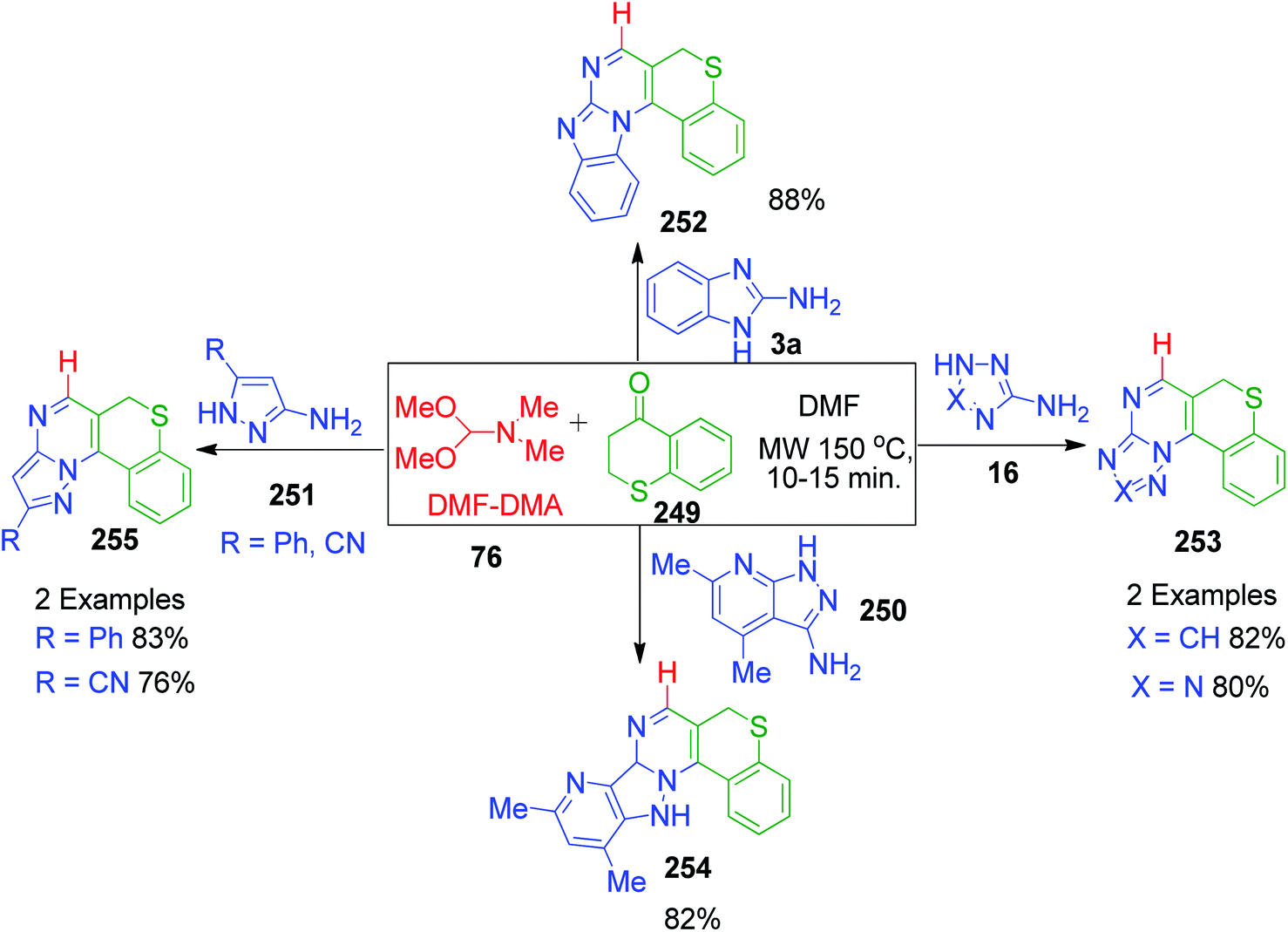
E. M. H. Abbas et al. developed an approach where dimethylformamide-dimethylacetal 76 (DMF-DMA) was utilized as an alternative to formaldehyde in the MCR synthesis of novel poly-heterocyclic ring systems 252, 253, 254 and 255. Various azole-fused pyrimidine systems 16, 3a, 250 and 251 were obtained by the three-component reactions between 1-benzothiopyran-4-ones 249, various aminoazoles and DMF-DMA 76 performed in refluxing DMF under controlled microwave heating (Scheme 78).158
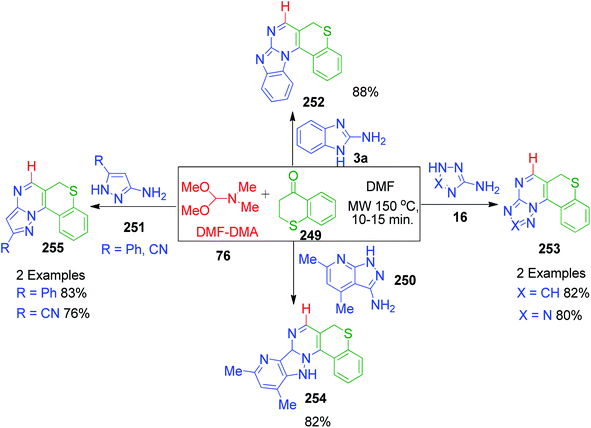 |
| | Scheme 78 Microwave-assisted synthesis of various azole-fused pyrimidines. | |
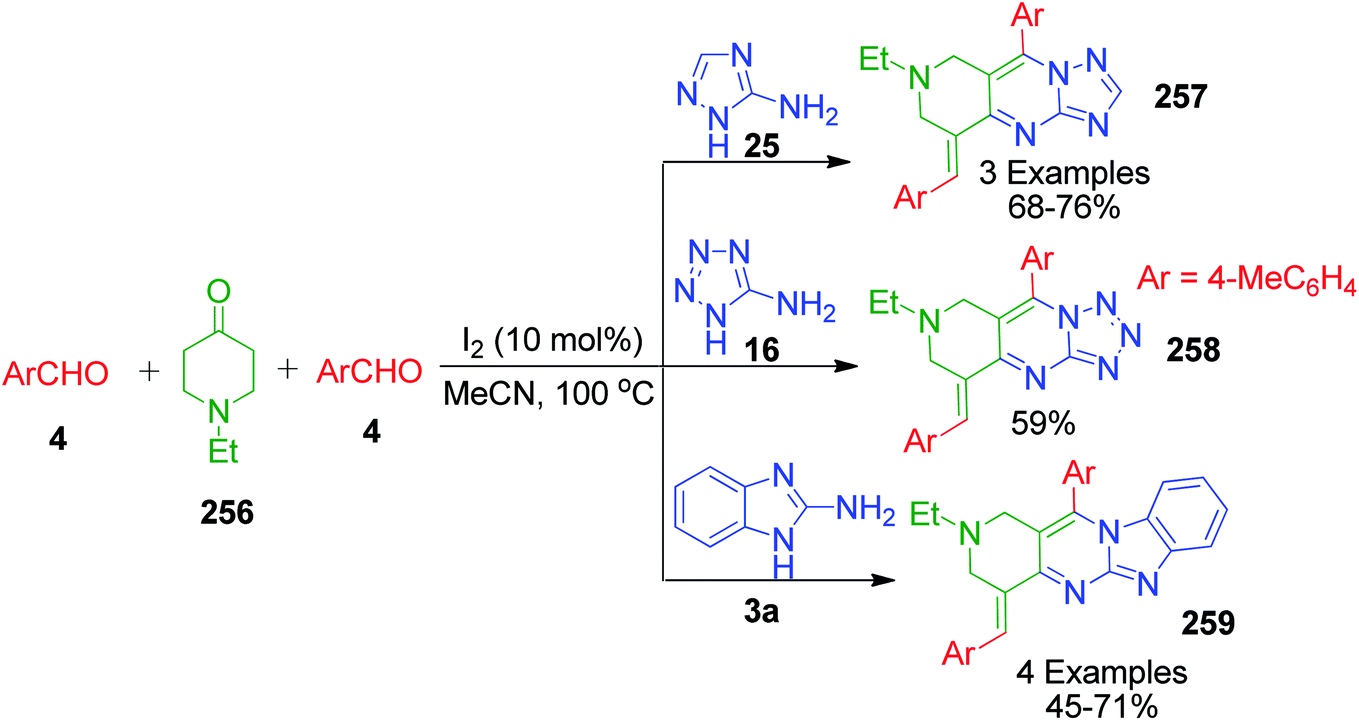
The same research group also reported a direct and efficient approach for the synthesis of pyrido[4,3-d]triazolo[1′,5′-a]pyrimidines 257, pyrido[4,3-d]tetrazolo[1′,5′-a]pyrimidine 258 and pyrido[4,3-4′,5']pyrimido[1′,2′-a]benzimidazoles 259. Condensation of 1-ethyl-4-piperidone 256 and two equivalents of appropriate aromatic aldehyde 4 with 3-amino[1,2,4]triazole/5-aminotetrazole 25 and 16 in the presence of iodine under refluxing acetonitrile afforded the corresponding triazolo/tetrazolopyrimidines 257 and 258. Reaction with 2-aminobenzimidazole 3a under the same conditions afforded the corresponding 1,2,3,4-tetrahydro-pyrido[4,3-d]benzoimidazolo[1,2-a]pyrimidines 259 (Scheme 80). When the reaction was carried out with 2 equivalents of 4-methyl benzaldehyde as the aldehydic component together with 2-aminobenzimidazole 3a and 1-ethyl-4-piperidone, it yielded a mixture of an intermediate and the final product. The intermediate was then transformed into the final pyrimidine derivative via in situ oxidation (Scheme 79).159
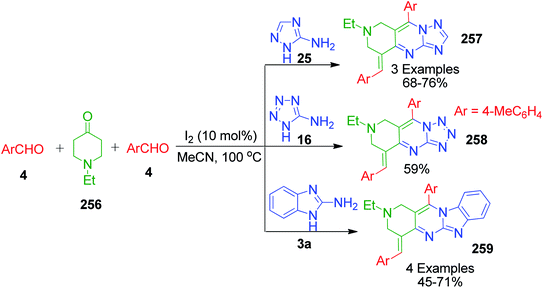 |
| | Scheme 79 Iodine-catalyzed synthesis of various azole fused pyrimidines. | |
 |
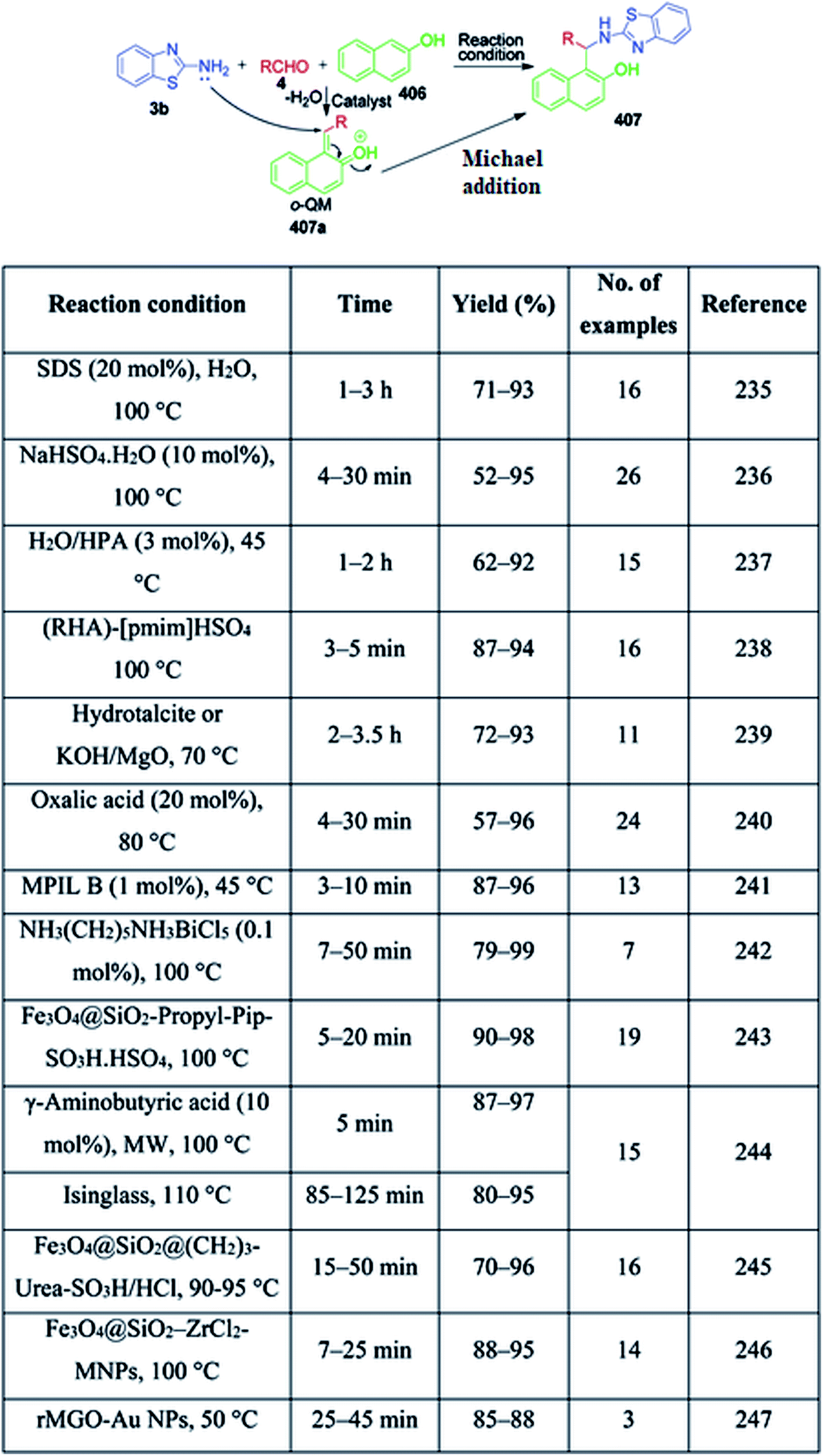
| | Scheme 80 Synthesis of novel tetrazolo-fused pyrimidines under microwave heating. | |
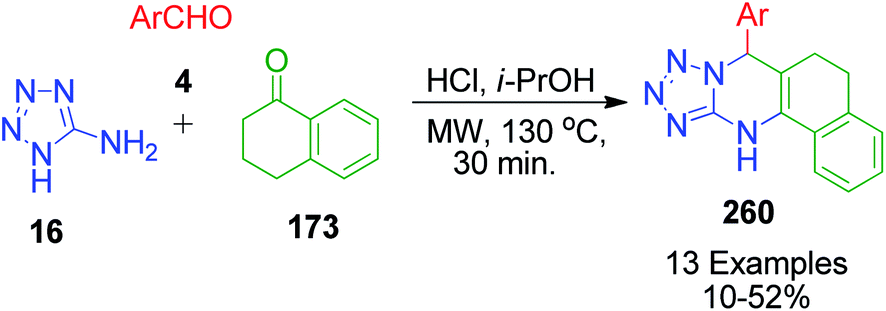
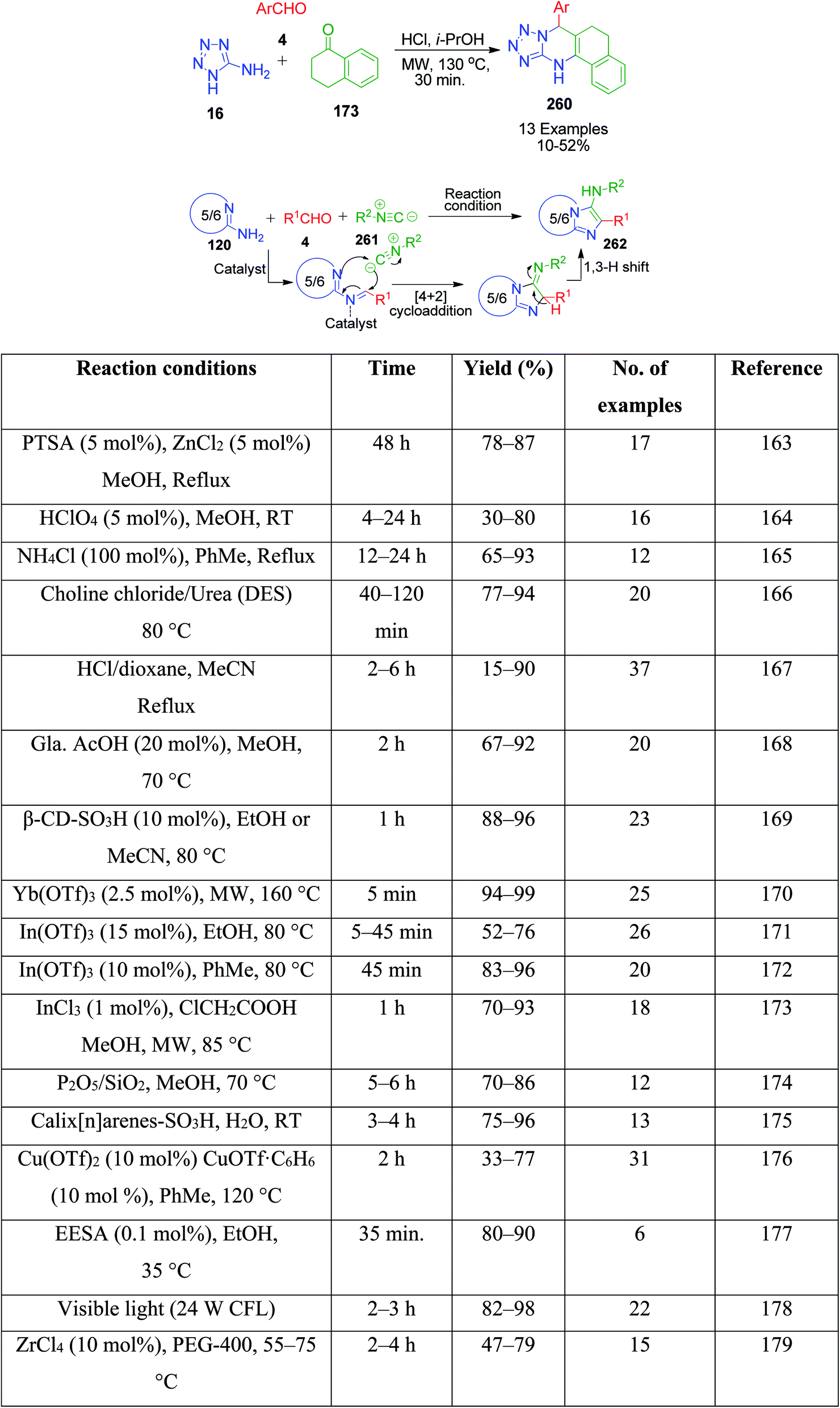
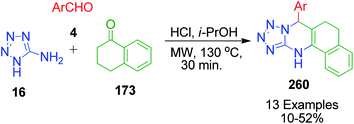
In continuation of these developments, G. P. Kantin and M. Krasavin demonstrated an acid-catalysed MCR synthesis of a series of novel tetrazolo-fused pyrimidines 260. A reaction mixture containing α-tetralone 173, 1H-tetrazol-5-amine 16, and aromatic aldehyde 4 in isopropanol was heated under microwave irradiation at 130 °C using hydrochloric acid as a catalyst. The corresponding 7-aryl-5,6,7,12-tetrahydrobenzo[h]tetrazolo[5,1-b]quinazolines 260 were obtained after 30 min in 10–52% yield (Scheme 81).160
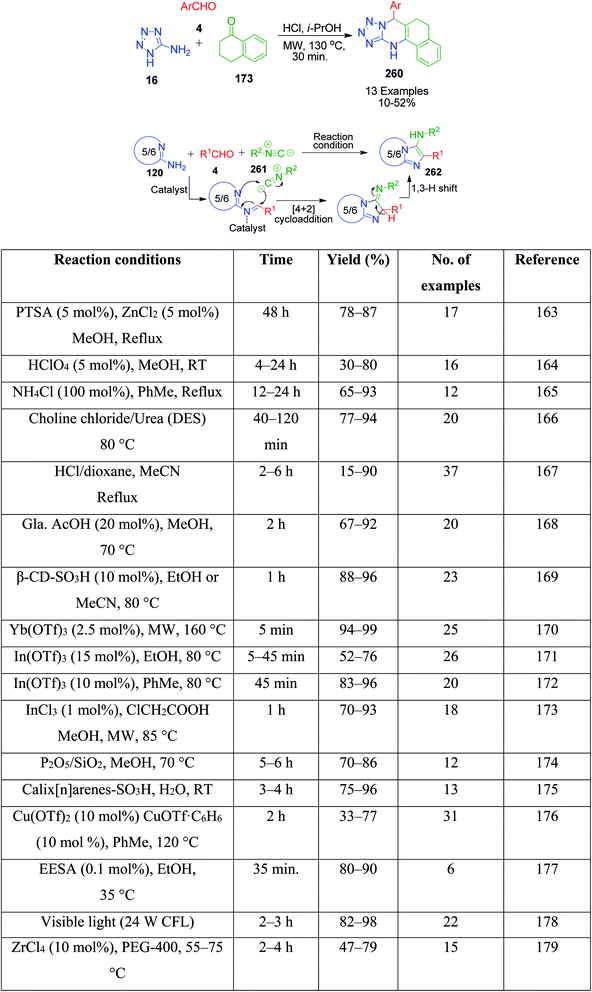 |
| | Scheme 81 Reported GBB methods utilizing various catalysts. | |
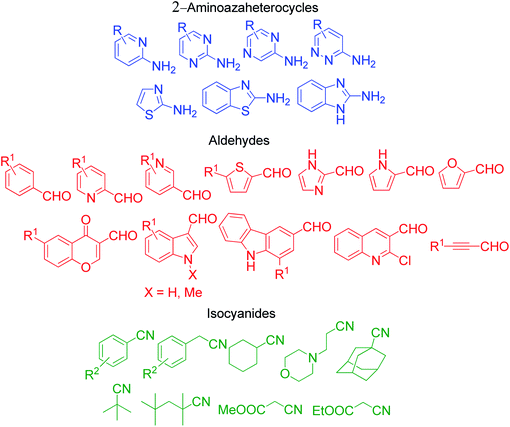 |
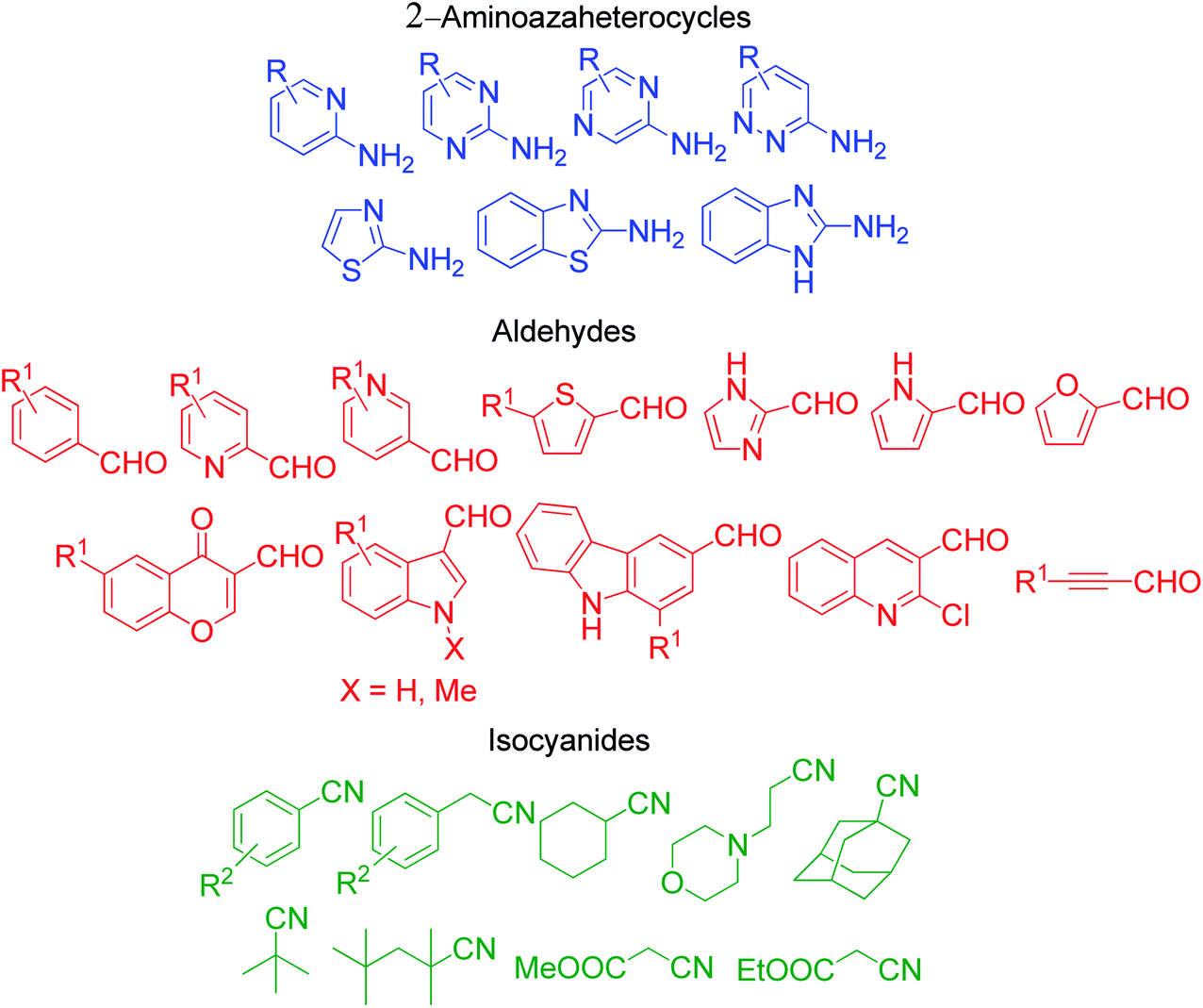
| | Fig. 6 Various derivatives of aminoazaheterocycles, aldehydes and isocyanides used in the GBB reaction. | |
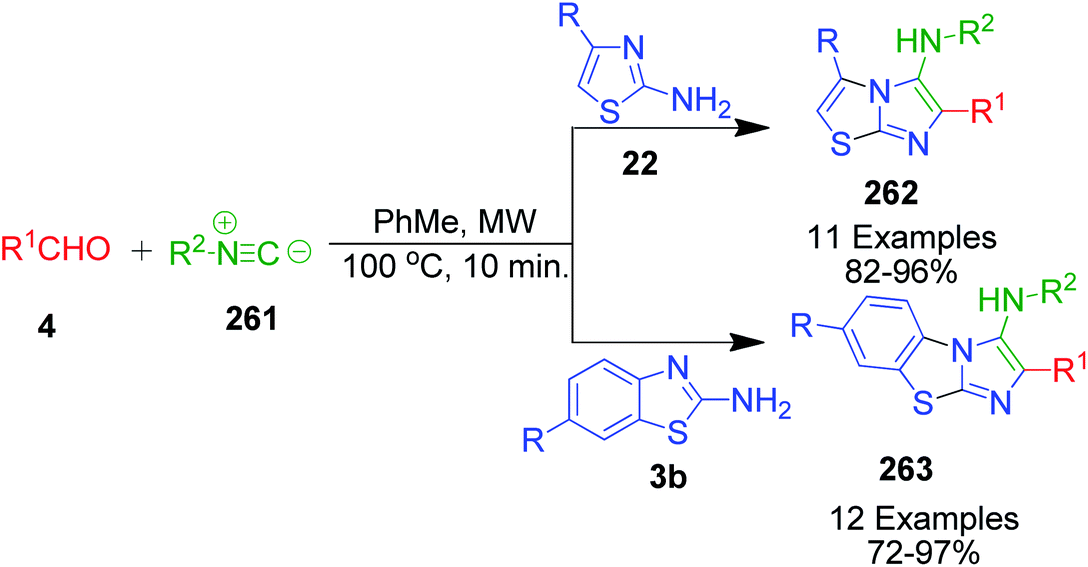
A series of twenty-three novel unsymmetrical bis-heterocycles having either imidazo[2,1-b]thiazole 262 or benzo[d]imidazo[2,1-b]thiazole 263 frameworks bound with chromone, quinoline or julolidine was obtained in good to excellent yields (82–97%) by an acid-free GBB reaction under microwave-heating conditions (Scheme 82).180 The density functional theory (DFT) approach was used to study the mechanism of this reaction (via concerted or non-concerted pathways) with the PCM(Toluene)[M06-2X/6-311+G(d,p)//M06-2X/6-311G(d)] level of theory and it was observed that only the non-concerted pathway is followed. The mechanism starts with the condensation of 2-aminoazoles 22 with aldehyde 264 to form an imine intermediate 265. Subsequently, the reaction can follow concerted and non-concerted pathways. Concerted pathway A involves the [4 + 1] cycloaddition between imine and isocyanide followed by a prototropic shift to give imidazo[2,1-b]thiazole-chromone 264. In non-concerted pathway B, the nucleophilic addition of isocyanide to imine takes place to form nitrilium ion 266, which undergoes sequentially a 5-exo-dig cyclization and a prototropic shift to generate the target product 264 (Scheme 83).
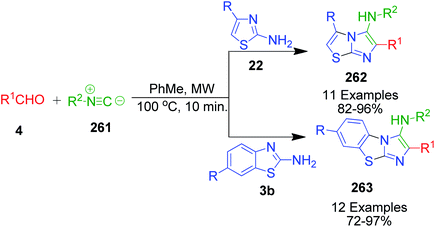 |
| | Scheme 82 Catalyst-free microwave-assisted GBB reaction. | |
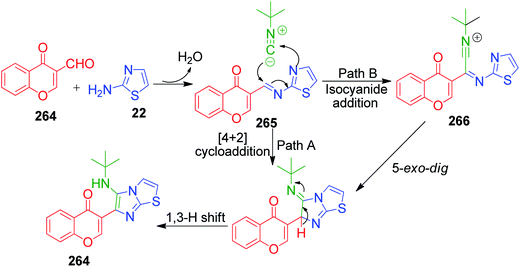 |
| | Scheme 83 Reaction mechanism to explain the synthesis of GBB product. | |
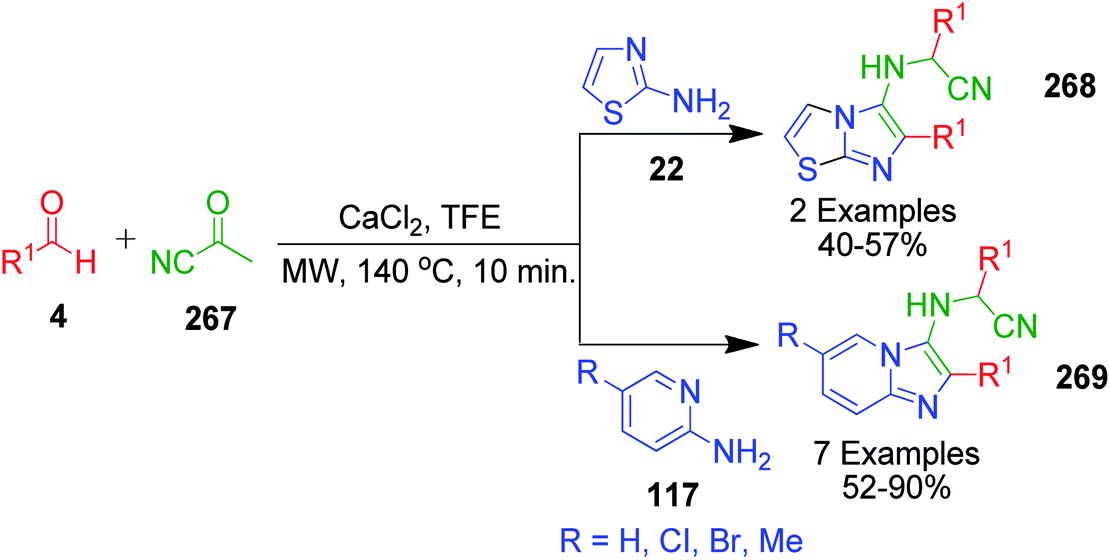
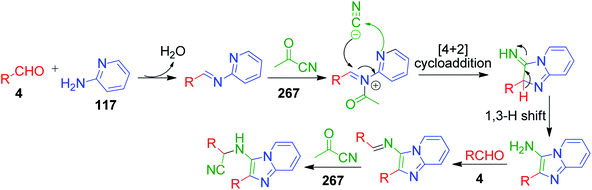
A concise one-pot three-component cascade approach was reported by G. Martinez-Ariza et al. to achieve biologically important imidazo[1,2-a]heterocycles 268 and 269 via the reaction of 2-aminothiazole/2-aminopyridine 22 and 117, two equivalents of each aldehyde 4 and acetyl cyanide 267 in the presence of calcium chloride at 140 °C (Scheme 84).181 This methodology utilizes acetyl cyanide as a non-classical isocyanide replacement for TMSCN in a microwave-assisted GBB reaction followed by Strecker reaction under catalyst-free conditions (Scheme 85).
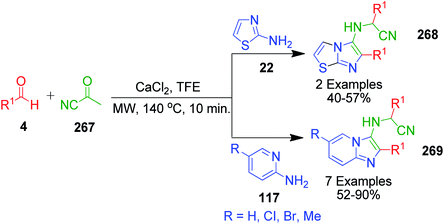 |
| | Scheme 84 Microwave-assisted GBB reaction to synthesize substituted imidazoles. | |
 |
| | Scheme 85 Proposed mechanism involving the GBB reaction followed by Strecker reaction. | |
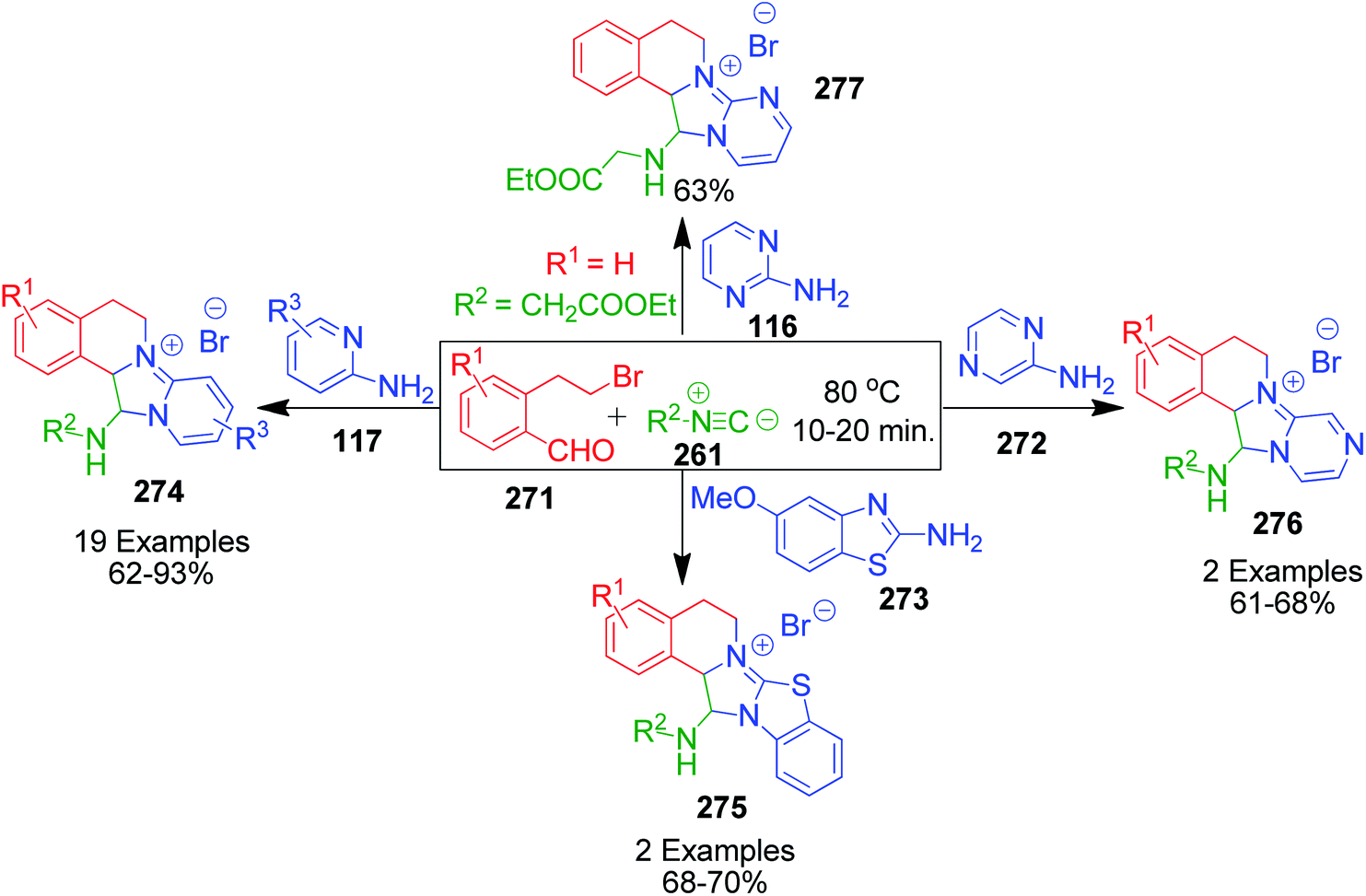
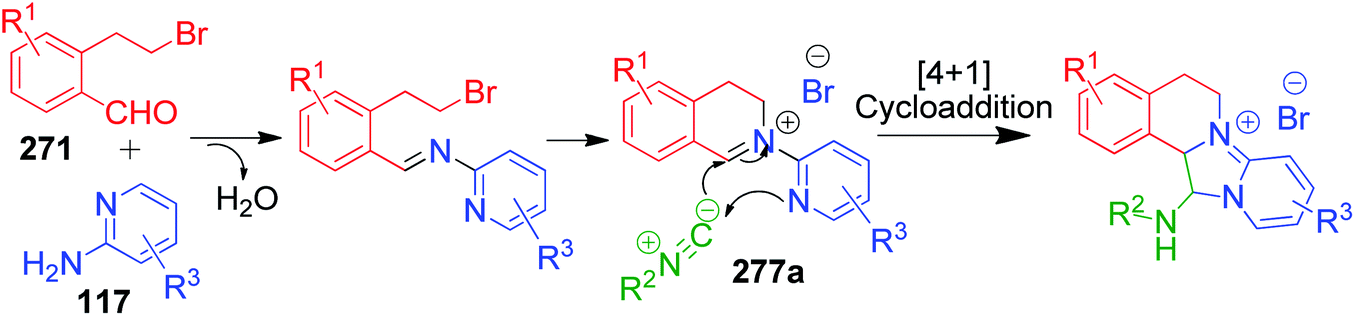
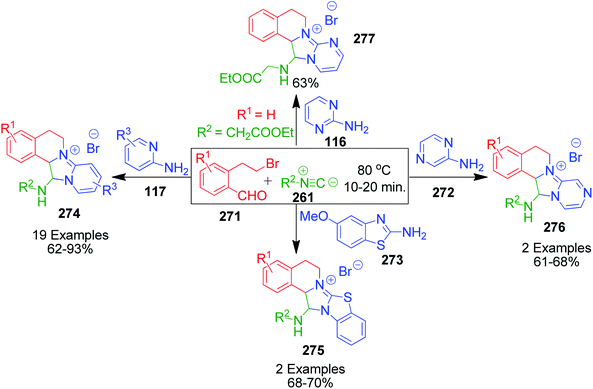
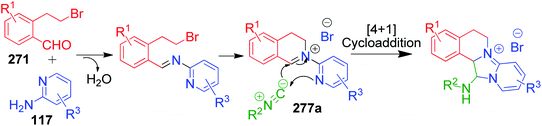
A. Sagar et al. developed an interesting one-pot GBB double annulation cascade process to synthesize diverse dihydroisoquinoline (DHIQ) salts 274, 275, 276 and 277. In this catalyst- and solvent-free protocol, a reaction mixture containing various heteroaromatic amines 116, 117, 272 and 273, 2-(2-bromoethyl)benzaldehyde derivatives 271 and isocyanides 261 was heated at 80 °C for 10–20 min, leading to the construction of two privileged heterocyclic rings (Scheme 86).182 Mechanistically, the formation of pyridoimidazo-DHIQ salts 274–277 starts with the reactive cyclic iminium ion 277a generated in situ from heteroaromatic amines and 2-(2-bromoethyl)benzaldehyde derivatives, which undergoes [4 + 1] cycloaddition with isocyanides (Scheme 87).
 |
| | Scheme 86 Catalyst- and solvent-free synthesis of pyridoimidazo-DHIQ salts. | |
 |
| | Scheme 87 Proposed mechanism involving a reactive cyclic iminium-induced GBB double annulation cascade route. | |
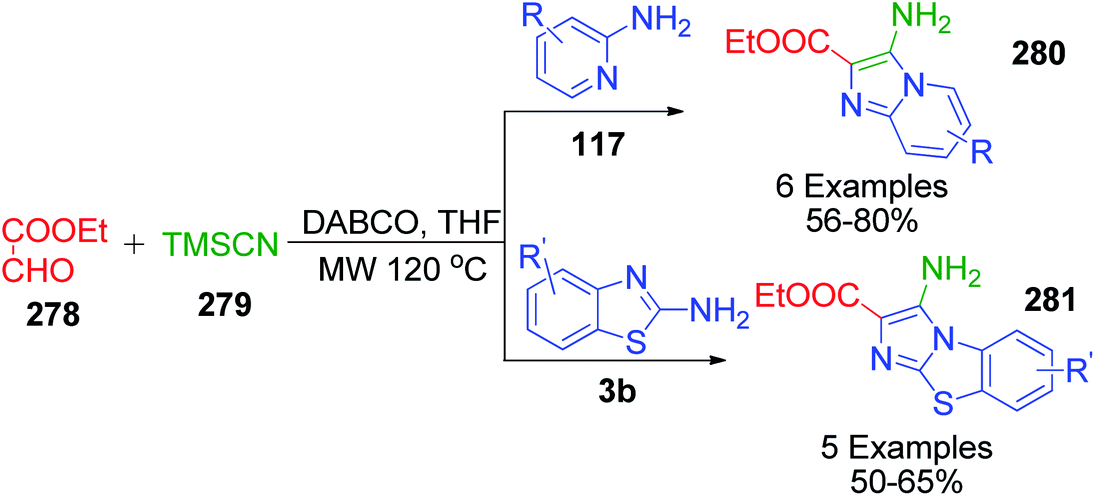
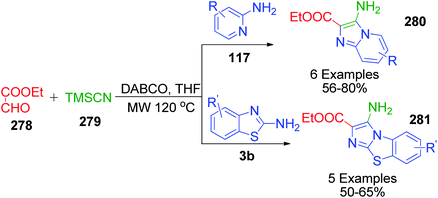
Furthermore, a chemoselective Strecker–Ugi-type reaction was developed by S. K. Guchhait and V. Chaudhary, wherein TMSCN 279, a isonitrile equivalent, ethyl glyoxalate 278 and 2-aminopyridine/2-ainobenzothiazole 117 and 3b were combined in a DABCO-THF solvent system under microwave irradiation and maintained at 120 °C (Scheme 88).183 Various 3-amine and 2-carboxyethyl functionalized N-fused imidazoles were proposed to be obtained through a reaction pathway involving the desilylative activation of TMSCN in DABCO-THF. Its flexibility for various derivatives of 2-aminopyridine/2-aminobenzothiazole 280 and 281 and the incorporation of ethyl glyoxalate as a viable aldehyde substrate make this method a very important Strecker–Ugi reaction using TMSCN 279.
 |
| | Scheme 88 DABCO-catalyzed synthesis of various 3-amino-2-carboxyethyl-linked fused imidazoles. | |
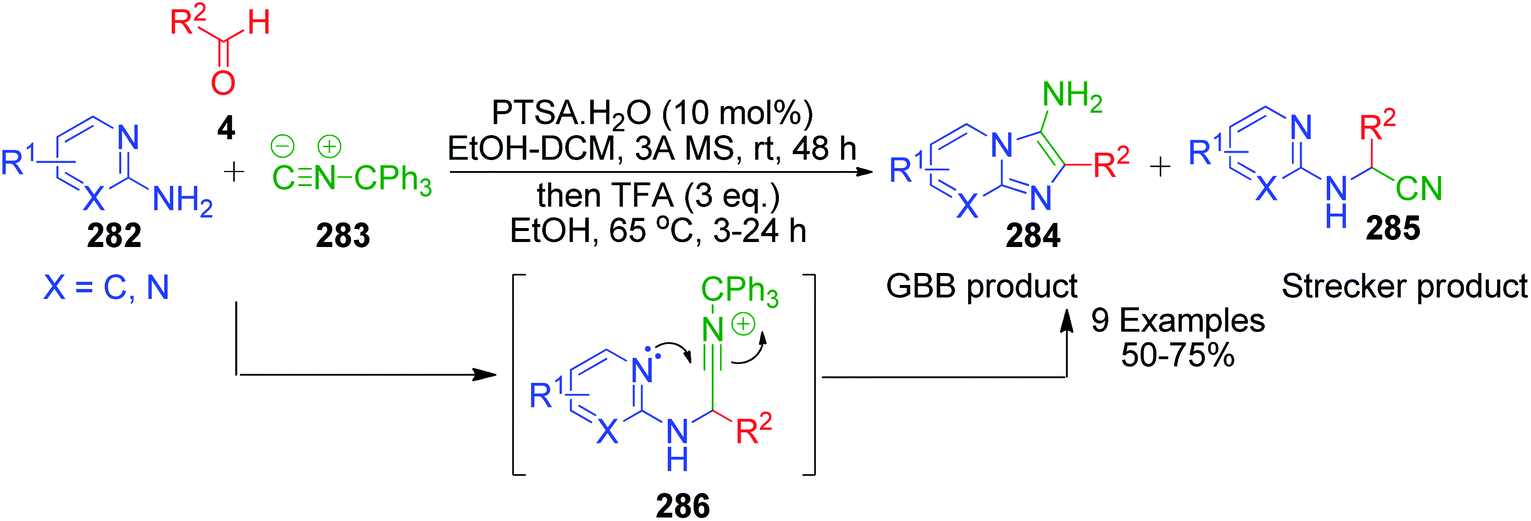
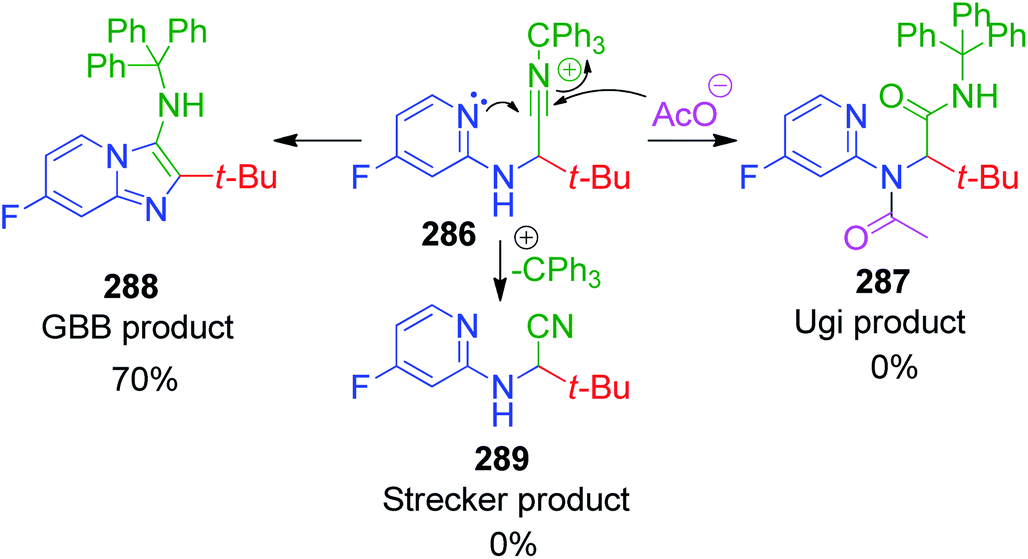
R. C. Cioc et al. reported a similar reaction, which describes the application of trityl isocyanide, as new isonitrile equivalent in a combinatorial approach for the first time. The Strecker reaction of 6-chloro-2-aminopyridine 282 with trityl isocyanide 283 and aldehyde 4 under the reaction conditions exclusively provided α-amino nitrile over the imidazo[1,2-a]pyridine 285 (Scheme 89).184 Application of the optimized Strecker protocol to a series of 2-aminopyridines and aromatic/aliphatic aldehydes showed that the cyclization to the GBB products 284 is favored. The success of this strategy relies on the fate of the nitrilium ion intermediate 286, which should undergo intramolecular trapping by the pyridine nitrogen rather than fragmentation. Significant fragmentation of the nitrilium ion (GBB product/Strecker product ratio = 5:1) was also observed for the electron-deficient 5-fluoro-2-aminopyridine. The detritylation of the GBB products was observed to be facile with 3 equivalents of TFA at 65 °C.
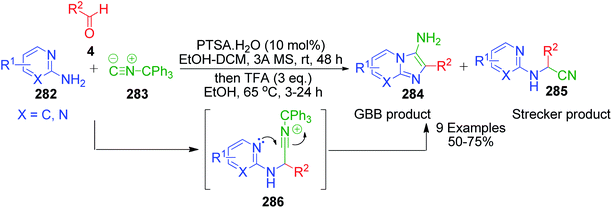 |
| | Scheme 89 Synthesis of imidazo[1,2-a]pyridin-3-amines through GBB 3-CR condensation/deprotection protocol. | |
They also performed a reaction in which all three pathways, Strecker, Ugi and GBB, are possible to investigate which product predominates. Accordingly, Ugi 4-CR with 5-fluoro-2-aminopyridine, pivalaldehyde, acetic acid, and trityl isocyanide was performed under the optimized conditions. Based on the NMR analysis, the selective formation of the GBB product clearly indicated the complete predominance of this pathway as no evidence of the Strecker and Ugi products was found. As expected, the addition of an external nucleophile (acetate) did not compete with the intramolecular trapping of the nitrilium ion by the pyridine nitrogen, even though it is relatively electron deficient. Furthermore, the mild activation of the imine by a carboxylic acid instead of a strong Brønsted acid (possibly through hydrogen bonding rather than protonation) completely suppressed the nitrilium ion fragmentation towards the Strecker product 289 (Scheme 90).
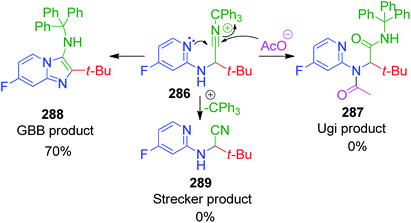 |
| | Scheme 90 Fate of nitrilium ion intermediate in the Ugi, Strecker and GBB reactions. | |
The well-known aminoazoles, 3-amino-5-methylisoxazole 291 and 5-amino-N-aryl-1H-pyrazole-4-carboxamides 287, were studied by M. V. Murlykina et al. as amine components in the Ugi and GBB multicomponent reactions (Scheme 91).185 Particularly, 5-amino-N-aryl-1H-pyrazole-4-carboxamide 287 reacted as 1,3-binucleophile with aromatic aldehydes 4 and alkylisocyanides 292 with the formation of 3-(alkylamino)-N,2-diaryl-1H-imidazo[1,2-b]pyrazole-7-carboxamides 294 (GBB reaction) (Scheme 92). In contrast, 3-amino-5-methylisoxazole 291 acted as a primary amine in the Ugi four-component reaction with aromatic aldehydes 4, phenylpropiolic acid and tert-butylisocyanide 292, giving N-(1-arylethyl-2-(tert-butylamino)-2-oxo)-N-(5-methylisoxazol-3-yl)-3-phenylpropiolamides 293.
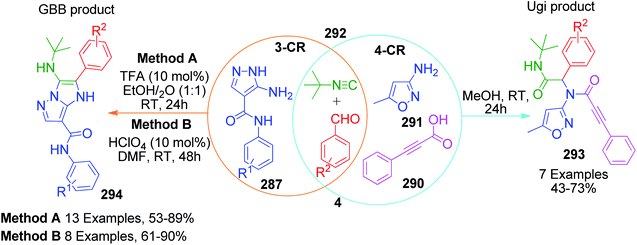 |
| | Scheme 91 Behaviour of 3-amino-5-methylisoxazole and 5-amino-N-aryl-1H-pyrazole-4-carboxamides in multicomponent reactions. | |
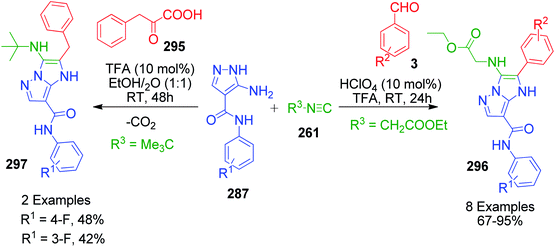 |
| | Scheme 92 GBB reaction with 5-amino-N-aryl-1H-pyrazole-4-carboxamide under two different conditions. | |
Y. K. Tailor et al. presented a modified form of the isocyanide-based GBB protocol, wherein aldehyde is replaced with ketones such as isatins 142 and cyclic carbonyl compounds 173 and 300. The synthesis of structurally diverse spiroheterocycles 301, 302 and 303, spiroannulated with imidazothiazole and imidazothiadiazole was achieved by the three-component reaction between 2-aminobenzothiazole/2-amino-1,3,4-thiadiazole 3a and 299, cyclohexyl/tert-butyl isocyanides 261 and isatins/cyclic carbonyl compounds in the presence of TiO2 nanoparticles using an ethanol/water solvent system at 90 °C (Scheme 93).186
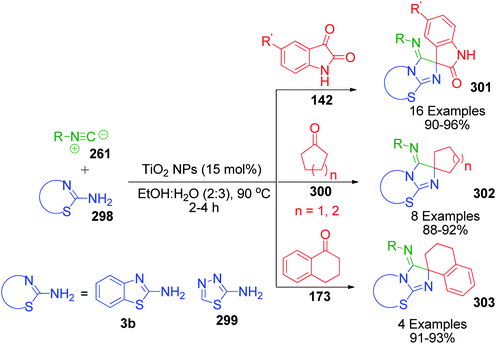 |
| | Scheme 93 Synthesis of structurally diverse spiroheterocycles. | |
The same authors recently developed an interesting and diversity oriented synthetic strategy for spirooxindoles spiroannulated with imidazo[4,5-c]isoquinolines 306, imidazo[4,5-c][2,7]naphthyridines 307 and imidazo[4,5-b]thieno[2,3-d]pyridines 308. This isocyanide-based multicomponent reaction involves GBB followed by the Pictet–Spengler reaction. A reaction mixture containing 2-aminobenzothiazole/2-amino-1,3,4-thiadiazole 3b/299, isocyanides 261, isatins 142 and aromatic/heteroaromatic aldehydes 304, 43 and 305 was treated with 10 mol% para-toluene sulfonic acid-modified TiO2 nanoparticles in aqueous medium (Scheme 94).187
 |
| | Scheme 94 Synthesis of spirooxindoles via GBB and Pictet–Spengler reaction. | |
B. Yang et al. developed a novel one-pot three-component protocol for biologically and pharmaceutically important tetracyclic-fused imidazo[1,2-a]pyridines 308.188 This method involves the GBB coupling of 2-aminopyridines 117, various isatins 142 and isocyanides 261 in the presence of HClO4 followed by a retro-aza-ene reaction and subsequent intramolecular nucleophilic reaction (path a) in butanol medium under reflux conditions to afford the title products in moderate yields (Scheme 95). The formation of benzodiazepinone-fused imidazo[1,2-a]pyridine (through path b) was not observed, which may be due to the strain of the seven-membered ring.
 |
| | Scheme 95 Synthesis of tetracyclic-fused imidazo[1,2-a]pyridines via sequential GBB, retro-aza-ene and intramolecular nucleophilic reaction pathways. | |
An ultrasound-assisted GBB reaction-based protocol was recently developed by M. Á. Claudio-Catalan and coworkers to synthesize bound-type fused bis-heterocycle imidazo or benzo[d]imidazo[2,1-b]thiazoles and 1,5-disubstituted tetrazole (1,5-DsT)-containing quinoline moiety 312.189 This solvent- and catalyst-free approach utilizes 2-aminothiazoles/2- aminobenzothiazoles 309, 2-chloro-3-formylquinoline 311, isocyanides 261 and trimethylsylilazide 311 under mild green conditions to generate two types of fused heterocycles in one step, which proceeds through the GBB reaction/SNAr/ring-chain azido-tautomerization strategy (Scheme 96). The synthesized compounds were also evaluated for antibacterial and antiamebic activities against various Gram-positive and Gram-negative bacteria.
 |
| | Scheme 96 Ultrasound-assisted synthesis of imidazo[2,1-b]thiazoles and benzo[d]imidazo[2,1-b]thiazoles. | |
In organic synthesis, the application of aldehydes, amines and alkyns for the MCR synthesis of propargyl amines is termed A3-coupling. Cascade processes involving cyclization by the Cu(I)-activated triple bond in propargyl amine afford versatile molecular complex compounds. Significant work has been reported based on multicomponent cascade reactions involving the A3-coupling of 5/6 membered 2-aminoazaheteroaromatic compounds with aldehydes and alkynes through 5-exo-dig/6-endo-dig cycloisomerization. A general and highly efficient method for the synthesis of imidazopyridine derivatives was reported by N. Chernyak and V. Gevorgyan using the copper-catalyzed three-component coupling of 2-aminopyridines with aryl/heteroaryl/alkyl aldehydes and terminal alkynes through 5-exo-dig cycloisomerization using CuCl and Cu(OTf)2.190 The employment of 2-aminoquinoline and 2-aminoisoquinoline as coupling partners in this transformation led to imidazoquinoline and imidazoisoquinoline frameworks in good yields. Similarly, P. Liu et al. reported a novel three-component reaction towards the synthesis of imidazo[1,2a]pyridines, which was independently developed based on CuSO4/TsOH-catalyzed three-component reaction using 2-aminopyridines, aldehydes and alkynes through 5-exo-dig cycloisomerization.191 S. K. Guchhait et al. explored the catalytic efficiency of a mixed Cu(I)–Cu(II) system by partial reduction of in situ-generated CuSO4 with glucose in ethanol (non-anhydrous) under open air in the A3-coupling of various 2-aminoazaheteroaromatic compounds with aldehydes and alkyne through an MCR cascade reaction. The corresponding fused imidazoles were generated via 5-exo-dig cycloisomerization and prototropic shift (Scheme 97, path a).192
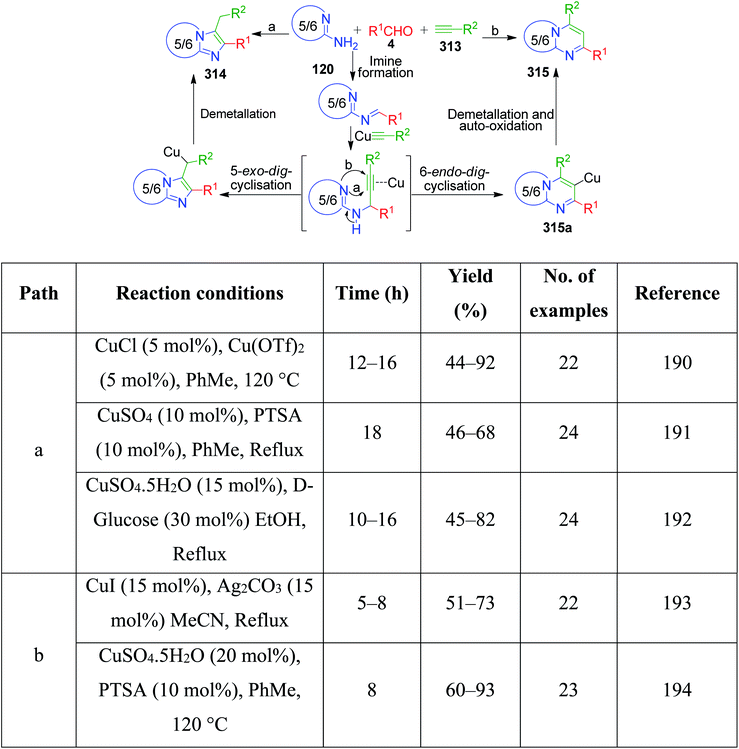 |
| | Scheme 97 Cu-catalyzed synthesis of substituted imidazoles and pyrimidines via 5-exo-dig and 6-endo-dig-cyclisation, respectively. | |
Conversely, A. Kumar and coworkers demonstrated an efficient regioselective cascade synthesis of pyrimidine ring 315 (ref. 193) via a transition-metal (copper/silver) catalyzed 6-endo-dig cyclization reaction. Herein, the coupling between 2-aminobenzimidazole 120, aldehydes 4, and alkynes 313 was carried out, leading to the formation of propargylamine intermediate 315a, which regioselectively undergoes 6-endo-dig cyclization through intramolecular N–H bond activation-mediated C–N bond formation. A recent report on the synthesis of nitrogen ring junction pyrimido-indazoles via 6-endo-dig cyclization was published by J. Palaniraja et al., wherein the A3 coupling reaction between 1H-indazol-3-amine, aromatic aldehydes and alkynes was achieved in the CuSO4-PTSA catalytic system. Conversion to the highly functionalized pyrimido[1,2-b]indazoles proceeded through 6-endo-dig cyclization (Scheme 97, path b).194 Fig. 7 shows the structure of various 5/6 membered 2-aminoazaheteroaromatic compounds, aldehydes and alkynes frequently employed in these cascade reactions.
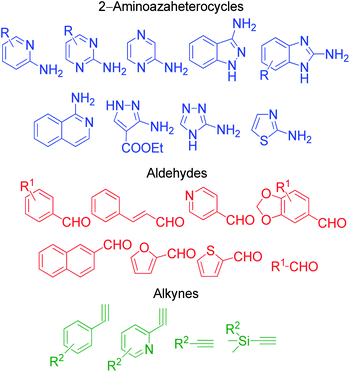 |
| | Fig. 7 Various 5/6 membered 2-aminoazaheteroaromatic compounds, aldehydes and alkynes used in the Cu-catalyzed 5-exo-dig/6-endo-dig-cyclisation process. | |
Next, facile access to various fused pyrimidine and azoles such as 2,3-, 2,4-disubstituted pyrimido[1,2-a]benzimidazoles, and 3-disubstituted imidazo[2,1-b]benzothiazoles was described by J. Wu et al. via reactions between heterocyclic azoles 3, aldehydes 4 and alkynecarboxylic acids 316.195 The CuI and K2CO3-catalyzed MCR synthesis of pyrimido[1,2-a]benzimidazoles and imidazo [2,1-b]benzothiazoles proceeds through a 6-endo-dig and 5-exo-dig cyclization, respectively (Scheme 98).
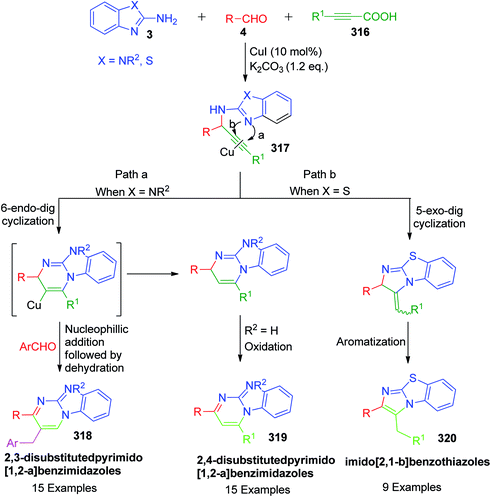 |
| | Scheme 98 Synthesis of pyrimido [1,2-a]benzimidazoles and imidazo [2,1-b]benzothiazoles via decarboxylic MCRs. | |
W. Sun et al. reported a one-pot multicomponent protocol utilizing magnetic Cu0@HAP@γ-Fe2O3 hybrid to catalyze the synthesis of imidazo[1,2-a]pyridine derivatives 322 from 2-aminopyridine derivatives 117, aldehydes 4/glyoxylic acids 321, and alkynes/alkynyl carboxylic acid 317.196 This method involves combining 2-aminopyridine derivatives either with a mixture of aldehydes with alkynes or with glyoxylic acids and alkynyl carboxylic acid together with 10 mol% catalyst in isopropanol at 100 °C (Scheme 99). The magnetic nanocatalyst could be easily separated from the reaction mixture using an external magnet and reused up to three runs with a slight loss in activity.
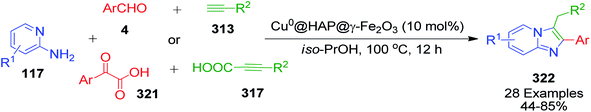 |
| | Scheme 99 Magnetic nanocatalyst-catalyzed synthesis of imidazo[1,2-a]pyridine derivatives via two different routes. | |
An interesting multicomponent approach to access imidazole-derived heterocycles 322b was reported by X. Li et al. through an oxidative cascade reaction.197 This Cu(OTf)2 and Li2CO3-catalyzed reaction was conducted using terminal alkyne 313, 2-amino N-heterocycle 120, benzyl/allylic bromide 322a and TEMPO (as an oxidant) in toluene under an oxygenated environment at 100 °C (Scheme 100). After 10 h, the target products, densely functionalized imidazo-fused heterocycles 322b, were obtained in moderate to good yields.
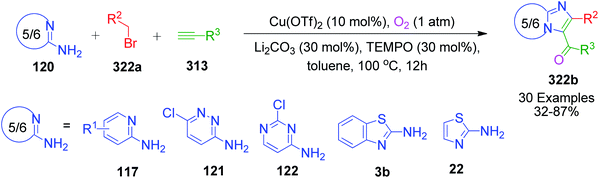 |
| | Scheme 100 Synthesis of imidazo-fused heterocycles through an oxidative cascade reaction. | |
Furthermore, the novel synthesis of dimethyl 4,5-dihydro-5-aryl-[1,2,4]triazolo[1,5-a]pyrimidine-6,7-dicarboxylates 324 was reported by B. Karami and coworkers through the one-pot condensation of 3-amino-1H-1,2,4-triazole 25, dimethyl acetylenedicarboxylate 323, and aryl aldehydes 4 using silica sodium carbonate as a solid base catalyst (Scheme 101).198
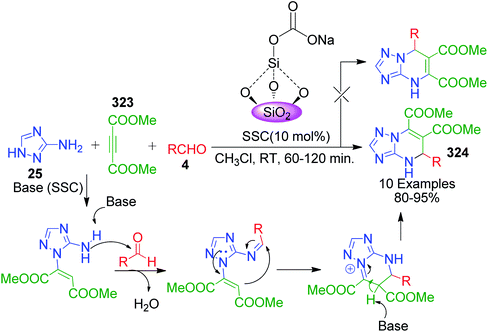 |
| | Scheme 101 Regioselective synthesis of fused imidazo[1,2-a]pyrimidines using a solid base catalyst. | |
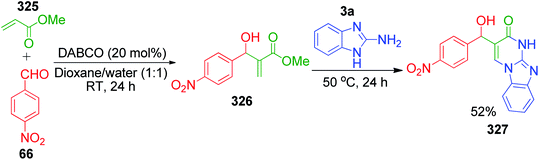 |
| | Scheme 102 Sequential three-component synthesis of benzimidazo[1,2-a]pyrimidinone based on Baylis–Hillman reaction. | |
A fused heterocyclic ring possessing three nitrogen atoms was synthesized by R. Moradivalikboni through an MCR. The catalyst benzene sulphonamide dibromide was synthesized and employed in the one-pot condensation of 2-amino thiadiazol 328, aromatic aldehydes 4 and acetamide 329 in toluene under reflux conditions, which afforded thiadiazole-fused triazine derivatives 330 in excellent yields (Scheme 103).200
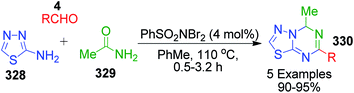 |
| | Scheme 103 Synthesis of thiadiazolotriazine derivatives using benzene sulphonamide dibromide as a catalyst. | |
A unique approach to benzothiazole-fused imidazoles 332 was developed by S. G. Balwe and Y. T. Jeong via the iron-catalyzed three-component cascade coupling of 2-aminobenzothiazole 3b, aldehydes 4 and nitroalkane 331 in air.201 The mechanistic aspect of this unprecedented formation of fused imidazoles includes a sequential aza-Henry reaction and subsequent intramolecular cyclization, followed by denitration. A variety of substituted benzo[d]imidazo[2,1-b]thiazole 332 was obtained after 6–8 h in excellent yields (Scheme 104).
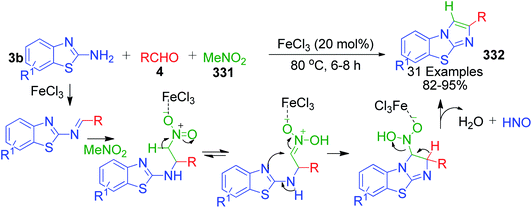 |
| | Scheme 104 FeCl3-catalyzed solvent-free synthesis of benzo[d]imidazo[2,1-b]thiazole derivatives. | |
M. W. Powner et al. proposed a totally different type of three-component pathway to access novel pyrimidine scaffolds 336 is aqueous media.202 The interesting feature of this approach involves the in situ generation of 2-aminothiazole 22 from β-mercapto-acetaldehyde 333 and cyanamide 334 in water at neutral pH, which then undergoes a three-component reaction with 4-amino-imidazole 5-carboxamide 335 and various aldehydes 4 in water at pH around 5.0 under an argon atmosphere (Scheme 105).
 |
| | Scheme 105 Synthesis of thiazole-fused pyrimidines involving the in situ generation of 2-aminothiazole. | |
D. N. Lyapustin et al. employed morpholino-nitroalkenes 337 in a reaction with aminoazoles 16 and aromatic aldehydes 4 to report the multicomponent synthesis of 4,7-dihydro-5-R-7-aryl-6-nitroazolo[1,5-a]pyrimidines 338. The reaction was performed with 1.5 equivalents of boron trifluoride etherate in butanol at 120 °C for 1.5–6 h.203 The proposed pathway for this multicomponent transformation involves the conversion of morpholino-nitroalkenes 337 into the corresponding nitroalkynes 337a under the effect of catalyst, followed by the formation of azolyl-nitroalkene 337b (Scheme 106).
 |
| | Scheme 106 Synthesis of 4,7-dihydro-5-R-7-aryl-6-nitroazolo[1,5-a]pyrimidines. | |
2.1.2 Aromatic α-aminoazaheterocycles as 1,1-binucleophiles. MCRs involving aromatic 2-aminoazaaromaticheterocycles as 1,1-binucleophiles are based on the participation of the exocyclic NH2-group exclusively. The usual products of these multicomponent interactions are five/six membered nitrogen-containing heterocycles having azaheterocycles as a substituent. The literature reveals that β-dicarbonyl compounds, organic anhydrides especially isatoic anhydride, alkyl acetylene dicarboxylate, mercaptoacetic acid, etc. have been employed to study the MCRs of this category.
2.1.2.1 MCRs involving β-dicarbonyl compounds. Dimedone, Meldrum's acid, 1,3-indanedione, and barbituric acid and its N,N-dialkyl derivatives are the frequently used β-dicarbonyl compounds in MCRs as they possess acidic hydrogens between two strong electron withdrawing group. M. Kumar's group conducted significant research on exploring MCRs involving these β-dicarbonyl compounds, AAHs and aldehydes/ketones, where the 1,1-binucleophilic reactivity of AAH is necessary. They synthesized diverse benzothiazolylquinoline-2,5-diones 341 and their spiro analogues 342 upon the one-pot four-component combination reaction of Meldrum's acid 339, 2-aminobenzothiazoles 340, dimedone 81 and isatin/carbonyl compound (aldehydes and ketones) 142/4 in the presence of sulfamic acid (Scheme 107).204
 |
| | Scheme 107 One-pot four-component synthesis of benzothiazolylquinoline-2,5-diones and their spiro analogues. | |
The plausible mechanistic path as suggested by the authors involves the initial Knoevenagel condensation reaction between Meldrum's acid 339 and carbonyl compound 4 and 142 followed by nucleophilic attack by dimedone 81, leading to the generation of a linker between two β-dicarbonyls. The intermediate formed after the addition of 2-aminobenzothiazole 340 to the dimedone ring with the removal of a water molecule undergoes intramolecular attack by the exocyclic amino group of benzothiazole at the carbonyl group of Meldrum's acid, which further on heating rearranges to the final heterocyclic product 342 with the elimination of carbon dioxide and acetone as gaseous side products (Scheme 108).
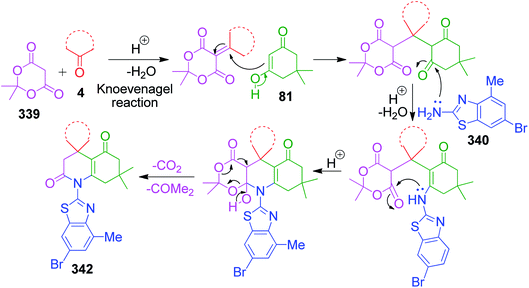 |
| | Scheme 108 Proposed reaction mechanism to benzothiazolylquinoline-2,5-diones and their spiro analogues. | |
On a similar basis, they described a diversity oriented synthetic protocol to access unsymmetrically annulated 1,4-dihydropyridine spiroheterocycles 345 via the four-component domino reaction of 2-aminobenzothiazoles 344, isatin 143 and two different cyclic β-diketones 123 using the Brønsted acidic imidazolium salt 3-methyl-1-(butyl-4-sulfonyl)imidazolium hydrogen sulphate (SFIL) as an ionic liquid in aqueous medium (Scheme 109, Method A).205 It was reported that the presence of the ionic liquid/water combination is crucial as the reaction medium for the formation of the target compound as no progress in the reaction was detected in the absence of both entities even after heating the reacting components for a long time. However, the yield of the products was dependent on the ratio of ionic liquid and water employed. Optimization of the ionic liquid/water ratio demonstrated that the best result was achieved with a 1:1 ratio in terms of yield and reaction time. Easy separation of the ionic liquid after completion of the reaction led to its reuse for at least five cycles without any appreciable loss in activity. After two years, the same group demonstrated a modified version of this approach, where a heteroaromatic aldehyde replaces isatin and the corresponding non-spiroheterocyclic ring was constructed in the presence of an organic catalyst. Structurally diverse unsymmetrically annulated 1,4-dihydropyridines 343 were obtained when an ethanolic solution of 2-aminobenzothiazole 344, thiophene-2-carbaldehyde 43 and various cyclic β-dicarbonyl compounds 122 was heated under reflux conditions in the presence of L-proline as an organocatalyst for 15–38 minutes.206 For the construction of these fused heterosystems, they utilized a variety of cyclic β-dicarbonyl compounds 122 except Meldrum's acid 339, which usually undergoes decomposition at moderate temperature, leading to the elimination of a small molecule and the formation of one-sided fused pyridine derivatives (Scheme 109, Method B).
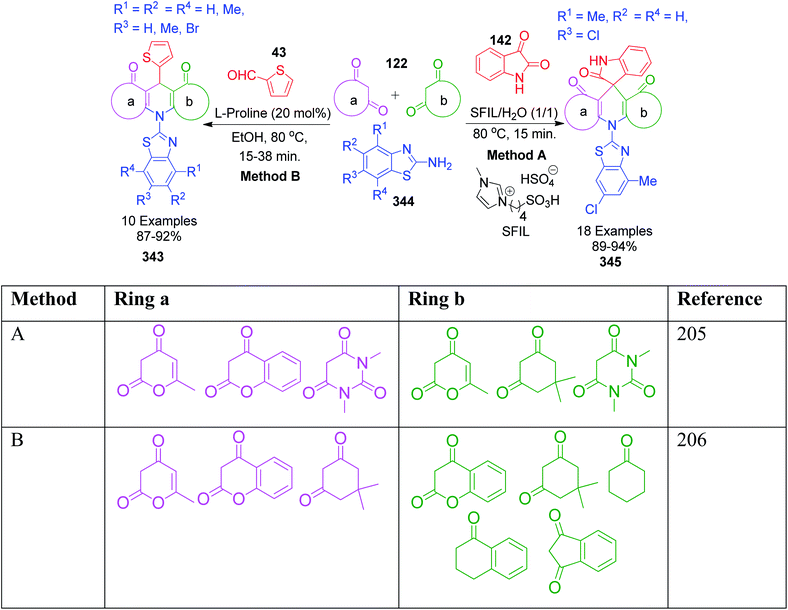 |
| | Scheme 109 Synthesis of unsymmetrically annulated 1,4-dihydropyridine spiro- and non-spiroheterocycles tethered with benzothiazole derivatives. | |
An interesting and catalyst-free method was developed by K. V. Sashidhara et al. to carry out the one-pot synthesis of substituted isoindolinones 348 utilizing dimedone 81 as a 1,3-dicarbonyl component, various AAH, and formylbenzoic acid 346.207
In this method, three different types, namely C–C, C–N, and C–S bonds, are formed in a single operation upon heating an ethanolic mixture of reacting components under microwave irradiation. The postulated mechanism starts with the in situ formation of imine from 2-formylbenzoic acid 346 and AAH 347 (Scheme 110). The Michael addition product formed after nucleophilic attack of the enol form of diketone on imine undergoes intramolecular nucleophilic cyclization of the amine in the subsequent step, leading to the desired isoindolinone 348.
 |
| | Scheme 110 Microwave-assisted catalyst-free synthesis of substituted isoindolinones. | |
Pyruvic acid and its derivatives also function as active methylene compounds in the reaction, where AAH acts as a 1,1-binucleophile, and the article concerning this was published by S. V. Ryabukhin and coworkers, which revealed the parallel synthesis of a series of 3-hydroxy-1,5-dihydro-2H-pyrrol-2-one derivatives 350. Pyruvic acid derivatives 349 were allowed to undergo a three-component condensation with aromatic aldehydes 4 and various 2-aminoaza aromatic heterocycles 347. This transformation was performed under two different reaction conditions, namely Me3SiCl as the reaction promoter in DMF and acetic acid as the reaction medium (Scheme 111, Method A).208 It was observed that the use of Me3SiCl in DMF is suitable in terms of providing the corresponding products in 73–93% yield compared to 38–73% in the case of the reaction in acetic acid. In 2014, an analogous reaction was performed with methyl 2-heteroylpyruvates 349, 2-aminothiazole 347 and substituted aldehydes 4 by V. L. Gein et al. to access 5-aryl-4-(2-heteroyl)-3-hydroxy-1-(2-thiazolyl)-3-pyrrolin-2-ones 350.209 The reaction mixture containing the starting materials was heated in acetic acid under reflux for 5–10 min. Given that the authors utilized a selected number of starting components, a few examples were possible by various permutations and combinations (Scheme 111, Method B).
 |
| | Scheme 111 Two different methods to synthesize polysubstituted 3-hydroxy-1,5-dihydro-2H-pyrrol-2-ones through a three-component condensation reaction. | |
H. R. Shaterian et al. described a one-pot three-component cyclocondensation reaction between isatoic anhydride 351, 2-aminobenzothiazole 3 and aldehydes 4, leading to the formation of quinazolinone derivatives 352. The reaction, which was catalyzed by a heterogeneous catalyst, [Al(H2PO4)3], under thermal solvent-free conditions, worked well with a variety of aryl aldehydes, including those bearing electron-withdrawing and electron-donating groups such as OMe, Cl, Br, and NO2, and the desired compounds were obtained in good to excellent yields. Conversely, aliphatic aldehydes could not be incorporated in the product under the same reaction conditions.210 The same research group further explored this method, where the synthesis of these pyrimidine-type heterocyclic compounds was carried out by employing alumina-supported phosphoric acid211 and cellulose-supported sulfonic acid212 as solid heterogeneous catalysts under solvent-free conditions. It was also demonstrated that all the solid acid catalysts could be recycled and reused for at least three times without any significant loss in activity. An analogous method was reported by L. Wu, where condensation of the starting materials was effected using Zr(HSO4)4 as an acid promoter under solvent-free conditions.213 M. Esmaeilpour et al. developed an efficient Fe3O4@SiO2-imid-PMAn nanocatalyst for the synthesis of quinazolinone derivatives by the condensation of isatoic anhydride with 2-aminoazaaromatic heterocycles and aldehydes under ultrasonic irradiation or reflux conditions.214 A brief comparison of the reported methods in terms of reaction time and yields is shown in Scheme 112.
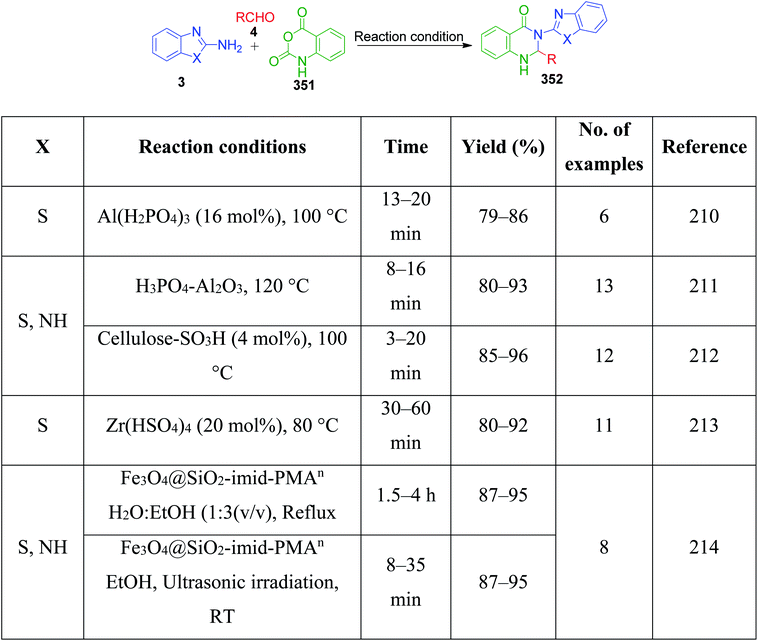 |
| | Scheme 112 Synthesis of 3-(2′-benzothiazolyl)-2,3-dihydroquinazolin-4(1H)-ones under different reaction conditions. | |
According to the authors, these methods follow a common mechanistic pathway, in which an acid–base interaction between catalyst and isatoic anhydride 351 is suggested as the initial step to generate reactive intermediate 352a, which undergoes nucleophilic attack on the carbonyl carbon by AAH 3. Intermediate 352c obtained after the decarboxylation of 2-amino-N-substituted-amide 352b reacts with the activated carbonyl group of aldehydes 4 to form imine intermediate 352d with the removal of a water molecule. Intramolecular nucleophilic attack of the amide nitrogen on the activated imine carbon leads to the cyclized structure, which then affords the desired quinazolinone derivatives 352 through a 1,5-proton transfer rearrangement reaction (Scheme 113).
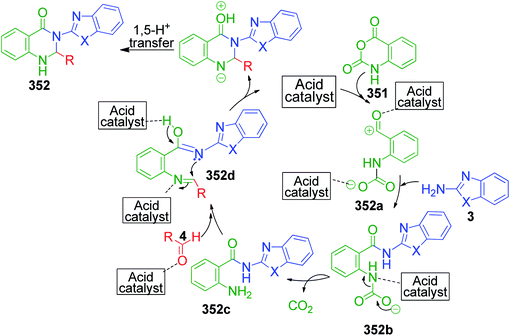 |
| | Scheme 113 Common mechanistic route to 3-(2′-benzothiazolyl)-2,3-dihydroquinazolin-4(1H)- ones. | |
In contrast to the acid-catalyzed reactions discussed above, a catalyst-free approach for the synthesis of these heterocyclic compounds was developed by J. M. Khurana and S. Kumar. The strategy involved heating a mixture containing the starting materials in water–ethanol 1:1 (v/v) system at 80 °C for 30 min.215 The corresponding products, 2,3-dihydro quinazolin-4(1H)-ones 354, were produced with excellent yields in the range of 81–94%. The versatility of this methodology can be inferred from the smooth incorporation of a range of aromatic, aliphatic and heteroaromatic aldehydes 4, as well as substituted and unsubstituted isatoic anhydrides 353 in the multi-component condensation reaction (Scheme 114).
 |
| | Scheme 114 Catalyst-free synthesis of 2,3-dihydro quinazolin-4(1H)-ones. | |
A relevant but less saturated quinazolinone ring was constructed by A. Khalafi-Nezhad et al. through a different method, which employed acetic anhydride 355, anthranilic acid 356 and various AAH 347. The use of anthranilic acid plays a crucial role in this three-component reaction as it possess the desired amino functionalities at the ortho position, which is essential for the cyclisation step in quinazolinone ring formation. In the presence of a catalytic amount of titanium dioxide nanoparticles, the condensation reaction among the starting components under thermal and solvent-free conditions for 8–10 h afforded quinazolinone derivatives 357 bearing different azaheterocycles (Scheme 115).216 Nano-TiO2 exhibited recyclable property and this property did not fade up to four consecutive runs.
 |
| | Scheme 115 Nano-TiO2-catalysed synthesis of tetrazolopyrimidine and proposed reaction pathway. | |
S. Koroji et al. developed a microwave-assisted silica sulfuric acid-catalyzed one-pot synthesis of novel 3-(benzo[d]thiazol-2-yl)-2-alkyl or 3-(2-benzimidazolyl)-2-alkyl quinazolin-4(3H)-one 360. This three-component reaction was performed by heating a solventless mixture of 2-aminobenzothiazole/2-aminobenzimidazole 3, anthranilic acid 359, and orthoesters 358 under microwave irradiation operating at 600 W (Scheme 116).217
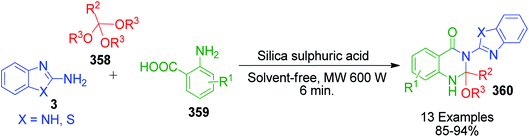 |
| | Scheme 116 Microwave-assisted synthesis of quinazolinones tethered with benzimidazole/benzothiazole. | |
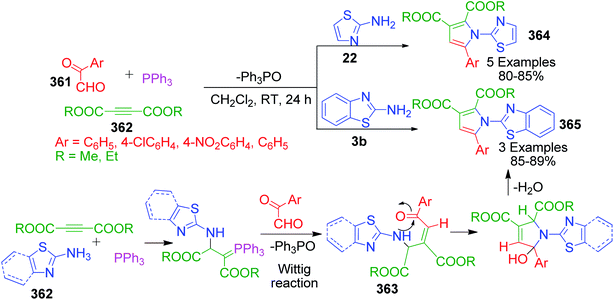 |
| | Scheme 117 One-pot synthesis of polysubstituted pyrrole derivatives and proposed mechanistic route. | |
M. A. Ghasemzadeh et al. developed an inexpensive and green protocol to access polyfunctionalized 2-pyrrolidinones 367 via a four-component reaction using a heterogeneous magnetic nanocatalyst.219 When a solvent-free mixture containing 2-aminobezothiazole 3b, dimethyl acetylenedicarboxylate 323, aromatic aldehyde 4 and piperidine/morpholine 366 was heated in the presence of Fe3O4/L-arginine nanoparticles at 80 °C, 2-pyrrolidinones 367 were obtained in excellent yields (Scheme 118). As a magnetic nanocatalyst, Fe3O4/L-arginine can be easily retrievable, and therefore reusable for many times.
 |
| | Scheme 118 Synthesis of pyrrolidinones via four-component reactions. | |
H. Gao et al. developed a method involving an intermediate, also known as a Huisgen 1,4-dipole after the name of the scientist who first described it, formed by the addition reactions of nitrogen-containing heterocycles to electron-deficient alkynes. The applicability of this intermediate was shown by synthesizing functionalized 2-pyrrolidinones 368 and morpholinium/piperidinium 2-pyrrolidinon-3-olates 369 via the four-component reaction of 2-aminobenzothiazole 3b, aromatic aldehydes 4, acetylenedicarboxylate 362 and piperidine/morpholine 366.220 Firstly, the Huisgen 1,4-dipole is obtained by mixing acetylenedicarboxylate 362 and piperidine/morpholine 366 at room temperature in ethanol, and then the reaction is further conducted with the remaining reactants toward the target compounds at moderate temperature. The reaction was stirred for two days to get both products in satisfactory yields. Moreover, the acid catalyst PTSA was required for the synthesis of morpholinium/piperidinium 2-pyrrolidinon-3-olates, showing the path-dependent nature of the reaction (Scheme 119).
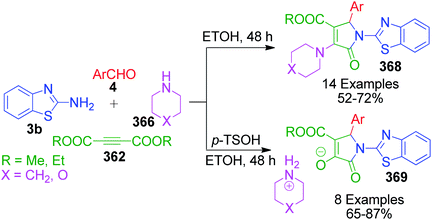 |
| | Scheme 119 Synthesis of pyrrolidinones via four-component reactions. | |
The mechanism proposed by the authors to explain the reaction pathway is based on the initial formation of 1,3-dipolar intermediate 368a from secondary amine 366 and DAAD 362 and imine intermediate 368b from aldehyde 4 and 2-aminobenzothiazole 3b. Intermediate 368c formed by the nucleophilic addition of 1,3-dipolar intermediate 368a to imine 368b experiences intramolecular nucleophilic attack from the amino group to carbonyl carbon, which produces polysubstituted pyrrolidinone ring 368. The enamine functionality of pyrrolidinone undergoes hydrolysis in the presence of PTSA to yield pyrrolidinedione 368d, which rearranges through keto–enol tautomerism to the more stable enol-form, connecting both the ester and amide groups. Being acidic in nature the hydroxyl proton of the enol form is deprotonated by piperidine in solution to give piperidinium 2-pyrrolidinon-3-olate 369 as the final product (Scheme 120).
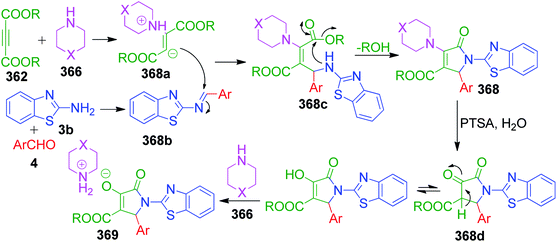 |
| | Scheme 120 Proposed mechanism for the formation of pyrrolidinones. | |
A closely related reaction based on the use of the 1,3-dipolar intermediate, which allows the construction of a fused imidazole ring, was reported by S. Arab-Salmanabadi and coworkers. Their approach gave a way to access a series of novel functionalized quinolones and isoquinoline-fused dihydroimidazoles 372 and 373 in excellent yields via the three-component reaction of isoquinoline 370 or quinoline 371 derivatives, DAAD 362 and 2-aminobenzothiazole 3b in dichloromethane at ambient temperature under catalyst-free conditions.221 The plausible rationalized path deduced to explain the product formation was assumed to begin with the formation of the zwitterionic intermediate or 1,3-dipolar intermediate 373a from isoquinoline 371 and DAAD 362. A proton from the amino group of 2-aminobenzothiazole 3b is then transferred to neutralize the negative charge of the zwitterion. Amide ion 373b formed from 2-aminobenzothiazole 3a adds to the isoquinolinium 373c system to generate intermediate 373d, which upon cyclisation and subsequent removal of alcohol affords the desired five-membered nitrogen-containing heterocycles 373 (Scheme 121).
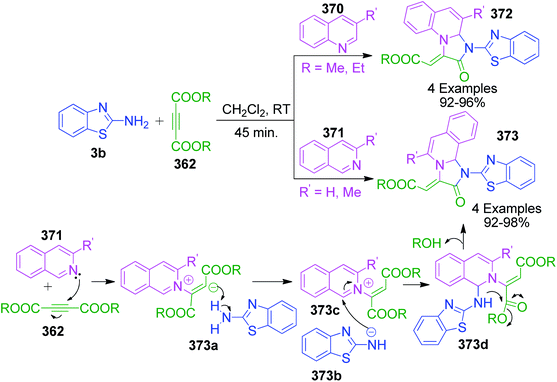 |
| | Scheme 121 Synthesis of novel functionalized dihydroimidazo[2,1-a]isoquinolines and dihydroimidazo[2,1-a] quinolines. | |
A totally different approach for the synthesis of seven-membered nitrogen-containing heterocycles involving a 1,3-dipolar intermediate was described by K. Ramesh et al., in which dimethyl/diethyl acetylenedicarboxylate 362, 2-aminopyridine 117 and 2,5-dimethoxytetrahydrofuran 374 were mixed in neutral water.222 Various cyclodextrins such as α-cyclodextrin, β-cyclodextrin, γ-cyclodextrin, 2-hydroxy propyl β-cyclodextrin and methyl-β-cyclodextrin were examined for their efficiency as promoters. However, the corresponding azepine 375 rings were formed in excellent yields with β-cyclodextrin (β-CD) and expensive γ-cyclodextrin (γ-CD), which were discarded as the reaction promoter. This approach was not extended to other AAH as use of 2-aminobenzathiazole 3b instead of 2-aminopyridine 117 did not yield any product. Further, only a small loss in activity of β-CD was observed after three successive runs, which indicated its recyclable and reusable nature as the reaction promoter (Scheme 122). The formation of substituted azepine 375 in the presence of β-CD was evidently suggested by authors through the inclusion complex between 2-aminopyridine 117 and β-CD, which was facilitated by the hydrophobic environment of the cyclodextrin. The primary and secondary-OH groups of CDs help in stabilizing the carbanion of 1,3-dipolar-type intermediate 375a, which further attacks 2,5-dimethoxytetrahydrofuran, leading to the opening of the furan ring. The succeeding step involves intramolecular cyclisation followed by the elimination of a methanol molecule, affording the desired substituted azepine ring.
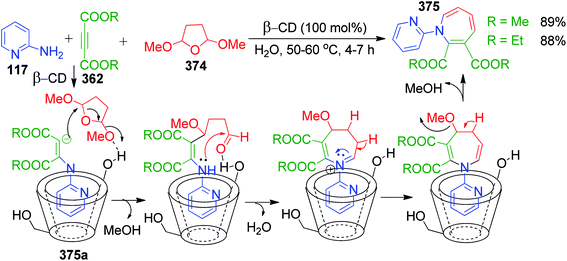 |
| | Scheme 122 Construction of substituted azepine ring promoted by β-CD. | |
Next, P. Wadhwa's group developed a catalyst- and solvent-free approach for the diversity-oriented synthesis of thiazole linked thiazin derivatives 377 from the three-component reaction of 2-aminobenzothiazoles 3b, various isothiocyanates 376 and DAAD 362 irradiated under microwave heating at 120 °C for 30 min.223 Conversely, when the same combination of reactants was heated in the conventional way, the products were formed after 10 h with low yields. With microwave heating, the quality of their approach was enhanced greatly both in terms of reaction time and product yield. The wide scope of this protocol was highlighted by the use of various substituted derivatives of 2-aminobenzothiazole 3b and isothiocyanates 376. The reaction progress as suggested by the authors starts with the formation of N,N′-substituted thiourea intermediate 377a by nucleophilic attack of 2-aminobenzothiazoles 3b on the electrophilic carbon center of isothiocyanate 376, which further attacks the C–C triple bond of dialkylacetylenedicarboxylates 362 through the soft sulfur terminal, followed by intramolecular cyclisation with the elimination of an alcohol molecule (Scheme 123).
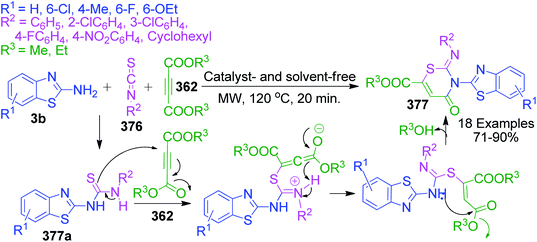 |
| | Scheme 123 Catalyst- and solvent-free synthesis of (Z)-alkyl 3-(benzo[d]thiazol-2-yl)-4-oxo-2-(cyclohexyl/arylimino)-3,4-dihydro-2H-1,3-thiazine-6-carboxylates under microwave irradiation. | |
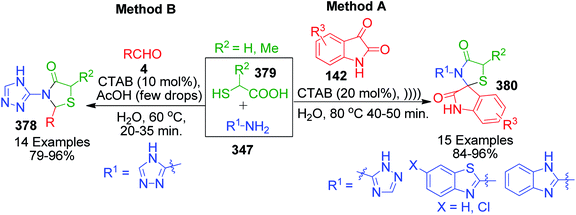 |
| | Scheme 124 Ultrasonic wave-assisted synthesis of spiro[indole-3,2¢-thiazolidinone]-2,4-diones. | |
The mechanistic course of these CTAB-based reactions is shown in Scheme 125. The mechanism starts with the formation of Schiff bases 380a in aqueous micellar medium from heterocyclic amine 25 and carbonyl compounds 4 (aldehydes and isatin), which experience in situ nucleophilic attack from the sulfur atom of α-mercaptocarboxylic acid 379 followed by intramolecular cyclization with the elimination of a water to afford the desired thiazolidinone derivatives 380. All the steps of the reaction sequence in the formation of the triazole-linked thiazolidinone hybrids were triggered by acetic acid, while ultrasonic waves were utilized to promote the synthesis of spiro linked thiazolidinones.
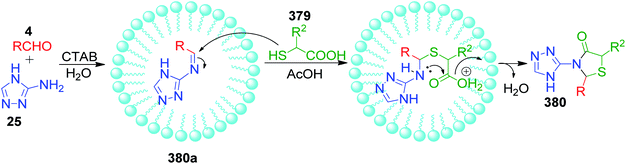 |
| | Scheme 125 Proposed mechanism for the formation of triazole-linked thiazolidinones. | |
A closely related MCR approach that allows the construction of non-spiro thiazolidinone and spiro triazole rings was described by W. S. Hamama et al. under suitable reaction conditions. The MCR for the synthesis of thiazolidinone 383 was conducted by heating 2-amino-1,3,4-thiadiazole 108, a specific aldehyde, 1,3-diphenyl-1H-pyrazole-5-carbaldehyde 382 and mercaptoacetic acid 381 in pyridine at reflux for 19 h.226 Conversely, when p-methoxybenzaldehyde 384 was used as aldehydic component, while retaining the other two components intact in toluene, the reaction led to the formation of thiazolidinone 385 via intermediate 386. The spiro-linked triazole moiety was constituted by refluxing 2-amino-1,3,4-thiadiazole 109, isatin 143 and thiosemicarbazide 387 in an ethanol/acetic acid mixture in a 9:1 proportion for 20 h. The formation of spiro compound 389 was assumed to proceed via ketimine intermediate 388 obtained from the condensation of 2-amino-1,3,4-thiadiazole 108 with isatin 142. This ketimine intermediate 388 is then attacked by thiosemicarbazide 387 and subsequent cyclocondensation affords intermediate 388a, the acetylation of which gives the final product (Scheme 126).
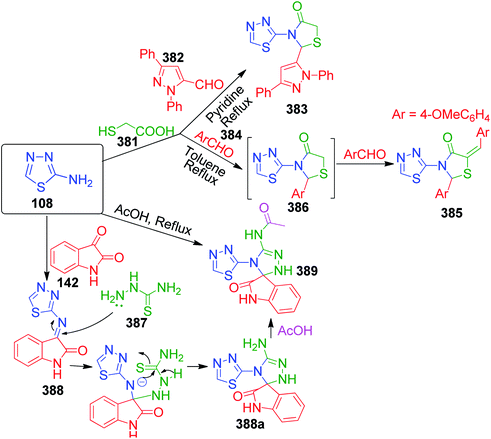 |
| | Scheme 126 Construction of non-spiro thiazolidinones and spiro-linked triazole ring. | |
Further progress in the field of utilizing mercaptoacetic acid 381 in the multicomponent synthesis of thiazolidinone ring system 391 was made by D. Kumar and coworkers. They investigated various catalytic potential solid supported protic acids for the one-pot tandem condensation–cyclisation reaction involving benzaldehyde 4, five- or six-membered azaheterocyclic amine 347 and thioglycolic acid 381 (Scheme 127, Method A).227 The use of silica gel with a mesh size of 230–400 as a solid support revealed that their relative activity follows the order of HClO4–SiO2 > TfOH–SiO ≫ H2SO4–SiO2 > p-TsOH–SiO2 > MsOH–SiO2 ∼ HBF4–SiO2 > TFA–SiO2 ∼ HOAc–SiO2. Further, they designed and performed some reactions to make a clear distinction between the superiority of two heterogeneous catalyst systems, HClO4–SiO2 and TfOH-SiO2, which appear closely related in activity order. The superior catalytic power of the former compared to that of the latter in terms of providing product yield was established from two sets of reactions run for 5 h, one with 2-aminopyridine and the other with 2-aminobenzothiazole, while benzaldehyde 4 and mercaptoacetic acid 381 were common to both. With HClO4–SiO2, the products were obtained in 70% and 69% yield in comparison to 52% and 35% yield when TfOH–SiO2 was employed, respectively. In 2016, the same reaction, which was catalyzed by ammonium persulfate (APS), was utilized by S. Ebrahimi for the preparation of thiazolidinone ring 390-bearing triazole and p-nitrophenyl substituents via heating a reaction mixture containing 3-amino-1,2,4-triazole, p-nitrobenzaldehydes and mercaptoacetic acid 381 at 90 °C under solvent-free conditions for 60 min (Scheme 127, Method B).228
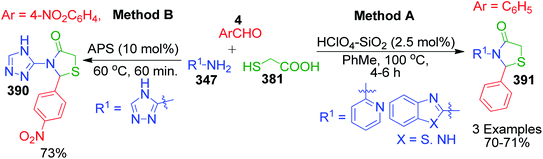 |
| | Scheme 127 Two different methods to synthesize thiazolidinone ring system. | |
Next, the synthesis of chromone-based thiazolidinone ring 393 was reported in the article published by N. M. Drosos et al., wherein a mixture containing mercaptoacetic acid 379 and its 2-methyl derivative with various 3-formylchromones 392 and 2-aminobenzimidazole 3a was irradiated with microwaves at 100 °C. The corresponding products were obtained after 80 min in moderate to good yields (Scheme 128).229 They also constructed the same five-membered sulfur-containing ring but with low yield (33–51%) through a two-step synthetic route and a comparison between the two approaches was established.
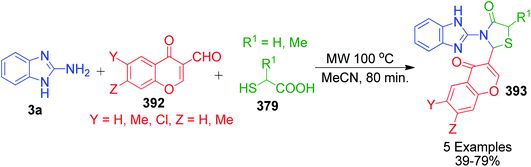 |
| | Scheme 128 Microwave-assisted synthesis of chromone-based thiazolidinones. | |
Recently, R. Singh et al. synthesized new spiro[indeno[1,2-b]quinoxaline-[11,20]-thiazolidine]-40-one 395 via a multi-component approach.230 The reaction between indeno[1,2-b]quinoxalinone, α-mercaptocarboxylic acid 381 and 2-aminobenzothiazole 3a was conducted in urea-choline chloride 394 as a green deep eutectic solvent using carbon-SO3H as a solid acid catalyst, resulting in the formation of a thiazolidine ring attached to indeno[1,2-b]quinoxaline 395 through spiro carbon in 90% yield (Scheme 129). Both the catalyst and DES could be recovered from the reaction mixture quantitatively, and thus reused several times without a significant loss in activity.
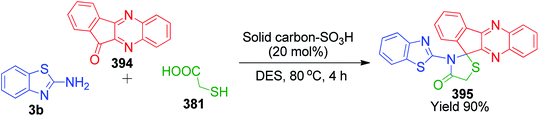 |
| | Scheme 129 Synthesis of thiazolidine ring-linked indeno[1,2-b]quinoxaline. | |
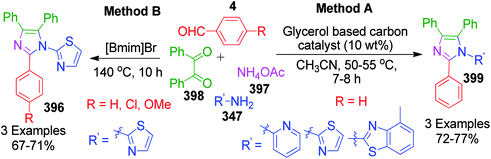 |
| | Scheme 130 Four-component synthesis of tetra-substituted imidazoles under different conditions. | |
A common mechanism for the synthesis of highly substituted imidazoles 396 is shown in Scheme 131. It was suggested that the reaction proceeds via diamine intermediate 396a, which is formed by the addition of ammonium acetate 397 and heterocyclic amine 347 to the activated carbonyl carbon of aldehyde 4. The desired product is obtained from this diamine intermediate 396a, which is condensed with 1,2-diketone 398 with the removal of two molecules of water followed by rearrangement.
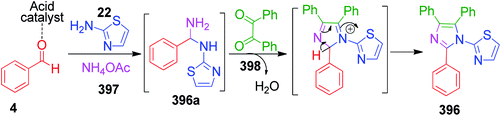 |
| | Scheme 131 Proposed mechanistic route to tetra-substituted imidazoles. | |
M. Kumar et al. synthesized structurally diverse 3-benzothiazolyl-2-styrylquinazolinones through a one-pot three-component sequential synthetic method by taking advantage of ionic liquids in terms of their unique solvating, catalytic and recycling abilities. Initially, 2-methyl-3,1-benzoxazin-4-one 400 and 2-aminobenzothiazoles 344 were combined with the help of SO3H-functionalized ionic liquids (SFILs I and II) at 80 °C, and the further addition of aromatic aldehyde 4 was done after 4–7 h when the reaction mixture showed the spot of 3-benzothiazolyl-substituted quinazolinone derivatives 402 on a TLC plate (Scheme 132).233 The mixture was maintained at 80 °C until the condensation of aldehyde, leading to the benzothiazole and styryl-linked quinazolinones as the final product. The investigation pertaining to the recyclability of SFIL I and II in terms of providing yield and overall reaction time revealed that no appreciable loss in activity there occurred for least consecutive five cycles. The authors also suggested a mechanism to explain the synthesis of 3-benzothiazolyl-2-styrylquinazolinones, as shown in Scheme 133.
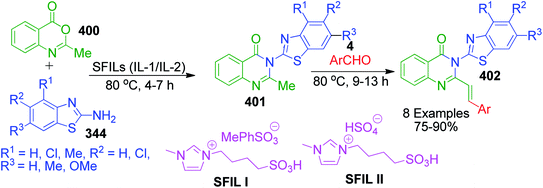 |
| | Scheme 132 Synthesis of 3-benzothiazolyl-2-styrylquinazolin-4(3H)-ones through sequential one-pot three-component reaction. | |
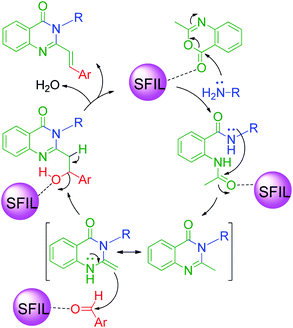 |
| | Scheme 133 Plausible mechanism for the formation of quinazolinones. | |
The simplest aldehyde, formaldehyde 403, was used in a three-component reaction with 2-aminobenzothiazole 3b and phenol 404 to construct benzothiazole tethered benzoxazine ring 405 by F. Shan and coworkers. The mixture containing the reactants was heated under solvent-free conditions at 120 °C for 2 h to afford the desired product in 16% yield (Scheme 134).234 The same reaction when carried out with 2-aminothiazole was found to be unsuccessful.
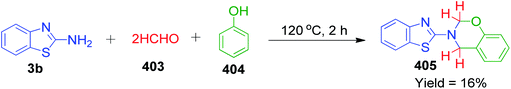 |
| | Scheme 134 Solvent-free synthesis of benzothiazole-tethered benzoxazine ring. | |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a