
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamically stable structure of hydroxy alkane sulfonate surfactant monomers in water achieving high water solubility and a low CMC†
Yukiko Tabuchi and
Takaya Sakai*
and
Takaya Sakai*
Material Science Research Laboratories, Kao Corporation, 1334, Minato, Wakayama-shi, Wakayama, Japan. E-mail: sakai.takaya@kao.com; Fax: +81-73-426-5065
First published on 1st June 2021
Abstract
Although sodium internal olefin sulfonates (IOSs) derived from petrochemicals are known to act as anionic surfactants, these compounds have few practical applications and their interfacial properties have not been examined in detail because they have complex compositions and are challenging to prepare. However, IOSs obtained from vegetable oils have recently been applied to detergents. The present work represents the first study of the IOS sodium 5-hydroxyhexadecane-4-sulfonate (C16-4S-5OH) as a pure substance. The properties of C16-4S-5OH in aqueous solution were assessed by differential scanning calorimetry, equilibrium surface tension measurements, Fourier transform infrared spectroscopy and proton nuclear magnetic resonance spectroscopy. This compound was found to have a low Krafft point and a low CMC, both of which are necessary for a modern sustainable surfactant. Although these two characteristics are normally difficult to achieve simultaneously, this work demonstrated that C16-4S-5OH in aqueous solution has a highly fixed stereostructure that enables both properties to be achieved. The C16 main carbon chain of this compound acts as a C12 and a C4 chain both oriented in the same direction, while the hydrophilic sulfo and hydroxy head groups form a rigid cyclic moiety including two carbons of the C16 alkyl chain based on hydrogen bonding. This highly fixed molecular structure of the C16-4S-5OH in water is maintained in the monomer and micellar states below and above the CMC, respectively.
Introduction
Surfactants have numerous uses in industry. In particular, anionic surfactants are able to act as inexpensive cleaning agents. Although the raw materials of most general anionic surfactants are mainly biobased, synthetic ones are also used. Therefore, the development of completely biobased anionic surfactants is urgently required to satisfy global sustainability demands.1 Sodium internal olefin sulfonates (IOSs), which can be synthesized by the sulfonation of alkenes with internal double bonds produced by the petrochemical industry, has been known as an anionic surfactant since the 1970s (Fig. 1).2 Those analogues in which the sulfo group is instead located at a terminal carbon of the alkyl chain are termed sodium α-olefin sulfonates, and there have been many reports concerning the properties of these compounds in aqueous solution.3,4 Conversely, there have been only a few studies on the aqueous solution properties of IOS3–5 presumably because these are typically complex mixtures (Fig. 1). Specifically, as-synthesized IOSs generally exhibit a wide distribution of sulfo group positions and also typically comprise a combination of hydroxy alkane sulfonates (1a) and alkene sulfonates (1b). IOSs have not found applications in industry, likely because it is challenging to prepare colorless IOSs,5 although a high quality IOS product was recently obtained from vegetable oils.6,7 Because this biobased IOS comprised linear alkenes derived from C16 and C18 saturated fatty acids (which have not been extensively employed industrially) rather than from C12 and C14 fatty acids that are currently widely used as raw materials for surfactants, it may represent a sustainable anionic surfactant.8 This IOS may also allow the effective utilization of surplus saturated long-chain fats.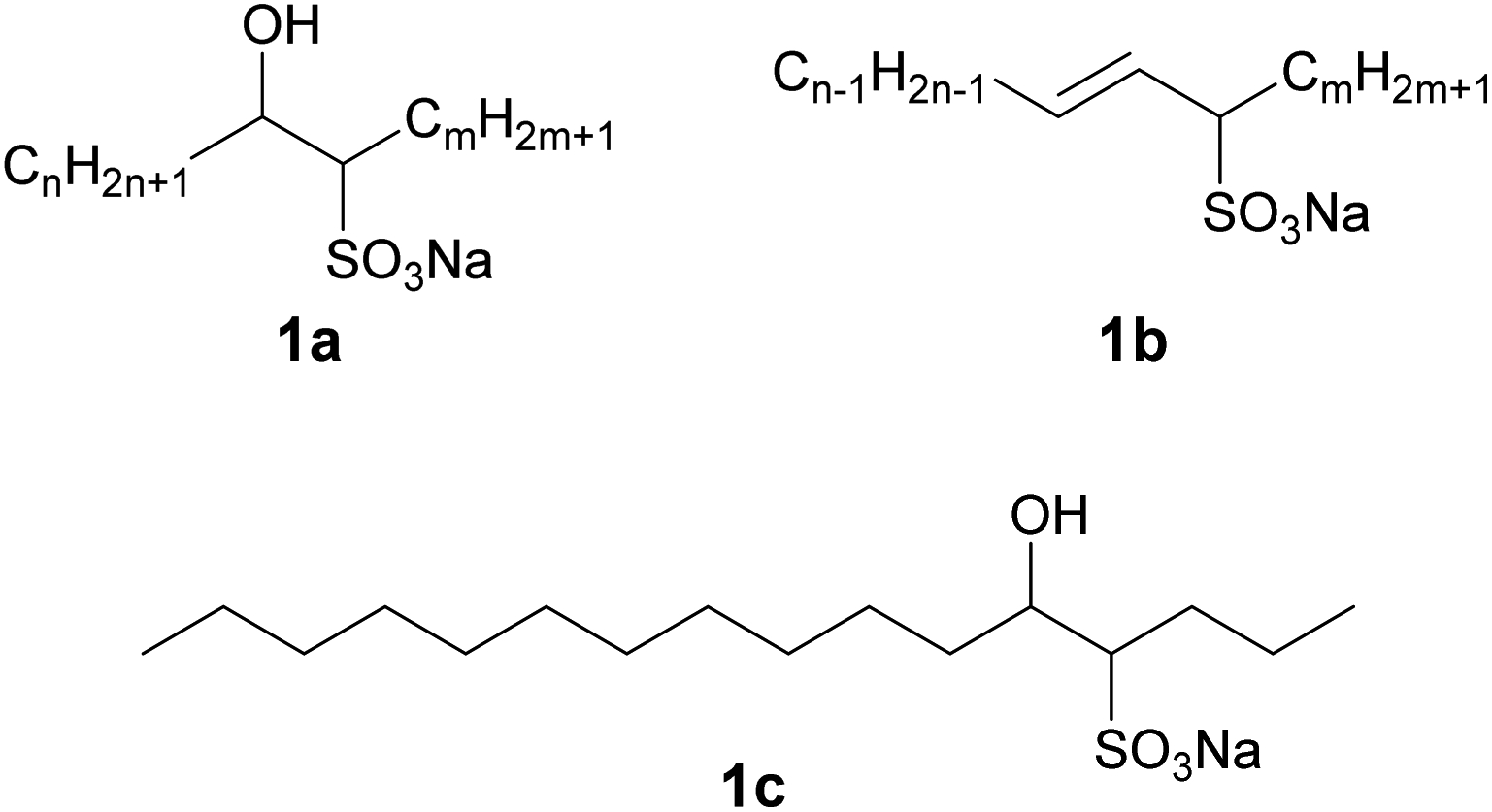 | ||
| Fig. 1 The molecular structures (1a and 1b) of typical IOSs, where n + m + 2 is the total carbon number and a hydrophilic head group is located at some point in the carbon chain. In this study, 1c (1a with n = 11, m = 3; C16-4S-5OH; sodium 5-hydroxyhexadecane-4-sulfonate) was utilized. | ||
The practical application of a surfactant requires high solubility in water and a low critical micelle concentration (CMC). The former is necessary to dissolve in water while the latter reduces the amount of surfactant required. Even so, it is very difficult to simultaneously obtain both properties, because the former requires high hydrophilicity while the latter necessitates significant hydrophobicity. In addition, an ideal surfactant will function under various conditions (such as at low temperatures or high water hardness values) and has to work with less amount from the viewpoint of sustainability. Therefore, the development of a design strategy for surfactant molecules that can simultaneously satisfy the aforementioned apparently incompatible requirements is desirable.
In the present study, the aqueous solution properties of IOS were subsequently assessed using the pure sodium 5-hydroxyhexadecane-4-sulfonate (C16-4S-5OH) (1c), which is one of the most representative components included in the biobased IOS showing a low Krafft point (Kp) and low CMC. It is the first-ever investigation of a completely pure single-component IOS. The molecular structure of this IOS is characterized by two hydrophilic groups (that is, sulfo and hydroxy groups) located at the 4- and 5-carbons of the alkyl chain. The relationship between molecular structure and interfacial properties is one of the most interesting and widely researched aspects of such compounds. However, the stereostructures of amphiphiles in solution must be examined not only based on their theoretical molecular structures but also by considering their actual forms induced by hydrophobic effects or hydration of the hydrophilic groups.9–12 Therefore, the actual state and stereostructure of C16-4S-5OH in water were studied in detail. Moreover, the relationship between the stereostructure of the surfactant in water and the aqueous solution properties was examined.
Experimental
Materials
In this study, the raw material for the synthesis of the C16-4S-5OH (4-hexadecene) was first made. During this process, 1432.6 g butyltriphenylphosphonium bromide (3.59 mol; Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) was dissolved in 4.50 L tetrahydrofuran (FUJIFILM Wako Pure Chemical Corp., Osaka, Japan) by stirring at 0 °C. Following this, 382.6 g potassium tert-butoxide (3.41 mol; FUJIFILM Wako) in 1.50 L tetrahydrofuran was added and the mixture was cooled to −10 °C and stirred for 1.50 h. Subsequently, the reaction mixture was further cooled to −60.0 °C and 507.6 g dodecanal (2.75 mol; Tokyo Chemical Industry) in 0.50 L tetrahydrofuran was added dropwise and the mixture was stirred for 3.00 h at room temperature. Following this, the reaction was quenched by adding a saturated aqueous solution of ammonium chloride (FUJIFILM Wako). The organic phase was washed with brine and dried with magnesium sulfate (FUJIFILM Wako), after which the solvent was removed by evaporation. The resulting residue was purified by column chromatography utilizing silica gel (60 N; 63–200 μm; Kanto Chemical Co., Inc., Tokyo, Japan) and hexane (FUJIFILM Wako). As in prior studies, gas chromatography (GC) was used to confirm that the 4-hexadecene was a mixture of stereoisomers (cis/trans = 97.1/2.9).13,14C16-4S-5OH was obtained by the sulfonation of 4-hexadecene followed by neutralization with sodium hydroxide.7 The reaction product was recrystallized from ethanol (>99.5%, FUJIFILM Wako) three times to give the high-purity C16-4S-5OH monohydrate (purity, >99.9%). The concentrations of impurities such as inorganic salts and residual 4-hexadecene were confirmed to be below the detection limits of the respective analytical techniques during analyses by ion chromatography and gas chromatography. The molecular structure of the product was determined using proton nuclear magnetic resonance (1H NMR) (Fig. S1, ESI†) and liquid chromatography with tandem mass spectrometry (Fig. S2, ESI†). Additionally, the purity of the material was confirmed by elemental analysis (Table S1, ESI†).
All the aqueous solution samples utilized for the subsequent experiments were prepared with ultrapure water obtained from an RFD250RB system (Advantec Toyo Kaisha, Ltd., Tokyo, Japan). The NMR and Fourier transform infrared (FT-IR) analyses utilized deuterium oxide (FUJIFILM Wako) and sodium isethionate (Combi-Blocks, Inc., San Diego, USA) as received without further purification.
Methods
Results and discussion
Aqueous solution properties of C16-4S-5OH


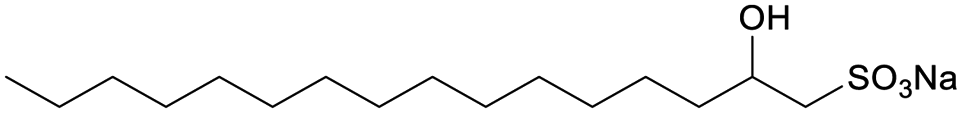

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C (where C is the total concentration) for a series of aqueous C16-4S-5OH solutions at 26.0 °C (just above the Kp value). The γ values are seen to have decreased linearly as C increased up to the point where they eventually plateaued, corresponding to the CMC. Interestingly, the plot displays a small minimum point close to the CMC. This phenomenon is typically attributed to the presence of a low concentration of hydrophobic impurities, which in the present case might be a byproduct (1b in Fig. 1) or positional isomers in which the hydroxy and/or sulfo groups are at different carbons. However, these impurities were not detected by the analyses described above and the elemental analysis data were in good agreement with the theoretical values. Thus, any such impurities are thought to have been present at negligible levels. The linear decrease in γ with increasing concentration in the more dilute regime corresponds to the Gibbs adsorption equation:
C (where C is the total concentration) for a series of aqueous C16-4S-5OH solutions at 26.0 °C (just above the Kp value). The γ values are seen to have decreased linearly as C increased up to the point where they eventually plateaued, corresponding to the CMC. Interestingly, the plot displays a small minimum point close to the CMC. This phenomenon is typically attributed to the presence of a low concentration of hydrophobic impurities, which in the present case might be a byproduct (1b in Fig. 1) or positional isomers in which the hydroxy and/or sulfo groups are at different carbons. However, these impurities were not detected by the analyses described above and the elemental analysis data were in good agreement with the theoretical values. Thus, any such impurities are thought to have been present at negligible levels. The linear decrease in γ with increasing concentration in the more dilute regime corresponds to the Gibbs adsorption equation:
 | (1) |
 | (2) |
 | ||
| Fig. 2 EST versus the concentration of aqueous C16-4S-5OH solutions at 26.0 °C. The straight line represents the Gibbs adsorption isotherm. | ||
| CMC/mmol dm−3 | γCMC/mN m−1 | Γmax/10−6 mol m−2 | Amin/nm2 | |
|---|---|---|---|---|
| a At 26 °C.b At 25 °C.c At 45 °C. The values for C16-4S-5OH were calculated using eqn (1) and (2) employing the experimental data in Fig. 2. Cn AS: CnH2n+1–OSO3Na. | ||||
| C16-4S-5OHa | 3.00 | 37.8 | 1.89 | 0.88 |
| C12 ASb | 7.94 (ref. 18) | 39.9 (ref. 19) | 3.16 (ref. 19) | 0.53 (ref. 19) |
| C14 ASb | 2.05 (ref. 18) | 35.2 (ref. 20) | 3.70 (ref. 20) | 0.45 (ref. 20) |
| C16 ASc | 0.50 (ref. 21) | |||
The CMC for C16-4S-5OH was determined to be 3.00 × 10−3 mol dm−3, which was lower than that of C12 AS, almost the same as that of C14 AS and much higher than that of C16 AS. These results clearly suggest that C16-4S-5OH can be considered as comprising asymmetric C12 and C4 chains rather than a single C16 alkyl chain. For double chain surfactants, empirical data show that the subchains are expected to exert almost half the effect of the main chain on the CMC values proportional to the carbon number.22 On this basis, the CMC of C16-4S-5OH should be almost equal to that of C14 AS. Although a C4 alkyl chain is too short to have a hydrophobic effect,10,23 the observed decrease in the CMC demonstrates that the C4 chain in this compound was able to function as a hydrophobic chain together with the C12 chain. The γCMC of C16-4S-5OH was intermediate between those of C12 AS and C14 AS, as was also the case for the CMC values. In contrast, the Amin value for C16-4S-5OH was 0.88 nm2, which was much larger than those for C12 AS and C14 AS (Table 2). This large Amin is not attributed to the presence of the C12 and C4 chains because the Amin value of the representative double-tailed anionic surfactant sodium 4-(dodecan-4-yl)benzenesulfonate is smaller (0.55 nm2).24 It is well known that surfactants reduce surface tension as a result of surface adsorption and coverage.22 Interestingly, this comparison of the Amin values suggests the opposite of the anticipated result, since C16-4S-5OH would be expected to form a more densely packed monolayer than C12 AS at the air–water interface.
As noted in the Introduction, a high degree of solubility in water and a low CMC are essential for modern surfactants. Although these two properties are normally not achieved simultaneously, the C16-4S-5OH exhibits both a low Kp and CMC. Common qualitative correlations, including that of Amin, between theoretical molecular structure and interfacial properties cannot adequately explain this result. Thus, the stereostructure of this compound in aqueous media was studied, as discussed in the next sub-section.
Analysis of the stereostructure of C16-4S-5OH in water
The actual states of the hydrophobic and hydrophilic portions of the C16-4S-5OH in water were studied to clarify the true stereostructure, which might be responsible for its various characteristics of interest. | ||
| Fig. 3 1H NMR spectra showing peaks related to the (a) carbon 4 and (b) carbon 5 protons of C16-4S-5OH. The position numbers are indicated in the molecular structure. | ||
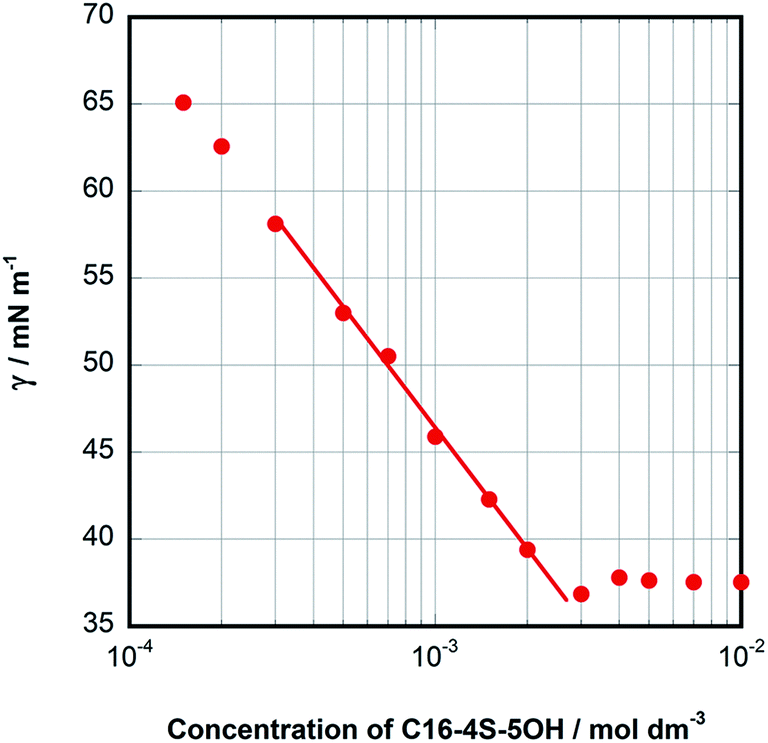
The effects of the solution concentration on this characteristic stereostructure of the monomer were also studied. In particular, the changes in the 1H NMR spectra associated with micelle formation were examined by comparing spectra acquired below and above the CMC. In Fig. 4, the difference in the δ values of the peaks for the methyl protons on the terminal carbons (that is, 1- and 16-H) are plotted against the inverse C16-4S-5OH concentration at values around the CMC. Although both peaks exhibited little change with variations in concentration below the CMC, they suddenly shifted downfield above the CMC, both to the same extent. In the case of DBS, which has two alkyl chains, the variations in this δ difference for the terminal methyl groups below and above the CMC have been studied in detail.12,15 Prior work showed that the peak generated by the methyl proton of the longer alkyl chain moved upfield by approximately 0.03 ppm as micelles were formed and aggregated in water. In contrast, the peak related to the shorter alkyl chain moved downfield by about 0.04 ppm. It has been established that the opposite movements of these methyl protons reflect the different positions of the methyl groups in the micelle aggregates. Specifically, the methyl group of the longer alkyl chain is located in the hydrocarbon core while the other is near the hydrophilic groups (that is, the palisade layer). However, the 1H NMR results in Fig. 4 show that the peaks for both the C12 and C4 chains of C16-4S-5OH moved downfield as micelles formed. This result suggests two possibilities concerning stereostructure in the micelles and the positions of the terminal methyl groups. One explanation is that the highly fixed stereostructures of the monomers below the CMC were maintained when the micelles were formed above the CMC. In this case, the short C4 subchains would function as hydrophobic groups, as was discussed above in relation to CMC values. Another possibility is that the terminal methyl groups continued to experience the deshielding effect of the highly electron-withdrawing sulfo groups, since they were close to these hydrophilic head groups. Downfield shifts of the terminal methyl group peaks were observed in conjunction with the formation of micelles of surfactants having mixed alkyl chain lengths.15 It has been proposed that this phenomenon occurs when the alkyl chains curl up or bend such that they approach the head groups in the micelles. Although the C16-4S-5OH in the present work was a single compound, the eight carbon difference in the alkyl chain lengths was large enough that these chains might be expected to bend and loop to fill the spaces in the micelle cores. The δ values for the 4- and 5-protons around the head groups were almost unaffected by increases in the solution concentration. This finding provides support for the hypothesis that the downfield shifts of the peaks for the two terminal methyl protons were caused by their environment in the micelle.
 | ||
| Fig. 4 Difference in the 1H NMR chemical shifts of the terminal methyl protons of C4 alkyl (○) and C12 alkyl (■) chains of C16-4S-5OH versus the inverse concentration of aqueous C16-4S-5OH solutions at 26 °C. | ||
H/D exchange experiments were employed to evaluate the presence of hydrogen bonding and its effect on stability in solvents.25–27 Initially, the H/D exchange of the hydroxy proton of C16-4S-5OH in D2O was investigated. Fig. 5(a) presents the FT-IR spectra of solid C16-4S-5OH samples obtained by lyophilizing solutions of C16-4S-5OH in D2O at various concentrations. The O–H and O–D stretching bands were observed at approximately 3000–3500 and 2500–3000 cm−1, respectively. The strong, sharp peaks in the range of 2800–3000 cm−1 were assigned to C–H stretching. The C16-4S-5OH samples obtained from D2O solutions at 60 °C with concentrations from 1.00 × 10−3 mol dm−3 (below the CMC) to 1.00 × 10−2 mol dm−3 (above the CMC) all generated O–H stretching bands but not O–D stretching bands. The 5-hydroxy proton of the C16-4S-5OH was evidently inactive during the H/D exchange regardless of the solution concentration (and therefore the aggregation state). The formation of a stable hydrogen bond between the hydroxy proton and sulfonyl oxygen could possibly have inhibited the H/D exchange of this hydrogen.
 | ||
| Fig. 5 Evidence for H/D exchange in the FT-IR spectra of C16-4S-5H and sodium isethionate (26 °C). (a) Effect of the concentration of C16-4S-5OH on H/D exchange. (b) Change in the FT-IR spectra of sodium isethionate before and after the H/D exchange treatment. | ||
The same H/D exchange experiments were repeated using sodium isethionate, which has a molecular structure that corresponds to the hydrophilic groups of C16-4S-5OH without the hydrophobic alkyl chains (Fig. 5). In these trials, a 1.00 × 10−2 mol dm−3 solution was employed and the FT-IR spectra acquired before and after the D2O treatment are shown in Fig. 5(b). In these spectra, the O–H stretching band around 3400 cm−1 is seen to have decreased in intensity while an O–D stretching band around 2500 cm−1 appeared after the D2O treatment. These results confirmed that an H/D exchange proceeded. The very different behaviors of the C16-4S-5OH and sodium isethionate can likely be ascribed to the presence of long alkyl chains in the former. It was inferred from the 1H NMR results that the long and short alkyl chains of C16-4S-5OH were highly restricted and also both oriented in the same direction in water. This fixed orientation of the two chains may have promoted the intramolecular cyclization to form a ring comprising the 4-CH, 5-CH, the oxygen and hydrogen of the hydroxy group, and the sulfur and oxygen of the sulfo group (Fig. 5(a)). Conversely, the cyclization of the head groups might have fixed the orientation of the two alkyl chains.
The H/D exchange of C16-4S-5OH in D2O was inhibited even in the monomer and micelle states. In aqueous solutions below the CMC, the C16-4S-5OH existed in its monomeric form. Because the distance between monomers was relatively large at these concentrations, intermolecular interactions could not readily proceed. Conversely, there intermolecular interactions occurred easily above the CMC. The minimal H/D exchange at both concentrations strongly suggests that the molecular structure was able to independently stabilize the hydroxy proton by intramolecular cyclization regardless of the aggregation state.
Actual stereostructure of C16-4S-5OH in water
As discussed above, the three aspects of the true molecular structure of C16-4S-5OH in water were clarified. Firstly, the geometries of the C12 and C4 alkyl chains and of the hydrophilic groups were strictly fixed. Secondly, the hydroxy protons were highly stabilized by intramolecular hydrogen bonding. Lastly, the hydroxy protons were stabilized by the two alkyl chains. The actual stereostructure of the two alkyl chains in water was determined to be a cis configuration (4R, 5S or 4S, 5R) based on the 1H NMR coupling constants (Fig. 6). Thus, C16-4S-5OH in water appears to consist of a hydrophobic unit containing long and short alkyl chains with highly ordered orientations and a large cyclic hydrophilic head unit containing the sulfo and hydroxy groups.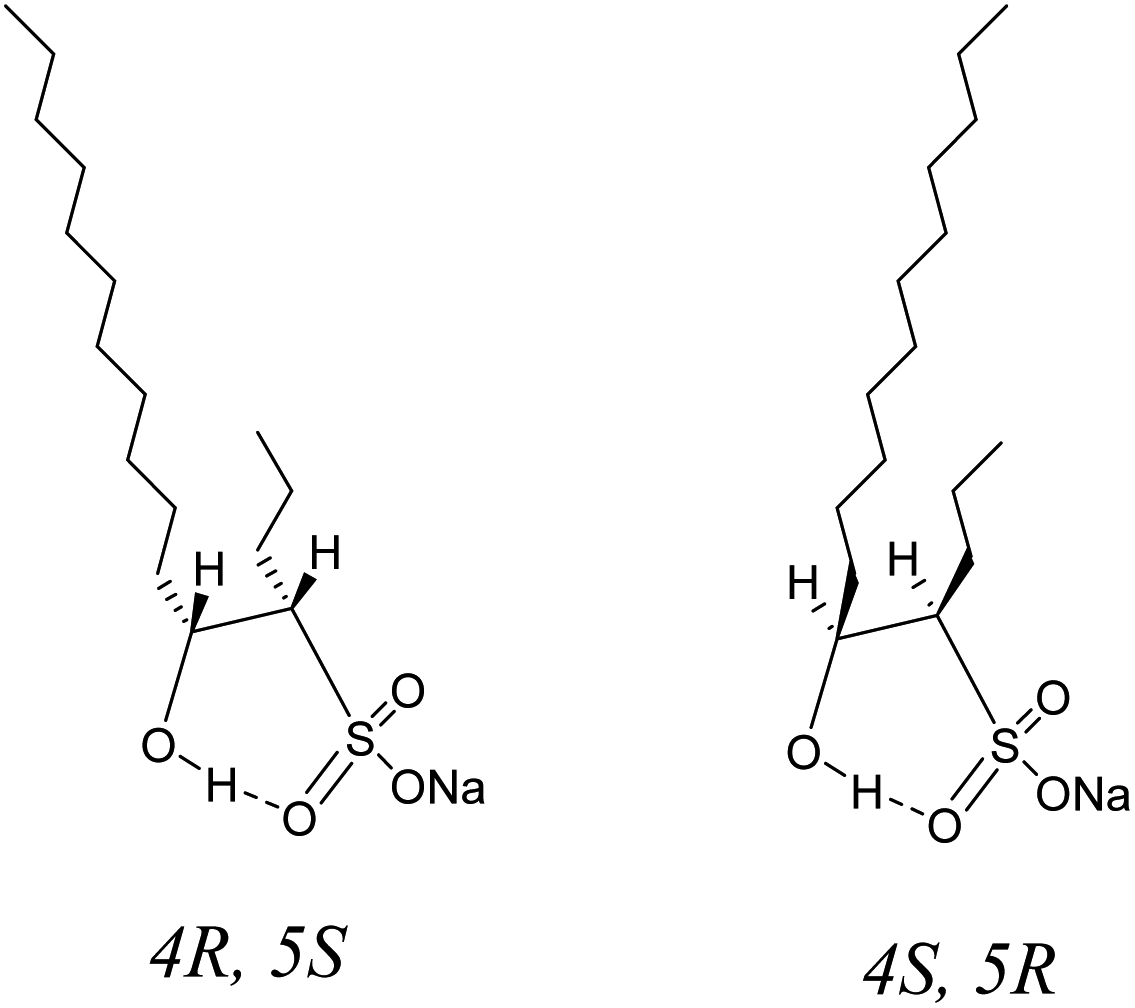 | ||
| Fig. 6 Possible molecular structures of sodium 5-hydroxyhexadecane-4-sulfonate (C16-4S-5OH) in water. | ||
The difference between C16-4S-5OH and a heterogemini surfactant
Gemini surfactants, which comprise two surfactants linked by a spacer unit, have attracted much attention because of their exceptional surface properties, such as low CMCs, good surface tension reduction characteristics and low Kp values. However, these compounds do exhibit large Amin compared with common surfactants possessing both a single chain and single head group.29–31 Those surfactants exhibiting asymmetry in their hydrophilic groups and/or alkyl chains are termed heterogemini surfactants.32–36 There are three important factors related to these compounds: the dissymmetry between the two alkyl chain lengths, the spacer chain length, and the type of hydrophilic head group. Although there have been many reports regarding heterogemini surfactants, such prior work has always involved subchains longer than six carbons, possibly because shorter chains may not be hydrophobic.10,23 Most previously reported spacer chains were also longer than two carbons because it is challenging to prepare gemini surfactants with shorter spacers. Moreover, such compounds might not be water insoluble because of the strong hydrophobic characteristics of the two alkyl chains in proximity to one another. As an example, even aqueous solutions of gemini surfactants possessing C2 spacers have exhibited very high viscosities at relatively low concentrations.37 There have also been a few reports of heterogemini surfactants, such as C16-4S-5OH, with shorter spacers (C1 or C0).38–41 Additionally, surfactants in which both the chain lengths and hydrophilic groups were different have been examined.38,39,41C16-4S-5OH could be easily regarded as a kind of heterogemini surfactant based on a casual analysis, although its alkyl subchain is too short to be considered as a hydrophobic moiety. Moreover, the spacer is only a C–C bond (that is, C0) while the two head groups are different and form an intramolecular ring structure. In fact, a heterogemini surfactant with the same molecular structure as C16-4S-5OH around the two head groups has been reported.38 However, unlike C16-4S-5OH, this compound possesses two long hydrophobic chains that are believed to be in a trans configuration as a result of the steric repulsion between them. These two long chains likely prevent the two head groups from approaching one another to the extent necessary to initiate hydrogen bonding. The Amin value for this surfactant is reported to be above 1.2 nm2, which is average for a gemini surfactant but much larger than that of C16-4S-5OH (0.88 nm2). Thus, although the Amin of C16-4S-5OH is greater than that of most common surfactants, it is very compact compared with gemini surfactants. Therefore, the highly fixed molecular structure of C16-4S-5OH could result from both the dissymmetric chains, which do not undergo steric repulsion, and the formation of the intramolecular ring by the two head groups. As such, C16-4S-5OH should not be regarded as a gemini surfactant but rather as an asymmetric double chain surfactant with a large head group.
Origin of the low Kp and CMC of C16-4S-5OH
Conclusions
An IOS derived from a C16 oleo resource exhibited both excellent water solubility and a low CMC, which are typically trade-off properties, despite possessing a long hydrophobic chain. Therefore, this compound is expected to have applications as a sustainable anionic surfactant. The C16-4S-5OH IOS synthesized in this work was isolated and purified and its actual stereostructure in water and the effect of its molecular structure on its physicochemical properties were investigated. The highly fixed stereostructure of this molecule was found to be maintained in aqueous solutions both below and above its CMC. The formation of highly oriented and largely hydrophobic asymmetric C12 and C4 chains was confirmed. In addition, the generation of a stable cyclic hydrophilic unit consisting of two carbons of the C16 alkyl chain was shown to occur via hydrogen bonding. The actual molecular structure of this surfactant lowered the packing density of the two alkyl chains and reduced its Kp, leading to exceptional water solubility. Conversely, only 16 carbons contributed to the hydrophobic effect during micelle formation, thereby affording a low CMC value. In this manner, the trade-off between water solubility and a low CMC was circumvented. The nature of these IOS characteristics depended not only on the heterogemini structure of the surfactant but also on the formation of the cyclic hydrophilic unit. The information gained in this work regarding the molecular design of such compounds could contribute to the development of sustainable surfactants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors did not receive a specific grant from any funding agency in the public, commercial, or not-for-profit sectors. The authors thank the staff of the Kao Corporation who assisted in the development of the biobased IOS.References
- V. B. Fainerman, D. Mobius and R. Miller, Surfactants: Chemistry, Interfacial Properties, Applications, 2001 Search PubMed.
- B. V. Vora, G. A. Peterson, S. W. Sohn and M. G. Riley, Handbook of Detergents Part F: Production, CRC Press, 2009 Search PubMed.
- H. W. Stache, Anionic Surfactants: Organic Chemistry, Marcel Dekker, 1996 Search PubMed.
- M. E. Tuvell, G. O. Kuehnhanss, G. D. Heidebrecht, P. O. Hu and A. D. Zielinski, J. Am. Oil Chem. Soc., 1978, 55, 70–80 CrossRef CAS.
- J. Stapersma, H. H. Deuling and R. van Ginkel, J. Am. Oil Chem. Soc., 1992, 69, 39–43 CrossRef CAS.
- S. Takada, WIPO (PCT), WO2011052732A1, 2011.
- H. Hori, Y. Yoshikawa, Y. Mitsuda, T. Fujioka and Y. Nishimoto, WIPO (PCT), WO2015098415A1, 2015.
- Y. Tabuchi, 11th World Surfactant Congress, Munich, 2019 Search PubMed.
- R. Vijay, A. B. Mandal and G. Baskar, J. Phys. Chem. B, 2010, 114, 13691–13702 CrossRef CAS PubMed.
- S. Z. Can, D. D. Mago, O. Esenturk and R. A. Walker, J. Phys. Chem. C, 2007, 111, 8739–8748 CrossRef CAS.
- Y. Hou, Y. Han, M. Deng, J. Xiang and Y. Wang, Langmuir, 2010, 26, 28–33 CrossRef CAS PubMed.
- S. Das, R. G. Bhirud, N. Nayyar, K. S. Narayan and V. V. Kumar, J. Phys. Chem., 1992, 96, 7454–7457 CrossRef CAS.
- W. G. Francis, Chem. Phys. Lipids, 1981, 29, 369–374 CrossRef.
- A. Shibahara, K. Yamamoto, T. Nakayama and G. Kajimoto, J. Jpn. Oil Chem. Soc., 1985, 34, 618–625 CrossRef CAS.
- P. Goon, S. Das, C. J. Clemett, G. J. T. Tiddy and V. V. Kumar, Langmuir, 1997, 13, 5577–5582 CrossRef CAS.
- T. Gu and J. Sjöblom, Acta Chem. Scand., 1991, 45, 762–765 CrossRef CAS.
- S. H. F. Püschel and B. Strobel, Tenside, 1967, 4, 349–352 Search PubMed.
- B. D. Flockhart, J. Colloid Sci., 1961, 16, 484–492 CrossRef CAS.
- M. Dahanayake, A. W. Cohen and M. J. Rosen, J. Phys. Chem., 1986, 90, 2413–2418 CrossRef CAS.
- H. Lange and M. J. Schwuger, Kolloid-Z. Z. Polym., 1968, 223, 145–149 CrossRef CAS.
- I. J. Lin and P. J. Somasundaran, J. Colloid Interface Sci., 1971, 37, 731–743 CrossRef CAS.
- M. J. Rosen and J. T. KunJappu, Surfactants and Interfacial Phenomena, Wiley, 4th edn, 2012 Search PubMed.
- J. A. Mondal, V. Namboodiri, P. Mathi and A. K. Singh, J. Phys. Chem. Lett., 2017, 8, 1637–1644 CrossRef CAS PubMed.
- J. G. Ma, B. J. Boyd and C. J. Drummond, Langmuir, 2006, 22, 8646–8654 CrossRef CAS PubMed.
- C. S. Maier and M. L. Deinzer, Methods Enzymol., 2005, 402, 312–360 CAS.
- M. M. G. Krishna, L. Hoang, Y. Lin and S. W. Englander, Methods, 2004, 34, 51–64 CrossRef CAS PubMed.
- S. W. Englander, N. W. Downer and H. Teitelbaum, Annu. Rev. Biochem., 1972, 41, 903–924 CrossRef CAS PubMed.
- E. Caponetti, R. Trilo, C. H. O. Patience, J. S. Johnson, L. J. Magid, P. Butler and K. A. Payne, J. Colloid Interface Sci., 1987, 116, 200–210 CrossRef CAS.
- F. M. Menger and C. Littau, J. Am. Chem. Soc., 1991, 113, 1451–1452 CrossRef CAS.
- R. Zana, Adv. Colloid Interface Sci., 2002, 97, 205–253 CrossRef CAS.
- M. J. Rosen, Chem. Tech., 1993, 23, 30–33 CAS.
- R. Oda, I. Huc and S. J. Candau, Chem. Commun., 1997, 21, 2105–2106 RSC.
- G. Bai, J. Wang, Y. Wang, H. Yan and R. K. Thomas, J. Phys. Chem. B, 2002, 106, 6614–6616 CrossRef CAS.
- D. A. Jaeger, B. Li and T. Clark, Langmuir, 1996, 12, 4314–4316 CrossRef CAS.
- E. Alami, K. Holmberg and J. Eastoe, J. Colloid Interface Sci., 2002, 247, 447–455 CrossRef CAS PubMed.
- E. Alami and K. Holmberg, Adv. Colloid Interface Sci., 2003, 100, 13–46 CrossRef.
- R. Zana, J. Colloid Interface Sci., 2002, 248, 203–220 CrossRef CAS PubMed.
- Y. Takamatsu, N. Iwata, K. Tsubone, K. Torigoe, T. Endo, K. Sakai, H. Sakai and M. Abe, J. Colloid Interface Sci., 2009, 338, 229–235 CrossRef CAS PubMed.
- K. Sakai, Y. Sangawa, Y. Takamatsu and T. Kawai, J. Oleo Sci., 2010, 59, 541–548 CrossRef CAS PubMed.
- K. Sakai, N. Umemoto, W. Matsuda, Y. Takamatsu, M. Matsumoto, H. Sakai and M. Abe, J. Oleo Sci., 2011, 60, 411–417 CrossRef CAS PubMed.
- S. Liu, X. Wang, L. Chen, L. Hou and T. Zhou, Soft Matter, 2014, 10, 9177–9186 RSC.
- G. Cevc, Biochemistry, 1991, 30, 7186–7193 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01046h |
| This journal is © The Royal Society of Chemistry 2021 |