
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuanidine functionalized core–shell structured magnetic cobalt-ferrite: an efficient nanocatalyst for sonochemical synthesis of spirooxindoles in water†
Mahla Dadaei and
Hossein Naeimi *
*
Department of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, 87317, I. R. Iran. E-mail: naeimi@kashanu.ac.ir; Fax: +98 3615912397; Tel: +98 3615912388
First published on 10th May 2021
Abstract
Core/shell nanoparticles have a wide range of applications in the science of chemistry and biomedicine. The core–shell material can be different and modified by changing the ingredients or the ratio of core to the shell. In this research, a CoFe2O4@SiO2-guanidine nanocomposite was prepared and identified as an efficient catalyst for the one-pot synthesis of spirooxindole derivatives in water under ultrasonic irradiation conditions. The advantages of this method are in its simplicity, saving costs and energy, high yields, short reaction times, environmental friendliness, reusability and easy recovery of the catalyst using an external magnet. The catalyst was characterized by XRD, SEM, TEM, EDX, FT-IR, TGA and VSM techniques.
1. Introduction
Spirooxindole alkaloid compounds were first extracted from plants of the Apocynaceae and Rubiaceae families.1 Synthesis of spiroxindole compounds among organic chemists is particularly important because these heterocyclic compounds have a wide range of pharmacological activities.2 These spirocycles possess anticancer and antimicrobial activities.3 They are also known to exhibit analgesic, anticonvulsant,4 and fungicidal5 activities.Recently, few synthetic methods for the synthesis of spiroxindoles have been reported in the literature. Some of the catalysts that have been utilized in these methods are; PPh3,6 HAuCl4·3H2O,7 Incl3/SiO2,8 sodium stearate,9 and ethylenediamine diacetate (EDDA).10
However, there are disadvantages to these reported methods, such as; a poor yield, the use of toxic solvents, protracted reaction time and reusability of the catalyst is hardship. Although, each of the known methods for the synthesis of spirooxindole compounds has its competency, however further studies are still necessary for the efficient, environmental and economical multicomponent methodology for the synthesis of these heterocyclic compounds.
The use of ultrasonic waves in organic synthesis has been boosted in recent years.11,12 This technique has many advantages including saving costs and energy, nontoxic, environmentally friendly solvent13,14 and reduce of side products.15 The power of ultrasound radiation is (20 kHz to 10 MHz).16 Ultrasound irradiation could be positive effect on increasing yields and rate of the reaction due to the mechanical effect of shock waves. The main event in sonochemistry is the creation, growth and collapse of a bubble that is formed in the liquid. The step leading to the growth of the bubble occurs through the diffusion of solute vapor into the volume of the bubble. The last stage is the crash of the bubble, which occurs when the bubble size reaches its maximum value.
Core/shell nanoparticles have a wide range of applications in the science of chemistry, biomedical, pharmaceutical, electronics and catalysis. Therefore, these nanoparticles have attracted the attention of researchers. The core–shell material can be different and modified by changing the ingredients or the ratio of core to the shell.17 Due to the coating of the shell material, the reactivity of core decreases and the thermal stability changes. The purpose of the core cover surface modification, reduce the consumption of precious metals and increase the efficiency of functional groups. In recent years, the applications of core–shell structured MNPs in organic reactions have developed.18–24
Herein, we would like to report the synthesis and application of guanidine immobilized on CoFe2O4@SiO2-CPTES nanocomposites as an environmentally benign, highly efficient and reusable catalyst for the preparation of spirooxindole derivatives via one-pot three component of isatin or 5-chloroisatin, malononitrile or cyanoacetic esters and 1,3-dicarbonyl compounds in water under ultrasonic irradiation.
2. Experimental
2.1. General information
The chemicals were purchased from Fluka and Merck Chemical Companies and used without purification. 1H NMR was recorded in DMSO-d6 solvent on a Bruker DRX-400 spectrometer with tetramethylsilane as internal reference. FT-IR spectra were obtained as KBr pellets on a Perkin-Elmer 781 spectrophotometer and on an impact 400 Nicolet FT-IR spectrophotometer. The nanostructures were characterized using a Holland Philips Xpert X-ray powder diffraction (XRD) diffractometer (CuK, radiation, k ¼ 0.154056 nm), at a scanning speed of 2° min−1 from 100 to 100° (2Ø). The morphological study of the nanocomposites was investigated by scanning electron microscopy (SEM, SIGMA VP). The Electron Dispersive X-ray (EDX) analysis of the catalyst was performed on Oxford Instrument Company. The nanocomposite was investigated by transmission electron microscopy (TEM, Zeiss-EM10C-100 kV). Thermogravimetric analysis (TGA) was performed on a mettler TA4000 system TG-50 at a heating rate of 10 K min−1 under N2 atmosphere. The magnetic properties of nanoparticles have been measured with a vibrating sample magnetometer (VSM, PPMS-9T) at 300 K in Iran (Kashan University). Melting points were obtained with a Yanagimoto micro melting point apparatus and are uncorrected. The purity determination of the substrates and reaction monitoring were accomplished by TLC on silica-gel polygram SILG/UV 254 plates (from Merck Company).2.2. Catalyst preparation
2-Amino-2′,5-dioxo-5H-spiro[indeno[1,2-b]pyran-4,3′-indoline]-3-carbonitrile. Yellow solid, mp 207–210 °C. IR (KBr) ν(cm−1): 3316, 3193, 2201, 1731, 1669, 1602, 1470, 1336, 1212. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.87 (d, 1H, Ar–H, J = 8.0 Hz), 6.95 (t, 1H, Ar–H, J = 8.0 Hz), 7.21 (d, 1H, Ar–H, J = 4.0 Hz), 7.24 (s, 1H, Ar–H), 7.29 (d, 1H, Ar–H, J = 4.0 Hz), 7.36 (d, 1H, Ar–H, J = 8.0 Hz), 7.43 (t, 1H, Ar–H, J = 8.0 Hz), 7.56 (t, 1H, Ar–H, J = 8.0 Hz), 7.70 (s, 2H, NH2), 10.68 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 45.69, 57.08, 107.17, 109.71, 117.34, 118.78, 122.17, 122.23, 124.57, 129.12, 130.43, 131.30, 131.98, 133.64, 135.07, 141.77, 160.41, 167.47, 177.34, 189.34.
7′-Amino-2,4′-dioxo-2′-thioxo-1′,2′,3′,4′-tetrahydrospiro [indoline-3,5′-pyrano[2,3-d]pyrimidine]-6′-carbonitrile. White solid, mp 232–235 °C, lit. 27 (mp 240–241 °C). IR (KBr) ν(cm−1): 3309, 3156, 2196, 1718, 1675, 1571, 1470, 1395, 1339, 1104. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.77 (d, 1H, Ar–H, J = 8.0 Hz), 6.89 (t, 1H, Ar–H, J = 8.0 Hz), 7.13 (t, 1H, Ar–H, J = 2.0 Hz), 7.15 (d, 1H, Ar–H, J = 8.0 Hz), 7.33 (s, 2H, NH2), 10.48 (s, 1H, NH), 12.15 (s, 1H, NH), 13.85 (s, 1H, NH).
7′-Amino-2,2′,4′-trioxo-1′,2′,3′,4′-tetrahydrospiro[indoline-3,5′-pyrano[2,3-d]pyrimidine]-6′-carbonitrile. White solid, mp 275–277 °C, lit. 28 (mp 274–276 °C). IR (KBr) ν(cm−1): 3353, 3304, 3145, 2829, 2203, 1723, 1673, 1395, 1616, 1531, 1468, 1337, 1111. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.77 (d, 1H, Ar–H, J = 8.0 Hz), 6.89 (t, 1H, Ar–H, J = 6.0 Hz), 7.12 (d, 1H, Ar–H, J = 8.0 Hz), 7.15 (d, 1H, Ar–H, J = 4.0 Hz), 7.37 (s, 2H, NH2), 10.47 (s, 1H, NH), 11.12 (s, 1H, NH), 12.30 (s, 1H, NH).
7′-Amino-5-chloro-2,4′-dioxo-2′-thioxo-1′,2′,3′,4′-tetrahydro spiro[indoline-3,5′-pyrano[2,3-d]pyrimidine]-6′-carbonitrile. White solid, mp 228–230 °C, lit. 28 (mp 224–226 °C). IR (KBr) ν(cm−1): 3358, 3278, 3150, 2849, 2196, 1679, 1570, 1476, 1340, 1172, 1110, 1066. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.79 (d, 1H, Ar–H, J = 8.0 Hz), 7.19–7.22 (dd, 1H, Ar–H, J = 12.0 Hz, J = 4.0 Hz), 7.37 (d, 1H, Ar–H, J = 4.0 Hz), 7.47 (s, 2H, NH2), 10.65 (s, 1H, NH), 12.43 (s, 1H, NH), 13.86 (s, 1H, NH).
7′-Amino-5-chloro-2,2′,4′-trioxo-1′,2′,3′,4′-tetrahydrospiro [indoline-3,5′-pyrano[2,3-d]pyrimidine]-6′-carbonitrile. White solid, mp 242–245 °C, lit. 29 (mp 250–258 °C). IR (KBr) ν(cm−1): 3296, 3163, 2821, 2200, 1693, 1644, 1537, 1401, 1336, 1121, 999. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.78 (d, 1H, Ar–H, J = 8.0 Hz), 7.18–7.21 (dd, 1H, Ar–H, J = 12.0 Hz, J = 4.0 Hz), 7.31 (d, 1H, Ar–H, J = 8.0 Hz), 7.43 (s, 2H, NH2), 10.60 (s, 1H, NH), 11.12 (s, 1H, NH), 12.32 (s, 1H, NH).
2-Amino-5′-chloro-2′,5-dioxo-5H-spiro[indeno[1,2-b]pyran-4,3′-indoline]-3-carbonitrile. Yellow solid, mp 219–222 °C. IR (KBr) ν(cm−1): 3316, 3241, 3199, 2196, 1738, 1667, 1600, 1472, 1334, 1209, 923. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.89 (d, 1H, Ar–H, J = 8 Hz), 7.26 (d, 1H, Ar–H, J = 4.0 Hz), 7.30 (d, 1H, Ar–H, J = 8.0 Hz), 7.37 (d, 1H, Ar–H, J = 8.0 Hz), 7.43 (d, 1H, Ar–H, J = 8.0 Hz), 7.45 (d, 1H, Ar–H, J = 8.0 Hz), 7.56 (t, 1H, Ar–H, J = 6.0 Hz), 7.78 (s, 2H, NH2), 10.83 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 46.55, 56.96, 106.47, 112.36, 117.68, 118.87, 122.20, 124.98, 126.61, 129.06, 130.51, 131.33, 133.62, 134.37, 135.16, 140.70, 160.55, 167.83, 176.25, 189.35.
Ethyl-2-amino-5′-chloro-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro[chromene-4,3′-indoline]-3-carboxylate. White solid, mp 278–281 °C, lit. 30 (mp 275–278 °C). IR (KBr) ν(cm−1): 3384, 3276, 2958, 1725, 1687, 1650, 1514, 1477, 1349, 1294, 1052. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.81 (t, 3H, CH3, J = 6.0 Hz), 0.95 (s, 3H, CH3), 0.99 (s, 3H, CH3), 2.04–2.15 (q, 2H, CH2, J = 16.0 Hz), 2.50 (t, 2H, CH2, J = 2.0 Hz), 3.67–3.70 (m, 2H, CH2), 6.67 (d, 1H, Ar–H, J = 8.0 Hz), 6.89 (d, 1H, Ar–H, J = 4.0 Hz), 7.07–7.09 (dd, 1H, Ar–H, J = 8.0 Hz, J = 4 Hz), 7.93 (s, 2H, NH2), 10.31 (s, 1H, NH).
Ethyl-2-amino-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro [chromene-4,3′-indoline]-3-carboxylate. White solid, mp 252–254 °C, lit. 30 (mp 256–258 °C). IR (KBr) ν(cm−1): 3369, 3178, 2958, 1715, 1688, 1613, 1525, 1473, 1316, 1259. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.78 (t, 3H, CH3, J = 6.0 Hz), 0.93 (s, 3H, CH3), 0.97 (s, 3H, CH3), 2.00 (d, 1H, CH, J = 16.0 Hz), 2.14 (d, 1H, CH, J = 16.0 Hz), 2.47 (d, 1H, CH, J = 16.0 Hz), 2.57 (d, 1H, CH, J = 16.0 Hz), 3.68–3.70 (m, 2H, CH2), 6.66 (d, 1H, Ar–H, J = 8.0 Hz), 6.75 (t, 1H, Ar–H, J = 6.0 Hz), 6.82 (d, 1H, Ar–H, J = 8.0 Hz), 7.03 (t, 1H, Ar–H, J = 8.0 Hz), 7.85 (s, 2H, NH2), 10.13 (s, 1H, NH).
2-Amino-5′-chloro-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydro spiro[chromene-4,3′-indoline]-3-carbonitrile. White solid, mp 292–294 °C, lit. 30 (mp > 300 °C). IR (KBr) ν(cm−1): 3286, 3154, 2958, 2192, 1726, 1652, 1602, 1475, 1350, 1222, 1056. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1.01 (s, 6H, 2CH3), 2.14 (s, 2H, CH2), 2.54 (d, 2H, CH2, J = 12.0 Hz), 6.79 (d, 1H, Ar–H, J = 8.0 Hz), 7.09 (d, 1H, Ar–H, J = 4.0 Hz), 7.17–7.19 (dd, 1H, Ar–H, J = 8.0 Hz, J = 4.0 Hz), 7.33 (s, 2H, NH2), 10.54 (s, 1H, NH).
3′-Acetyl-6′-amino-5-chloro-2′-methyl-2-oxo-spiro[indoline-3,4′-pyran]-5′-carbonitrile. White solid, mp 280–282 °C. IR (KBr) ν(cm−1): 3356, 3237, 2192, 1705, 1653, 1592, 1479, 1298, 1214, 1054. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.22 (s, 3H, CH3), 2.38 (s, 3H, CH3), 6.77 (d, 1H, Ar–H, J = 8.0 Hz), 7.10 (d, 1H, Ar–H, J = 4.0 Hz), 7.16 (d, 1H, Ar–H, J = 4.0 Hz), 7.20 (s, 2H, NH2), 10.49 (s, 1H, NH). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 19.80, 31.53, 56.13, 110.67, 114.41, 117.36, 123.19, 125.61, 128.15, 136.69, 140.88, 158.05, 159.01, 178.33, 197.03.
2-Amino-7,7-dimethyl-2′,5-dioxo-5,6,7,8-tetrahydrospiro [chromene-4,3′-indoline]-3-carbonitrile. White solid, mp 288–290 °C, lit. 30 (mp 290–292 °C). IR (KBr) ν(cm−1): 3375, 3312, 3142, 2960, 2192, 1722, 1655, 1604, 1469, 1349, 1220, 1054. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 0.99 (s, 3H, CH3), 1.02 (s, 3H, CH3), 2.06–2.18 (q, 2H, CH2, J = 16 Hz), 2.54 (d, 2H, CH2, J = 4.0 Hz), 6.77 (d, 1H, Ar–H, J = 8.0 Hz), 6.88 (t, 1H, Ar–H, J = 8.0 Hz), 6.97 (d, 1H, Ar–H, J = 8.0 Hz), 7.13 (t, 1H, Ar–H, J = 8.0 Hz), 7.23 (s, 2H, NH2), 10.39 (s, 1H, NH).
3′-Acetyl-6′-amino-2′-methyl-2-oxo-spiro[indoline-3,4′-pyran]-5′-carbonitrile. White solid, mp 240–243 °C, lit. 31 (mp 243–245 °C). IR (KBr) ν(cm−1): 3378, 3346, 3120, 2188, 1709, 1680, 1653, 1467, 1296, 1211. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.13 (s, 3H, CH3), 2.28 (s, 3H, CH3), 6.76 (d, 1H, Ar–H, J = 8.0 Hz), 6.90 (t, 1H, Ar–H, J = 8.0 Hz), 7.02 (d, 1H, Ar–H, J = 8.0 Hz), 7.12 (s, 2H, NH2), 7.15 (d, 1H, Ar–H, J = 8.0 Hz), 10.38 (s, 1H, NH).
3. Results and discussion
3.1. Preparation and characterization of catalyst
To prepare the catalyst, we have chosen a mixture of CoCl2·6H2O, FeCl3·6H2O and NaOH dissolved in deionized water under reflux condition according to the procedure reported in the literature.18 In the next step, the nanomagnetic was coated with silica. Then, the resulting silica-coated CoFe2O4 nanoparticles were allowed to react under vigorous stirring with an appropriate concentration of 3-chloropropyltriethoxysilane.25,26 Finally, the reaction of CoFe2O4@SiO2 carried out with guanidine in toluene under reflux condition for 30 h to give amino-functionalized silica-coated nanomagnetic (CoFe2O4@SiO2-guanidine). The synthetic path for preparation of the catalyst is shown in Scheme 1. In order to characterize the catalyst structure, the synthesized nanocatalyst was analysed using XRD, SEM, EDX, TEM, FT-IR, TGA and VSM techniques.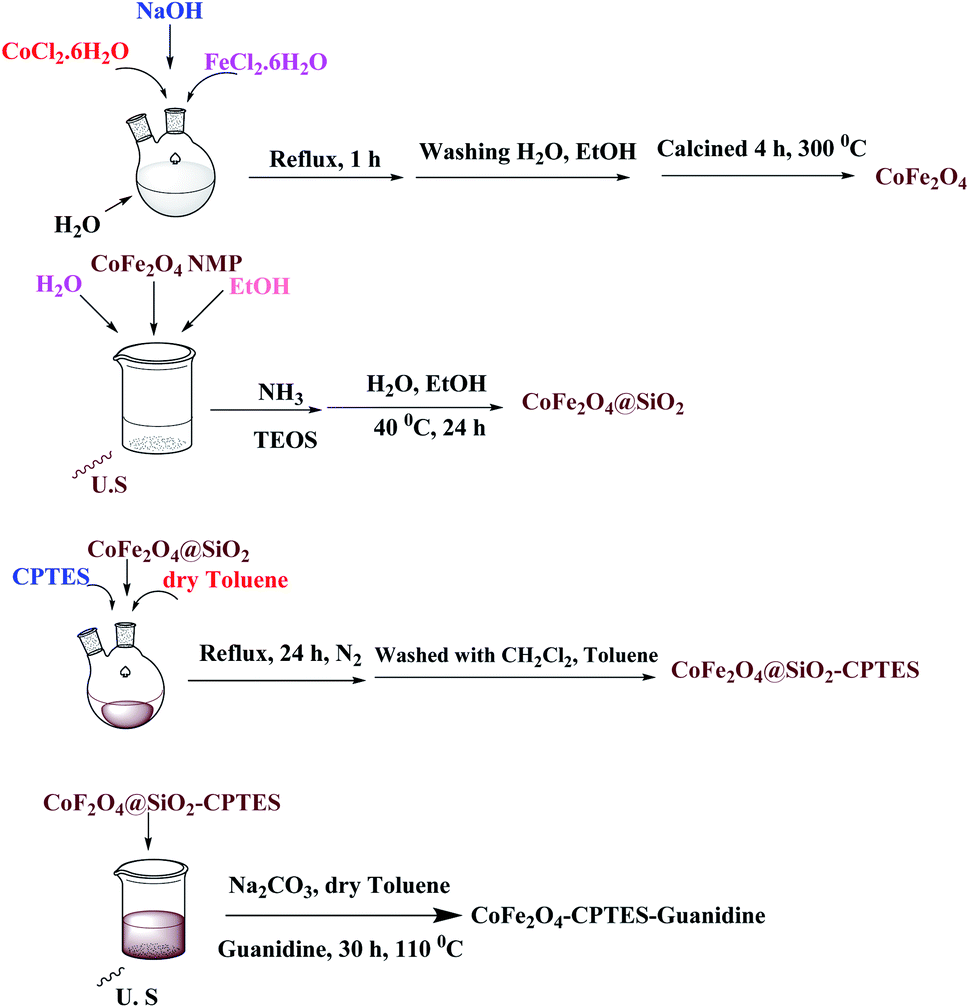 | ||
| Scheme 1 Synthesis of the CoFe2O4@SiO2-guanidine catalyst. | ||
The structure of the magnetic nanocatalyst was characterized by XRD as shown in Fig. 1. There are six diffraction peaks at 2θ about 30.26, 35.61, 43.24, 53.73, 57.16 and 62.72 corresponding to the (220), (311), (400), (422), (511) and (440) planes in the CoFe2O4 which is correspond to the standard pattern. As shown in Fig. 1, the positions of all peaks in the XRD pattern of CoFe2O4@SiO2-guanidine were according to standard XRD pattern of CoFe2O4.
 | ||
| Fig. 1 XRD spectrum of (a) CoFe2O4, (b) CoFe2O4@SiO2-guanidine. | ||
The morphology of the CoFe2O4@SiO2-guanidine magnetic nanocomposite was determined by SEM. The average size of nanoparticles on the support determined from Fig. 2 was about 54 nm.
 | ||
| Fig. 2 SEM images of CoFe2O4@SiO2-guanidine. | ||
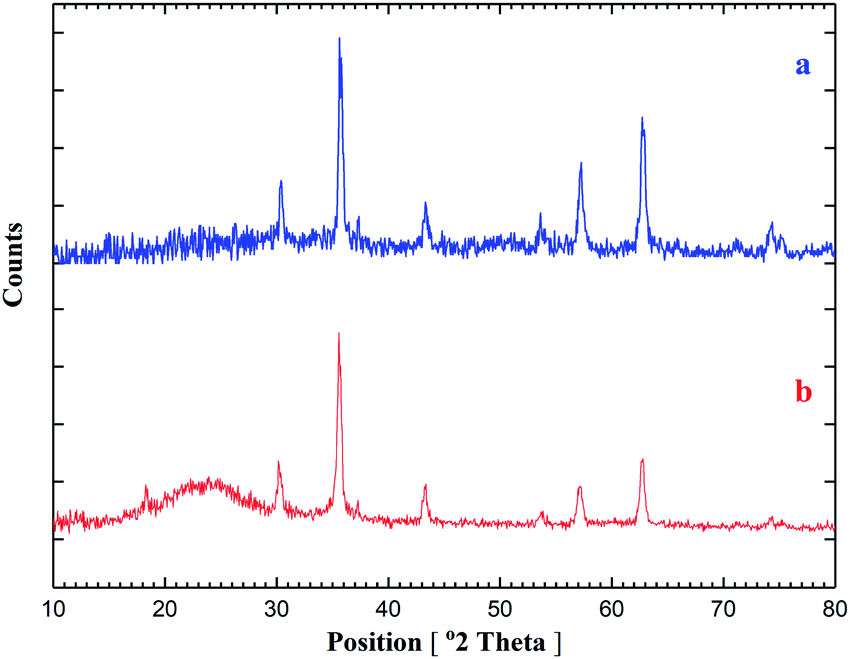
The EDX pattern shown in Fig. 3 confirms the signals for carbon, iron, cobalt, oxygen, nitrogen and silicon elements. The results of EDX, XRD and SEM for CoFe2O4@SiO2-guanidine nanocomposite confirm that nanoparticles were exactly fabricated on the surface.
 | ||
| Fig. 3 The EDX pattern of the CoFe2O4@SiO2-guanidine. | ||
The TEM image of the nanocomposite was shown in Fig. 4. This image displays the formation of CoFe2O4@SiO2-guanidine nanostructure that contains core–shell nature.
 | ||
| Fig. 4 The TEM image of the CoFe2O4@SiO2-guanidine. | ||
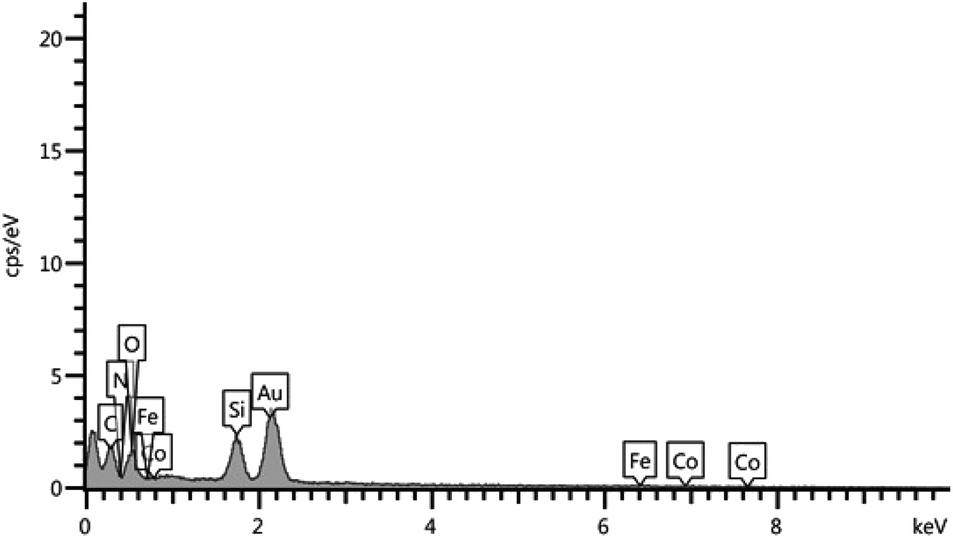
Furthermore, the Fig. 5a–d shows the FT-IR spectra of CoFe2O4 MNPs, CoFe2O4@SiO2, CoFe2O4@SiO2-CPTES and CoFe2O4@SiO2-guanidine, respectively. The FT-IR spectrum of CoFe2O4 MNPs shows peaks characteristic of Co–O at 603 cm−1 and the broad band at 3428 cm−1 is due to the O–H of adsorbed water molecule and silica surface (Fig. 5a). Fig. 1b shows the FT-IR spectrum of CoFe2O4@SiO2 MNPs. The band at 1089 cm−1 is attributed to the vibration of Si–O–Si bonds (Fig. 5b). Fig. 5c shows the FT-IR spectrum of the CoFe2O4@SiO2-CPTES. The FT-IR spectrum shows two peaks at 2924 and 2854 cm−1 which are related to C–H stretching vibrations. In comparison to CoFe2O4@SiO2-CPTES, the presence of new bands in CoFe2O4@SiO2-guanidine is observed and the guanidine is recognized by the presence of stretching vibrations of N–H bonds at 3402 cm−1. The stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds at 1664 cm−1 are indicated (Fig. 5d).
N bonds at 1664 cm−1 are indicated (Fig. 5d).
 | ||
| Fig. 5 FT-IR spectra of (a) CoFe2O4 nanoparticles, (b) CoFe2O4@SiO2, (c) CoFe2O4@SiO2-CPTES, (d) CoFe2O4@SiO2-guanidine. | ||
In Fig. 6, the TG analysis of CoFe2O4@SiO2-guanidine are displayed. The primary small weight loss about 5% below 200 °C which corresponds to the removal of physically adsorbed solvent and surface hydroxyl groups. The weight loss observed about 15% between 200–800 °C in TGA curve of CoFe2O4@SiO2-guanidine is mainly related to the decomposition of supported organic components on the nanoparticles (Fig. 6).
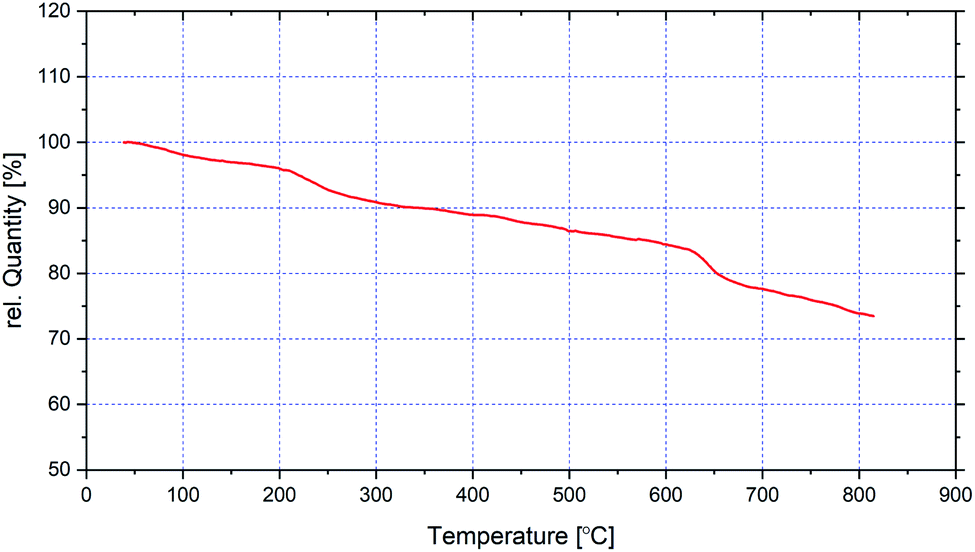 | ||
| Fig. 6 TG analysis of CoFe2O4@SiO2-guanidine. | ||
Magnetic properties of CoFe2O4 nanoparticles, CoFe2O4@SiO2-guanidine were investigated by VSM technique at room temperature. As shown in Fig. 7, MNPs to 15 emu g−1 in the final CoFe2O4@SiO2-guanidine catalyst. These consequences show that the magnetization property reduces via coating and functionalization surface of the CoFe2O4 nanoparticles. But the advantage of the nanocomposite is that can be easily separated by an external magnet from the mixture reaction after the catalytic reaction.
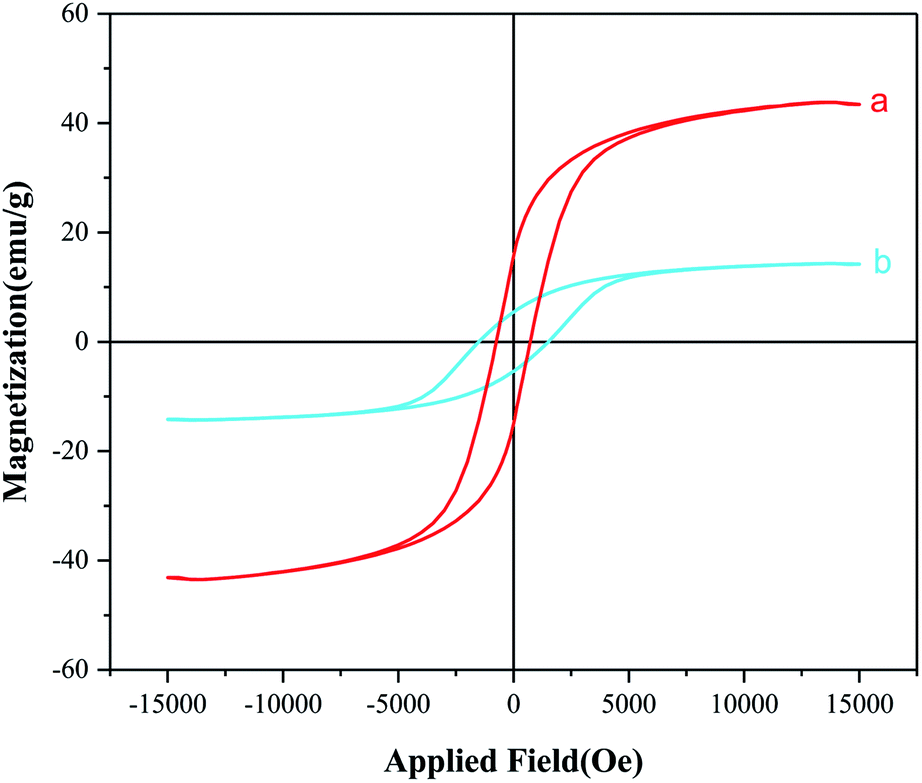 | ||
| Fig. 7 Magnetization curves for (a) CoFe2O4 and (b) CoFe2O4@SiO2-guanidine. | ||
Finally, the prepared CoFe2O4@SiO2-guanidine was used as heterogeneous catalyst in the reaction of isatin or 5-chloroisatin, malononitrile or cyanoacetic ester and 1,3-dicarbonyl compounds in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mole ratio under ultrasonic irradiation and water as a solvent (Scheme 2).
:1:1 mole ratio under ultrasonic irradiation and water as a solvent (Scheme 2).
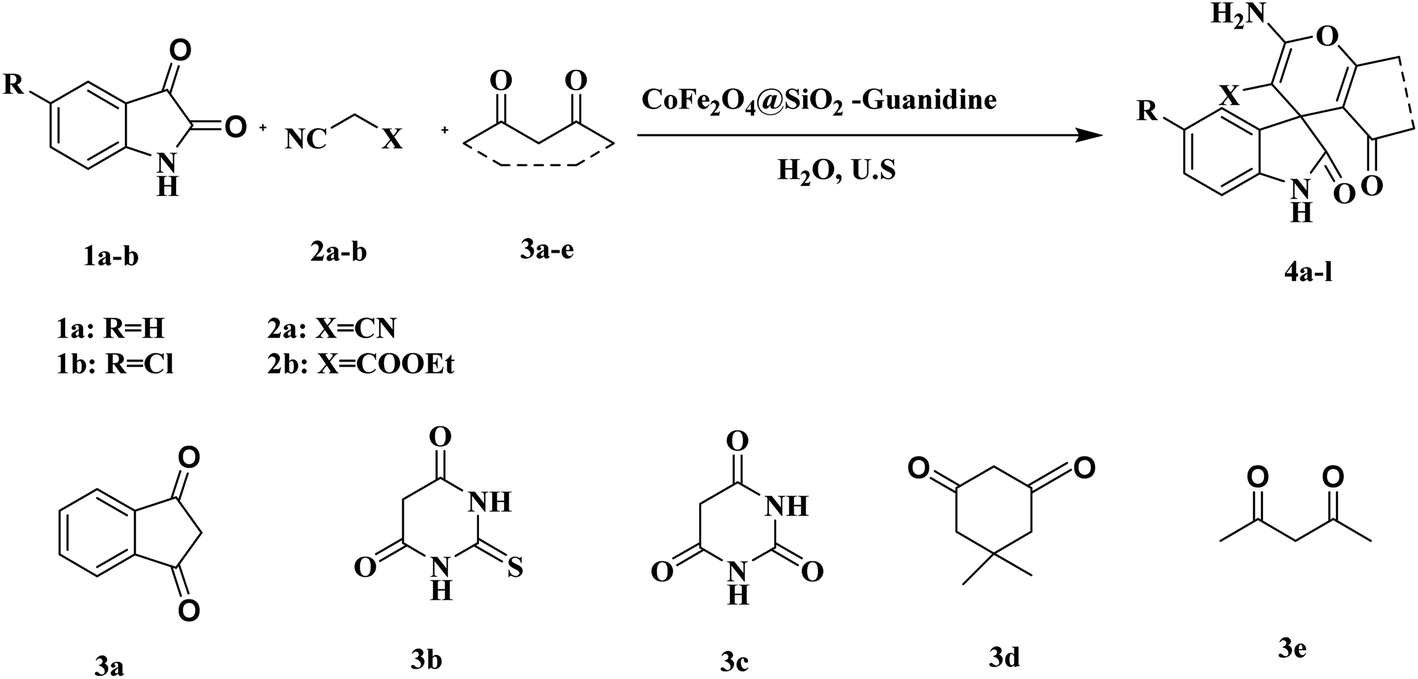 | ||
| Scheme 2 The preparation of spirooxindole derivatives using nanocomposites as catalyst. | ||
To optimize the reaction conditions in amount of catalyst, condensation of isatin (1 mmol), malononitrile (1 mmol), dimedone (1 mmol) was selected as a model reaction and studied under different conditions.
In order to obtain the optimal amount of the catalyst, the reaction was carried out in the presence of different amounts of catalyst under ultrasonic irradiation and water as a solvent (Table 1).
As can be seen in Table 1, the reaction was efficiently performed using 8 mg of the catalyst under ultrasonic irradiation and water as a solvent and the desired product was obtained in high yield within a short reaction time (Table 1, entry 5).
After the optimization of the catalyst amount, the reaction was carried out in water under ultrasonic irradiation at different powers (Table 2). In the same way, the highest yield of product was resulted from the reaction run under ultrasonic irradiation with a power of 40 W (Table 2, entry 3).
| Entry | Power (W) | Time (min) | Yield (%) |
|---|---|---|---|
| a Reaction condition: isatin (1 mmol), malononitrile (1 mmol), dimedone (1 mmol) under ultrasonic irradiation and water as a solvent. | |||
| 1 | 30 | 10 | 70 |
| 2 | 35 | 10 | 84 |
| 3 | 40 | 10 | 90 |
| 4 | 45 | 10 | 90 |
In continuation, the reaction of isatin (1 mmol), malononitrile (1 mmol), dimedone (1 mmol) and CoFe2O4@SiO2-guanidine (8 mg) as catalyst and power of 40 W was carried out in various solvents (Table 3). The best results were obtained using water as a solvent (Table 3, entry 2).
After optimization of the catalyst amount, ultrasonic power and optimization of solvents, synthesis of spirooxindole derivatives were carried out. The corresponding results are depicted in Table 4.
| Entry | Isatin | Nitril | Carbonyl compound | Product | Time (min) | Yieldb (%) |
|---|---|---|---|---|---|---|
| a Reaction condition: isatin/5-chloroisatin (1 mmol), malononitrile/cyanoacetic esters (1 mmol), 1,3-dicarbonyl compounds and CoFe2O4@SiO2-guanidine (8 mg) under ultrasound irradiation (40 W), water as a solvent condition.b Isolated yields. | ||||||
| 1 | 1a | 2a | 3a |  |
15 | 94 |
| 2 | 1a | 2a | 3b | 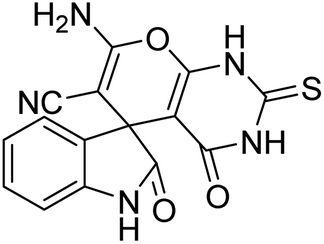 |
10 | 92 |
| 3 | 1a | 2a | 3c | 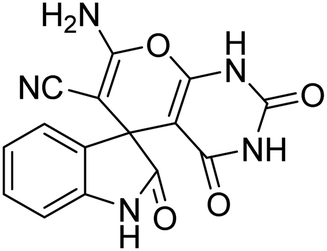 |
8 | 92 |
| 4 | 1b | 2a | 3b | 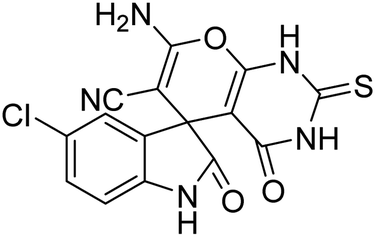 |
10 | 91 |
| 5 | 1b | 2a | 3c | 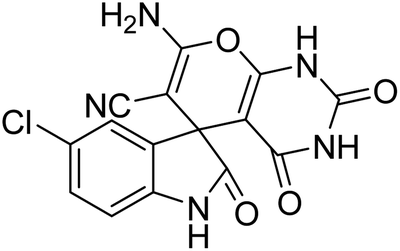 |
10 | 93 |
| 6 | 1b | 2a | 3a | 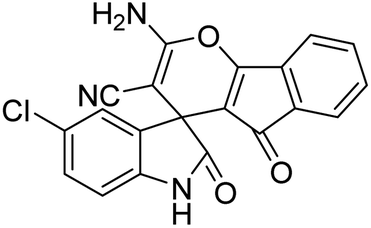 |
12 | 92 |
| 7 | 1b | 2b | 3d | 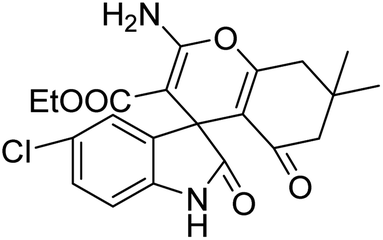 |
15 | 88 |
| 8 | 1a | 2b | 3d | 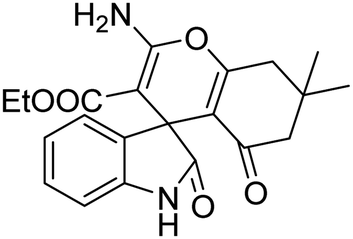 |
15 | 90 |
| 9 | 1b | 2a | 3d | 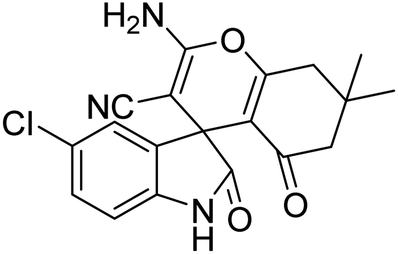 |
10 | 87 |
| 10 | 1b | 2a | 3e | 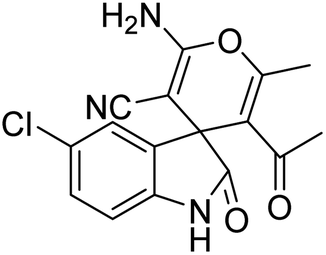 |
10 | 92 |
| 11 | 1a | 2a | 3d | 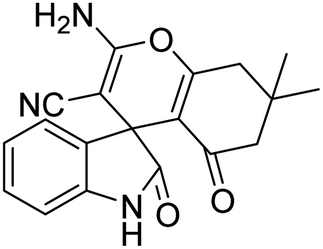 |
10 | 90 |
| 12 | 1a | 2a | 3e | 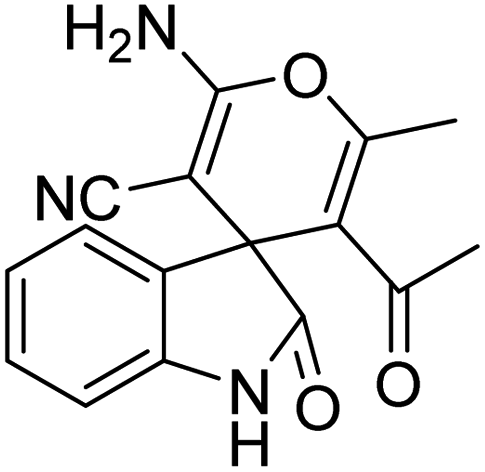 |
8 | 92 |
The present work was compared with the previously reported works for synthesis of spirooxindoles and the results are shown in Table 5. It was found that the present work was the better than the other works on the basis of reaction times, product yields and used catalyst amounts for the reactions (Table 5, entry 6 vs. entries 1–5).
In a plausible mechanism, at first, malononitrile is activated by catalyst and attacks to the carbonyl group of isatin and removes of H2O. Then, in second step, 1,3-dicarbonyl compounds is activated by catalyst and attacks to the α carbon atom of isatin. Then a tautomer is performed and the hydroxyl group attack on the carbon of nitrile and the final product is produced (Scheme 3).
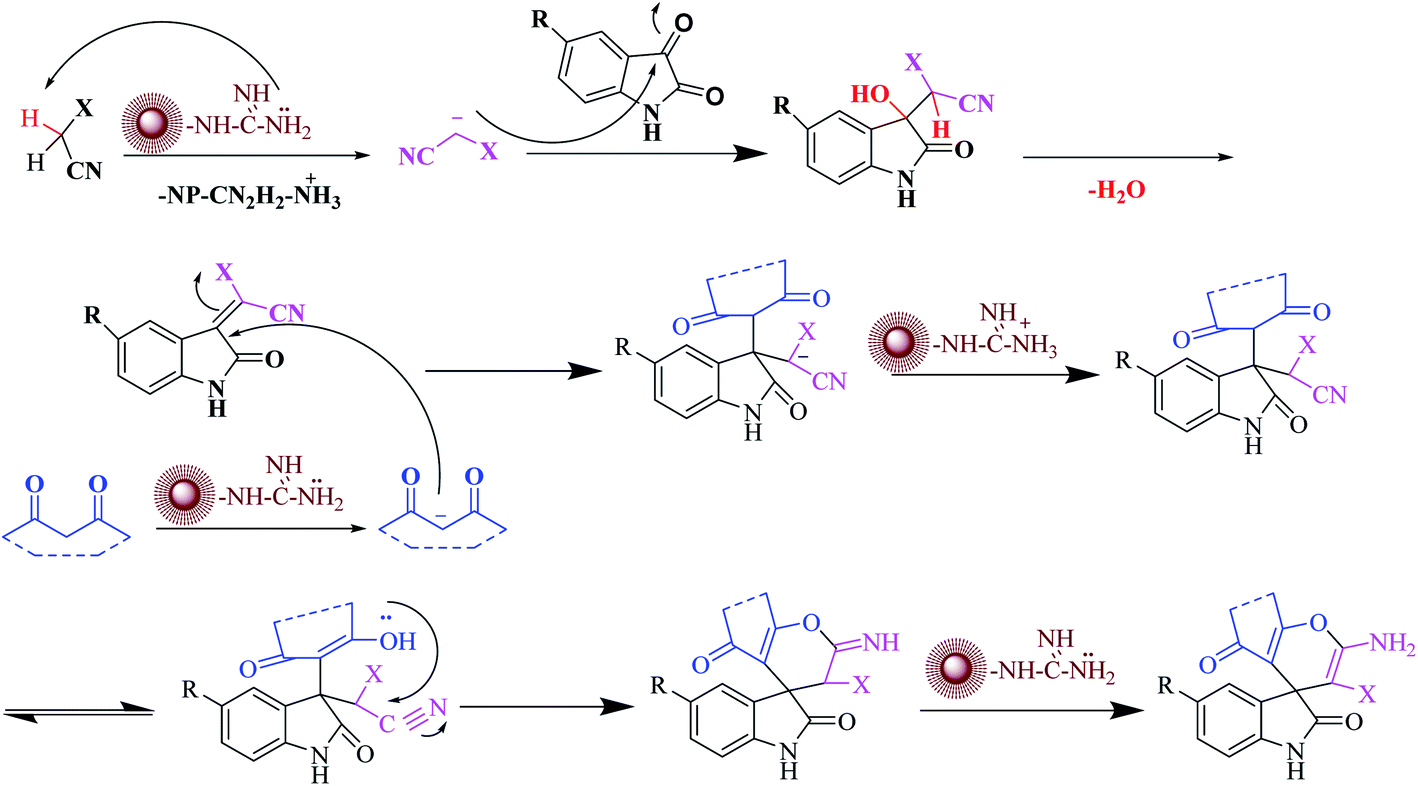 | ||
| Scheme 3 Proposed reaction mechanism for spirooxindole derivatives by CoFe2O4@SiO2-guanidine nanocomposite as a catalyst. | ||
3.2. Catalyst reusability
As shown in Fig. 8, it was investigated the reusability of CoFe2O4@SiO2-guanidine as heterogeneous catalyst for the reaction of isatin, malononitrile, indandion as a model reaction. After the completion of the reaction, the catalyst was separated from the reaction mixture using an external magnet and the residual catalyst was washed with ethyl acetate and dried in an oven at 50 °C. The recycled catalyst could be reused for three times without considerable loss of its catalytic activity.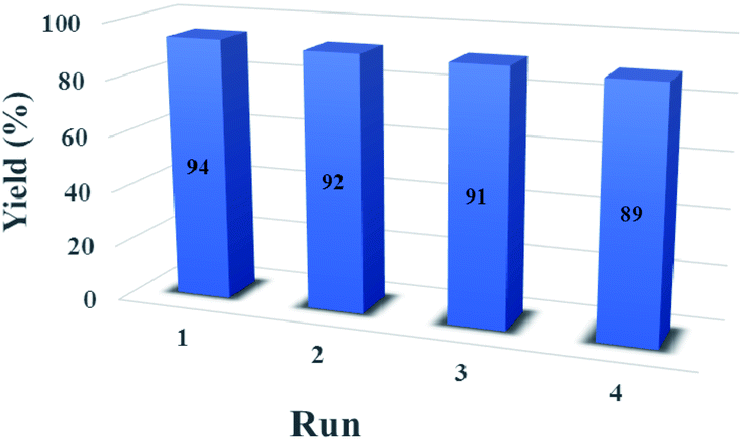 | ||
| Fig. 8 The reusability of the CoFe2O4@SiO2-guanidine catalyst. | ||
4. Conclusion
In summary, this paper proposed a new protocol for the synthesis of spirooxindole derivatives in the presence of CoFe2O4@SiO2-guanidine nanocomposites (core–shell) as a nanocatalyst in water as a solvent under ultrasonic irradiation. The main advantages of this present procedure are the easy recovery of catalyst, non-toxic, small amount of used catalyst and given products with high yields and short reaction times.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to University of Kashan for supporting this work by Grant No. 159148/85.References
- J. S. Bindra, The Alkaloides: Chemistry and Physiology, 1973, vol. 14, pp. 83–121 Search PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748 CrossRef CAS PubMed.
- P. B. Alper, C. Meyers, A. Lerehner, D. R. Siegel and E. M. Carreira, Angew. Chem., Int. Ed., 1999, 38, 3186 CrossRef CAS.
- W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, Y. Wang, J. Zhao, S. Jia, J. Herich, D. Labreque, R. Storer, K. Meerovitch, D. Bouffard, R. Rej, R. Denis, C. Blais, S. Lamothe, G. Attardo, H. Gourdeau, B. Tseng, S. Kasilbhatla and S. X. Cai, J. Med. Chem., 2004, 47, 6299 CrossRef CAS.
- A. H. Abdel-Rahman, E. M. Keshk, M. A. Hanna and Sh. M. El-Bady, Bioorg. Med. Chem., 2004, 12, 2483 CrossRef CAS PubMed.
- S. Riyaz, A. Naidu and P. K. Dubey, Lett. Org. Chem., 2012, 9, 101–105 CrossRef CAS.
- M. Kidwai, A. Jahan and N. K. Mishra, Appl. Catal., A, 2012, 425–426 Search PubMed.
- G. Shanthi, G. Subbulakshmi and P. T. Perumal, Tetrahedron, 2007, 63, 2057–2063 CrossRef CAS.
- L. M. Wang, N. Jiao, J. Qiu, J. J. Yu, J. Q. Liu, F. L. Guo and Y. Liu, Tetrahedron, 2010, 66, 339–343 CrossRef CAS.
- G. S. Hari and Y. R. Lee, Synthesis, 2010, 3, 453–464 Search PubMed.
- F. Kiani and H. Naeimi, Ultrason. Sonochem., 2018, 48, 267–274 CrossRef CAS PubMed.
- H. Naeimi and R. Shaabani, Ultrason. Sonochem., 2017, 34, 246–254 CrossRef CAS PubMed.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- T. J. Mason, Ultrason. Sonochem., 2003, 10, 175–179 CrossRef CAS.
- C. L. Ni, X. H. Song, H. Yan, X. Q. Song and R. G. Zhong, Ultrason. Sonochem., 2010, 17, 367–369 CrossRef CAS PubMed.
- K. S. Suslick, S. B. Choe, A. A. Cichowlas and M. W. Grinstaff, Nature, 1991, 353, 414 CrossRef CAS.
- S. J. Oldenberg, R. D. Averitt, S. L. Westcott and N. Halas, J. Phys. Chem. Lett., 1998, 288, 243 CrossRef.
- M. Shaker and D. Elhamifar, New J. Chem., 2020, 44, 3445–3454 RSC.
- M. Norouzi and D. Elhamifar, Composites, Part B, 2019, 176, 107308 CrossRef CAS.
- M. Neysi, A. Zarnegaryan and D. Elhamifar, New J. Chem., 2019, 43, 12283–12291 RSC.
- M. Norouzi, D. Elhamifar and R. Mirbagheri, Microporous Mesoporous Mater., 2019, 278, 251–256 CrossRef CAS.
- R. Mirbagheri and D. Elhamifar, J. Alloys Compd., 2019, 790, 783–791 CrossRef CAS.
- Z. Ramazani, D. Elhamifar, M. Norouzi and R. Mirbagheri, Composites, Part B, 2019, 164, 10–17 CrossRef CAS.
- S. Abaeezadeh, D. Elhamifar, M. Norouzi and M. Shaker, Appl. Organomet. Chem., 2019, 33, e4862 CrossRef.
- P. H. Li, B. L. Li, Z. M. An, L. P. Mo, Z. S. Cui and Z. H. Zhang, Adv. Synth. Catal., 2013, 355, 2952–2959 CrossRef CAS.
- L. Heidari and L. Shiri, Appl. Organomet. Chem., 2019, 33, 4636 CrossRef.
- R. Jamatia, A. Gupta and A. K. Pal, RSC Adv., 2016, 6, 20994–21000 RSC.
- D. R. Chandam, A. G. Mulik, D. R. Patil and M. B. Deshmukh, Res. Chem. Intermed., 2016, 42, 1411–1423 CrossRef CAS.
- A. Khalafi-Nezhad, E. Shaikhi-Shahidzadeh, S. Sarikhani and F. Panahi, J. Mol. Catal. A: Chem., 2013, 379, 1–8 CrossRef CAS.
- Y. Li, H. Chen, C. Shi, D. Shi and S. Ji, J. Comb. Chem., 2010, 12, 231–237 CrossRef CAS PubMed.
- F. Mohamadpour, M. T. Maghsoodlou, M. Lashkari, R. Heydari and N. Hazeri, Org. Prep. Proced. Int., 2019, 51, 456–476 CrossRef CAS.
- M. Esmaeilpour, J. Javidi and M. Divar, J. Magn. Magn Mater., 2017, 423, 232–240 CrossRef CAS.
- G. M. Ziarani, A. Badiei, S. Mousavi, N. Lashgari and A. Shahbazi, Chin. J. Catal., 2012, 33, 1832–1839 CrossRef.
- A. Dandia, V. Parewa, A. Kumar Jaina and K. S. Rathore, Green Chem., 2011, 13, 2135 RSC.
- B. Karmakar, A. Nayak and J. Banerji, Tetrahedron Lett., 2012, 53, 5004–5007 CrossRef CAS.
- S. Pradhan and B. G. Mishra, J. Mol. Catal., 2018, 446, 58–71 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00967b |
| This journal is © The Royal Society of Chemistry 2021 |