DOI:
10.1039/D1RA00865J
(Paper)
RSC Adv., 2021,
11, 23365-23373
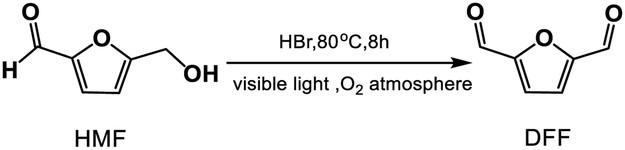
Highly efficient and tunable visible-light-catalytic synthesis of 2,5-diformylfuran using HBr and molecular oxygen†
Received
1st February 2021
, Accepted 12th May 2021
First published on 2nd July 2021
Abstract
This paper discloses that inexpensive hydrobromic acid (HBr) is active and highly selective to the photo-oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-diformylfuran (DFF) with dioxygen (O2) or even with water under visible light illumination, which can achieve the highest 89.1% DFF yield in DMSO at 80 °C under pure O2 atmosphere. More importantly, under bifunctional acid-photooxidation catalysis of HBr, fructose can be directly converted to DFF and its two-step cascade conversion in DMSO provides a far higher DFF yield (80.2%) than the one-step cascade conversion in MeCN (42.1%). The results of HMF photooxidation catalyzed by hydrohalic acids, free radical quenching tests and EPR spectrum support that the Br atom and superoxide (O2−˙) anion radicals generated by HBr photolysis in O2 are active species for the oxidation of HMF to DFF and their activities are adjusted by the reaction medium. This photo-synthetic protocol is very simple and practical, especially with low operating costs, showing a good industrial application prospect.
Introduction
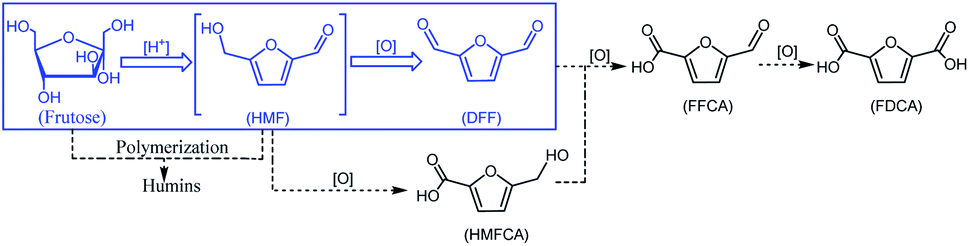
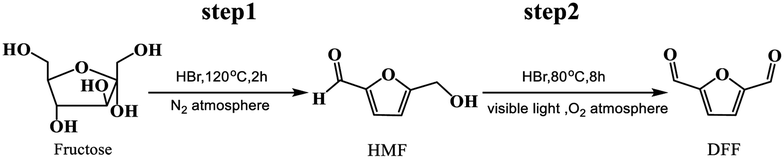
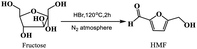
With growing concerns about diminishing fossil, global warming and environmental pollution, the development of new and clean technical routes to produce chemicals from renewable and abundant biomass has attracted worldwide attention.1–5 5-Hydroxymethylfurfural (HMF), a “bridge” molecule, has been widely recognized as one of the top bio-based platform chemicals in the future chemical industry, not only its derivatives can serve as fuels, but can also replace oil-based building blocks in the production of polymers.6,7 2,5-diformylfuran (DFF), one of the HMF-derived oxygenated products, also is an important platform chemical and has been used to synthesize many useful compounds and new polymer materials through hydrogenation, oxidation, polymerization, hydrolysis and other chemical reactions. These DFF-derived chemicals have extensive potential applications in medicine, furan-based biopolymers, organic conductors, fluorescent whitening agents and macrocyclic ligands.8–10 In general, DFF is easily synthesized in high yield by direct selective oxidation of HMF under thermo-catalytic conditions, and the reported some homogeneous11,12 or heterogeneous13 catalysts are very efficient for this selective oxidation. In addition to the traditional thermo-catalytic process, some new technologies, such as bio-catalysis14 and electro-catalysis,15,16 have been applied for this selective oxidation. However, it is hard to recover the used homogeneous catalyst from the reaction system. In addition, this synthesis protocol is still limited to the fact that HMF is not readily available and it is easy to perform polycondensation at high concentration.17 Therefore, the direct synthesis of DFF from easily available carbohydrates such as fructose and glucose is highly appreciated and has received widespread attention.18–21 The direct synthesis of DFF from fructose, as shown in Scheme 1, is a two-step tandem reaction that typically involves fructose dehydration to HMF catalyzed by an acid22–24 and subsequent HMF oxidation to DFF catalyzed by a redox catalyst.25–27 It is expected that this direct synthesis protocol for the fructose-derived DFF, the above HMF polycondensation is significantly reduced due to its very low concentration in situ.
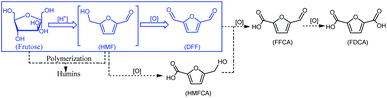 |
| | Scheme 1 Synthesis of DFF from fructose. | |
Some efficient catalytic protocols using molecular oxygen as an oxidant, which are summarized in Table 1, have been developed to directly synthesize DFF from fructose by one-pot, two-steps28–36 or one-step,37–43 affording medium to high DFF yields of 49–87%. However, these synthesis protocols usually employ relatively complex catalysts and high reaction temperatures above 110 °C. The two-step synthesis protocol usually requires the stepwise addition of acidic and oxidative catalysts in order to improve the DFF yield. The use of the toxic V-containing Cs3HPMo11V1O40, α-CuV2O6, PMo11V1, and PMoV2@CP-3.5-400 catalysts does not conform to the current subject of environmental protection. The metal-containing catalysts often give a relatively high DFF yield, such as f-Ce9Mo1Oδ, Au0.5Ru2.5/rGO, MoO3–ZrO2 and Co–Al hydrotalcites. However, the transition metal and especially precious-metal-based catalysts applied in tandem transformations are usually expensive and contaminative. Some metal-free catalysts such as GO, NaBr, GN-NS and CC-SO3H–NH2 are appreciated, but a high reaction temperature (140–150 °C) and long reaction time (19–24 h) are required to achieve medium DFF yields, thus leading to large energy consumption in industrial production. These defects described above are hampering their future industrial applications. Therefore, it is still highly desirable to develop a direct synthesis protocol of DFF from readily available fructose, O2 and metal-free catalysts under mild conditions.
Table 1 One-pot synthesis of 2,5-diformylfuran from fructose using various methods
| Catalyst |
Procedure |
Reaction conditions |
DFF yield (%) |
Ref. |
| Amberlyst-15 and Ru/HT |
Two-step |
Step 1: 100 °C, 3 h, O2 (20 mL min−1) with Amberlyst-15 |
49.0 |
28 |
| Step 2: 100 °C, 6 h, O2 (20 mL min−1) with Ru/HT |
| Cs3HPMo11V1O40 |
Two-step |
Step 1: 110 °C, 2 h, N2 (0.1 MPa) |
60.0 |
29 |
| Step 2: 120 °C, 6 h, O2 (0.1 MPa) |
| H2SO4/V-CP |
Two-step |
Step 1: 140 °C, 20 min, air with H2SO4 |
68.4 |
30 |
| Step 2: 140 °C; 6 h, O2 (40 mL min−1) with V-CP |
| CC-SO3H–NH2 |
Two-step |
Step 1: 140 °C, 2 h, N2 atmosphere |
69.0 |
31 |
| Step 2: 140 °C, 17 h, O2 (20 mL min−1) |
| GO |
Two-step |
Step 1: 140 °C, 2 h, N2 (20 mL min−1) |
72.5 |
32 |
| Step 2: 140 °C, 22 h, O2 (20 mL min−1) |
| f-Ce9Mo1Oδ |
Two-step |
Step 1: 120 °C, 2 h, N2 (10 mL min−1) |
74.0 |
33 |
| Step 2: 120 °C, 10 h, O2 (10 mL min−1) |
| Fe3O4-SBA-SO3H and K-OMS-2 |
Two-step |
Step 1: 110 °C, 2 h, air with Fe3O4-SBA-SO3H |
80.0 |
34 |
| Step 2: 110 °C, 6 h, O2 (10 mL min−1) with K-OMS-2 |
| Au0.5Ru2.5/rGO |
Two-step |
Step 1: 110 °C, 2 h, air with visible light |
86.0 |
35 |
| Step 2: 110 °C; 6 h, O2 (5 bar) with visible light |
| Co–Al hydrotalcites |
Two-step |
Step 1: 120 °C, 2 h, N2 (1 bar) |
87.0 |
36 |
| Step 2: 120 °C; 8 h, O2 (3 bar) |
| P(EVPI-Br)/α-CuV2O6 |
One-step |
135 °C, 3.5 h, O2 (1 bar) |
63.1 |
37 |
| NaBr |
One-step |
150 °C, 23 h, O2 atmosphere |
67.0 |
38 |
| GN-NS |
One-step |
150 °C, 25 h, O2 (20 mL min−1) |
70.3 |
39 |
| MoO3–ZrO2 |
One-step |
150 °C, 10 h, O2 (20 mL min−1) |
74.0 |
40 |
| Cs0.5H2.5PMo12 |
One-step |
160 °C, 2 h, air |
69.3 |
41 |
| PMA-MIL-101 |
One-step |
150 °C, 7 h, O2 (20 mL min−1) |
75.1 |
42 |
| PMoV2@CP-3.5-400 |
One-step |
120 °C, 4 h, O2 atmosphere |
87.3 |
43 |
| HBr |
One-step |
50 °C, 12 h, O2 (1 atm), light, MeCN |
42.1 |
This work |
| HBr-Amberlyst-15 |
Two-step |
Step 1: 120 °C, 2 h, air, Amberlyst-15, DMSO |
79.7 |
| Step 2: 80 °C, O2 (1 atm), 8 h, visible light with HBr |
| HBr |
Two-step |
Step 1: 120 °C, 2 h, N2 atmosphere, DMSO |
80.2 |
| Step 2: 80 °C, 8 h, O2 (1 atm), visible light |
Photocatalysis techniques have received enormous attention during recent decades because of their potential applications in environmental treatment44,45 and the synthesis of fuel and fine chemicals.46–48 Semiconductor materials have outstanding advantages in cost, stability and ease of fine-tuning optical properties,49,50 which have been widely used in heterogeneous photo-catalysis processes. Among them, g-C3N4, Nb2O5, and TiO2 have been reported to efficiently photo-catalyze HMF oxidation with O2, achieving ca. 52–91% conversion of HMF and 16–40% yield to DFF51–53 but their heterogeneous photocatalytic properties usually give relatively low quantum efficiency and are accompanied by serious by-reactions, such as deep oxidation,53 which results in their photocatalytic efficiency is much lower than most of the above thermal catalytic systems. Hydrobromic acid (HBr) is not only an inexpensive and easy to handle strong acid catalyst but is also a good homogeneous photo-catalyst, which can efficiently achieve the aerobic oxidation of alcohols to the corresponding aldehydes under visible light.54–56 We suggest that HBr combined O2 and visible light may construct a very simple and practical synthesis protocol for achieving the direct transformation of fructose to DFF under relatively mild conditions. Here, we report the initial results obtained using this photoreaction protocol.
Experimental
Reagents
The main reagents used in this work were of analytical grade, which included hydrobromic acid (40%, HBr), hydrochloric acid (40%, HCl), hydroiodic acid (47.0%, HI), commercial Amberlyst-15 (SO3H density, 5 mmol g−1, diameter in a range of 0.15–0.053 mm), fructose (C6H12O6), 5-hydroxymethyl furfural (HMF), acetonitrile (MeCN), dimethyl sulfoxide (DMSO, dehydrated with molecular sieve 3A), N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA). Distilled water was used throughout the experiment.
Aerobic oxidation of HMF to DFF
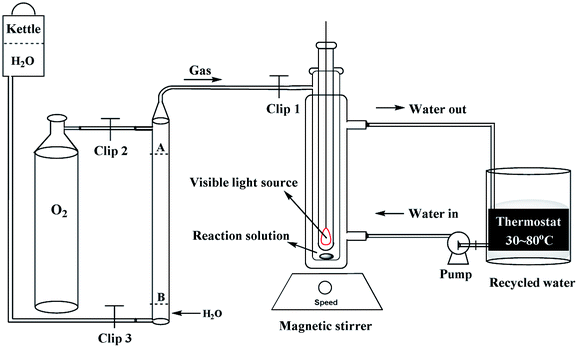
The photoreaction for the synthesis of DFF from HMF was carried out in a self-assembly photo-reactor equipped with a constant temperature water circulation device and an oxygen storage vessel (1 atm, see Fig. 1). A 35 W tungsten-bromine lamp (with the UV light filter, light intensity, 535 mW cm−2) was used as a built-in light source. The whole photocatalytic reactions were operated in the closed reactor under constant temperature and normal pressure. Briefly, HMF (18.0 mg, 0.1 mmol), HBr (40 wt%, 0.3 mmol) and DMSO or MeCN (5 mL) were mixed in the self-assembly photo-reactor. The mixture was incubated at 80 °C (DMSO solvent, 8 h) or 50 °C (MeCN solvent, 12 h) with continuous stirring under visible light illumination and O2 atmosphere. After the reaction, the mixture was gradually cooled to room temperature.
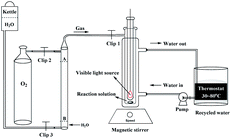 |
| | Fig. 1 Schematic diagram of the photoirradiation reaction system. | |
Direct synthesis of DFF from fructose by one step or two steps
One step. Direct synthesis of DFF from fructose by one step was carried out using the above photo-reaction device. In a typical reaction, fructose (18 mg, 0.1 mmol), HBr (40 wt%, 2.5 mmol), water (0.3 mL) and MeCN (5 mL) were mixed in the photo-reactor and the mixture was incubated at 50 °C for 12 h under continuous stirring under visible light illumination and O2 atmosphere. After the reaction, the mixture was gradually cooled to room temperature.
Two steps. Dehydration of fructose to HMF was carried out in a two-necked flask under heating and stirring conditions. In a typical reaction, fructose (306 mg, 1.7 mmol), HBr (40 wt% 0.085 mmol) or Amberlyst-15 (0.02 g), and DMSO (10 g) were added to a two-necked flask (25 mL) equipped with a magnetic stirrer bar. The mixture was heated at 120 °C with vigorous stirring for 2 h. After the reaction, the mixture was treated with activated carbon (AC, 4.2 g) at 80 °C for 4 h, then filtered and diluted with DMSO to obtain 0.02 M HMF solution. The following photo-catalytic oxidation conditions for the synthesis of DFF from fructose-derived HMF were the same as described in Aerobic oxidation of HMF to DFF.
Analysis of products
The quantitative analysis of fructose-derived products was performed on a high-performance liquid chromatography (Alltech1500 HPLC) instrument equipped with a C18 AQ column and an ultraviolet detector at 280 nm. The mixture of acetonitrile and water (containing 0.1 wt% acetic acid) whose volume ratio was 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40, was used as the mobile phase at a rate of 0.5 mL min−1, and column oven temperature was maintained at 30 °C. The analyses were done in triplicate to get more accurate results. The obtained results were shown as average values ± standard deviations. The yield of products are defined as follows:
:40, was used as the mobile phase at a rate of 0.5 mL min−1, and column oven temperature was maintained at 30 °C. The analyses were done in triplicate to get more accurate results. The obtained results were shown as average values ± standard deviations. The yield of products are defined as follows:
| Yield of HMF (%) = (mole of HMF produced)/(mole of starting fructose) × 100% |
| Yield of DFF (%) = (mole of DFF produced)/(mole of starting fructose or HMF) × 100% |
Results and discussion
Aerobic oxidation of HMF to DFF under different conditions
Table 2 lists the data for the synthesis of DFF from HMF under various reaction conditions. Entry 1 shows that the HBr-based photo-synthetic protocol was very efficient at the photo-oxidation of HMF to DFF and could achieve 90.8% HMF conversion and up to 89.1% DFF yield under the optimal standard reaction conditions. The data listed in parentheses of entry 1 illuminate that dry HBr gas dissolved in anhydrous DMSO also gave almost the same photo-catalytic efficiency as that of 40% HBr solution. Two control experiments illuminate that HMF photo-oxidation could not proceed in the absence of HBr (entry 2) or without visible light irradiation (entry 3), but the consumption (or conversion) of HMF caused by its polymerization was up to 37.4 and 7.8%, respectively. Entries 4 and 5 further show that HF and HCl were not active for the photo-oxidation of HMF, except that compared to entry 2, HMF consumption was slightly reduced by 5–8%. HI only achieved a very low 4.4% DFF yield and 4.6% HMF consumption (entry 6). These findings support that HBr and visible light irradiation are the key influencing elements for the photo-oxidation of HMF. On the other hand, when the photo-oxidation of HMF hardly proceeds, its polymerization side reaction occurs easily and becomes more serious under light irradiation than under the darkness, but this side reaction can be gradually and efficiently restrained with a change of adding acid from HF, HCl to HBr or HI. Notably, the selective oxidation of HMF photocatalyzed by HBr also proceeded well under air, even N2 atmosphere, providing 73.3 and 71.8% DFF yields, respectively (entries 7 and 8). The above experimental results strongly support that the active Br atom generated by HBr photolysis likely plays a vital role in achieving HMF oxidation with O2. The introduced water may provide an efficient oxygen source for the HBr-catalyzed HMF photooxidation under the N2 atmosphere. Entries 9 and 10 show that using DMA or especially DMF instead of DMSO as a solvent, the yield of DFF was drastically reduced to 5.6 and 0.4%, respectively, along with a severe polymerization side reaction. Even if only adding 0.02 mmol DMF or DMA in entry 1 also resulted in DFF yield decreasing by 6 or 10% (entries 11 and 12). Notably, HBr was very active in the MeCN medium and could efficiently function at a mild temperature of 50 °C and even 25 °C although DFF yield decreased to 56.7% (entry 13) and 40.3% (entry 14), respectively, owing to severe polymerization of HMF. The HBr-based photo-synthetic protocol in MeCN could realize a one-step cascade conversion of fructose, affording 42.1% DFF yield under optimal conditions (entry 15 and Fig. S1†). But it could not proceed in DMSO even at 80 °C. These findings support that the photocatalytic performances of HBr in the synthesis of DFF from HMF and fructose are drastically affected by solvents. Fig. S2† illuminates that the UV-vis spectral curve of HBr in MeCN and especially DMSO solvents clearly displays a tail extending out into the visible region of 400–550 nm, but its absorption in this visible region was negligible in DMF and DMA solvents, supporting that the photolysis of HBr can proceed more effectively in MeCN and DMSO than in DMA and DMF. On the other hand, the Br atom radicals generated by HBr photolysis may be very active in MeCN, but they become relatively stable in DMSO and even inert in DMA and DMF. Additionally, the HBr-catalyzed fructose dehydration to HMF is drastically affected by reaction media, HBr in MeCN shows a very high acid-catalytic activity for this dehydration at even room temperature (Table S1†), accompanied by the polycondensation of product HMF, but this dehydration catalyzed by HBr cannot occur in DMSO at even 80 °C (see the following Table 3).
Table 2 Synthesis of DFF from HMF photo-oxidation under different conditions
|
| Entry |
Change from standard conditions |
Conversion of HMF (%) |
Yield of DFF (%) |
| Standard reaction condition: HMF, 0.1 mmol; 40% HBr, 0.3 mmol; DMSO, 5 mL; O2, 1 atm; temperature, 80 °C; time, 8 h. Using dry HBr gas dissolved in anhydrous DMSO. HMF, 0.1 mmol; 40% HBr, 0.2 mmol; MeCN, 5 mL; O2, 1 atm; time, 12 h. Fructose, 0.1 mmol; 40% HBr, 2.5 mmol; H2O, 0.3 mL; MeCN, 5 mL; O2, 1 atm; temperature, 50 °C; time, 12 h. Fructose, 0.1 mmol; 40% HBr, 0.3 mmol; DMSO, 5 mL; temperature, 80 °C; time, 8 h; O2, 1 atm. Yield of HMF. |
| 1a |
Standard |
90.8(89.1)b |
89.1(88.6)b |
| 2 |
Without HBr |
37.4 |
0 |
| 3 |
Without light illumination |
7.8 |
0 |
| 4 |
HF instead of HBr |
32.4 |
0 |
| 5 |
HCl instead of HBr |
29.3 |
0 |
| 6 |
HI instead of HBr |
4.6 |
4.4 |
| 7 |
Air instead of O2 |
75.2 |
73.3 |
| 8 |
N2 instead of O2 |
74.0 |
71.8 |
| 9 |
DMA instead of DMSO |
41.0 |
5.6 |
| 10 |
DMF instead of DMSO |
39.6 |
0.4 |
| 11 |
Adding 0.2 mmol DMA |
83.9 |
83.8 |
| 12 |
Adding 0.2 mmol DMF |
78.9 |
78.8 |
| 13c |
MeCN instead of DMSO at 50 °C |
83.4 |
56.7 |
| 14c |
MeCN instead of DMSO at 25 °C |
56.4 |
40.3 |
| 15d |
Fructose in MeCN at 50 °C |
10.3f |
42.1 |
| 16e |
Fructose in DMSO at 80 °C |
0f |
0 |
Table 3 Dehydration of fructose to HMF under different conditionsa
|
| Entry |
HBr (mmol) |
Temperature (°C) |
Time (h) |
Yield of HMF (%) |
| Reaction condition: fructose, 1.7 mmol; DMSO, 10 g. 0.7% yield of DFF was detected out. Using 20 mg Amberlyst-15 as a catalyst. |
| 1 |
0.340 |
100 |
2 |
66.9 |
| 2 |
0.170 |
100 |
2 |
80.4 |
| 3 |
0.085 |
100 |
2 |
86.9 |
| 4 |
0.085 |
100 |
4 |
90.4 |
| 5b |
0.085 |
100 |
6 |
87.8 |
| 6 |
0.340 |
120 |
2 |
50.9 |
| 7 |
0.170 |
120 |
2 |
76.2 |
| 8 |
0.085 |
120 |
2 |
95.4 |
| 9 |
0.085 |
80 |
4 |
0 |
| 10c |
Amberlyst-15 |
120 |
2 |
99.6 |
Effect of various variables
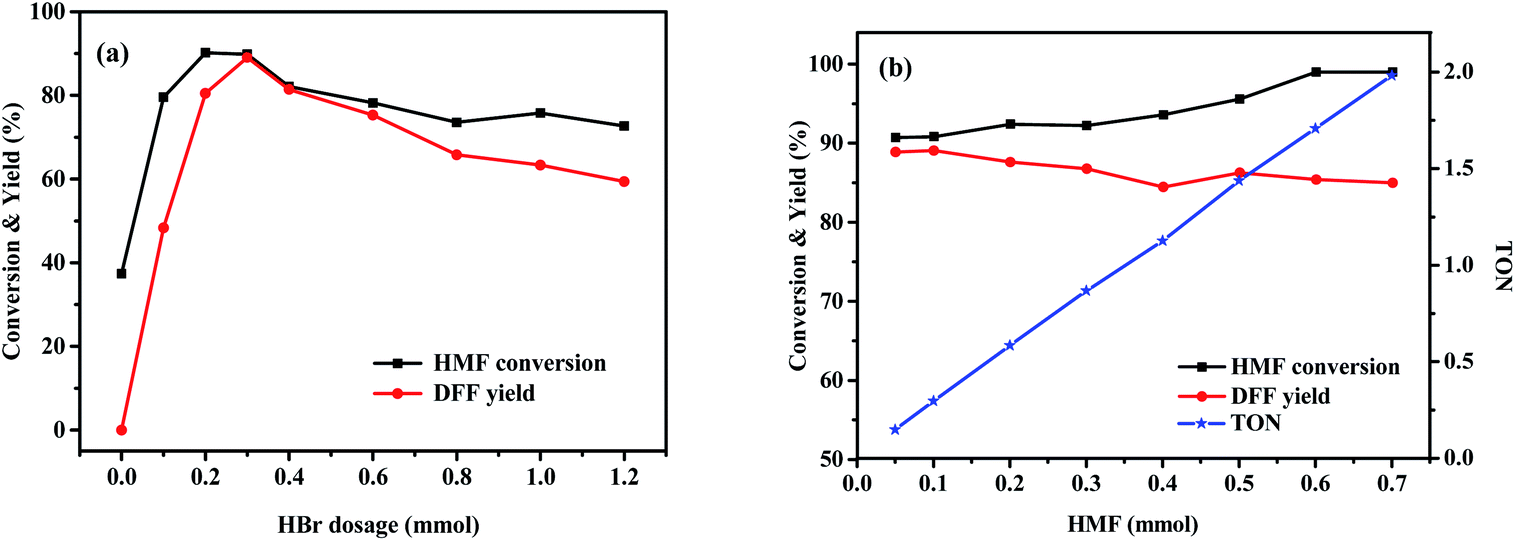
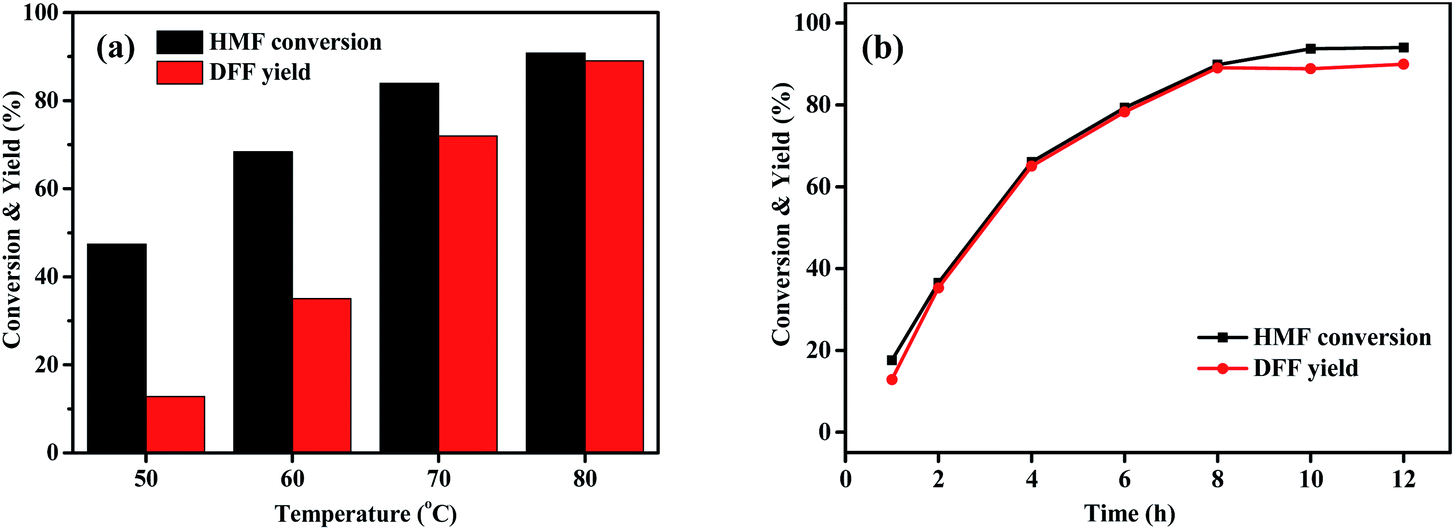
The effects of various variables such as HBr and HMF dosages, temperature and time on the photo-oxidation of HMF by O2 were further investigated using the best DMSO as a solvent and the obtained results are shown in Fig. 2 and 3. Fig. 2a shows that when the amount of HBr was enhanced from 0 to 0.3 mmol, HMF conversion and DFF yield gradually and drastically increased from 0 to 90.8% and from 0 to 89.1%, respectively, which was indicative of an excellent photo-catalytic function of HBr. Attempting to further increase the amount reversely resulted in a significant decrease in photo-oxidation efficiency (73.5% HMF conversion and 59.4% DFF yield), which may be due to the following reasons: based on a fact that, HMF conversion at a high HBr amount was 14% higher than DFF yield, we propose that the polymerization side reaction of HMF occurs more easily under the excessive use of HBr57,58 and the by-products seem to slow down the photo-oxidation based on the subsequent experiments. On the other hand, the control test of adding NaBr to the standard photoreaction system, as shown in Table S2,† supports that the introduction of excessive Br− ions play an inhibiting role in the photo-oxidation of HMF to DFF. Fig. 2b shows that DFF yield was almost unchanged (about 89.0%) when HMF dosage was enhanced from 0.05 to 0.10 mmol. Thereafter, it gradually and slightly reduced with increasing HMF amount to 0.4 mmol and then stayed almost 86% when HMF amount was between 0.4 and 0.6 mmol. However, HMF conversion always increased continuously from 90.8 to 99.6% with enhancing HMF amounted from 0.05 to 0.6 mmol. This is likely due to the fact that HMF polymerization would occur at a higher frequency as its dosage increases, thus leading to a slight decrease in its photo-oxidation efficiency. It is worth noting that the turnover number (TON, the molar ratio of DFF to HBr) in Fig. 2b was almost linearly enhanced with the increase of HMF dosage, which can be indicative of a continuous improvement in the photocatalytic efficiency of HBr. Fig. 3a displays that as the photoreaction temperature rose from 50 to 80 °C, HMF conversion and DFF yield were continuously improved from 47.4 to 90.3% and from 12.8 to 89.1%, respectively, presenting a gradual approaching trend for both. This likely implies that the activation energy of HMF photooxidation is higher than that of its polymerization and the probability of its occurrence gradually increases upon elevating the temperature. Fig. 3b displays the influence of illumination time on HBr-photocatalyzed HMF to DFF. In that, HMF conversion and DFF yield almost presented a rapid and synchronous increase upon prolonging the time, respectively, achieving 90.8 and 89.1% at 8 h. Thereafter, as the photo-reaction progressed, DFF yield almost no longer increased but HMF conversion was slightly enhanced to 94.5% at 10–12 h, indicating that only HMF polymerization could proceed at this reaction stage, but its photo-oxidation is perhaps completely blocked by cumulative polymers.
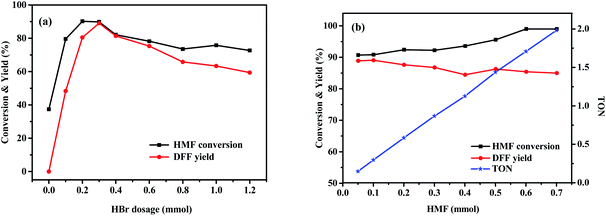 |
| | Fig. 2 Influence of HBr (a) or HMF dosage (b) on the HBr-photocatalyzed HMF oxidation to DFF (by the use of the standard reaction conditions of Table 2 except for HBr or HMF dosage). | |
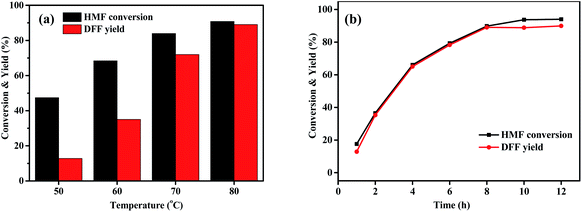 |
| | Fig. 3 Influence of reaction temperature (a) and reaction time (b) on the HBr-photocatalyzed HMF oxidation to DFF (by use of the standard reaction conditions of Table 2 except for temperature or time). | |
Synthesis of DFF from fructose by two steps
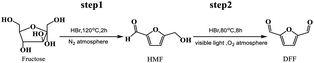
As described above, the synthesis of DFF from fructose in the HBr–MeCN photoreaction system could be achieved by one step under a relatively mild temperature, but providing a low 42.1% DFF yield owing to severe polymerization for the generated HMF. Also, this one-step approach using DMSO as a medium has failed even if the temperature rises to a maximum of 80 °C that the current photo-reactors can withstand, this is because the dehydration of fructose to HMF catalyzed by HBr in DMSO cannot occur efficiently at 80 °C, which can be confirmed from the following experiments (Table 3, entry 9). In order to overcome this obstacle, the synthesis of DFF from fructose was carried out by the following two steps.
In the first step, the dehydration of fructose to HMF was carried out using HBr or Amberlyst-15 as a catalyst. As shown in Table 3, HBr showed good activity for this dehydration at 100–120 °C, achieving about 50–95% HMF yield (entries 1–8), but it became invalid at 80 °C (entry 9). The HMF yield at 100 and especially 120 °C was gradually and significantly reduced upon increasing HBr dosage from 0.085 to 0.34 mmol, this abnormal inverse relationship between HMF yield and catalyst dosage may be because the use of excessive HBr easily causes serious polymerization of fructose with dehydrated product HMF and especially HMF itself,59 and these polymerizations becomes more serious at a higher temperature of 120 °C. The effect of reaction time on the dehydration of fructose at 100 °C was also checked using 0.085 mmol HBr. As shown in entries 3–5, HMF yield achieved a maximum of 90.4% at 4 h and a further prolonging the time led to the slight reduction of HMF yield owing to its subsequent oxidation to DFF (about 0.73%) and polymerization.
The highest HMF yield of 95.4% could be obtained using 0.085 mmol HBr at 120 °C for 2 h. Besides, Amberlyst-15, as a solid acid, even achieved 99.6% HMF yield under the same reaction conditions (entry 10), in consistence with the result reported by Shimizu et al.60
In the second step, the above two dehydration solutions obtained in entries 8 and 10 of Table 3 were diluted to 0.02 M HMF solutions with DMSO and then these two diluted solutions were oxidized to DFF under the standard photoreaction conditions of Table 2 and the results are listed in Table 4. Entries 1 and 2 show that the photo-oxidation of two fructose-derived HMF solutions were also efficiently catalyzed by HBr, but achieving a relatively low DFF yield (72–74%) compared to the use of analytical pure HMF (89% DFF yield), which may be due to the inhibition of trace byproducts produced during fructose dehydration,61 as supported in the following control tests. After the two dehydrated stock solutions were treated with a large amount of active carbon (AC) to remove the possible soluble polymers as much as possible, their diluted solutions (0.02 M HMF) exhibited more reactivity than the corresponding ones in entries 1 and 2, which could afford DFF yields of 84.2 and 80.2%, respectively (entries 3 and 4), thus providing about 80% yield of fructose-derived DFF from two-step conversions.
Table 4 Direct conversion of fructose to DFF by two steps
|
| Entry |
Step 1: yield of HMF (%) |
Step 2a: yield of DFF (%) |
Yield of DFF from fructose (%) |
| Using standard reaction conditions of Table 2. Using fructose-derived HMF in entry 8 of Table 3. Using fructose-derived HMF in entry 10 of Table 3. Using fructose-derived HMF treated with AC at 80 °C. |
| 1b |
95.4 |
72.1 |
68.7 |
| 2c |
99.6 |
74.6 |
74.2 |
| 3b,d |
95.4 |
84.2 |
80.2 |
| 4c,d |
99.4 |
80.2 |
79.7 |
The proposed photo-catalytic mechanism for the conversion of HMF to DFF
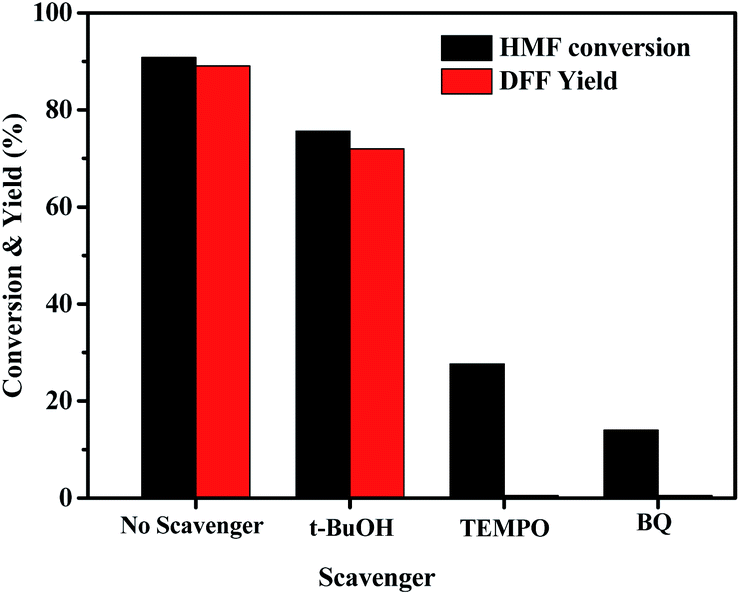
Three radical scavengers were used to confirm the active species for the HBr-photocatalytic oxidation of HMF by O2 and the results are shown in Fig. 4. In the presence of t-BuOH as a hydroxyl radical (˙OH) scavenger,62 this photo-catalytic oxidation could still efficiently proceed although the DFF yield was slightly reduced. But it hardly happened in the presence of TEMPO (a quencher of superoxide anion radical (O2−˙) and ˙OH)63, especially benzoquinone (BQ, a quencher of O2−˙)64, suggesting that the O2−˙ radicals, perhaps also including Br atom radicals, are the dominant species in the HBr-based photo-catalytic oxidation.
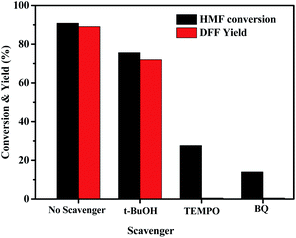 |
| | Fig. 4 Effect of various scavengers (0.2 mmol) on HBr-photocatalyzed HMF oxidation to DFF under standard reaction conditions of Table 2. | |
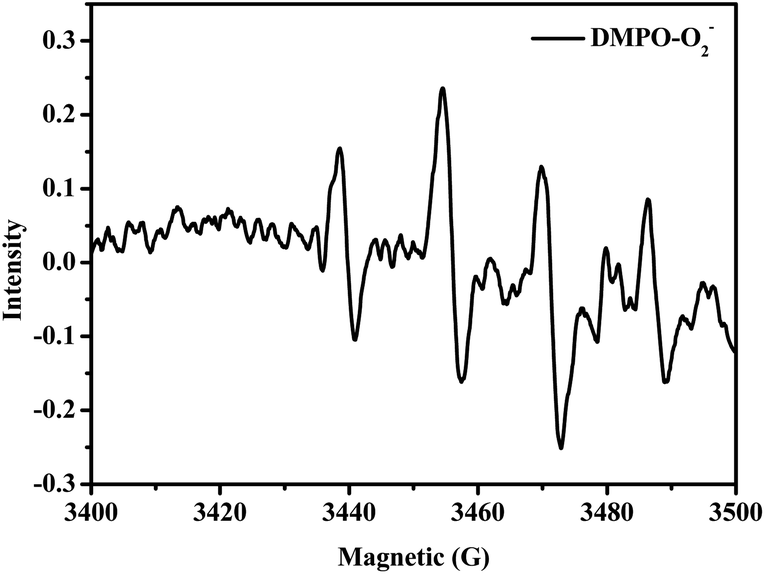
In addition, Fig. 5 shows that after the HBr-based photo-catalytic solution with DMPO (50 mM) was stirred for 5 min with illumination, its EPR spectrum clearly displayed the DMPO-O2˙− EPR signal. This provides direct proof for the existence of O2−˙ species in the photooxidation protocol.
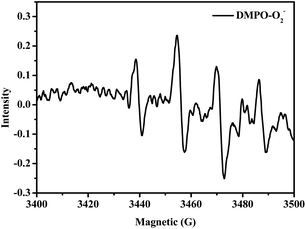 |
| | Fig. 5 EPR spectra of DMPO-captured O2−˙ in HBr-photocatalyzed HMF oxidation. | |
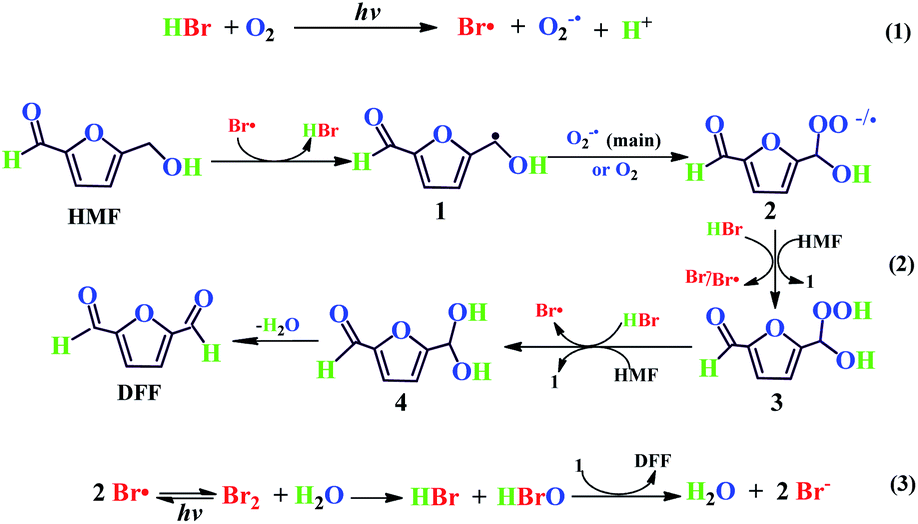
From the present experimental results and previous literature,54,55,65–68 a plausible reaction mechanism has been proposed for the selective synthesis of DFF from HMF. As shown in Scheme 2. The photolysis of HBr in O2 under visible light can generate the active Br atom and the derived superoxide anion (O2−˙) radicals54,55 (eqn (1)). Then, the Br atom radical may abstract one hydrogen atom of HMF to a benzyl radical species (1), along with the regeneration of HBr. Followed by that, the species 1 trap O2−˙ (the main pathway35) or O2 (minor pathway65) to afford its peroxy anion or peroxy radical (2), and the latter may combine with a proton of HBr or react with HBr/HMF to give peroxygen species (3), subsequently, the species 3 further reacts to achieve the regeneration of the Br atom or benzyl radical species. Species 4 are eventually converted into the goal product DFF through intramolecular dehydration66 (eqn (2)). Notably, the introduced water, as supported in the HBr-photocatalyzed HMF oxidation under N2 (entry 8 of Table 2), may provide an efficient oxygen source in the form of hypobromous acid (HBrO) through the following reactions: the combination of two Br atoms generates a Br2 molecule and the latter easily reacts with water to produce HBrO and HBr, then, HBrO, as a strong oxidant, may directly deprive the hydroxyl hydrogen atom of the species 1 to yield HBr and DFF (eqn (3)). Based on a fact that the mass spectrum of DFF obtained from the isotope-tracing test of H218O under N2 lacks the 18O-labeled fragment peaks (see Fig. S3†), we suggest that the generated O2 by decomposition of HBrO under visible light is likely to be ruled out as another way for water to participate in HMF oxidation. It is reasonable that the O2−˙ anion radical combines with benzyl radical (1) much faster than O2, thus accelerating to generate benzyl peroxy anion as the main pathway,35 as supported by the radical quenching experiment conducted by BQ. The photo-generated Br atom and O2−˙ anion radicals are likely to play a synergistic role in the selective oxidation of HMF to DFF. Namely, the Br atom dominates the activation of HMF to benzyl radicals and O2−˙ anion accelerates the conversion of benzyl radical to benzyl with HBr/HMF to yield species 4. These two redox processes can peroxy anion, thus significantly improving the photo-oxidation efficiency. This synergistic mechanism can reasonably explain the photo-catalytic behaviors of the other two hydrohalic acids HCl and HI in HMF oxidation. The reason why HCl lacks photo-catalytic activity should be due to its poor reducibility so that it is difficult to produce the active Cl atom and the derived O2−˙ anion radicals through the photolysis pathway of HCl in eqn (1). In sharp contrast, due to the strong reducibility, HI should be easy to generate I atom and O2−˙ anion radicals via its photolysis pathway in O2. But the relatively inert I atom radical should hardly initiate the selective oxidation of HMF and even if the derived O2−˙ anion radical in the photolysis is also difficult to initiate this reaction, otherwise, the good DFF yield should be obtained in a HI-based photocatalytic system.
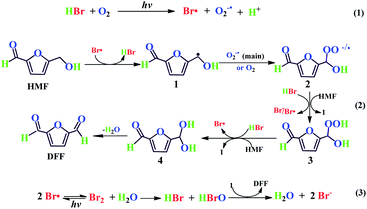 |
| | Scheme 2 Possible mechanism for HBr-photocatalyzed HMF oxidation to DFF under visible light irradiation. (eqn (1): The generation of Free radical; eqn (2): Selective oxidation of HMF to DFF; eqn (3): Possible reaction pathways under N2 atmosphere). | |
Conclusion
In conclusion, for the first time, we have developed a simple and efficient photo-reaction protocol for the synthesis of DFF from HMF and especially fructose under relatively mild conditions, which has the following advantages: (i) this protocol uses readily available HBr as a catalyst and molecular oxygen/or even water as a green oxygen donor. (ii) This protocol can function well under visible light, which achieves a very good efficiency for HMF photo-oxidation and 100% DFF selectivity in DMSO at 80 °C. (iii) This protocol can efficiently synthesize DFF (80% yield) from the two-step cascade conversion of fructose in DMSO. With such kind of robust photo-reaction protocol in hand, we are interested in further exploring its potential applications for highly efficient synthesis of DFF from the one-step conversion of fructose or two-step conversion of glucose, sucrose, cellobiose and cellulose.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the financial support for this work by the National Natural Science Fund of China (21546010, 21676079, 22008062), and the Natural Science Fund of Hunan Province (2018JJ3335, 2020JJ4425). The Innovation Platform Open Fund of Hunan College (18K016), Hunan 2011 Collaborative Innovation Center of Chemical Engineering & Technology with Environmental Benignity and Effective Resource Utilization, Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Hunan Province.
References
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- C. Liu, C. Zhang, K. Liu, Y. Wang, G. Fan, S. Sun, J. Xu, Y. Zhu and Y. Li, Biomass Bioenergy, 2015, 72, 189–199 CrossRef CAS.
- J. Song, H. Fan, J. Ma and B. Han, Green Chem., 2013, 15, 2619–2635 RSC.
- X. Tong, Y. Ma and Y. Li, Carbohydr. Res., 2010, 345, 1698–1701 CrossRef CAS PubMed.
- S. Hu, Z. Zhang, Y. Zhou, B. Han, H. Fan, W. Li, J. Song and Y. Xie, Green Chem., 2008, 10, 1280–1283 RSC.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754–793 RSC.
- T. S. Hansen, I. Sádaba, E. J. García-Suárez and A. Riisager, Appl. Catal., A, 2013, 456, 44–50 CrossRef CAS.
- M. Del Poeta, W. A. Schell, C. C. Dykstra, S. Jones, R. R. Tidwell, A. Czarny, M. Bajic, M. Bajic, A. Kumar, D. Boykin and J. R. Perfect, Antimicrob. Agents Chemother., 1998, 42, 2495 CrossRef CAS PubMed.
- K. T. Hopkins, W. D. Wilson, B. C. Bender, D. R. McCurdy, J. E. Hall, R. R. Tidwell, A. Kumar, M. Bajic and D. W. Boykin, J. Med. Chem., 1998, 41, 3872–3878 CrossRef CAS PubMed.
- F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728–737 CrossRef CAS PubMed.
- Y. Yan, K. Li, J. Zhao, W. Cai, Y. Yang and J.-M. Lee, Appl. Catal., B, 2017, 207, 358–365 CrossRef CAS.
- J. Zhao, X. Chen, Y. Du, Y. Yang and J.-M. Lee, Appl. Catal., A, 2018, 568, 16–22 CrossRef CAS.
- Z. Du, J. Ma, F. Wang, J. Liu and J. Xu, Green Chem., 2011, 13, 554–557 RSC.
- R. Qu, W. Zhang, N. Liu, Q. Zhang, Y. Liu, X. Li, Y. Wei and L. Feng, ACS Sustainable Chem. Eng., 2018, 6, 8019–8028 CrossRef CAS.
- T. Cao, M. Wu, V. V. Ordomsky, X. Xin, H. Wang, P. Métivier and M. Pera-Titus, ChemSusChem, 2017, 10, 4851–4854 CrossRef CAS PubMed.
- P. Prabhu, Y. Wan and J.-M. Lee, Matter, 2020, 3, 1162–1177 CrossRef.
- L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou and J. Xu, ACS Sustainable Chem. Eng., 2018, 6, 15915–15935 CrossRef CAS.
- V. P. Kashparova, Russ. Chem. Bull., 2015, 64, 1069–1073 CrossRef CAS.
- W. Zhang, T. Meng, J. Tang, W. Zhuang, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2017, 5, 10029–10037 CrossRef CAS.
- J. Zhao, A. Jayakumar, Z.-T. Hu, Y. Yan, Y. Yang and J.-M. Lee, ACS Sustainable Chem. Eng., 2017, 6, 284–291 CrossRef.
- Q. Girka, B. Estrine, N. Hoffmann, J. Le Bras, S. Marinković and J. Muzart, React. Chem. Eng., 2016, 1, 176–182 RSC.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- K. Tsutsumi, N. Kurata, E. Takata, K. Furuichi, M. Nagano and K. Tabata, Appl. Catal., B, 2014, 147, 1009–1014 CrossRef CAS.
- Y. Xiao and Y.-F. Song, Appl. Catal., A, 2014, 484, 74–78 CrossRef CAS.
- J. Ma, Z. Du, J. Xu, Q. Chu and Y. Pang, ChemSusChem, 2011, 4, 51–54 CrossRef CAS PubMed.
- A. S. Amarasekara, D. Green and E. McMillan, Catal. Commun., 2008, 9, 286–288 CrossRef CAS.
- J. Artz, S. Mallmann and R. Palkovits, ChemSusChem, 2015, 8, 672–679 CrossRef CAS PubMed.
- A. Takagaki, M. Takahashi, S. Nishimura and K. Ebitani, ACS Catal., 2011, 1, 1562–1565 CrossRef CAS.
- R. Liu, J. Chen, L. Chen, Y. Guo and J. Zhong, ChemPlusChem, 2014, 79, 1448–1454 CrossRef CAS.
- M. Cui, R. Huang, W. Qi, R. Su and Z. He, RSC Adv., 2017, 7, 7560–7566 RSC.
- P. V. Rathod, S. D. Nale and V. H. Jadhav, ACS Sustainable Chem. Eng., 2016, 5, 701–707 CrossRef.
- G. Lv, H. Wang, Y. Yang, T. Deng, C. Chen, Y. Zhu and X. Hou, Green Chem., 2016, 18, 2302–2307 RSC.
- Z. Yang, W. Qi, R. Su and Z. He, ACS Sustainable Chem. Eng., 2017, 5, 4179–4187 CrossRef CAS.
- Z.-Z. Yang, J. Deng, T. Pan, Q.-X. Guo and Y. Fu, Green Chem., 2012, 14, 2986–2989 RSC.
- B. Ma, Y. Wang, X. Guo, X. Tong, C. Liu, Y. Wang and X. Guo, Appl. Catal., A, 2018, 552, 70–76 CrossRef CAS.
- A. B. Raut and B. M. Bhanage, ChemistrySelect, 2018, 3, 11388–11397 CrossRef CAS.
- W. Hou, Q. Wang, Z. Guo, J. Li, Y. Zhou and J. Wang, Catal. Sci. Technol., 2017, 7, 1006–1016 RSC.
- C. Laugel, B. Estrine, J. Le Bras, N. Hoffmann, S. Marinkovic and J. Muzart, ChemCatChem, 2014, 1195–1198 CAS.
- J. Zhao, Y. Yan, Z.-T. Hu, V. Jose, X. Chen and J.-M. Lee, Catal. Sci. Technol., 2020, 10, 4179–4183 RSC.
- J. Zhao, A. Jayakumar and J.-M. Lee, ACS Sustainable Chem. Eng., 2018, 6, 2976–2982 CrossRef CAS.
- Y. Liu, L. Zhu, J. Tang, M. Liu, R. Cheng and C. Hu, ChemSusChem, 2014, 7, 3541–3547 CrossRef CAS PubMed.
- J. Zhao, J. Anjali, Y. Yan and J.-M. Lee, ChemCatChem, 2017, 9, 1187–1191 CrossRef CAS.
- Q. Wang, W. Hou, T. Meng, Q. Hou, Y. Zhou and J. Wang, Catal. Today, 2019, 319, 57–65 CrossRef CAS.
- X. Li, G. Chen, Y. Po-Lock and C. Kutal, J. Chem. Technol. Biotechnol., 2003, 78, 1246–1251 CrossRef CAS.
- A. Zertal, J. Environ. Sci. Eng., 2012, 844–8522 Search PubMed.
- A. Maldotti, A. Molinari and R. Amadelli, Chem. Rev., 2002, 102, 3811–3836 CrossRef CAS PubMed.
- S. Tang, J. She, Z. Fu, S. Zhang, Z. Tang, C. Zhang, Y. Liu, D. Yin and J. Li, Appl. Catal., B, 2017, 214, 89–99 CrossRef CAS.
- B. Yang, Z. Fu, A. Su, J. She, M. Chen, S. Tang, W. Hu, C. Zhang and Y. Liu, Appl. Catal., B, 2019, 242, 249–257 CrossRef CAS.
- H. Wu, H. L. Tan, C. Y. Toe, J. Scott, L. Wang, R. Amal and Y. H. Ng, Adv. Mater., 2020, 32, 1–21 Search PubMed.
- H. L. Tan, F. F. Abdi and Y. H. Ng, Chem. Soc. Rev., 2019, 48, 1255–1271 RSC.
- I. Krivtsov, E. I. García-López, G. Marcì, L. Palmisano, Z. Amghouz, J. R. García, S. Ordóñez and E. Díaz, Appl. Catal., B, 2017, 204, 430–439 CrossRef CAS.
- H. Zhang, Q. Wu, C. Guo, Y. Wu and T. Wu, ACS Sustainable Chem. Eng., 2017, 5, 3517–3523 CrossRef CAS.
- S. Yurdakal, B. S. Tek, O. Alagöz, V. Augugliaro, V. Loddo, G. Palmisano and L. Palmisano, ACS Sustainable Chem. Eng., 2013, 1, 456–461 CrossRef CAS.
- S. Gazi and R. Ananthakrishnan, RSC Adv., 2012, 2, 7781–7891 RSC.
- S.-i. Hirashima, S. Hashimoto, Y. Masaki and A. Itoh, Tetrahedron, 2006, 62, 7887–7891 CrossRef CAS.
- N. Tada, K. Ban, T. Ishigami, T. Nobuta, T. Miura and A. Itoh, Tetrahedron Lett., 2011, 52, 3821–3824 CrossRef CAS.
- I. K. M. Yu, D. C. W. Tsang, A. C. K. Yip, S. S. Chen, L. Wang, Y. S. Ok and C. S. Poon, Bioresour. Technol., 2017, 237, 222–230 CrossRef CAS PubMed.
- X. Zhang, B. B. Hewetson and N. S. Mosier, Energy Fuels, 2015, 29, 2387–2393 CrossRef CAS.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS PubMed.
- K.-i. Shimizu, R. Uozumi and A. Satsuma, Catal. Commun., 2009, 10, 1849–1853 CrossRef CAS.
- G. Sampath and S. Kannan, Catal. Commun., 2013, 37, 41–44 CrossRef CAS.
- Q. Wu, Y. He, H. Zhang, Z. Feng, Y. Wu and T. Wu, Mol. Catal., 2017, 436, 10–18 CrossRef CAS.
- Q. Chen, Y. Huang, X. Wang, J. Wu and G. Yu, Org. Biomol. Chem., 2018, 16, 1713–1719 RSC.
- H. Zhang, Z. Feng, Y. Zhu, Y. Wu and T. Wu, J. Photochem. Photobiol., A, 2019, 371, 1–9 CrossRef CAS.
- A. Itoh and S.-i. Hirashima, Synthesis, 2006, 2006, 1757–1759 CrossRef.
- N. Tada, K. Ban, M. Yoshida, S.-i. Hirashima, T. Miura and A. Itoh, Tetrahedron Lett., 2010, 51, 6098–6100 CrossRef CAS.
- J. Sun, Y. Wang, L. Han, D. Xu, Y. Chen, X. Peng and H. Guo, Org. Chem. Front., 2014, 1, 1201–1204 RSC.
- A. Itoh, N. Tada, K. Ban, T. Nobuta, S.-i. Hirashima and T. Miura, Synlett, 2011, 2011, 1381–1384 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The HBr-photocatalyzed fructose to DFF in MeCN, UV-vis spectrum of HBr in different solvents, synthesis of HMF from fructose using HBr and isotope tracing test of heavy oxygen water (H218O). See DOI: 10.1039/d1ra00865j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Bo Yanga,
Huanhuan Zhanga and
Dabo Jianga
*a,
Bo Yanga,
Huanhuan Zhanga and
Dabo Jianga