DOI:
10.1039/D1RA00841B
(Paper)
RSC Adv., 2021,
11, 14755-14768
Stereoselective synthesis of spirocyclic pyrrolidines/pyrrolizidines/pyrrolothiazolidines using L-proline functionalized manganese ferrite nanorods as a novel heterogeneous catalyst†
Received
31st January 2021
, Accepted 8th March 2021
First published on 20th April 2021
Abstract
An efficient, green, one-pot, and three-component protocol has been reported for the stereoselective synthesis of a new class of spiro thiazolidines. A series of spiro-heterocycle derivatives were produced stereoselectively in high yields by the reaction of 5-arylidene thiazolidine-2,4-diones, isatin, and secondary amino acids in the presence of MnCoCuFe2O4@L-proline (MCCFe2O4@L-proline) magnetic nanorods as a novel nanocatalyst. The synthesized catalyst was fully characterized for thermal stability, magnetic properties, and other physicochemical properties via numerous techniques. It was applied as an efficient and reusable catalyst for the synthesis of endo-isomers of spirocyclic pyrrolidine/pyrrolizidine/pyrrolothiazolidine derivatives in high yield. The regioselectivity and stereochemistry of these heterocyclic spiro-compounds were established by 1H, 13C, HMBC, HSQC, and COSY NMR spectroscopy techniques. The main attractive characteristics of the presented protocol are high yield, high level of diastereoselectivity, and easy recovery of catalyst without significant loss of its catalytic activity.
Introduction
Multicomponent 1,3-dipolar cycloaddition reactions play a key role in the synthesis of five-membered heterocyclic compounds; also, they are one category of the most important reactions in organic chemistry.1,2 Multicomponent 1,3-dipolar cycloaddition reactions have found extensive application as a high-performance and efficient stereocontrolled and regiocontrolled method for the synthesis of many new heterocyclic five-membered nitrogen spiro scaffolds.2 Heterocyclic spirooxindole systems contain two rings sharing one sp3 carbon, and they are frequently used as important construction blocks in organic synthesis. Placing the ylide dipole and the alkene dipolarophile in the same molecule provides direct access to bicyclic products of considerable complexity.3 The proximity of the reactants and conformational constraints often lead to ready cycloaddition with very high or complete selectivity.4 For the preparation of five-membered cyclic amines, in particular pyrrolidines, pyrrolizidines, and dihydropyrroles, cycloaddition of azomethine ylides with alkenes is very effective.2–6 Azomethine ylides have four π electrons spread over the three-atom C–N–C unit, of which the most common representation has a positive charge located on the nitrogen atom and a negative charge distributed over the two carbon atoms.7 The cycloaddition of an azomethine ylide with a π-system involves a total of six π electrons [π4s + π2s] and takes place by a thermally allowed, suprafacial process in which two carbon–carbon bonds are formed on the same face of the azomethine ylide and on the same face of the dipolarophile.8 It is generally accepted that the cycloaddition involves both carbon–carbon π-bonds being formed at the same time, although not necessarily to the same extent. In terms of frontier molecular orbital (FMO) theory, in which a reaction takes place by maximizing the overlap of the HOMO and the LUMO, azomethine ylides can be considered to be electron-rich, and the dominant interaction involves the HOMO of the azomethine ylide with the LUMO of the π-system. This is borne out by the general preference for reaction of azomethine ylides with electron-poor alkenes.9–11
Our previous theoretical study was performed on the 1,3-dipolar cycloaddition between the HOMO of an azomethine and the LUMO of an alkene using density functional theory (DFT) in combination with the 6-311++G(d,p) basis set.11 Our results showed that a combination of FMO theory, atomic Fukui indices, Gibbs activation free energies (ΔG#) and the reaction constant, ρ, explains that the reactivity in the 1,3-dipolar cycloaddition was affected by electron-withdrawing substituents on the dipolar component and enhanced the reaction constant (ρ > 0), especially in more polar aprotic solvents. The early transition state for these substituents can be inferred based on the slope of the Hammett plot and the similarity of the reactants and transition state structures in various solvents.11
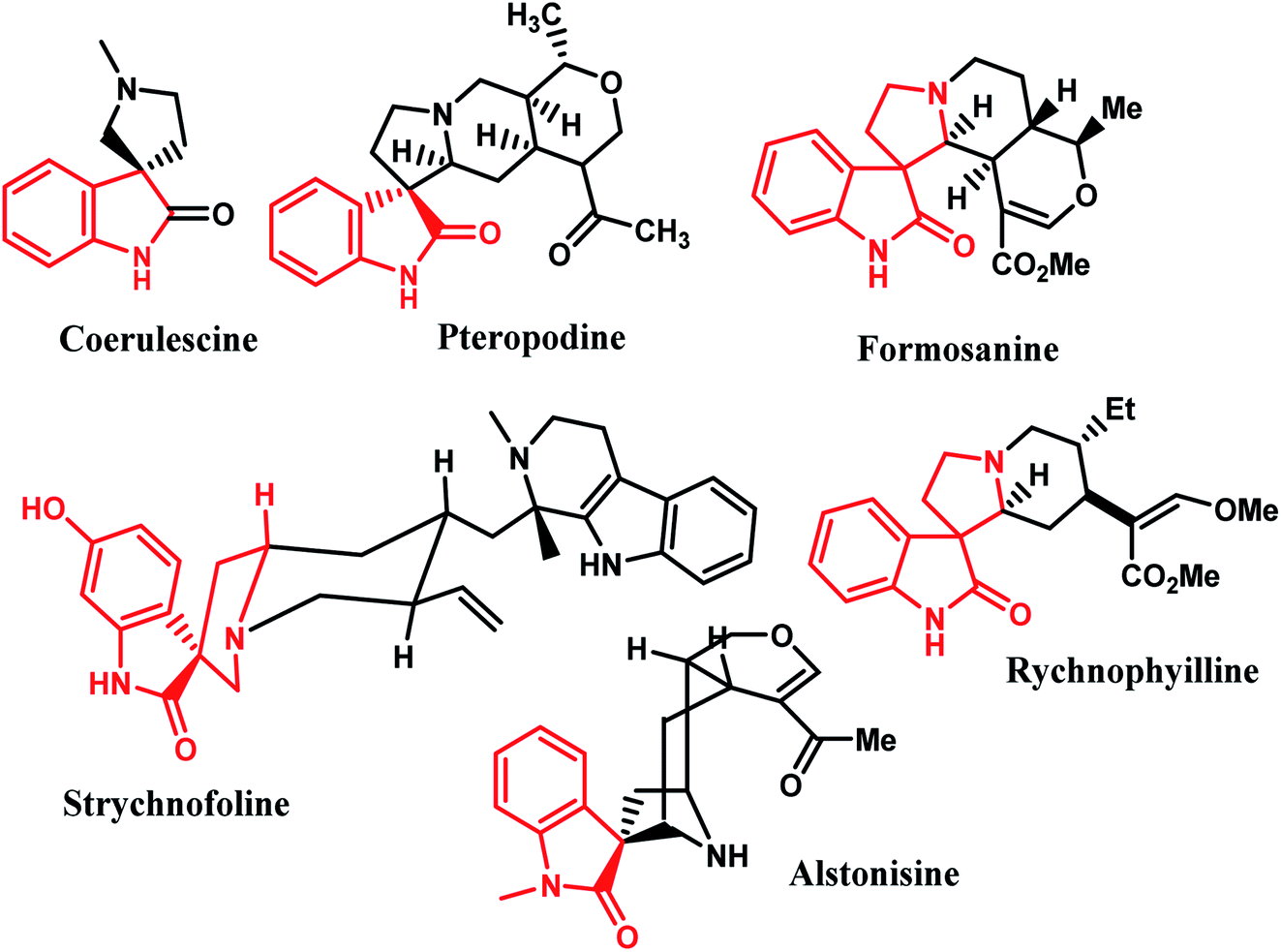
Due to the constrained structure of spiro compounds, the existence of the chiral central spiro carbon and the indole core represents an interesting pharmacophore, and these compounds show a wide range of biological and pharmacological activities.9 Furthermore, a broad range of natural alkaloids contain well-known spirooxindole moieties, such as formosanine,4 pteropodine,5 coerulescine,6 strychnofoline,7 rychnophyilline,4 and alstonisine,8 with highly pronounced proved pharmacological properties (Fig. 1). For instance, spirooxindoles exhibit a range of biological properties, including antimicrobial, antitumoral, and antibiotic properties, as well as inhibition of human NK-1 receptor.12–14
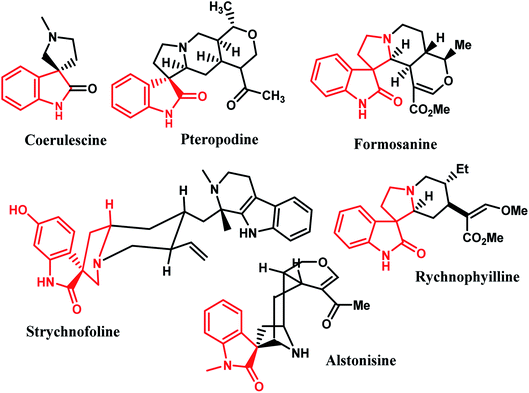 |
| | Fig. 1 Representative naturally occurring spiro-oxindole alkaloids. | |
In medicinal chemistry and drug design, TZD-containing compound derivatives in medicine are also well recognized for their anti-inflammatory15,16 and antihypertensive17,18 activities. Therefore, according to the significant biological importance associated with the above heterocyclic core, we became interested in the synthesis of hybrid spiro structure heterocycles with five heterostructural motifs, including TZD and oxindole.19–22
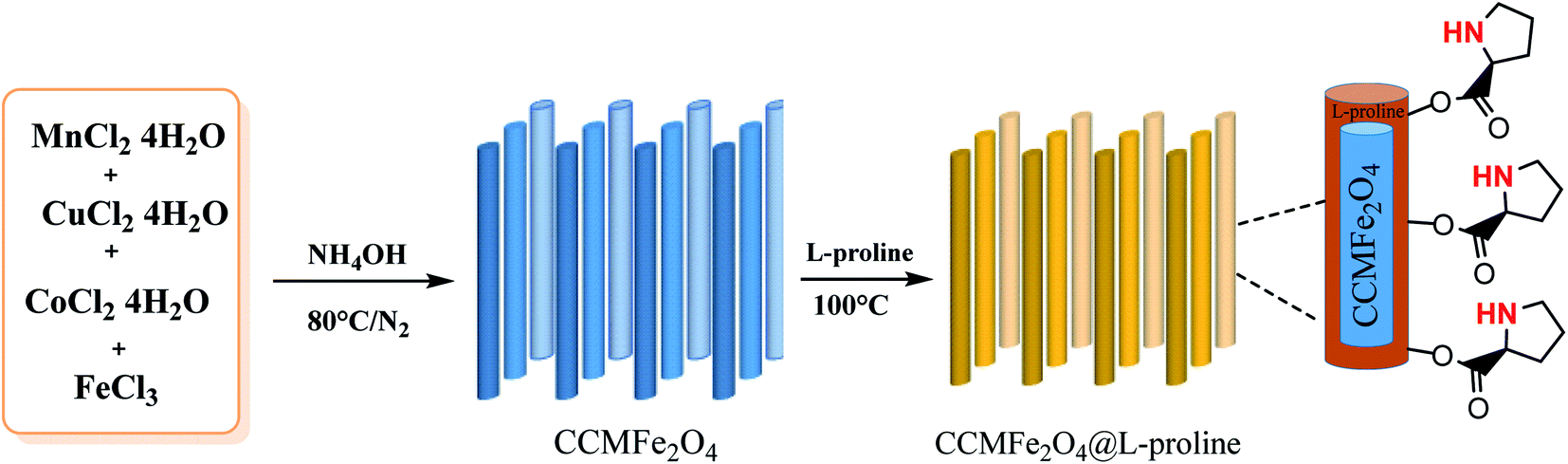
Heterogeneous catalysts are an important class of soft magnetic materials which have high magnetic permeability, saturation magnetization, electrical resistivity and low power losses.23 Because of the efficient recyclability and facile separation of the products, heterogeneous catalysts have gained prominent attention in economical and environmentally benign chemical processes.24 MNRs have recently received special and significant attention that has led to many advancements, such as in drug delivery25 and magnetic resonance imaging (MRI).25–27 Moreover, L-proline MNR-supported catalysts which were synthesized by coating MnCoCu ferrite nanorods with proline showed high surface areas, high catalyst loading capacity, reusability, dispersion, excellent yields and superior stability.28–30 In this work, a novel MCCFe2O4@L-proline recyclable heterogeneous nanorod catalyst was prepared; it is promising due to its easy operation, low cost, low temperature, and controllable conditions, the high reactivity of the obtained products and the prevention of undesired side-product formation (Scheme 1).
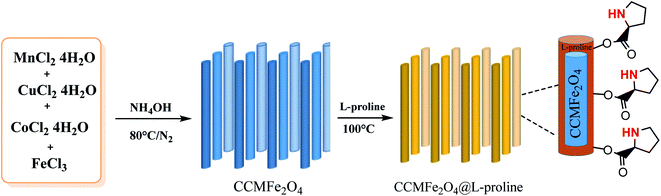 |
| | Scheme 1 Preparation of CoCuMnFe2O4@L-proline MNRs. | |
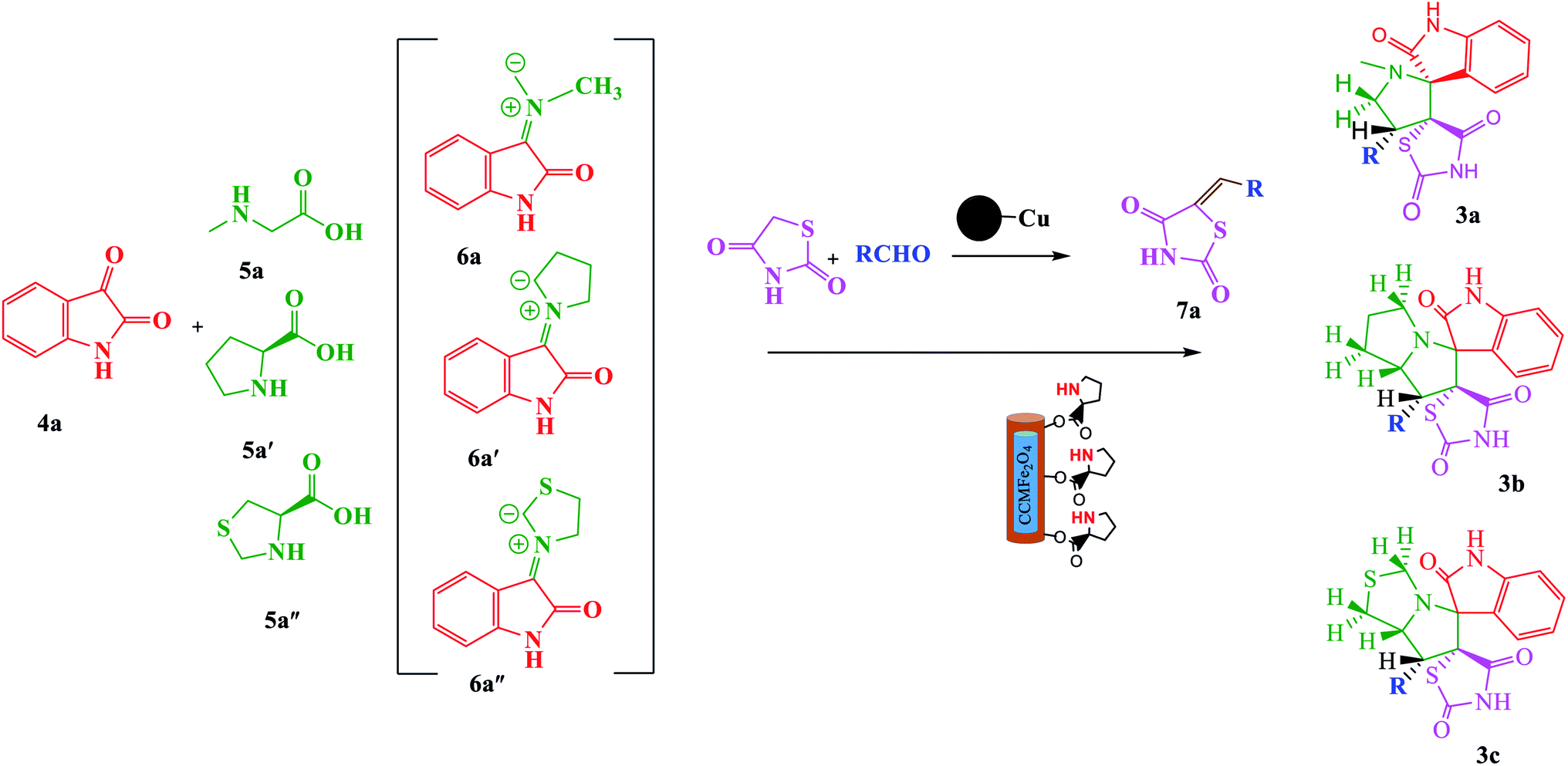
In continuation of our research and considering the above reasons related to multicomponent 1,3-dipolar cycloaddition reactions and our ongoing program for the synthesis of complex organic compounds based on green chemistry,31–37 herein, we were fascinated by the possibility of using green and nano chemistry to design an efficient, reusable and diastereoselective catalyst for the synthesis of spiro-pyrrolidines/pyrrolizidines and pyrrolothiazole derivatives via a one-pot process involving azomethine ylides using MCCFe2O4@L-proline MNRs (Scheme 2).
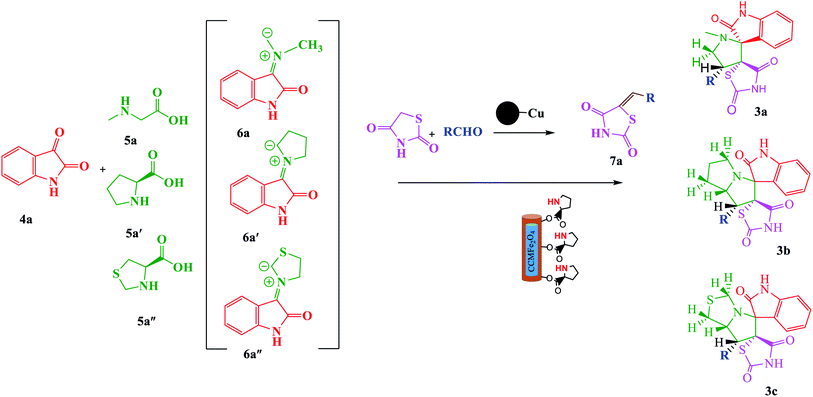 |
| | Scheme 2 Green one-pot, three-component asymmetric 1,3-dipolar cycloaddition catalyzed by the CCMFe2O4@L-proline MNRs catalyst. | |
Results and discussion
Synthesis and characterization of the catalyst
According to Scheme 1, we report a facile preparation of MCCFe2O4@L-proline MNRs as a new, reusable and recoverable heterogeneous catalyst. This newly synthesized catalyst was fully characterized by various techniques, such as powder X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDAX), vibrating sample magnetometry (VSM), and thermogravimetric (TGA) analysis. The results obtained from these techniques confirmed the successful preparation of this novel catalyst.
Structural properties of the MCCFe2O4@L-proline MNRs
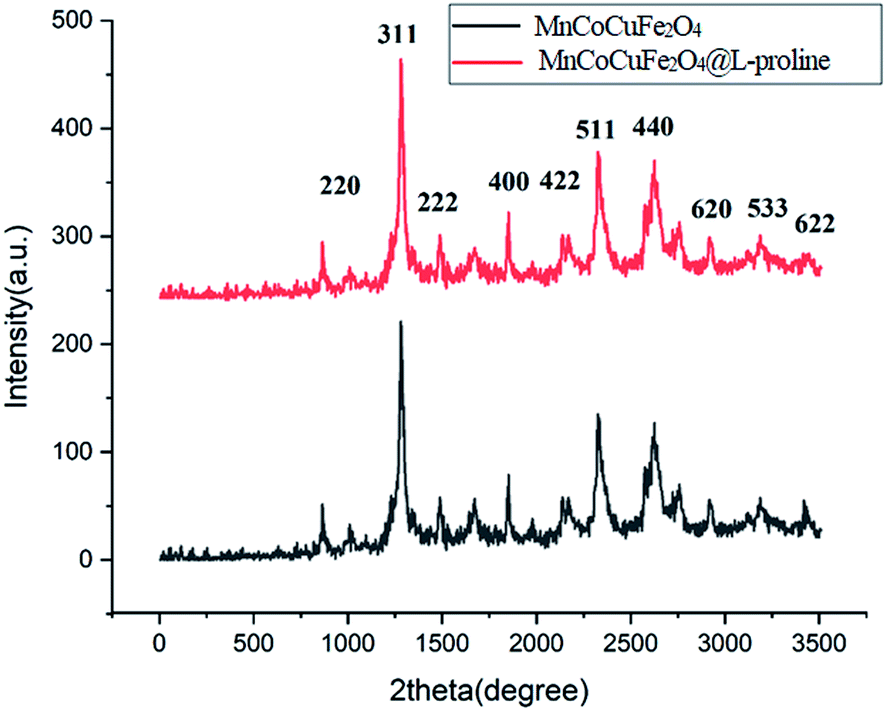
XRD analysis of the MCCFe2O4@L-proline MNRs. The crystalline structures of the MCCFe2O4 and MCCFe2O4@L-proline MNRs were investigated by means of XRD (Fig. 2). The positions and relative intensities of the diffraction peaks in the XRD spectra show invers spinel structures for the nanoparticles. All the peaks can be indexed to the (2 2 0), (3 1 1), (222), (4 0 0), (4 2 2), (5 1 1), (440), (620), (533) and (622) reflections, which agree well with the cubic structure of the pure spinel structure (JCPDS 74-2401).
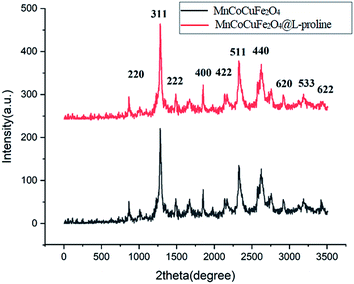 |
| | Fig. 2 XRD patterns of the MnCoCuFe2O4@L-proline MNRs. | |
Morphological studies of the MCCFe2O4@L-proline MNRs. A SEM image of the MCCFe2O4@L-proline MNRs is demonstrated in Fig. 3, which provides sufficient data on the morphology features and sizes of the nanoparticles. The SEM image clearly demonstrates that the nanoparticles possess a rod-like morphology with 30–60 nm width and 150–300 nm length. It can be concluded that the shape of MCCFe2O4@L-proline is nanorods.
 |
| | Fig. 3 SEM images of the MnCoCuFe2O4@L-proline MNRs. | |
Fig. 4 shows TEM images of the uniform-sized particles with rod shapes with an average size of 30 nm. It is noticeable that the average diameter obtained from the TEM measurements is smaller than the sizes obtained from the SEM measurements.
 |
| | Fig. 4 TEM images of the MnCoCuFe2O4@L-proline MNRs. | |
Chemical compositions of the MCCFe2O4@L-proline MNRs
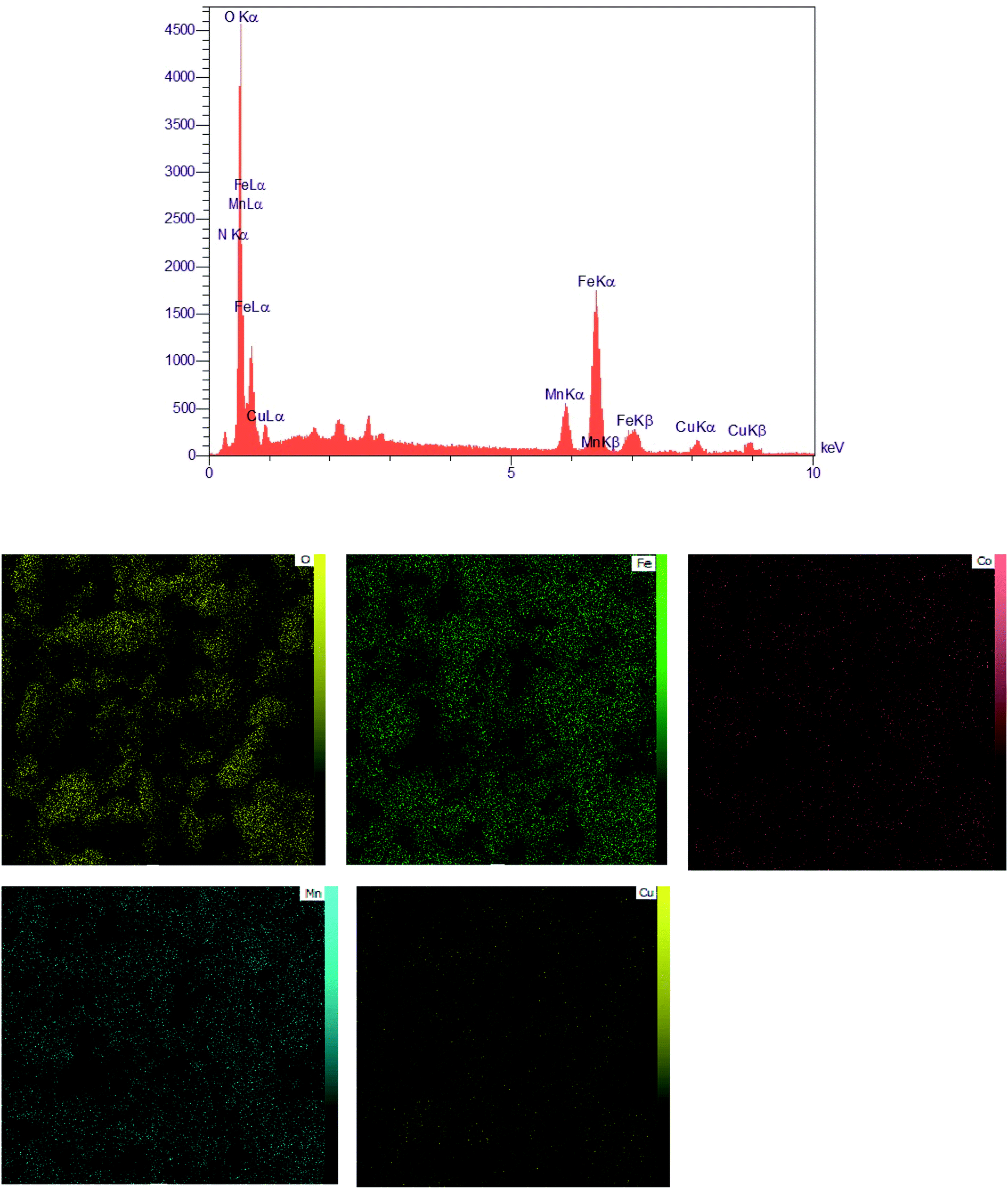
EDX spectra and elemental mapping of the MCCFe2O4@L-proline MNRs. The EDAX analysis of the MCCFe2O4@L-proline MNRs is depicted in Fig. 5. The surface layer consists of manganese, zinc, iron, copper, and oxygen; therefore, the N signal is present in the EDAX spectrum. According to the EDX analysis, it can be deduced that MCCFe2O4@L-proline was successfully synthesized. In the EDX spectrum, three peaks were observed for each metal and one peak each for C and O. The EDX elemental mapping from the SEM analysis clearly confirms that MCCFe2O4@L-proline consists of Mn, Co, Cu, C, O and Fe; also, the absence of other elements indicates that MCCFe2O4@L-proline has a high purity level.
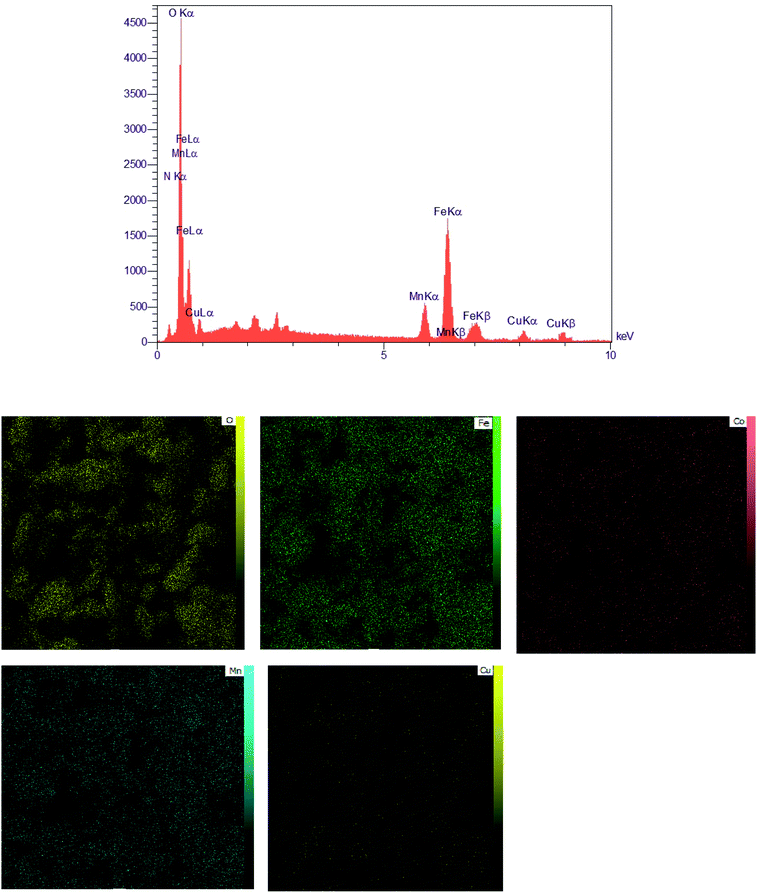 |
| | Fig. 5 EDAX spectrum of the MCCFe2O4@L-proline MNRs and the corresponding EDX elemental mapping pattern results of the MCCFe2O4@L-proline MNRs. | |
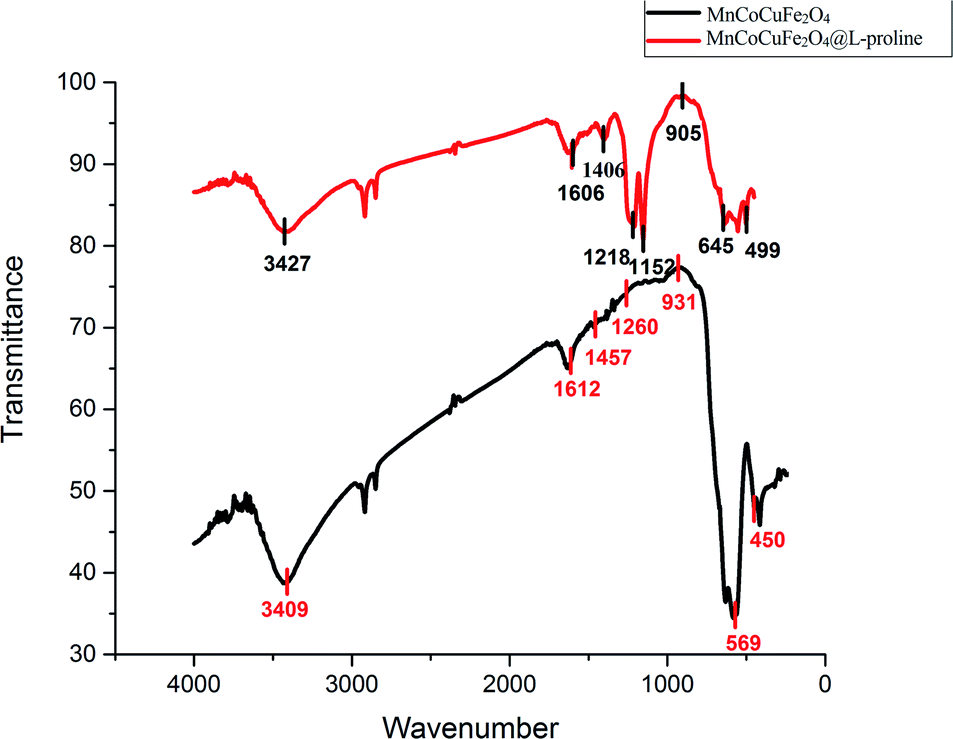
FT-IR spectroscopy of the MCCFe2O4@L-proline MNRs. The FT-IR spectra of MCCFe2O4 and the MCCFe2O4@L-proline MNRs in the wavenumber range of 4000–400 cm−1 are depicted in Fig. 6. The magnetic nanoparticles were proved by the two significant absorption bands at 450 and 569 cm−1, which are related to the stretching vibration of metal–oxygen. Furthermore, the broad peaks in the region of 3100–3600 cm−1 concern O–H groups on the surface of the MCCFe2O4 nanoparticles, which can be attributed to absorbed water molecules. For the L-proline functional group, the band perceived at 1647 cm−1, which is due to the COO–Fe band, proves the complexation between the carboxylate moiety of L-proline and the ions on the surface of the magnetic nanoparticles.
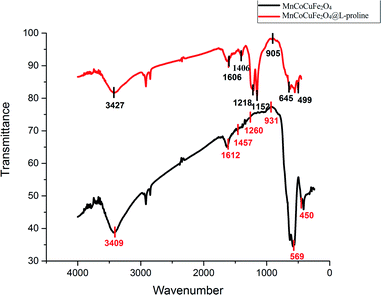 |
| | Fig. 6 FTIR spectra of the CCMFe2O4 and MnCoCuFe2O4@L-proline MNRs. | |
Physical properties
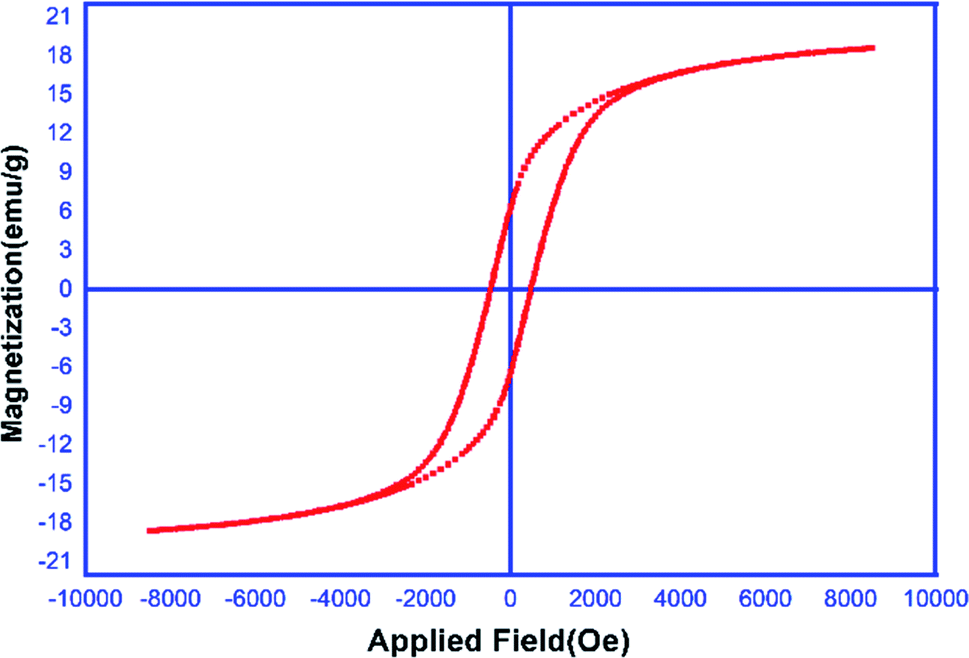
Magnetic properties of the MCCFe2O4@L-proline MNRs. The magnetic properties of the prepared MNRs-supported catalyst were studied using VSM (20 kOe magnetic field) at room temperature (Fig. 7). According to the magnetization curves, the saturation magnetization quantity of the MCCFe2O4@L-proline MNRs was measured to be 18 emu g−1.
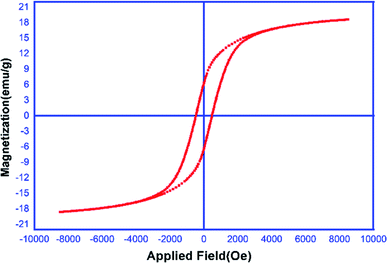 |
| | Fig. 7 Magnetization curves of the MCCFe2O4@L-proline MNRs. | |
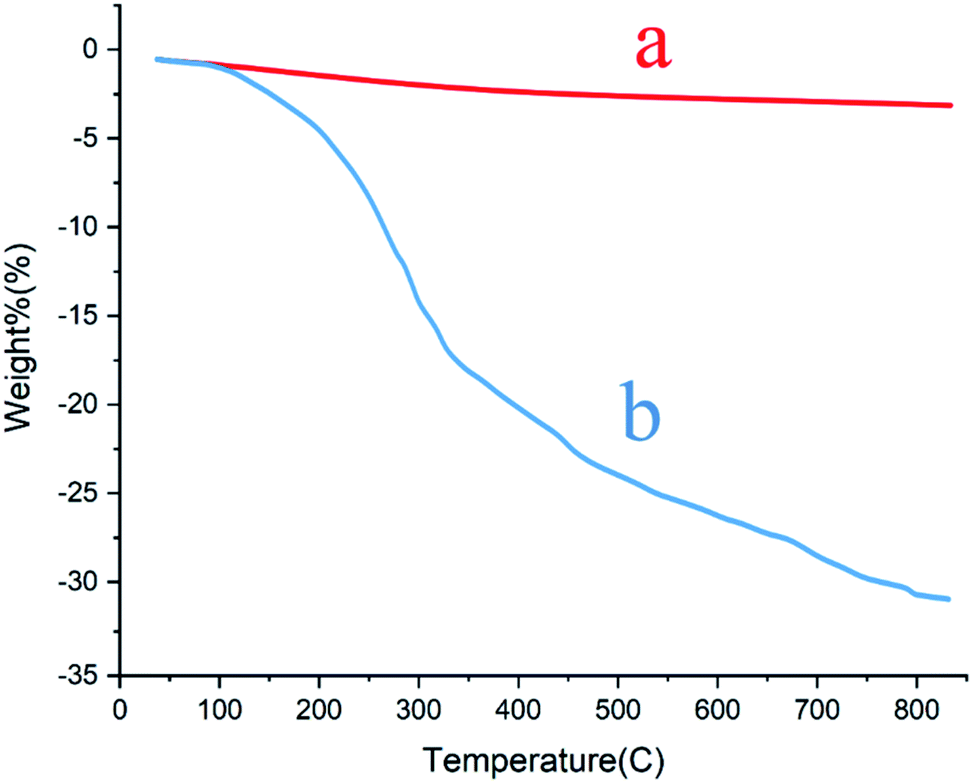
Thermal gravimetric analysis of the MCCFe2O4@L-proline MNRs. A thermogravimetric analyzer (TGA) was used to confirm and evaluate the stability of the MCCFe2O4 MNPs (Fig. 8a) and the content of organic functional groups on the surface of the MCCFe2O4@L-proline MNRs (Fig. 8b). The mass changes under the temperature scan from 30 °C to 1300 °C at a heating rate of 20 °C min−1 and under a nitrogen flow were monitored and recorded. Thermogravimetric analysis (TGA) gives the thermal stability of a catalyst. Therefore, the main changes in the masses of the samples were monitored from 260 °C to 500 °C. The weight loss of 23.9% at 60–500 °C may be due to the removal of absorbed water and L-proline from the MCCFe2O4@L-proline MNR nanoparticles. As shown in the TGA curves of the MCCFe2O4@L-proline MNRs, a high amount of L-proline (2.01 mmol g−1) is immobilized on the magnetic nanoparticles (Fig. 8). Using carbon hydrogen nitrogen (CHN) analysis, the coating of L-proline onto the MCCFe2O4 MNRs was further evaluated, and the results indicated that the catalyst is composed of 3.19% nitrogen, 3.06% hydrogen and 13.31% carbon elements.
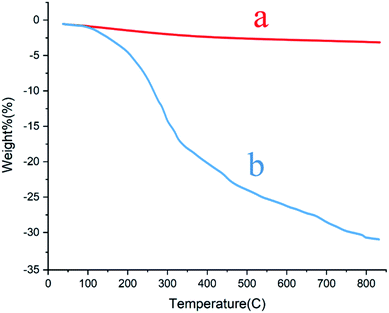 |
| | Fig. 8 TGA patterns of the (a) MnCoCuFe2O4 and (b) MnCoCuFe2O4@L-proline MNRs. | |
Catalytic properties of the MCCFe2O4@L-proline MNRs
Synthesis of 1,3-dipolar cycloaddition using MCCFe2O4@L-proline MNRs. After characterization of the MCCFe2O4@L-proline MNRs catalyst, we evaluated its catalytic activity and found an appropriate reaction medium for the green one-pot synthesis of spiro oxindole pyrrolidines/pyrrolizidines/pyrrolothiazoles. By applying 1,3-dipolar cycloaddition reaction conditions (Scheme 2), the catalyst in the reaction was investigated by performing a model reaction of isatin 4a (1.0 mmol), thiazolidine-4-carboxylic acid 5a′′ (1.0 mmol) and the dipolarophiles of 5-arylidene thiazolidine-2,4-diones 7a (1.0 mmol). The model reaction behavior was studied under different conditions, and the results are summarized in Table 1. It can be seen that when the model reaction was carried out at 100 °C in EtOH media, the yield was lower than 40% under catalyst-free conditions (Table 1, entry 1). By using the CCMFe2O4@L-proline MNRs in different reactions, one regioisomer with yields of up to 80–91% was obtained. Then, we investigated the effects of different solvents and amounts of catalyst on the model reaction (Table 1, entries 2–4). Further, studies established that ethanol was the best choice among the screened solvents (methanol, acetonitrile, and chloroform); it leads to a fast reaction rate, high isolated yield, and selectivity. In the presence of 4–18 mol% of catalyst in other solvents under reflux conditions, the reaction took a longer time to complete, and the yields were low in EtOH; applying lower amounts of the catalyst resulted in 70% and 85% yields of the product after a long time (Table 1, entries 6 and 7). The obtained results demonstrated that both variables affected the reaction. Using 14 mol% MNPs in EtOH especially increased the yield of the reaction to 91% and decreased the reaction time to 3 h with enhanced regioselectivity (Table 1, entry 5). The product selectivity was not influenced by the nature of the solvents.
Table 1 Optimization of the reaction conditions
| Entry |
Catalyst (mol%) |
Solvent |
T (°C) |
Time (h) |
Yielda (%) |
| Isolated yield. Recyclability experiments after five reactions. |
| 1 |
Catalyst-free |
EtOH |
80 |
24 |
40 |
| 2 |
14 |
MeOH |
|
9 |
67 |
| 3 |
14 |
CHCl3 |
|
48 |
43 |
| 4 |
14 |
CH3CN |
|
24 |
52 |
| 5 |
14 |
EtOH |
80 |
3 |
80–91 |
| 6 |
4 |
EtOH |
80 |
9 |
70 |
| 7 |
8 |
EtOH |
80 |
7 |
85 |
| 8 |
14b |
EtOH |
80 |
3 |
92, 92, 90, 90, 88 |
To assess the efficiency of the catalytic activity of CCMFe2O4@L-proline for the synthesis of spirocyclic pyrrolidines/pyrrolizidines/pyrrolothiazolidines and the methodology, the obtained results from the model reactions were compared with previously reported work (Table 2, entries 1–7). Recently, proline (Table 2, entry 2), 4 mol% Pd (CH3CN)2Cl2, 8 mol% CuCl, 14 mol% iPrQuinox, 40 mol% KHCO3, 50 equiv. (Table 2 entry 3), TsNH2 (0.5 mmol%), AgOTf (0.5 mmol%), Au(PPh3)Cl (0.5 mmol%) (Table 2, entry 4), Ni(COD)2 (10 mol%), PBu3 (20 mol%), Et3SiH (5 equiv.) (Table 2, entry 5), and [Cp2TiMe2] (Table 2, entry 6) were employed for the synthesis of spirocyclic compounds. All those methods require toxic organic solvents with high reaction times, whereas the present procedures afford environmental friendliness, a reusable and efficient magnetic catalyst, low catalyst loading, good to excellent yields, and short reaction times. These results clearly accentuate the efficiency of this applied methodology.
Table 2 Comparison of the catalytic activity of the CCMFe2O4@L-proline MNRs with that of other catalysts in the preparation of spirocyclic pyrrolidines/pyrrolizidines/pyrrolothiazolidines
| Entry |
Catalyst/conditions |
Time (min) |
Yield (%) |
Ref. |
| 1 |
Method A: silica/MW; B: BiCl3–silica/MW; C: TiO2–silica/MW |
6–8 |
52–80 |
38 |
| 2 |
L-Proline/toluene DMF CH3CN (1 equiv.) and rt |
1200 |
17–90 |
39 |
| 3 |
4 mol% Pd (CH3CN)2Cl2, 8 mol% CuCl, 14 mol% i-PrQuinox, 40 mol% KHCO3, 50 equiv. of MeOH, O2 balloon, rt, toluene/THF, 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 24 h :1, 24 h |
1440 |
53–72 |
40 |
| 4 |
TsNH2 (0.5 mmol%), AgOTf (0.5 mmol%), Au(PPh3)Cl (0.5 mmol%)/toluene and 85 °C |
360 |
72 |
41 |
| 5 |
Ni(COD)2 (10 mol%), PBu3 (20 mol%), Et3SiH (5 equiv.)/THF and 45–50 °C |
20–120 |
51–85 |
42 |
| 6 |
[Cp2TiMe2]2 (5.0 mol%)/toluene and 110 °C |
1200 |
83 |
43 |
| 7 |
CCMFe2O4@L-proline MNRs |
540 |
80–91 |
This work |
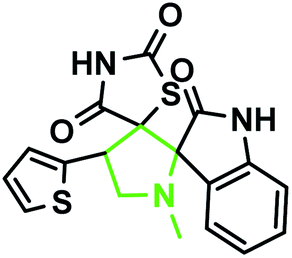
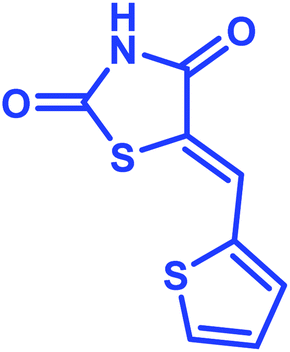
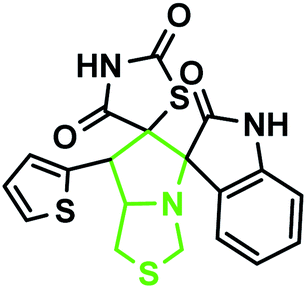
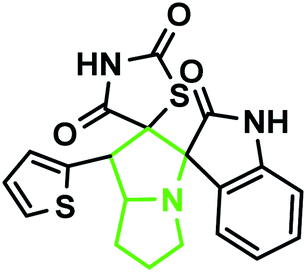
According to the optimized reaction conditions in Table 1, we extended this method to various secondary amino acids and 5-arylidenthiazolidine-2,4-dione derivatives (Table 3). In all cases, 1,3 dipolar cycloaddition was completed in a short time and spirooxindole derivatives were isolated in good to excellent yields. Using this methodology in the reaction with different secondary amino acids produced only one of the possible single isomers with an endo-configuration. The results are shown in Table 3 (entries 1–9), and they demonstrate that the reaction was successfully compatible with a broad range of substituted 5-arylidenthiazolidine-2,4-dione derivatives. In an attempt to expand the scope of our methodology, the possibility of performing the reaction with secondary amino acid derivatives was studied for the synthesis of three types of spiro compounds. From Table 3, it can be observed that the 1,3-dipolar cycloaddition reaction of the 5-arylidenthiazolidine-2,4-dione derivatives as potential dipolarophiles and isatin with sarcosine 5a/thiaproline 5a′′/L-proline 5a′ proceeded smoothly, and the corresponding pyrrolidine/pyrrolizidine/pyrrolothiazole derivatives were obtained in high yields (Table 3).
Table 3 Multicomponent one-pot synthesis of pyrrolidine/pyrrolizidine/thiapyrrolizidine derivatives using the MNR catalysts
| Entry |
Products |
Dipolarophiles |
Compound |
Yield (%) |
MP (°C) |
| Found |
Reported [lit.] |
| 1 |
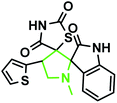 |
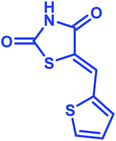 |
1a |
85 |
155–157 |
153 (ref. 22) |
| 2 |
 |
1b |
89 |
158–160 |
— |
| 3 |
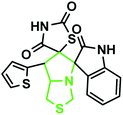
 |
1c |
78 |
195–197 |
— |
| 4 |
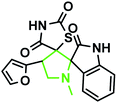 |
 |
2a |
88 |
160–162 |
— |
| 5 |
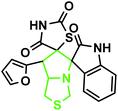 |
2b |
90 |
186–189 |
— |
| 6 |
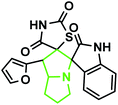 |
2c |
91 |
208–210 |
— |
| 7 |
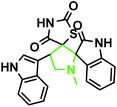 |
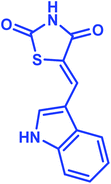 |
3a |
89 |
198–200 |
— |
| 8 |
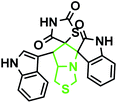 |
3b |
87 |
227–230 |
— |
| 9 |
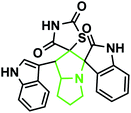 |
3c |
80 |
208–210 |
— |
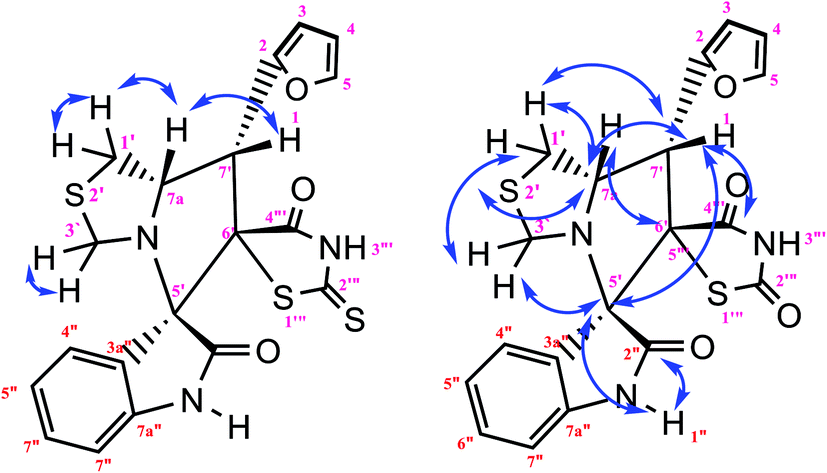
Identification of products. With this catalyst, a safe and efficient three-component reaction for the regioselective generation of spiro-oxindole derivatives has been developed. Initially, the required dipolarophiles were synthesized based on our previous study by the condensation reaction of thiazolidine-2,4-dione and rhodamine with thiophene-2-carboxaldehyde, fural-2-carboxaldehyde and indole-3-carbaldehyde using Fe3O4@SiO2–NH2/Cu(II) MNPs in EtOH31 (Scheme 2).The structures of all the products were established by IR, 1H NMR, 13C NMR and mass spectral analyses. These studies confirmed the spiroheterocyclic ring structures. The high stereoselectivity of this cycloaddition reaction was determined by HMBC, HSQC and COSY techniques. In continuation of the synthesis of novel spiro-oxindole derivatives, through a 1,3-dipolar cycloaddition, three new bonds and five concurrent stereogenic centers, including one quaternary carbon, were incorporated simultaneously in a single step. The cycloaddition reaction of azomethine ylides proceeded in situ via decarboxylative condensation of sarcosine (5a)/thiaproline (5a′′)/L-proline (5a′) with isatin (4a) to 5-arylidenthiazolidine-2,4-diones (7a) with a green catalyst, and a new class of spiro-substituted thiazolidine compounds was formed (Scheme 2). The IR spectrum of the spiro-pyrrolidine derivative (compound 1a) gave stretching at 3453 and 3142 cm−1 for the TZD and indole NH groups, 1765 and 1723 cm−1 for the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of thiazolidine-2,4-dione and 1284 cm−1 for the indole CO groups, respectively. The 1H NMR spectrum of this compound showed three singlets at δ 2.07, 8.03 and 10.75, which correspond to the N1′–CH3, indole N1′′–H and thiazolidine-2,4-dione N3′′′–H, respectively, and the peaks that appeared as a multiplet in the region of δ 7.01–7.66 are due to aromatic protons. Similarly, the 5′-CH2 pyrrolidine ring protons appeared as two triplets at δ 3.85 (J = 8.4 Hz) and 3.55 (J = 9.2 Hz) due to their coupling with benzylic protons Ha and diastereotopic protons Hb and Hc. In particular, the regiochemistry proposed for this product was decided on the basis of a doublet of doublets at δ 4.67 (J = 8.8 Hz) for the C4′–H proton due to its coupling with protons Hc and Hb. If another isomer had been formed, the C4–H would be expected to be a singlet instead of a doublet of doublets. The 13C NMR spectrum of this compound showed two peaks at δ 74.75 and 79.35, reflecting the two spiro carbons. The regiochemistry of this product was also confirmed by its 2D NMR spectrum. The peaks found at δ 35.14, 46.28 and 58.70 correspond to the N′–CH3, methine and methylene carbons, respectively, which was confirmed on the basis of HSQC and H-COSY correlations in the ESI files ( Fig. 5S1a and Fig. 6S1†). The other characteristic carbon peaks were found at δ 168.07, 170.58 and 176.71, which correspond to the C2′′, C4′′′ and C2′′′ carbonyl groups, respectively. The two-dimensional NMR spectra of HMBC and COSY correlations were useful in the signal assignment of 1a, and various characteristic signals are shown in Fig. 10. The H,H-COSY spectrum of this compound revealed two triplets at δ 3.85 (J = 8.4 Hz) and 3.55 (J = 9.2 Hz), two each for diastereotopic protons due to one of the diastereotopic protons and the methine proton, and a doublet of doublets at δ 4.67 (J = 8.8 Hz) for the methine proton due to diastereotopic protons. Within the HSQC spectrum, 35.14, 46.28 and 58.70 correspond to the N′–CH3, methine and methylene carbon, respectively. The selective long-range HMBC correlation and COSY correlation of compound 1a are given in Fig. 9.
O groups of thiazolidine-2,4-dione and 1284 cm−1 for the indole CO groups, respectively. The 1H NMR spectrum of this compound showed three singlets at δ 2.07, 8.03 and 10.75, which correspond to the N1′–CH3, indole N1′′–H and thiazolidine-2,4-dione N3′′′–H, respectively, and the peaks that appeared as a multiplet in the region of δ 7.01–7.66 are due to aromatic protons. Similarly, the 5′-CH2 pyrrolidine ring protons appeared as two triplets at δ 3.85 (J = 8.4 Hz) and 3.55 (J = 9.2 Hz) due to their coupling with benzylic protons Ha and diastereotopic protons Hb and Hc. In particular, the regiochemistry proposed for this product was decided on the basis of a doublet of doublets at δ 4.67 (J = 8.8 Hz) for the C4′–H proton due to its coupling with protons Hc and Hb. If another isomer had been formed, the C4–H would be expected to be a singlet instead of a doublet of doublets. The 13C NMR spectrum of this compound showed two peaks at δ 74.75 and 79.35, reflecting the two spiro carbons. The regiochemistry of this product was also confirmed by its 2D NMR spectrum. The peaks found at δ 35.14, 46.28 and 58.70 correspond to the N′–CH3, methine and methylene carbons, respectively, which was confirmed on the basis of HSQC and H-COSY correlations in the ESI files ( Fig. 5S1a and Fig. 6S1†). The other characteristic carbon peaks were found at δ 168.07, 170.58 and 176.71, which correspond to the C2′′, C4′′′ and C2′′′ carbonyl groups, respectively. The two-dimensional NMR spectra of HMBC and COSY correlations were useful in the signal assignment of 1a, and various characteristic signals are shown in Fig. 10. The H,H-COSY spectrum of this compound revealed two triplets at δ 3.85 (J = 8.4 Hz) and 3.55 (J = 9.2 Hz), two each for diastereotopic protons due to one of the diastereotopic protons and the methine proton, and a doublet of doublets at δ 4.67 (J = 8.8 Hz) for the methine proton due to diastereotopic protons. Within the HSQC spectrum, 35.14, 46.28 and 58.70 correspond to the N′–CH3, methine and methylene carbon, respectively. The selective long-range HMBC correlation and COSY correlation of compound 1a are given in Fig. 9.
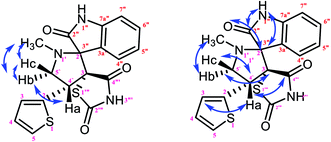 |
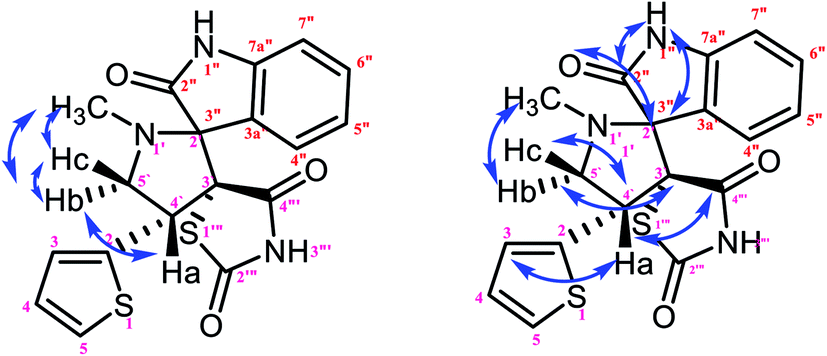
| | Fig. 9 Selected HMBC and H–H COSY correlations of compound 1a. | |
The characteristic methine proton (C4′–H) of the pyrrolidine ring showed an HMBC correlation with the C4′′′ (δ 170.58), C2 (δ 144.33), C4 (δ 137.97), C3 (δ 134.76), C3′ (δ 74.75) and C5′ (δ 58.70) carbons. The diastereotopic methylene protons (C5′–CH2) showed correlations with the C3 (δ 134.36), C3′ (δ 74.75), C2′ (δ 798.35) C4′ (δ 46.28), and N1′–CH3 (δ 35.14) carbons, and the N1′–CH3 protons showed correlations with the C2′ (δ 79.35) and C5′ (δ 58.70) carbons. Selected HMBC compounds from the combination are listed in Table 3.
Based on regioselectivity analysis, the observed regioisomer 1a via path A is more favourable because path A is plausibly favored by attractive π-interactions between aromatic rings, as depicted in Scheme 4. It was assumed that the reaction proceeds through the interactions of the highest occupied molecular orbital (HOMO) azomethine along with the lowest unoccupied molecular orbital (LUMO) of alkene. The approach of azomethine to alkene according to path A would lead to the formation of 1a.
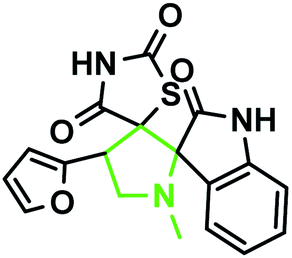
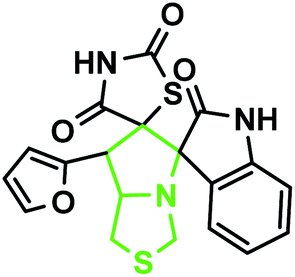
The reaction scope was extended to the synthesis of pyrrolothiazole-substituted thiazolidine-2,4-dione heterocycles using the 5-(thiophen-2-ylmethylene) thiazolidine-2,4-dione and azomethine ylides. The IR spectrum of the 2,4-dione substituted pyrrolothiazole 1b showed stretching at 3438, 3054, and 1703 and 1683 cm−1 for the TZD and indole NH groups and CO groups, respectively. The 1H NMR spectrum of this compound showed two singlets at δ 8.50 and 10.40 for the indole N2′′–H and TZD N3′′′–H, respectively. The C1′–CH2 protons were found at δ 2.70 (J = 8.8 Hz) as a triplet and δ 3.00 (J = 9.2 Hz, 5.6 Hz) as a doublet of doublets, respectively. The doublets at δ 3.76 (J = 5.6 Hz) and 4.17 (J = 6.0 Hz) were due to the C3′–CH2 protons. A doublet at δ 4.3 4 (J = 8.8 Hz) was due to the C7′–H proton, indicating the regioselectivity of the product. A multiple at δ 4.81–4.85 was due to the C7a′–H proton. The aromatic protons appeared in the chemical shift region of δ 6.88–7.67. The 13C NMR spectrum showed the presence of three carbonyl groups at 168.0, 2176.72, and 176.72 and two spiro carbons at 79.33 and 74.73. The structure was further confirmed on the basis of 2D NMR spectral studies. The HSQC correlation assigned the carbons C3′, C7′, and 7a′ to the C3′–CH2, C7′–CH, and C7a–CH protons, respectively. The H,H-COSY spectrum of the compound, shown in Fig. 10, revealed a doublet at δ 4.54 (J = 8.8 Hz), which confirmed only one nearby hydrogen in C7′; this indicated the regioselectivity of the compound. In the long-range HMBC correlation, the characteristic C7′–H was correlated with the methylene carbon (C1′) at 31.73 ppm, N-bonded methine carbon (C7a′) at 67.21 ppm, spiro carbon (C6′) at 78.38 ppm, and carbon (C4) at 138.99 ppm. The methylene protons (C3′–CH2) at δ 3.45 and 3.83 showed HMBC correlations to the methylene carbon (C1′) at 31.73 ppm, the N-bonded methane carbon at 67.21 ppm and one of the spiro carbons (C5′) at 75.02 ppm. Similarly, the methylene protons at δ 2.82 and 3.18 showed correlations with two methine carbons (C7′ and C7a′) at 30.25 ppm and 58.92 ppm, respectively. The selected HMBC correlation of compound 1b is shown in Table 4 and Fig. 10. Based on the above spectral details and the elemental analysis (Table 2), the structure was confirmed to be 7′-(thiophen-2-yl)-7′,7a′-dihydro-1′H,3′H-dispiro[indoline-3,5′-pyrrolo[1,2-c]thiazole-6′,5′′-thiazolidine]-2,2′′,4′′-trione (Table 5, entry 2) (1b).
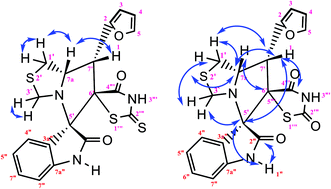 |
| | Fig. 10 Selected HMBC and H–H COSY correlations of compound 1b. | |
Table 4 Selected HMBC correlations of compound 1a
| S. no. |
Proton |
Correlated carbons |
| 1 |
N1′–CH3 (s, 3H) at δ 2.16 |
C5′ (54.48), C2′ (78.76) |
| 2 |
C4′–H (t, 1H, J = 8.8 Hz) at δ 4.92 |
C3′ (74.52), C5′ (54.48), C2 (151.38), C3 (130.55), C4 (139.07), C4′′′ (178.43) |
| 3 |
C5′–CH2 (t, 1H, J = 8.4 Hz) at δ 3.65 (t, 1H, J = 9.2 Hz) at δ 4.27 |
C2′ (78.76), C3′ (74.52), C4′ (46.52), N1′–CH3 (34.71), C3 (130.55) |
| 4 |
Indole N′′–H (s, 1H) at δ 10.84 |
C2′′ (176.18), C2′ (78.76) |
Table 5 Selected HMBC correlations of compound 1b
| S. no. |
Proton |
Correlated carbons |
| 1 |
C1′–CH2 (t, 1H, J = 8.8 Hz) at δ 2.82 (dd, 1H, J = 9.2 Hz, 5.6 Hz) at δ 3.18 |
C7′ (30.25), C7a′ (58.69) |
| 2 |
C3′–CH2 (d, 1H, J = 5.6 Hz) at δ 3.76 (d, 1H, J = 6.0 Hz) at δ 4.17 |
C1′ (35.15), C5′ (79.33), C7a′ (58.69) |
| 3 |
C7′–H (d, 1H, J = 8.8 Hz) at δ 4.34 |
C1′ (35.15), C6′ (74.73), C7a′ (58.69) |
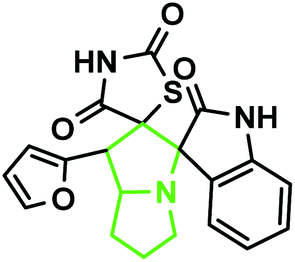
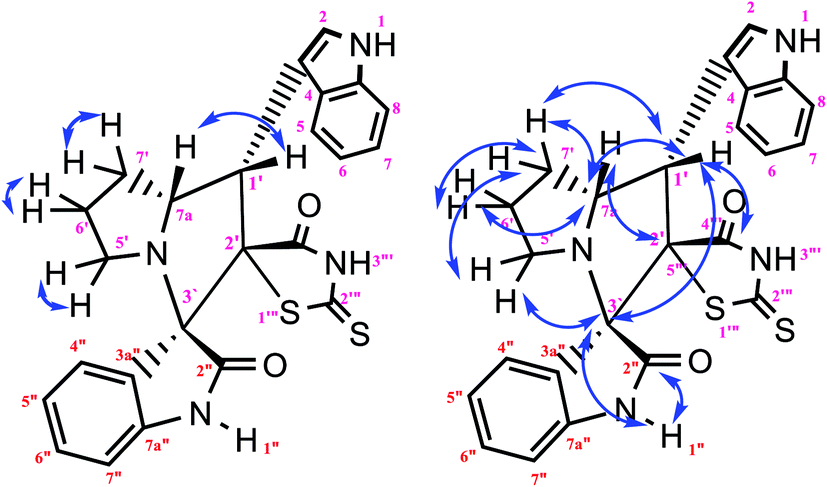
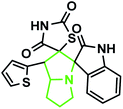
The IR spectrum of the thiazolidine-2,4-dione substituted pyrrolizidine 3c revealed peaks at 3097 cm−1 (N–H stretch), 1765 cm−1 (carbonyl stretch), 1645 cm−1 (amide CO stretch), 1505 cm−1 (N–O asymmetric stretch) and 1336 cm−1 (N–O symmetric stretch). The 1H NMR spectrum of 3c showed three singlets at δ 12.15, 10.59, and 8.29 for the TZD N3′′′–H, indole N2′′–H and C2–H, respectively. Nine aromatic protons in the region of δ 6.78–8.29 and one methane proton (C1′–CH) appeared as a doublet at δ 3.45 (3J = 9.5 Hz) and three methine protons as multiplets at δ 4.11–4.13 and δ 2.59–2.63, 2.30–2.39, and 1.70–1.75, corresponding to C7a′–CH, C5′–CH2, C7′–CH2, and C6′–CH2, respectively. The 13C NMR spectrum of this compound showed three methylene carbons that appeared at δ 46.56, 24.24, and 23.68 and three methane carbons that appeared at δ 66.97, 61.27 and 60.28. Three carbonyl carbons appeared at δ 185.44, 175.89 and 174.99, corresponding to the carbonyls of the oxindole and TZD ring, and nine aromatic carbons appeared in the region of δ 112.89–150.51. The heteronuclear single-quantum correlation assigned the carbons C5′, C6′, C7′, 7a, and C1′ to the C5′–CH2, C6′–CH2, C7′–CH2, C7a–CH, and C1′–CH protons, respectively. The 2D NMR spectra of the HMBC and COSY correlations are useful in the signal assignment of 3c, and various characteristic signals are shown in Fig. 11. From the selected long-range HMBC correlations of compound 3c shown in Table 6, the correlation between the proton (C7′–CH2) interacting with carbons C1′, C7a′, C5′ and protons (C5′–CH2) of the pyrrolizidine ring showed HMBC correlations with the C6′, C7′ and C3′ carbons. The correlation between the protons (C7′–CH2) interacting with carbons C7a′, C5′ and C1′ suggests that the formation of product 3c proceeds via path A (Scheme 3).
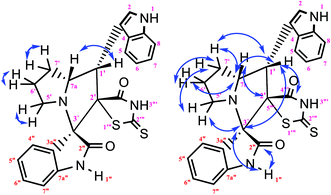 |
| | Fig. 11 Selected HMBC and H–H COSY correlations of compound 3c. | |
Table 6 Selected HMBC correlations of compound 3c
| S. no. |
Proton |
Correlated carbons |
| 1 |
C1′–CH (dd, 1H, J = 9.5 Hz) at δ 3.45 |
C7′ (23.68), C7a (60.28) |
| 2 |
C5′–CH2 (M, 1H) at 2.59–2.63 |
C1′ (24.24), C5′ (46.56), C7a (δ 60.28) |
| 3 |
C7′–CH2 (m, 2H) at δ 2.30–2.39 |
C1′ (δ 24.24), C6′ (δ 21.09), C7a (δ 60.28) |
| 4 |
C6′–CH2 (m, 2H) at δ 1.70–1.75 |
C7′ (23.68), C7a (60.28), C5′ (46.56) |
| 5 |
C7a–CH (m, 1H) at δ 4.11–4.13 |
C7′ (23.68), C1′ (δ 24.24), C3′ (δ 66.97), C6′ (δ 21.09) |
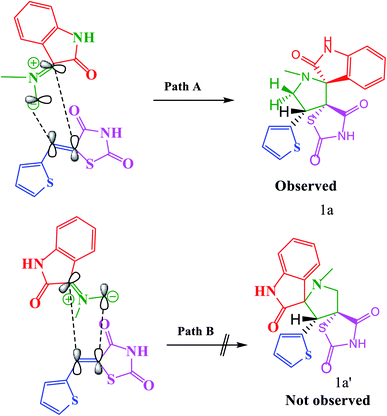 |
| | Scheme 3 Plausible transition state for the formation of 1a, 1a′. | |
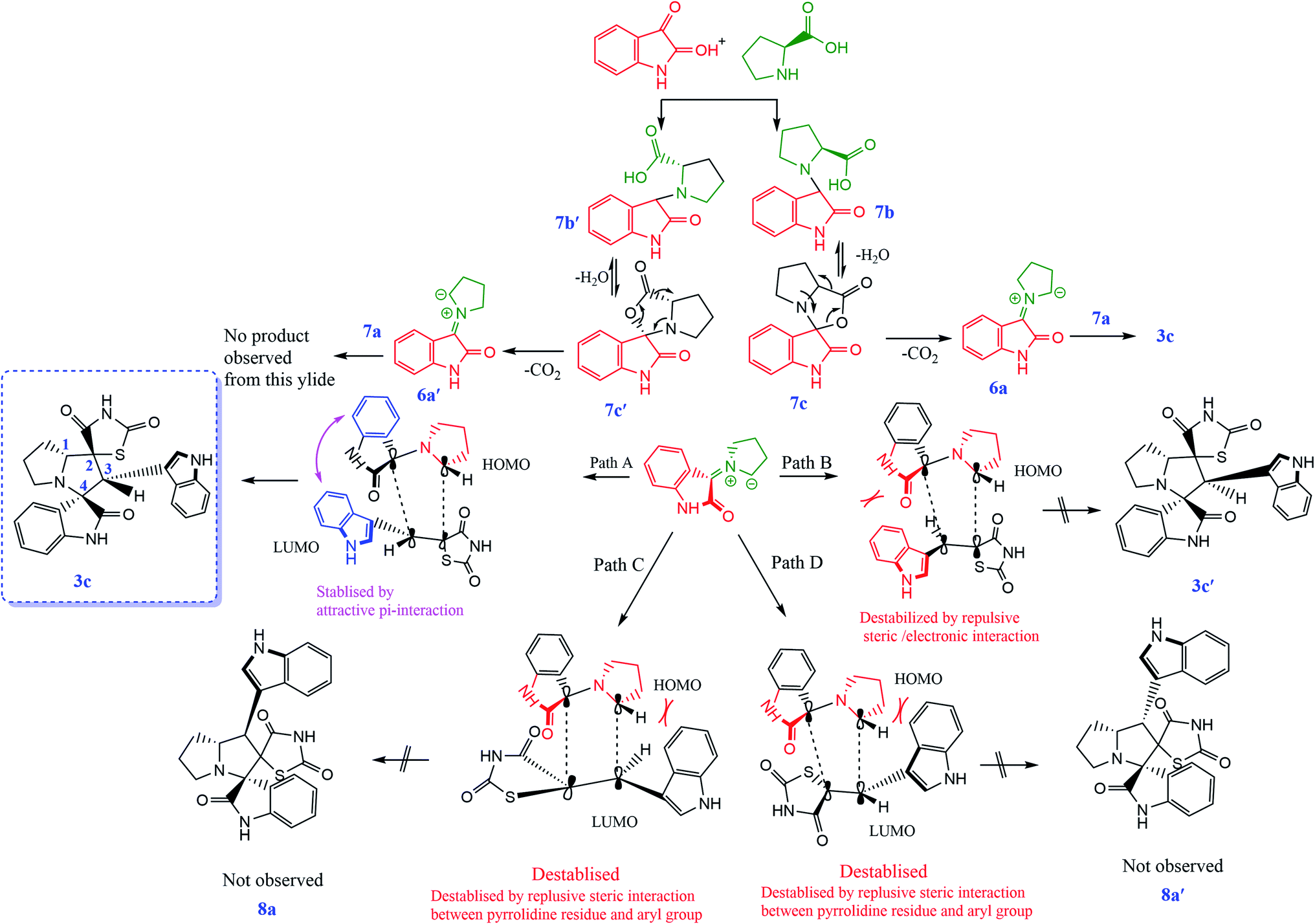
According to recent reports, 1,3-dipolar cycloaddition reactions of azomethine ylides with dipolarophiles proceed in a concerted manner rather than a step-wise diradical transition state.32 Product 3c has four stereocenters, indicating the possibility of 24 = 16 stereoisomers (eight pairs of diastereoisomers). Hence, the reaction is stereospecific, and the relative stereochemistry of the secondary amino acid (L-proline 5a′) and dipolarophile (5-arylidenthiazolidine-2,4-diones 7a) would influence the stereochemistry at the C-1, C-2, and C-3 stereocenters (the stereocenters of 3c are marked as 1, 2, 3, and 4 in Scheme 3). The reaction mechanism involves the formation of the corresponding azomethine ylide, which we have described in three steps. The first step, condensation of the secondary amino acid with the isatin, would lead to the formation of the intermediates 7b and 7b′, with a new C–N bond that can freely rotate. In the second step, one molecule of water is lost due to an intramolecular rearrangement, generating two possible intermediates 7c and 7c′ (Scheme 3).
The reaction of 7a with the ylide 7b leads to the formation of two different diastereomers, 3c and 3c′, depending upon how the ylide 6a approaches dipolarophile 7a (path A and path B). The reaction proceeds through the interactions of the highest occupied molecular orbital (HOMO) of azomethine ylide 6a with the lowest unoccupied molecular orbital (LUMO) of dipolarophile 7a. Following path A, the dipolarophile can approach 6a from both its faces with equal feasibility to 3c. Therefore, the 3c product with the more stabilized transition state will require less free energy for activation. Thus, the observation that 3c′ was not formed can be demonstrated by the plausible transition state, as depicted in path B. The approach of 6a to 7a through path B appears to be hindered by the steric hindrance between the aryl moiety and isatin core, which will result in electrostatic repulsion within these components, enhancing the free energy of activation for this transition state. Also, the observed regioselectivity can be explained by considering the HOMO–LUMO interactions of 6a and 7a. As shown in Scheme 3, either of the two possible pathways (paths C and D) leads to strong steric crowding between the aryl part of dipolarophile 7a and the pyrrolidine residue of 6a in the transition states and agrees well with the non-observation of regioisomers 8a and 8a′ (Scheme 4).
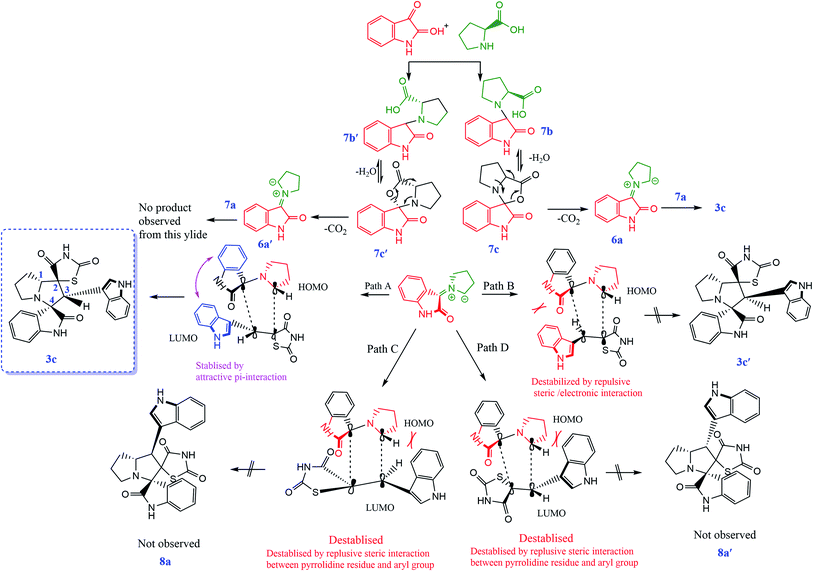 |
| | Scheme 4 Proposed transition states for the formation of 3c, 3c′, 8a and 8a′. | |
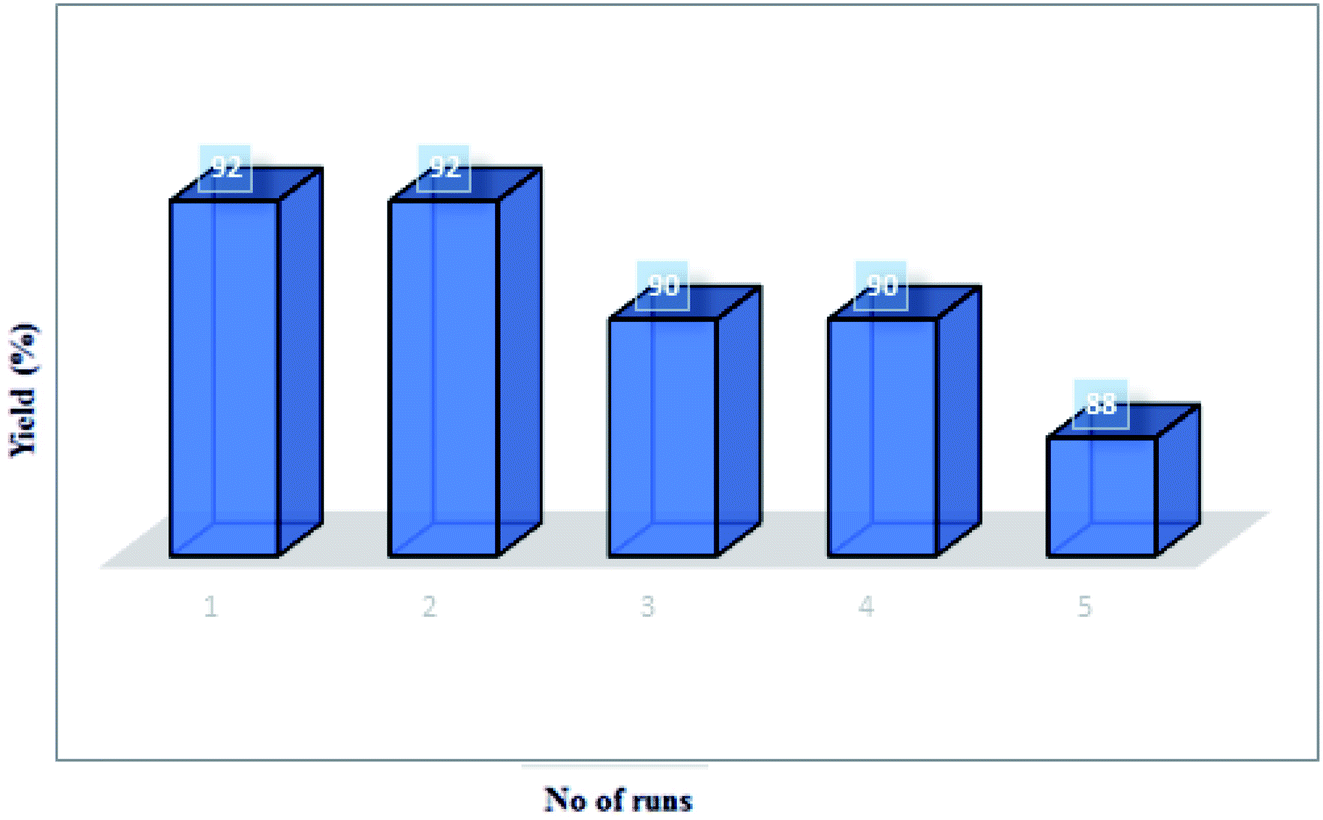
Stability of the MCCFe2O4@L-proline MNRs. The recycling of catalysts is important from practical and environmental perspectives. Therefore, we investigated the stability, high catalytic activity and easy recovery of the catalyst from this reaction. The reusability of the catalyst was investigated using a model reaction between isatin 4a (1.0 mmol), thiazolidine-4-carboxylic acid 5a′′ (1.0 mmol) and dipolarophiles of 5-arylidenthiazolidine-2,4-diones 7a in the presence of 0.03 g of the catalyst at 90 °C. The MNP-supported catalyst in the ethanol readily suspends and separates from the reaction mixture. This separation approach is not time-consuming in comparison with standard methods (centrifugation and filtration) and prevents the loss of heterogeneous catalyst in the process of separation. The separated catalyst was reused in the model reaction five times with minor variation of its catalytic activity, and no considerable decrease in the product yield was observed after repeated cycles of the reaction (run 1, 92%; run 2, 92%; run 3, 90%; run 4, 90%; run 5, 88%) (Fig. 12).
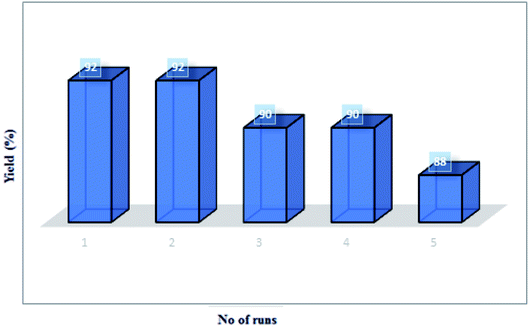 |
| | Fig. 12 The catalytic efficiency of the MCCFe2O4@L-proline MNRs in cycloaddition reactions. | |
Experimental
Materials
All reagents and solvents were purchased from Merck and were used without additional purification. The FT-IR spectra were obtained in the region of 400–4000 cm−1 as KBr pellets with a PerkinElmer 550 spectrometer.
Instrumental measurements
Melting points were recorded with an electrothermal 9200 apparatus and are not corrected. 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra were recorded with a Bruker Advance spectrometer with DMSO-d6 as solvent and TMS as an internal standard. TLC was performed on silica gel polygram SILG/UV 254 nm plates. The magnetite properties of the catalyst were analyzed using a vibrating sample magnetometer (VSM) with a varying magnetic field from −10000 to 10000 on a BHV-S5 instrument. Mass spectra were recorded with a CH7A Varian mat Bremen instrument at 70 eV electron impact ionization, in m/z (rel%). The X-ray powder (XRD) pattern of the catalyst was recorded with an X-Pert Pro instrument (Cu Kα radiation, λ = 1.54 A) in the region of 2Θ = 20–80°. Morphology was analyzed by SEM (Hitachi S4160 scanning electron microscope). Thermogravimetric analysis (TGA) curves were obtained using a PL-STA 1500 device manufactured by Thermal Sciences with a heating rate of 10 °C min−1 over a temperature limited area of 30–1300 °C under N2 atmosphere.
Preparation of L-proline-coated MCCFe2O4 magnetic nanorods
The nano-catalyst was prepared by taking advantage of chemical co-precipitation. Firstly, a solution of FeCl3·6H2O (0.541 g, 2 mmol), MnCl2·2H2O (0.0809 g, 0.5 mmol), CuCl2·2H2O (0.0426 g, 0.25 mmol), and CoCl2·6H2O (0.0595 g, 0.25 mmol) in 150 ml distilled water was prepared. Then, this solution was added to 100 ml NH4OH (3 mol L−1) solution dropwise at room temperature, and the solution was warmed to 95 °C. The obtained mixture was heated for 5 h with intense stirring. Eventually, 1 g of L-proline and NH4OH (25 wt%, 20 ml) were added to the mixture until the pH was raised to 11, and a black suspension was formed. The mixture was filtered, washed and dried at 70 °C for 12 h.
Synthesis of spiro-pyrrolidine, pyrrolizidine, and pyrrolothiazole derivatives
Isatin (1.0 mmol), sarcosine (1 mmol), and 5-arylidene-1,3-thiazolidine-2,4-dione (1 mmol) were placed in a 10 ml round-bottomed flask in EtOH (5 ml). Sequentially, MCCFe2O4@L-proline MNRs (4 mol%) were added, and the resulting mixture was stirred at 100 °C. The reaction progress was monitored by TLC. After completion of the reaction, the catalyst was separated with an external magnet and the solvent was evaporated. The separated catalyst was washed several times with EtOH and water, dried under vacuum and utilized four times for the same reaction. The final product was purified by column chromatography using n-hexane and ethyl acetate (3 to 2) as the eluent. All novel spiro-products were identified by physical and spectroscopic data.
Conclusions
In summary, we developed an efficient, green one-pot procedure and a new approach for the construction of pyrrolidine/pyrrolizidine/pyrrolothiazolidine skeletons based on tandem cycloaddition using new synthetic MCCFe2O4@L-proline nanorods. The nanocatalyst can be recycled and reused for the next run without any significant changes in its catalytic activity. This methodology is applicable to provide spiro thiazolidines with excellent regio- and stereoselectivity.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
This work was supported by the National Institute for Medical Research Development (NIMAD) [grant number 971338]. Also, the authors gratefully acknowledge the support for this work received from the Mazandaran University of Medical Sciences “Professor's Projects and Sabbatical Funds”.
References
- I. Coldham and R. Hufton, Chem. Rev., 2005, 105, 2765–2810 CrossRef CAS PubMed
 .
. - G. S. Kumar, R. Satheeshkumar, W. Kaminsky, J. Platts and K. J. R. Prasad, Tetrahedron Lett., 2014, 55, 5475–5480 CrossRef .
- G. Periyasami, R. Raghunathan, G. Surendiran and N. Mathivanan, Bioorg. Med. Chem. Lett., 2008, 18, 2342–2345 CrossRef CAS PubMed .
- C. Marti, Eur. J. Org. Chem., 2003, 2209–2229 CrossRef CAS .
- A. Ilenia Alfano, A. Zampella, E. Novellino, M. Brindisi and H. Lange, React. Chem. Eng., 2020, 5, 2091–2100 RSC .
- A. Dandia, S. Kumari, S. Maheshwari and P. Soni, Experimental and Theoretical Approach. 2017, vol. 3, p. 39 Search PubMed .
- L. E. Carloni, S. Mohnani and D. Bonifazi, Eur. J. Org. Chem., 2019, 44, 7322–7334 CrossRef .
- R. L. Garnick and P. W. Le Quesne, J. Am. Chem. Soc., 1978, 100, 4213–4219 CrossRef CAS .
- P. Saraswat, G. Jeyabalan, M. Z Hassan, M. U. Rahman and N. K. Nyola, Synth. Commun., 2016, 46, 1643–1664 CrossRef CAS .
- B. Yu, D. Q. Yu and H. M Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed .
- A. R. Bekhradnia, S. Arshadi and S. A. Siadati, Chem. Pap., 2014, 2, 283–290 Search PubMed .
- C. Mhiri, S. Boudriga, M. Askri, M. Knorr, D. Sriram, P. Yogeeswari and C. Strohmann, Bioorg. Med. Chem. Lett., 2015, 25, 4308–4313 CrossRef CAS PubMed .
- F. Rouatbi, M. Askri, F. Nana, G. Kirsch, D. Sriram and P. Yogeeswari, Tetrahedron Lett., 2016, 57, 163–167 CrossRef CAS .
- G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef CAS PubMed .
- M. Akhavan, N. Foroughifar, H. Pasdar and A. Bekhradnia, Green Synthesis, Combinatorial Chemistry & High Throughput Screening, 2019, vol. 22, pp. 716–727 Search PubMed .
- C. D. Barros, A. A. Amato, T. B. de Oliveira, K. B. R. Iannini, A. L. da Silva, T. G. da Silva and F. D. A. R. Neves, Bioorg. Med. Chem., 2010, 11, 3805–3815 CrossRef PubMed .
- V. Mamakou, I. Eleftheriadou, N. Katsiki, K. Makrilakis, K. Tsioufis and N. Tentolouris, Curr. Vasc. Pharmacol., 2018, 16(1), 70–78 CAS .
- R. Mishra, K. K. Jha, S. Kumar and I. Tomer, Der Pharma Chem., 2011, 3, 38–54 CAS .
- R. Murugan, S. Anbazhagan and S. S. Narayanan, Eur. J. Med. Chem., 2009, 44, 3272–3279 CrossRef CAS PubMed .
- A. Y. Barkov, N. S. Zimnitskiy, V. Y. Korotaev, I. B. Kutyashev, V. S. Moshkin and V. Y. Sosnovskikh, Tetrahedron, 2016, 72, 6825–6836 CrossRef CAS .
- S. U. Maheswari, K. Balamurugan, S. Perumal, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem. Lett., 2010, 24, 7278–7282 CrossRef PubMed .
- P. Dhanalakshmi, S. S. Babu, S. Thimmarayaperumal and S. Shanmugam, RSC Adv., 2015, 5, 33705–33719 RSC .
- M. Afradi, S. A. Pour, M. Dolat and A. Yazdani-Elah-Abadi, Appl. Organomet. Chem., 2018, 32, e4103 CrossRef .
- R. Jahanshahi and B. Akhlaghinia, RSC Adv., 2016, 6, 29210–29219 RSC .
- S. A. Pour, H. R. Shaterian, M. Afradi and A. Yazdani-Elah-Abadi, J. Magn. Magn. Mater., 2017, 438, 85–94 CrossRef .
- N. Lee and T. Hyeon, Chem. Soc. Rev., 2012, 41, 2575–2589 RSC .
- P. Thakur, R. Sharma, M. Kumar, S. C. Katyal, N. S. Negi, N. Thakur and P. Sharma, Mater. Res. Express, 2016, 3, 075001 CrossRef .
- A. Ponti and G. Molteni, Eur. J. Org. Chem., 2020, 39, 6173–6191 CrossRef .
- T. P. Loh, L. C. Feng, H. Y. Yang and J. Y. Yang, Tetrahedron Lett., 2002, 43, 8741–8743 CrossRef CAS .
- R. Arulmurugan, B. Jeyadevan, G. Vaidyanathan and S. Sendhilnathan, J. Magn. Magn. Mater., 2005, 288, 470–477 CrossRef CAS .
- M. Akhavan, N. Foroughifar, H. Pasdar, A. Khajeh-Amiri and A. Bekhradnia, Transition Met. Chem., 2017, 42, 543–552 CrossRef CAS .
- C. E. P. Galvis and V. V. Kouznetsov, Org. Biomol. Chem., 2013, 11, 7372–7386 RSC .
- Z. Esam, M. Akhavan, A. R. Bekhradnia, M. Mohammadi and S. Tourani, Catal. Lett., 2020, 8, 1–20 Search PubMed .
- Z. Esam, M. Akhavan and A. R. Bekhradnia, Appl. Organomet. Chem., 2021, 35 DOI:10.1002/aoc.6005 .
- S. N. Azizi, M. J. Chaichi, P. Shakeri and A. Bekhradnia, J. Fluoresc., 2013, 23, 227–235 CrossRef CAS PubMed .
- E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, Curr. Org. Synth., 2017, 14, 557–567 CrossRef CAS .
- T. Tamoradi, M. Masoumeh and M. Mohammadi, ChemistrySelect, 2020, 5, 5077–5081 CrossRef CAS .
- A. S. Babu and R. Raghunathan, Tetrahedron, 2007, 63, 8010–8016 CrossRef .
- I. Ibrahem, R. Rios, J. Vesely and A. Córdova, Tetrahedron Lett., 2007, 48, 6252–6257 CrossRef CAS .
- R. Jana, T. P. Pathak, K. H. Jensen and M. S. Sigman, Org. Lett., 2012, 14, 4074–4077 CrossRef CAS PubMed .
- M. Shi, L. P. Liu and J. Tang, Org. Lett., 2006, 8, 4043–4046 CrossRef CAS PubMed .
- X. Q. Tang and J. Montgomery, J. Am. Chem. Soc., 1999, 121, 6098–6099 CrossRef CAS .
- I. Bytschkov, H. Siebeneicher and S. Doye, Eur. J. Org. Chem., 2003, 2003, 2888–2902 CrossRef .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00841b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab