
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh pressure polymorphism of LiBH4 and of NaBH4†
Adrien Marizy a,
Grégory Genestea,
Gaston Garbarinob and
Paul Loubeyre*a
a,
Grégory Genestea,
Gaston Garbarinob and
Paul Loubeyre*a
aCEA, DAM, DIF, F-91297 Arpajon, France. E-mail: paul.loubeyre@cea.fr
bESRF, The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
First published on 21st July 2021
Abstract
The pressure-induced structural changes in LiBH4 and in NaBH4 have been investigated experimentally up to 290 GPa by coupling Raman spectroscopy, infrared absorption spectroscopy and synchrotron X-ray diffraction. This data set is also analysed in the light of Density Functional Theory calculations performed up to 600 GPa. The [BH4]− unit appears to be remarkably resistant under pressure. NaBH4 remains stable in the known Pnma γ-phase up to 200 GPa and calculations predict a transition to a metallic polymeric C2/c phase at about 480 GPa. LiBH4 is confirmed to exhibit a richer polymorphism. A new Pnma orthorhombic phase VI is found to be stable above 60 GPa and there are hints of a possible phase VII above 160 GPa. DFT calculations predict that two other high pressure LiBH4 phases should appear at about 290 and 428 GPa. A very slight solubility of H2 inside phases II, III and V of LiBH4 is observed. A NaBH4(H2)0.5 complex is predicted to be stable above 150 GPa.
Introduction
The unique potential of LiBH4 and NaBH4 for solid hydrogen storage is widely discussed in the literature, since these compounds could theoretically meet the 2025 DOE target for onboard hydrogen storage in terms of gravimetric and volumetric hydrogen densities (respectively 18.4 wt% and 122.5 kg H2 per m3 for LiBH4 and 10.6 wt% and 113.1 kg H2 per m3 for NaBH4). However, the strong covalent bond in the BH4− units of borohydrides requires a high decomposition temperature (>500 °C for NaBH4 (ref. 1) and 380 °C for LiBH4 (ref. 2)) which hampers their use in practical applications. A lot of chemical engineering work has already been done with some success,3–10 through catalyst addition or nanotailoring, to try to lower their decomposition temperature but near-room-temperature hydrogen desorption/absorption has not been achieved yet. Therefore, it is of tremendous importance to continue to explore the possible mechanisms of destabilization of the boron–hydrogen bonds.Recently, pressure has emerged as an effective means of producing new structures and materials, potentially interesting for hydrogen storage.11–14 Indeed, the application of high pressure (>GPa) significantly reduces interatomic distances and thus modifies the chemical bonding, the molecular configuration and the crystal structure. More specifically, for borohydrides, a high pressure study is an effective way of producing new polymorphs with increased coordination numbers, new [BH4]− anion charges, reduced H–H distances and mono/bi/tri-dentate configurations. Disclosing such effects should be useful to strengthen our understanding of the factors governing the B–H bond stability. Besides, the search for novel materials with targeted properties using Density Functional Theory (DFT) calculations has recently made tremendous progress. One way to evaluate the reliability of such calculations to explore the configuration space of the borohydrides family is to test them against the high pressure phase diagrams of selected compounds, such as LiBH4 and NaBH4. Finally, it has been demonstrated in many compounds that hydrogen uptake drastically increases under very high hydrogen pressure. Not only are interstitial hydrides more filled with hydrogen15 but new polyhydrides16,17 or hydrogen complexes such as NH3BH3(H2)1.5 are also created.18,19 Hence, is it possible to insert more hydrogen into LiBH4 and in NaBH4? Even if the new dense polymorphs/hydrides found under pressure cannot be recovered at atmospheric pressure, a chemical substitution could then be attempted to stabilize them at ambient pressure.
The present work aims to experimentally and numerically extend the known phase diagrams of NaBH4 and LiBH4 up to the metallization pressure and to numerically explore the possibility of including more hydrogen inside NaBH4. Sodium borohydride was studied at room temperature up to 90 GPa by X-ray diffraction and Raman spectroscopy with neon or hydrogen as a pressure medium and up to 200 GPa by infrared absorption spectroscopy without any pressure medium. LiBH4 was studied at room temperature up to 130 GPa by X-ray diffraction in a helium pressure medium, by Raman spectroscopy up to 70 GPa and by IR absorption up to 290 GPa without any pressure medium. Ab initio calculations were conducted to complement the experimental investigation. The use of molecular dynamics simulations, in particular, has enabled us to discover a potential NaBH4(H2)0.5 compound.
Current knowledge on the phase diagrams of LiBH4 and NaBH4
Many works have already explored the phase diagram of NaBH4 and LiBH4 up to the 50 GPa pressure range. At ambient pressure and temperature, NaBH4 has a cubic phase (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) in which the tetrahedral units [BH4]− are disordered due to thermal agitation.20–24 This structure was found to transform via a disorder–order transition into a tetragonal P
m) in which the tetrahedral units [BH4]− are disordered due to thermal agitation.20–24 This structure was found to transform via a disorder–order transition into a tetragonal P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c structure, either below 190 K at atmospheric pressure21,25,26 or above 6 GPa at ambient temperature. A tetragonal to orthorhombic transition (space group Pnma) is observed at 9 GPa.27–33 This experimental sequence of transitions was not initially predicted by DFT27 but subsequent numerical studies accurately described it.34–37 It should be noted that two nearly identical structures have been proposed experimentally for the tetragonal phase26,38 and that calculations show that the most stable tetragonal structure is described in the higher symmetry P42/nmc space group and not in the P21c space group.39 The last experimental work published on NaBH4 suggests a monoclinic transition near 20 GPa due to anomalies in the evolution versus pressure of the B–H Raman vibrons.32 Nevertheless, previous X-ray studies up to 30 GPa have not reported any monoclinic phase apparition.28,30,31
21c structure, either below 190 K at atmospheric pressure21,25,26 or above 6 GPa at ambient temperature. A tetragonal to orthorhombic transition (space group Pnma) is observed at 9 GPa.27–33 This experimental sequence of transitions was not initially predicted by DFT27 but subsequent numerical studies accurately described it.34–37 It should be noted that two nearly identical structures have been proposed experimentally for the tetragonal phase26,38 and that calculations show that the most stable tetragonal structure is described in the higher symmetry P42/nmc space group and not in the P21c space group.39 The last experimental work published on NaBH4 suggests a monoclinic transition near 20 GPa due to anomalies in the evolution versus pressure of the B–H Raman vibrons.32 Nevertheless, previous X-ray studies up to 30 GPa have not reported any monoclinic phase apparition.28,30,31
At room temperature and atmospheric pressure, the disordered cubic Fmm structure is shared by all other univalent borohydrides but LiBH4.40 LiBH4 has a richer polymorphism, as shown as early as 1974 by Pistorius41 and slightly revised and refined 30 years later.33,42–47 Its phase at ambient conditions (phase II) is a Pnma orthorhombic structure without disorder48 and with undistorted [BH4]− tetrahedra.44,49 This phase transforms with slight hydrogen desorption (0.3 wt% (ref. 50)) into a hexagonal one at 381 K (phase I, space group P63mc).44,48 Under pressure at ambient temperature, at 1 GPa, a new phase appears. It was first described in the Ama2 space group by Filinchuk et al.45 and lately better described in the I41/acd space group46,51 (phase III). This phase was proven to be metastable down to atmospheric pressure below 200 K.42,52 This phase III starts to transform into the well-known disordered cubic phase (phase V, Fmm) from 17 GPa at 300 K and at lower pressure if temperature is raised.43 Nakano et al. identified that this transition was achieved through an intermediate tetragonal but also disordered phase V′, between 17 GPa and 30 GPa at room temperature.46 The authors also pointed out that a significant weakening of the recorded XRD signal near 50 GPa probably portended a new phase transition. After this study, Yao & Klug used DFT calculations to predict two possible monoclinic candidate phases for phase VI with a C2/m and a C2/c (distorted NiAs) space group. LiBH4 was also predicted to be metallic in a new polymeric monoclinic phase VII at 425 GPa.51 Although the calculations accurately predicted that the tetrahedron would have an ideal geometry in phase II,53 they also predict other structures for phases I and II, which are clearly at variance with the experimental data54–57 or have later been contradicted.58 Besides, several recent DFT studies are not accurate at high pressure either because they did not take into account the stable I41/acd structure59,60 or because they did not have enough atoms in their supercells to find it.61
It has to be pointed out that historically a PT domain between phase I and III was assigned to a new phase IV by Pistorius et al.41 No new phase was found by synchrotron X-ray diffraction but a mixture of the two phases at the transition.43 Hence, phase IV does not exist but the numeration remained unchanged. Therefore, in this study, the usual numeration will be kept.
Methods
Sample preparation
NaBH4 and LiBH4 samples were obtained from commercial Sigma-Aldrich powder with a purity of respectively 99% and 95%. Manipulations of the two borohydrides were made inside a high-purity argon glove box (H2O < 1 ppm). After the loading of the samples inside the Diamond Anvil Cell (DAC), the DAC is manually closed tightly to avoid any water contamination outside the glovebox. Diamond anvil culets (300 μm, 150 μm, 100 μm or 40 μm in diameter) were used to cover various pressure domains. The rhenium gasket was covered with a 1000 Å thick layer of gold to ensure good airtightness prior to any loading with a pressure transmitting gas under 1400 bars. When no pressure medium was used (IR absorption measurements and Raman spectroscopy on LiBH4), no gold was used on the gasket. When the DACs were loaded with gas, neon and hydrogen were used with a moisture content inferior to 2 ppm (Air Products Premier Line) for NaBH4 and with helium with a moisture content inferior to 20 ppb (Air Products BIP Technology) for LiBH4. Several purges/pumping cycles were done to dry all the gas loading device before the opening of the DAC for gas loading, especially for LiBH4 because of its highly hygroscopic nature.Sample characterisations
LiBH4 and NaBH4 are weak X-ray scatterers, which renders their high pressure structural characterisation in a DAC particularly challenging. X-ray diffraction measurements on LiBH4 and NaBH4 were performed at the ID27 beamline of the European Synchrotron Radiation Facility with a wavelength of 0.3738 Å, an X-ray beam of around 5 μm and a MAR-CCD detector. An XRD pattern of LiBH4 at 68 GPa was also collected using a MAR555 detector on ID15b (0.4111 Å) for improved sensitivity. The pressure was measured using the equation of state of a small gold piece placed next to the sample for LiBH4 and with the hydrostatic ruby pressure scale for NaBH4. The X-ray diffraction patterns were analysed using DIOPTAS62 and the FULLPROF software.63 Uncertainties are estimated to be ±2% for pressure and ±0.2 Å3 for volume.Infrared absorption and Raman spectroscopy have been performed to complement the XRD structural investigation. First, discontinuities in the frequency shift versus pressure of the various excitation modes of the BH4 entity reflect the variation of the crystal field acting on it with high sensitivity. Therefore, they can reveal subtle structural changes that would be difficult to extract from XRD, such as a change of orientational order of the BH4 tetrahedra. Besides, for spectroscopic studies, the size of the sample is much less problematic than for XRD and so IR absorption can be used to extend the structural investigation up to 200 GPa.
Infrared absorption spectroscopy was performed at room temperature using the synchrotron radiation source at the SMIS beamline of the French SOLEIL Synchrotron. The IR experimental configuration already described previously64 includes a horizontal infrared microscope made up of two Schwarzschild objectives (×15, N.A. 0.5, W.D. = 43 mm) producing a 22 μm (FWHM) IR spot. Spectra were taken with a 4 cm−1 resolution with 512 accumulations. The high-frequency edge of the T2g Raman band of the diamond anvil was used to estimate the pressure according to Akahama's calibration.65
Confocal Raman spectroscopy was performed using an Alpha300M+ (Witec) spectroscopic system with a continuous Ar–Kr laser emitting at 488.0 nm. The Stokes Raman signal was collected in back-scattering geometry by a CCD coupled to a 600 g mm−1 grating.
Ab initio calculations
Ab initio calculations have been performed to guide the discussion of the experimental data with insights at a microscopic level. We have performed DFT calculations using the Generalized Gradient Approximation in the form proposed by Perdew, Burke and Ernzerhof (GGA-PBE).66 We have used the ABINIT code.67 Four kinds of calculations have been performed: first, structural optimizations under fixed pressure; second, phonon calculations in the framework of the Density-Functional Perturbative Theory (DFPT);68,69 third Molecular Dynamics (MD) runs in the isothermal–isobaric (NPT) ensemble; fourth constrained Ab Initio Random Structure Searching (AIRSS).70The structural optimizations have been done in the Projector Augmented Wave (PAW) framework, with a plane-wave (PW) cut-off of 40 Hartrees, and a 6 × 6 × 6 k-point mesh for insulating phases and 12 × 12 × 12 k-point mesh for metallic phases. All the non-disordered experimental or predicted stable polymorphs for LiBH4 were also adapted and optimized for NaBH4 and conversely.
DFPT calculations have been performed, at selected pressures, using both the PAW framework (PW cut-off 40 Ha) and ONCVPSP norm-conserving pseudopotentials (PW cut-off 50 Ha).71 In the latter case, the structures were re-optimized using ONCVPSP pseudopotentials prior to the DFPT calculation.
The MD runs have been performed on a cell of Pnma NaBH4 with additional hydrogen molecules, to inspect the possibility of hydrogen insertion in the borohydride, and the structure that may be formed in such conditions. MD runs in the (NPT) ensemble have been performed using a 192 atoms supercell of the disordered cubic phase V of LiBH4 over more than 5000 steps in order to obtain the equation of state at 300 K. For checking purpose (NVT) MD runs have been performed using the averaged volume obtained in (NPT) MD runs, providing the right pressure with a precision of 0.3 GPa.
AIRSS was used to search for the ground state of phase VI of LiBH4. The starting cell was given by the experiment. Six configurations were first generated with a random distribution of lithium and boron atoms. Then, four hydrogen atoms were randomly placed inside a shell around each boron atom. A total of 34 random configurations with 24 atoms were optimized by DFT at 68 GPa. This seemed sufficient to identify the crystal structure of phase VI.
Results and discussion
Pressure-induced structural changes in NaBH4
A previous Raman study of NaBH4 under pressure up to 30 GPa suggested that a new phase transition to a possible monoclinic α-LiAlH4 (P21/c)-type phase occurred at 14 GPa.32 To confirm this hypothesis, the Raman stretching modes of the B–H bonds were measured up to 60 GPa. The corresponding background-corrected spectra are represented in Fig. 1 along with the pressure evolution of the vibrons of the Pnma phase. Six peaks can be clearly followed up to 30 GPa but the signal becomes broader as the pressure increased, making the determination of the peak position less accurate even with the subtraction of the second-order contribution of the diamond. Nevertheless, the six peaks, sometimes in the forms of shoulders, have been followed up to 60 GPa. The evolution of their frequencies is sub-linear as a function of pressure and no new peak seems to emerge, in contrast to George et al.'s suggestion.32 It has to be noted that the broadening of the Raman lines is probably due to non-hydrostaticity, as the Raman spectra in George et al.'s study are broader than ours from 12 GPa on. Hence, from the Raman measurements alone, it is not possible to confirm the existence of a phase transition above 20 GPa. | ||
| Fig. 1 (a) Raman spectra of high pressure Pnma NaBH4 as a function of pressure (neon as a pressure medium). (b) Corresponding pressure evolution of the six Raman peaks related to B–H stretching vibrations. | ||
Diffraction on the Pnma phase had been already performed with silicone fluid as a pressure medium up to 30 GPa by Kumar et al.28 and also by Filinchuk et al.30 who decompressed it down to 7 GPa. We extended the X-ray diffraction study of NaBH4 up to 70 GPa in neon pressure transmitting medium. Our measurements are in very good agreement with both the already published data and with the calculated equation of state of the Pnma phase, as can be seen in Fig. 2a. A Vinet adjustment of a combination of the present data and the already published data gives a bulk modulus of 19.6 GPa with its pressure derivative of 4.6 and a volume at atmospheric pressure of 56.5 Å3. Fig. 2b presents a Le Bail refinement of the NaBH4 diffraction pattern in the Pnma phase under hydrogen at 63 GPa.
 | ||
Fig. 2 (a) Experimental and calculated equations of state of Pnma NaBH4 along with the evolution of the corresponding cell parameters under pressure (inset). The parameters of the Vinet fits for experimental and calculated data are respectively: K0 = 19.6 & 15.7 GPa;  = 4.6 & 5.2; V0 = 56.5 & 56.1 Å3. Error bars smaller than or equal to the symbol size if not shown. (b) Le Bail refinement of the recorded diffraction pattern at 63 GPa in hydrogen (a = 6.11 Å, b = 3.71 Å, c = 4.82 Å). = 4.6 & 5.2; V0 = 56.5 & 56.1 Å3. Error bars smaller than or equal to the symbol size if not shown. (b) Le Bail refinement of the recorded diffraction pattern at 63 GPa in hydrogen (a = 6.11 Å, b = 3.71 Å, c = 4.82 Å). | ||
 | ||
| Fig. 3 Enthalpy difference per formula unit as a function of pressure for various NaBH4 polymorphs. The Pnma NaBH4 structure is taken as a reference. A solid line indicates a stable polymorph. The black arrows indicate if the structure was optimized under increasing or decreasing pressure. The I41/acd structure adapted from phase III of LiBH4 (not shown here) was also optimized at low pressure (<10 GPa) and was found to be very unstable in this pressure range. Structural optimization of the structure adapted from C2/c LiBH4 gave rise to a new structure during pressure increase. This new polymorph was then optimized under decreasing pressure and turned out to be of polymeric and metallic type. | ||
Infrared absorption spectroscopy was carried out above 60 GPa and three IR bands centred around 3000 cm−1 corresponding to the B–H stretching modes have been followed up to 190 GPa (see ESI, Fig. S1†). Their evolution is linear when the pressure increases, which tends to show that no new phase transition nor decomposition occur under very high pressure. After structural optimizations of all the polymorphs tested, the observed phase II to phase III transition pressure could be numerically reproduced. The Pnma phase remained stable up to 474 GPa, above which NaBH4 is predicted to metallize in a new C2/c structure (Fig. 3). This structure appeared during pressure-increasing optimization of the adapted non-metallic C2/c LiBH4 polymorph found by Yao & Klug.51 This metallic sodium borohydride has a polymeric/layered feature without any more tetrahedral (see ESI, Fig. S5†). Each boron & hydrogen-containing layer is made of zigzag chains linked between them by a Bchain1–H–B–H–Bchain2 link. Each chain is composed by a pattern which alternates simple B–B bonds and B–2H–B bonds as in diborane(6) or maybe as in diborane(4) (plus terminal hydrogen atoms).
The phase diagram of NaBH4 can thus be extended and a new one is presented in Fig. 4.
 | ||
| Fig. 4 Temperature vs. pressure phase diagram of NaBH4 adapted and completed from Sundqvist.40 Green writing indicates a phase with disorder caused by tetrahedral reorientation. Na, B and H atoms are respectively represented by yellow, green and pink spheres. Beige background indicates the new high pressure phase diagram proposed by this work. | ||
Pressure-induced structural changes in LiBH4
X-ray diffraction was performed on LiBH4 embedded in helium as a hydrostatic pressure medium. As can be seen in Fig. 5, the transition pressures and measured volumes are in good agreement with Nakano et al.‘s previous study.46 Even the intermediate disordered phase V′ appears, while it was hypothesised that it would appear only in non-hydrostatic conditions. The volume of the disordered cubic phase V was also calculated by averaging the volume over the equilibrated isothermal–isobaric MD trajectories at 300 K and several pressures. This calculated volume agrees well with the experimental data within the experimental error. Starting from 45 GPa, the recorded X-ray signal of the cubic phase V slowly decreases in intensity as already seen by Nakano et al.46 and new peaks of a phase VI start to emerge. A first run with 300 μm culets leads to a mixture of phase V and VI up to 72 GPa, the highest pressure achieved in this run. The mixture was confirmed by Raman spectroscopy. The six new peaks detected at this pressure were not sufficient to determine a plausible cell for the new phase VI. Therefore, X-ray diffraction was then performed with a more sensitive detector (mar555) on the cell that was used for the Raman study here after. At 68 GPa, the LiBH4 sample without pressure medium was found totally transformed into phase VI and an orthorhombic cell containing four LiBH4 unit formula can be inferred from the first eleven reflections seen in the image plate (up to 2θ = 16°). The highest angle peaks are broad and weak but could also be described by this orthorhombic cell. As can be seen in (Fig. 5a), this cell corresponds to the computed Pnma phase VI with the same structure as the NaBH4 Pnma phase III. This structure is calculated to be thermodynamically stable from 29 GPa in comparison to the I41/acd structure and not the disordered Fmm phase V. Besides, to be sure of the hydrogen atoms orientation, more than thirty random draws of the atoms inside the experimental cell followed by structural optimization gave the NaBH4-like Pnma structure as the most stable configuration. Besides, the apparition of this phase for LiBH4 just after the Fmm disordered phase V is logical, as the structure is only a pressure-distorted version of the P42/nmc phase II of NaBH4 which appears right after the same disordered Fmm phase I of NaBH4.
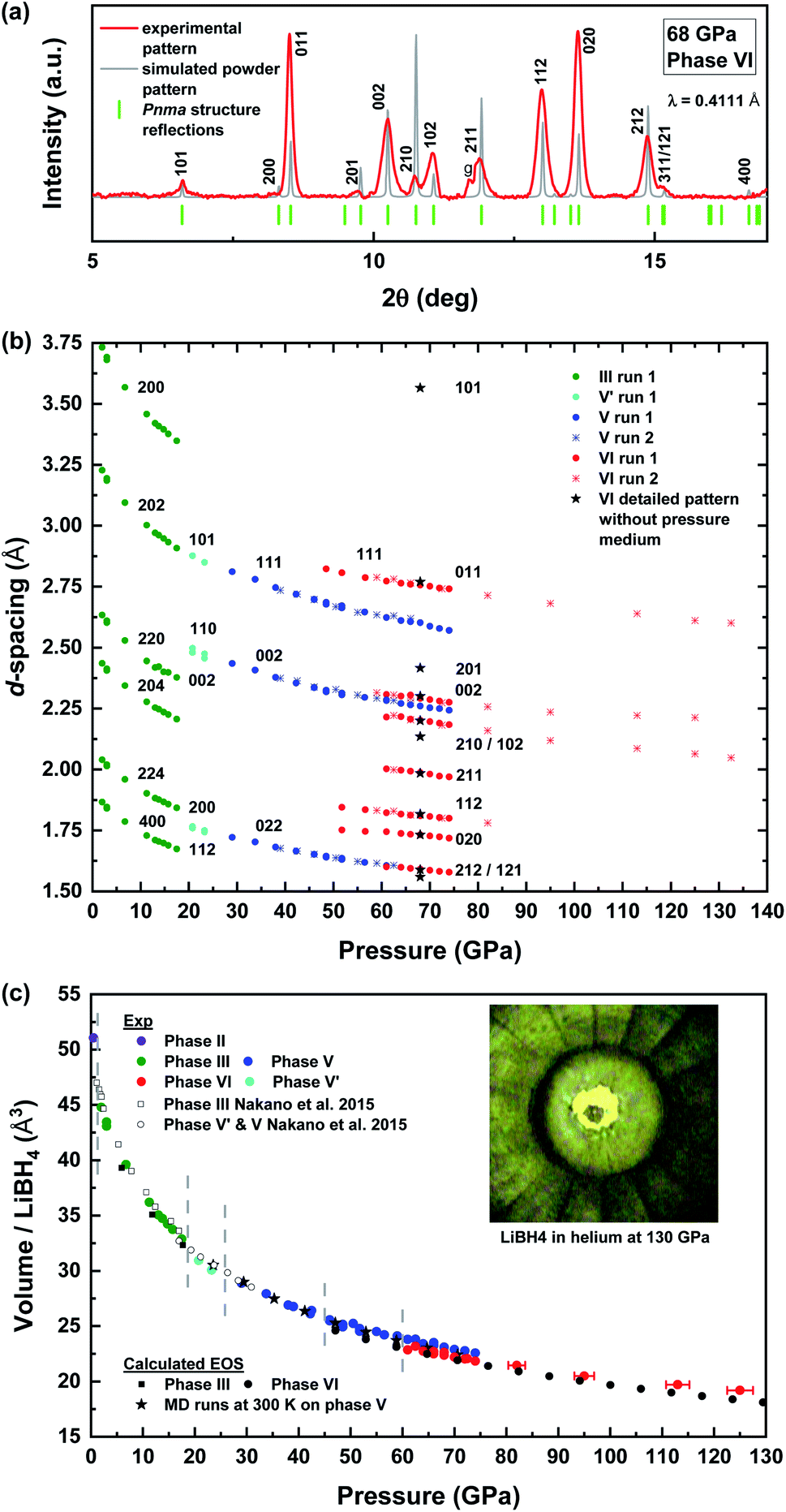 | ||
Fig. 5 (a) Diffraction pattern of the new phase VI recorded at 68 GPa without pressure medium along with a simulated powder pattern of the corresponding Pnma structure (a = 5.67 Å, b = 4.60 Å, c = 3.46 Å). “g” stands for gasket. The sample was not in a sufficient powder state to perform Rietveld refinement. (b) d-Spacing evolution in pressure for each phase of LiBH4. (c) Experimental and calculated equation of state of LiBH4 up to 130 GPa. Vertical grey lines materialize the pressure region of the phase diagram given in Fig. 9. All data points here were taken on LiBH4 under hydrostatic condition in helium which seems to favour the persistence of the disordered phase V above 60 GPa. The parameters of the Vinet fit for phase III are: K0 = 25 GPa; 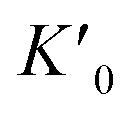 = 3.4; V0 = 48 Å3. For the sake of clarity, pressure error bars are only shown for data points above 80 GPa (volume error bars smaller than the symbol size). = 3.4; V0 = 48 Å3. For the sake of clarity, pressure error bars are only shown for data points above 80 GPa (volume error bars smaller than the symbol size). | ||
The X-ray study was completed by a second run in helium with a much smaller sample and a 100 μm culet, three peaks could be followed up to 130 GPa and have allowed us to extend the PV equation of state of LiBH4 at room temperature. At 74 GPa, the volume drops between phase V and the new phase VI is around 3%.
Typical Raman spectra without pressure medium are presented in Fig. 6. A striking quasi disappearance of the low frequency modes is recorded from 20 GPa on, when LiBH4 transforms from an ordered tetragonal I41/acd phase III to two disordered phases: a tetragonal and then a cubic phase (phases V′ and V). As the pressure increases, new low frequency vibrons start to appear at 45 GPa and are well defined near 55 GPa. Nevertheless, at this pressure, the Raman stretching modes in the 2500 cm−1 region indicate that a fraction of the cubic phase remains, as three peaks are distinguishable whereas from 64 GPa on, only two peaks are distinguishable. Hence Raman spectra clearly indicate a reordering of the [BH4]− tetrahedron leading to the new high pressure phase VI of LiBH4.
 | ||
| Fig. 6 Raman spectra of LiBH4 at selected pressures and 300 K (no pressure medium). Each different colour corresponds to a LiBH4 phase. Black: Pnma – phase II; green: I41/acd – phase III; light-blue: disordered tetragonal I4/mmm – phase V′; blue: disordered cubic Fmm – phase V; violet: mixture between phase V and VI; red: orthorhombic Pnma – phase VI. | ||
The infrared absorption study presented in Fig. 7 confirms the appearance of this new phase as well and has allowed us to extend the study LiBH4 under higher pressure. As can be seen, the transition between phase III and phase V′/V is clearly indicated by the disappearance of the denoted σ3 absorption band and a clear slope change in the evolution of the denoted σ2 (corresponding to a bending mode72) starting from 17 GPa. Other slope changes for σ2 occur: (1) near 25 GPa, indicating the complete transformation of the disordered tetragonal phase V′ into the disordered phase V, (2) near 45 GPa, corresponding to the coexistence zone between phase V and VI and (3) above 60 GPa, interpreted as the corresponding starting pressure at which only phase VI is present.
 | ||
| Fig. 7 Infrared frequencies versus pressure for LiBH4 without any pressure medium (only a few absorption bands could be followed). The Y axis of the data in red background is on the left (corresponding to a single run with 300 μm culets) and the Y axis of the data in green background is on the right (corresponding to two runs with 40 μm culets). The two bands followed with the 40 μm culets were saturated with the 300 μm culets. No pressure medium was used. Band no. 4 added (it was missing but presented in Fig. S3†). | ||
Higher pressure IR absorption study up to 290 GPa was also conducted with a 40 μm DAC by following the two main stretching bands which were saturated in the experiment with the 300 μm culet. A slight slope jump and a slope change are detected at 160 GPa which could be a very first indicator of a new phase transition to a potential phase VII (see Fig. S2 and S3 in ESI† for IR spectra). Should it be confirmed by XRD, this phase VII would probably not be one of the phases predicted by Yao & Klug, i.e. monoclinic C2/m or C2/c insulating phases, as they are computed to be unstable with respect to the new phase VI at this pressure (Fig. 8). One can hypothesize a slight monoclinic distortion of phase VI. At higher pressure, the insulating C2/c structure proposed by Yao & Klug51 is calculated to become thermodynamically favourable near 290 GPa. Then, LiBH4 is expected to metallize at 428 GPa by transforming into the polymeric C2/m structure predicted by Yao & Klug.51 This metallic phase, unlike the metallic phase of NaBH4, does not contain B–2H–B bonds but simple B–H–B or B–B bonds.
 | ||
| Fig. 8 Enthalpy difference per formula unit as a function of pressure for various LiBH4 polymorphs. The new Pnma phase VI is taken as a reference. The black arrows indicate if the structure was optimized under increasing or decreasing pressure. Structural optimization of the LiBH4 phase I and of the structure adapted from P21c NaBH4 gave rise to a new structure during pressure increase. These two new polymorphs were then optimized under decreasing pressure but none of them was found to be stable inside any pressure range. | ||
The phase diagram of LiBH4 can thus be extended and a new one is presented in Fig. 9.
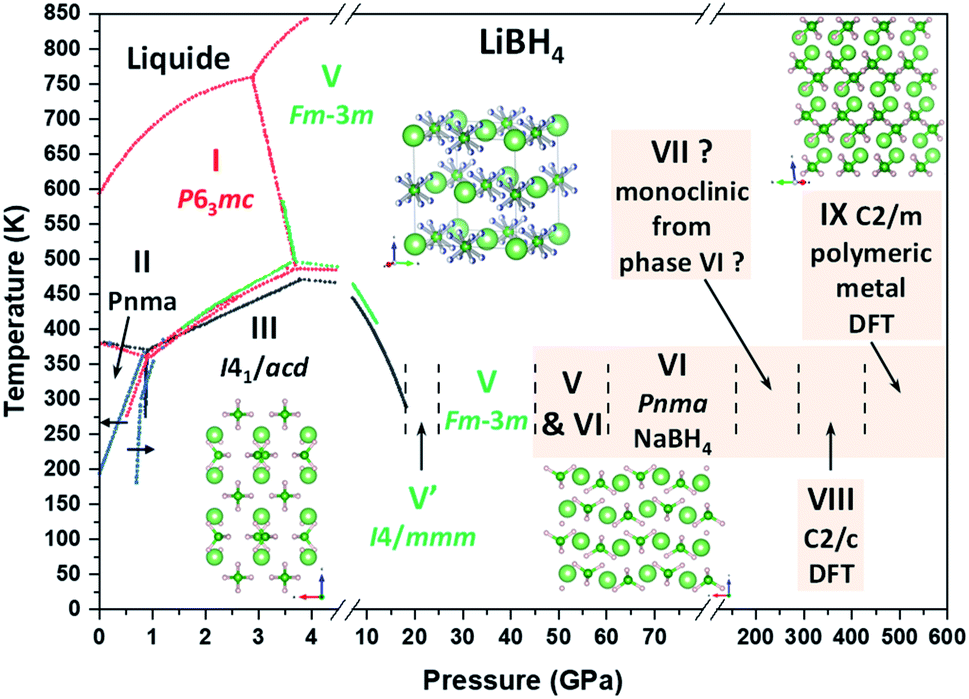 | ||
| Fig. 9 Temperature vs. pressure phase diagram of LiBH4 adapted and completed from past studies33,40,43,46,47,51 (dotted lines: red – Pistorius;41 black – Dmitriev et al.;43 blue – Sundqvist et al.;33 green – Yamawaki et al.47). Green writing indicates a phase with disorder caused by tetrahedral reorientation. Red writing indicates a phase with disorder caused by tetrahedral rotation along one of its axes. Pressure range for the coexistence of phase V and VI is given for non-hydrostatic conditions; for helium pressure transmitting medium, this zone starts at 60 GPa and probably terminates near 80 GPa. Note that phase V′ is considered as metastable46 and that there is unconfirmed theoretical debate for structure of phase I.73,74 Li, B and H atoms are respectively represented by light green, dark green and pink spheres. Phase VIII and IX are the phases from Yao & Klug for which the transition pressures were recalculated. Beige background indicates the new high pressure phase diagram proposed by this work. | ||
Hydrogen impurities in LiBH4
Infrared transmission measurements on lithium borohydride reveal the presence of a small absorption band near 4550 cm−1 in phase II. Its frequency is greater than the frequency of pure H2 and corresponds to the stretching vibration of few H2 molecules trapped inside the LiBH4 crystal lattice. As far as we know, this slight amount of hydrogen inside LiBH4 was never reported before; maybe because this trapped H2 is hardly detected by Raman spectroscopy (IR absorption measurements can be much more sensitive than Raman spectroscopy to detect a hydrogen vibron). Incidentally, it should be noted that the LiBH4 commercial batch was new and the glove box free of O2/H2O at a level inferior to 1 ppm and no water contamination was detected by IR absorption, which probably rules out a hypothetical hydrolysis reaction. Hence, the presence of H2 is thought to be due to impurities trapped in the LiBH4 structure during synthesis. Such impurities do not change the crystal structure of LiBH4 and probably only marginally change its volume, measured by XRD. Indeed, the experimental structures and volumes agree fairly well with the earlier literature studies and the DFT-calculated ones (Fig. 5).This H2 vibron has been followed during pressure increase (band denoted σ7 in Fig. 7). The intensity of the band increases during the transition from phase II to III (see ESI, Fig. S9†). This can be due to a higher polarisation of the H2 molecule in phase III or a slight decomposition during the phase change, even if such decomposition is more a temperature effect (as in the phase II to the high-temperature phase I transition at ambient pressure50) than a pressure effect. A frequency jump near 20 GPa is correlated with the phase III to phase V′/V transformation, and the breaks in slope at higher pressure are correlated to the coexistence of phases V and VI defined above. Hence, this small amount of H2 in the LiBH4 lattice turned to be a good probe of the structural transformation under pressure. During the phase V to VI transition, the H2 signal broadens and tends to vanish, indicating a low signal or a possible diffusion of the hydrogen into the gasket.
Hydrogen complexes with NaBH4
As we have seen, NaBH4 stays in its orthorhombic Pnma phase under high pressure which is not very interesting from a hydrogen storage perspective. Several NPT MD runs have been performed to try to find new stable NaBH4(H2)x complexes with high hydrogen content. The additional hydrogen was initially inserted inside the void in the NaBH4 Pnma structure at 50 GPa. Several stoichiometries have been tested with x = 1/2, 1, 3/2, 2. The total number of atoms in the cell was 28. On the four stoichiometries tested, only the one with x = 1/2 lead to a structure compatible with a possible insertion. The resulting structure, shown in Fig. 10, was then optimized and could be described in the P21/c space group (see ESI† for the Wyckoff positions). The lattice of the orthorhombic Pnma structure was distorted into a monoclinic one due to hydrogen insertion without any major reorganisation of the atoms. In this structure, H forms a bridge between the edges of two distinct tetrahedral. By comparing the enthalpies of pure NaBH4 + 0.5H2 and NaBH4(H2)0.5 as a function of pressure, we found that this new complex becomes stable above 153 GPa. For the reference phase of H2, we have used the C2/c-32 phase17 for phase I and the C2/c-24 phase which is the accepted candidate phase for the phase III of dense hydrogen.75 Phonon band structures of NaBH4 and NaBH4(H2)0.5 at 206 GPa can be found in ESI (Fig. S7 and S8†). Note that a slight dynamical instability is observed in the case of NaBH4(H2)0.5, close to the gamma point, proving that a slightly more stable structure may exist (however, its direct simulation would require the building of a supercell 10 times larger, which is currently beyond our numerical possibilities). The new compound remains an insulator and the stretching vibration frequency of the inserted H2 molecules is higher than that of pure hydrogen by 650 cm−1 at 200 GPa (H2 at 3750 cm−1 at 75 K and 200 GPa (ref. 76)). Experimentally, this higher vibration frequency should easily be detected thanks to Raman or IR spectroscopy. The volume is also slightly increased by 8% at 200 GPa (+1.57 Å3) compared to Pnma NaBH4. We have performed preliminary experiments on NaBH4 loaded with H2 in a DAC, and no such hints of H2 insertion have been detected up to 88 GPa during the room temperature pressure increase, which is in agreement with the calculations. | ||
| Fig. 10 Enthalpy difference between NaBH4 + 0.5H2 and NaBH4(H2)0.5 as a function of pressure. NC: Norm-Conserving method. PAW: Projector Augmented Wave method. View along the c axis of (a) the NaBH4 Pnma structure and (b) the NaBH4(H2)0.5 P21/c structure. View along the b axis of (c) the NaBH4 Pnma structure and (d) the NaBH4(H2)0.5 P21/c structure. | ||
Conclusion
The phase diagrams of both lithium and sodium borohydrides have been experimentally extended in pressure above 100 GPa and up to the metallization of these compounds by ab initio calculations. Their extended phase diagrams in the multi-Mbar range are shown in Fig. 9 and 4, respectively. The polymorphism of LiBH4 is richer than the one of NaBH4. Indeed, LiBH4 is transformed under pressure into five specific polymorphs at least. Previously, only the disordered Fmm cubic phase was known to be shared between the two compounds, but in the present study, we have found a shared high pressure Pnma structure. As suggested by the IR absorption data, this phase might undergo a monoclinic distortion under pressure for LiBH4 but not for NaBH4.
Moreover, the reliability of highly converged GGA-PBE calculations has been demonstrated in this study, both by a good replication of the phase diagram of the two borohydrides but also by a successful ab initio search for the structure of phase VI of LiBH4. Hence, machine learning/AIRSS search and design should be useful to discover new borohydrides or, more generally, new ternary hydrides for better solid hydrogen storage.
With the prediction of the stability of a new NaBH4(H2)0.5 hydride under pressure, the question of the possibility of inserting more hydrogen inside alkaline borohydrides which do not have a very open metal–organic framework is raised. Moreover, the detection of trapped H2 inside the polymorphs of LiBH4 under pressure calls for new numerical and experimental searches for LiBH4(H2)x complexes with high hydrogen content, as what was done for NaBH4, especially in phase VI which is shared between the two compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to very warmly thank Paul Dumas for his help on SMIS beamline and also Mrs Stéphanie Blanchandin and Mrs Karine Chaouchi for their precious help with the glovebox at the Soleil Synchrotron.Notes and references
- P. Martelli, R. Caputo, A. Remhof, P. Mauron, A. Borgschulte and A. Züttel, J. Phys. Chem. C, 2010, 114, 7173–7177 CrossRef CAS.
- A. Züttel, S. Rentsch, P. Fischer, P. Wenger, P. Sudan, P. Mauron and C. Emmenegger, J. Alloys Compd., 2003, 356–357, 515–520 CrossRef.
- A. Züttel, A. Borgschulte and S. I. Orimo, Scr. Mater., 2007, 56, 823–828 CrossRef.
- J. Manna, M. Vashistha and P. Sharma, Int. J. Energy a Clean Environ., 2010, 11, 65–97 CrossRef CAS.
- I. Saldan, Open Chem, 2011, 9, 761–775 CrossRef CAS.
- L. H. Rude, T. K. Nielsen, D. B. Ravnsbaek, U. Bösenberg, M. B. Ley, B. Richter, L. M. Arnbjerg, M. Dornheim, Y. Filinchuk, F. Besenbacher and T. R. Jensen, Phys. status solidi, 2011, 208, 1754–1773 CrossRef CAS.
- M. L. Christian and K.-F. Aguey-Zinsou, ACS Nano, 2012, 6, 7739–7751 CrossRef CAS PubMed.
- M. B. Ley, L. H. Jepsen, Y. Lee, Y. W. Cho, J. M. Bellosta von Colbe, M. Dornheim, M. Rokni, J. O. Jensen, M. Sloth, Y. Filinchuk, J. E. Jørgensen, F. Besenbacher and T. R. Jensen, Mater. Today, 2014, 17, 122–128 CrossRef CAS.
- J. Mao and D. Gregory, Energies, 2015, 8, 430–453 CrossRef CAS.
- M. Paskevicius, L. H. Jepsen, P. Schouwink, R. Černý, D. B. Ravnsbæk, Y. Filinchuk, M. Dornheim, F. Besenbacher and T. R. Jensen, Chem. Soc. Rev., 2017, 46, 1565–1634 RSC.
- Y. Song, Phys. Chem. Chem. Phys., 2013, 15, 14524–14547 RSC.
- V. Ban, A. V. Soloninin, A. V. Skripov, J. Hadermann, A. Abakumov and Y. Filinchuk, J. Phys. Chem. C, 2014, 118, 23402–23408 CrossRef CAS.
- N. A. Tumanov, E. Roedern, Z. Łodziana, D. B. Nielsen, T. R. Jensen, A. V. Talyzin, R. Černý, D. Chernyshov, V. Dmitriev, T. Palasyuk and Y. Filinchuk, Chem. Mater., 2016, 28, 274–283 CrossRef CAS.
- M. A. Rafiee, Phys. Chem. Res., 2018, 6, 657–666 CAS.
- A. Marizy, G. Geneste, P. Loubeyre, B. Guigue and G. Garbarino, Phys. Rev. B, 2018, 97, 184103 CrossRef CAS.
- C. M. Pépin, A. Dewaele, G. Geneste, P. Loubeyre and M. Mezouar, Phys. Rev. Lett., 2014, 113, 265504 CrossRef PubMed.
- C. M. Pépin, G. Geneste, A. Dewaele, M. Mezouar and P. Loubeyre, Science, 2017, 357, 382–385 CrossRef PubMed.
- R. S. Chellappa, M. Somayazulu, V. V. Struzhkin, T. Autrey and R. J. Hemley, J. Chem. Phys., 2009, 131, 224515 CrossRef PubMed.
- Y. Lin, E. Welchman, T. Thonhauser and W. L. Mao, J. Mater. Chem. A, 2017, 5, 7111–7117 RSC.
- A. M. Soldate, J. Am. Chem. Soc., 1947, 69, 987 CrossRef CAS.
- W. H. Stockmayer and C. C. Stephenson, J. Chem. Phys., 1953, 21, 1311–1312 CrossRef CAS.
- S. C. Abrahams and J. Kalnajs, J. Chem. Phys., 1954, 22, 434–436 CrossRef CAS.
- R. L. Davis and C. H. L. Kennard, J. Solid State Chem., 1985, 59, 393–396 CrossRef CAS.
- A. Remhof, Z. Łodziana, P. Martelli, O. Friedrichs, A. Züttel, A. Skripov, J. Embs and T. Strässle, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1–9 CrossRef.
- D. G. Allis and B. S. Hudson, Chem. Phys. Lett., 2004, 385, 166–172 CrossRef CAS.
- P. Fischer and A. Züttel, Mater. Sci. Forum, 2004, 443–444, 287–290 CAS.
- C. M. Araújo, R. Ahuja, a. V. Talyzin and B. Sundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 1–5 Search PubMed.
- R. S. Kumar and A. L. Cornelius, Appl. Phys. Lett., 2005, 87, 261916 CrossRef.
- B. Sundqvist and O. Andersson, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 2005–2007 CrossRef.
- Y. Filinchuk, A. V. Talyzin, D. Chernyshov and V. Dmitriev, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 7–10 CrossRef.
- E. Kim, R. Kumar, P. F. Weck, A. L. Cornelius, M. Nicol, S. C. Vogel, J. Zhang, M. Hartl, A. C. Stowe, L. Daemen and S. Carolina, J. Phys. Chem. B, 2007, 111, 13873–13876 CrossRef CAS PubMed.
- L. George, V. Drozd, H. Couvy, J. Chen and S. K. Saxena, J. Chem. Phys., 2009, 131, 1–7 CrossRef PubMed.
- B. Sundqvist and O. Andersson, Int. J. Thermophys., 2009, 30, 1118–1129 CrossRef CAS.
- G. Lee, J.-Y. Lee and J. S. Kim, Solid State Commun., 2006, 139, 516–521 CrossRef CAS.
- F. Yu, J.-X. Sun, R.-G. Tian, G.-F. Ji and W.-J. Zhu, Chem. Phys., 2009, 362, 135–139 CrossRef CAS.
- R. Caputo, A. Tekin, W. Sikora and A. Züttel, Chem. Phys. Lett., 2009, 480, 203–209 CrossRef CAS.
- C. Y. Zhu, Y. H. Liu, F. B. Tian and T. Cui, Phys. Status Solidi B, 2011, 248, 1139–1142 CrossRef CAS.
- Y. Filinchuk, D. Chemyshov and V. Dmitriev, Zeitschrift fur Krist, 2008, 223, 649–659 CrossRef CAS.
- R. Caputo and A. Tekin, J. Solid State Chem., 2012, 190, 310 CrossRef CAS.
- B. Sundqvist, Solid State Phenom, 2009, 150, 175–195 CAS.
- C. W. F. T. Pistorius, Zeitschrift für Phys. Chemie, 1974, 88, 253–263 CrossRef CAS.
- A. V. Talyzin, O. Andersson, B. Sundqvist, A. Kurnosov and L. Dubrovinsky, J. Solid State Chem., 2007, 180, 510–517 CrossRef CAS.
- V. Dmitriev, Y. Filinchuk, D. Chernyshov, A. V. Talyzin, A. Dzwilevski, O. Andersson, B. Sundqvist and A. Kurnosov, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 1–11 Search PubMed.
- Y. Filinchuk, D. Chernyshov and R. Cerny, J. Phys. Chem. C, 2008, 112, 10579–10584 CrossRef CAS.
- Y. Filinchuk, D. Chernyshov, A. Nevidomskyy and V. Dmitriev, Angew. Chem., Int. Ed., 2008, 47, 529–532 CrossRef CAS PubMed.
- S. Nakano, H. Fujihisa, H. Yamawaki and T. Kikegawa, J. Phys. Chem. C, 2015, 119, 3911–3917 CrossRef CAS.
- H. Yamawaki, H. Fujihisa, Y. Gotoh and S. Nakano, J. Phys. Chem. Solids, 2015, 76, 40–44 CrossRef CAS.
- J.-P. Soulié, G. Renaudin, R. Černý and K. Yvon, J. Alloys Compd., 2002, 346, 200–205 CrossRef.
- M. R. Hartman, J. J. Rush, T. J. Udovic, R. C. Bowman and S. J. Hwang, J. Solid State Chem., 2007, 180, 1298–1305 CrossRef CAS.
- A. Züttel, P. Wenger, S. Rentsch, P. Sudan, P. Mauron and C. Emmenegger, J. Power Sources, 2003, 118, 1–7 CrossRef.
- Y. Yao and D. D. Klug, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 064107 CrossRef.
- B. Sundqvist, A. V. Talyzin and O. Andersson, MRS Proc, 2006, 971, Z07–Z03 Search PubMed.
- K. Miwa, N. Ohba, S. Towata, Y. Nakamori and S. Orimo, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 245120 CrossRef.
- Z. Łodziana and T. Vegge, Phys. Rev. Lett., 2004, 93, 145501 CrossRef PubMed.
- T. J. Frankcombe, G.-J. Kroes and A. Züttel, Chem. Phys. Lett., 2005, 405, 73–78 CrossRef CAS.
- P. Vajeeston, P. Ravindran, A. Kjekshus and H. Fjellvåg, J. Alloys Compd., 2005, 387, 97–104 CrossRef CAS.
- A. Tekin, R. Caputo and A. Züttel, Phys. Rev. Lett., 2010, 104, 215501 CrossRef PubMed.
- S. Li, X. Ju and C. Wan, Int. J. Hydrogen Energy, 2014, 39, 9330–9338 CrossRef CAS.
- M. Santhosh and R. Rajeswarapalanichamy, J. Phys. Chem. Solids, 2016, 88, 68–77 CrossRef CAS.
- T. Ghellab, Z. Charifi, H. Baaziz, K. Bouferrache and B. Hamad, Int. J. Energy Res., 2019, 43, 3653–3667 CrossRef CAS.
- E. Tetik and U. Erdiven, Chin. J. Physiol., 2019, 59, 585–590 CrossRef CAS.
- C. Prescher and V. B. Prakapenka, High Press. Res., 2015, 35, 223–230 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
- P. Loubeyre, F. Occelli and P. Dumas, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 134101 CrossRef.
- Y. Akahama and H. Kawamura, J. Appl. Phys., 2006, 100, 043516 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- X. Gonze, G.-M. Rignanese, M. Verstraete, J.-M. Beuken, Y. Pouillon, R. Caracas, F. Jollet, M. Torrent, G. Zerah, M. Mikami, P. Ghosez, M. Veithen, J.-Y. Raty, V. Olevano, F. Bruneval, L. Reining, R. Godby, G. Onida, D. R. Hamann and D. C. Allan, Zeitschrift für Krist. –Cryst. Mater., 2005, 220, 558–562 CrossRef CAS.
- X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10337–10354 CrossRef CAS.
- X. Gonze and C. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 10355–10367 CrossRef CAS.
- C. J. Pickard and R. J. Needs, J. Phys. Condens. Matter, 2011, 23, 053201 CrossRef PubMed.
- D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085117 CrossRef.
- V. D'Anna, A. Spyratou, M. Sharma and H. Hagemann, Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2014, 128, 902–906 CrossRef PubMed.
- S. A. Shevlin, C. Cazorla and Z. X. Guo, J. Phys. Chem. C, 2012, 116, 13488–13496 CrossRef CAS.
- D. T. Shane, R. C. Bowman and M. S. Conradi, J. Phys. Chem. C, 2009, 113, 5039–5042 CrossRef CAS.
- C. J. Pickard and R. J. Needs, Nat. Phys., 2007, 3, 473–476 Search PubMed.
- P. Loubeyre, F. Occelli and R. LeToullec, Nature, 2002, 416, 613–617 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00816a |
| This journal is © The Royal Society of Chemistry 2021 |