
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of novel seven-membered carbasugars and evaluation of their glycosidase inhibition potentials†
Vignesh Athiyarath ,
Naveen J. Roy,
A. T. V. Vijil and
Kana M. Sureshan*
,
Naveen J. Roy,
A. T. V. Vijil and
Kana M. Sureshan*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Kerala-695551, India. E-mail: kms@iisertvm.ac.in
First published on 2nd March 2021
Abstract
Here, we report the synthesis of five novel seven-membered carbasugar analogs. We adopted a chiral-pool strategy starting from the cheap and readily available D-mannitol to synthesize these ring-expanded carbasugars. Apart from several regioselective protecting group manipulations, these syntheses involved Wittig olefination and ring-closing metathesis as the key steps. We observed an unprecedented deoxygenation reaction of an allylic benzyl ether upon treatment with H2/Pd during the synthesis. Preliminary biological evaluation of the carbasugars revealed that these ring expanded carbasugars act as inhibitors of various glycosidases. This study highlights the importance of the synthesis of novel ring expanded carbasugars and their biological exploration.
Introduction
Carbohydrate mimics1 are important tools to unravel complex signalling pathways involving various carbohydrates2 and carbohydrate-binding or metabolizing enzymes.3 They are also important for pharmaceutical interventions in diseases caused by the aberration in activities of these glycoenzymes.4 Carbasugars constitute an important family of glycomimetics wherein a one-carbon unit replaces the endocyclic oxygen of pyranoses.5 As carbasugars lack a hemiacetal or hemiketal functionality, they are stable towards glycosidases and often can be competitive inhibitors of these enzymes.6 Additionally, many of them show antiviral,7 antibiotic,8 antidiabetic9 and antitumor activities.10 More than 200 carbasugar natural products are known.11 Most of the natural carbasugars have a six-membered ring as their core, and they are structural mimics of pyranoses.11a Many of these natural pyranose mimics, and their unnatural derivatives have been synthesised.12 The natural products, namely calystegines,13 a group of cyclitols with a seven-membered ring as core show attractive biological properties. Furthermore, many synthetic ring-expanded sugar analogs such as septanose,14 seven-membered iminocyclitols15 etc. show interesting biological activities. Due to the wide range of biological activities of ring-expanded analogs, there is great interest in synthesising novel analogs of this class of carbasugars.16 Several strategies have been implemented for the synthesis of seven-membered carbasugar analogs. Both de novo synthesis from achiral fossil-derived fine chemicals involving a step of asymmetric induction17 and synthesis from naturally abundant chiral pool starting materials are known.18 From a practical point of sustainability, methods that use renewable natural chiral pools are attractive for synthesizing these densely hydroxylated products. We report the synthesis of five novel seven-membered carbasugars and their biological activities.We have previously reported the total syntheses of various carbasugars such as cyclophellitol, valienamine, lincitols, uvacalols, gabosines and others.19 We found that (−)-gabosine J (1) and its reduced derivatives 2 and 3 (ref. 19b) inhibit various glycosidases. Notably, (−)-gabosine J inhibits α-mannosidase three times stronger than the well-known mannosidase inhibitor deoxymannojirimycin. This result inspired us to synthesize the homologous seven-membered analogs 4, 5 and 6 (Fig. 1a).
 | ||
| Fig. 1 (a) Chemical structures of gabosine J (1), gabosinol J-α (2), gabosinol J-β (3) and corresponding seven-membered analogs 4, 5 and 6. (b) Retrosynthetic analysis of seven-membered analogs 4, 5 and 6. | ||
A retrosynthetic analysis suggested that cheap and readily available D-mannitol is an ideal chiral pool starting material for the synthesis of these derivatives. These three molecules can be obtained from the diene 7 through Grubb's ring closing metathesis. The diene 7 can be obtained from enal 8 by a Barbier type allylation.20 The enal 8 can be obtained from D-mannitol through several synthetic steps involving a Wittig olefination21 and several regio- and chemoselective protecting group manipulations (Fig. 1b).
Results and discussion
We synthesized aldehyde 10 in ten steps, starting from D-mannitol 9.19d The aldehyde 10 on Barbier allylation using metallic zinc and allyl bromide resulted in an inseparable mixture of diastereomers 11 in 70% yield. We could not calculate the ratio of isomers from the 1H NMR spectrum due to the extensive overlapping of the peaks. The mixture of diastereomers 11 was directly cyclized using Grubb's second generation catalyst, to give a mixture of cyclic diastereomers 12.The 1H NMR spectrum of this mixture was well resolved and we calculated the diastereomeric ratio as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3. Unfortunately, the mixture of diastereomers 12 was also chromatographically inseparable. Acetylation of the free hydroxyl of the diastereomers 12 resulted in a mixture of diastereomers, 13 and 14, which could be separated by column chromatography. We assigned the stereochemistry at C-4, of the major isomer 13, using NOESY experiment. H-4 of compound 13 showed a spatial interaction with H-5 and H-6, which confirms the syn relationship of H-4 with H-5 and H-6. For the minor isomer 14, peaks corresponding to H-5, H-6 and H-7 merged in the range 3.93–3.87 ppm of 1H NMR spectrum. Hence we could not confirm the stereochemistry at C-4 of this isomer. The major isomer 13 and the minor isomer 14 were deacetylated using sodium methoxide to get compounds 12β and 12α respectively.
:0.3. Unfortunately, the mixture of diastereomers 12 was also chromatographically inseparable. Acetylation of the free hydroxyl of the diastereomers 12 resulted in a mixture of diastereomers, 13 and 14, which could be separated by column chromatography. We assigned the stereochemistry at C-4, of the major isomer 13, using NOESY experiment. H-4 of compound 13 showed a spatial interaction with H-5 and H-6, which confirms the syn relationship of H-4 with H-5 and H-6. For the minor isomer 14, peaks corresponding to H-5, H-6 and H-7 merged in the range 3.93–3.87 ppm of 1H NMR spectrum. Hence we could not confirm the stereochemistry at C-4 of this isomer. The major isomer 13 and the minor isomer 14 were deacetylated using sodium methoxide to get compounds 12β and 12α respectively.
As per the NOESY spectrum of compound 12β, H-4 showed syn-relationship with H-5. For compound 12α, H-4 did not spatially interact with H-5 indicating the anti-relationship between H-4 and H-5. Compounds 12β and 12α were debenzylated using BCl3 to obtain pentaols 6 (84% yield) and 5 (65% yield) respectively (Scheme 1). We made the stereochemical assignments of both pentaols 5 and 6 using NOESY experiments. The cross peaks showing spatial interaction of H-4 with H-6 in the NOESY spectrum of the compound 6 indicate that they have syn-relationship. On the other hand, H-4 does not show cross peaks with H-6, in the NOESY spectrum of compound 5, and hence these two hydrogens have anti-relationship with each other.
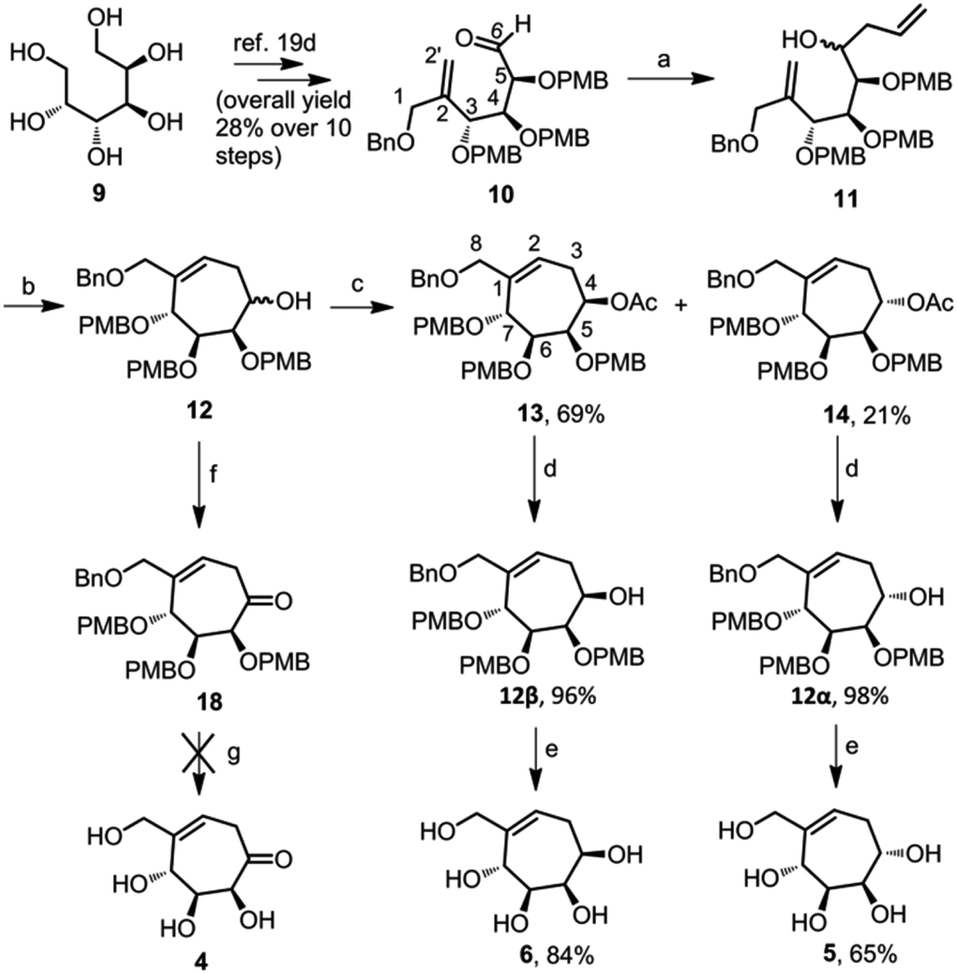 | ||
| Scheme 1 Synthesis of seven-membered carbasugars 5 and 6. Reagents and conditions: (a) Zn, allylbromide, THF, 0 °C, 30 min, 70%; (b) Grubb's 2nd generation catalyst, DCM, reflux, 12 h, 81%; (c) Ac2O, DMAP, pyridine, 3 h; (d) NaOMe, methanol, rt, 12 h; (e) BCl3, DCM, −78 °C, 3 h; (f) Dess–Martin periodinane, DCM, rt, 4 h, 96%; (g) BCl3 or BBr3 or FeCl3 (reagent conditions as per the literature).19b,23 | ||
The endocyclic double bond in these compounds in principle can be reduced to get a set of fully saturated cyclitols. To test this, we have treated compound 12β with H2 in the presence of Pd/C (Scheme 2). Surprisingly, the reaction resulted in a mixture of products, as evidenced by TLC analysis. In the TLC, there were two well-separated spots having RF 0.7 and 0.3, respectively when eluted using methanol–ethyl acetate (1:4, v/v). The polar spot corresponds to an inseparable mixture of two diastereomers 17 (27% yield) as evidenced by 1H NMR spectroscopy. The non-polar spot also corresponds to two diastereomers, which could be separated when eluted using methanol–chloroform (1:6, v/v, RF 0.3 and 0.25 respectively). We identified the non-polar compound among these two as compound 15 (36% yield) and the polar one as compound 16 (22% yield) by various NMR spectral studies, such as 1H, 13C, DEPT, COSY and HMQC spectra. We identified the peaks corresponding to the newly formed methyl groups at 0.96 ppm and 0.86 ppm for compounds 15 and 16 respectively.
 | ||
| Scheme 2 Hydrogenation of 12β resulting in unusual reduction products 15 and 16. | ||
Stereochemistry of the methyl groups of cyclitols 15 and 16 was confirmed using NOESY experiment. In compound 15, H-1 does not have spatial interaction with H-7, indicating that both have anti-relation with each other. Whereas in compound 16, H-1 has spatial interaction with H-7, indicating the syn-relation between H-7 and H-1. Single crystal X-ray diffraction analysis further confirmed the stereochemical assignment of compound 15 (Fig. 2a). The crystal structure also validated the stereochemical assignments at position C-4, done using NOESY experiments of compounds 12β and its precursor. In the crystal structure of compound 15, the hydroxyl substituents on C4, C6 and C7 are present in the equatorial position, and C-5 is in axial position. The bond angles and the torsional angles in the seven-membered ring matched with a twist-chair conformation of cycloheptane (Table S2†).22
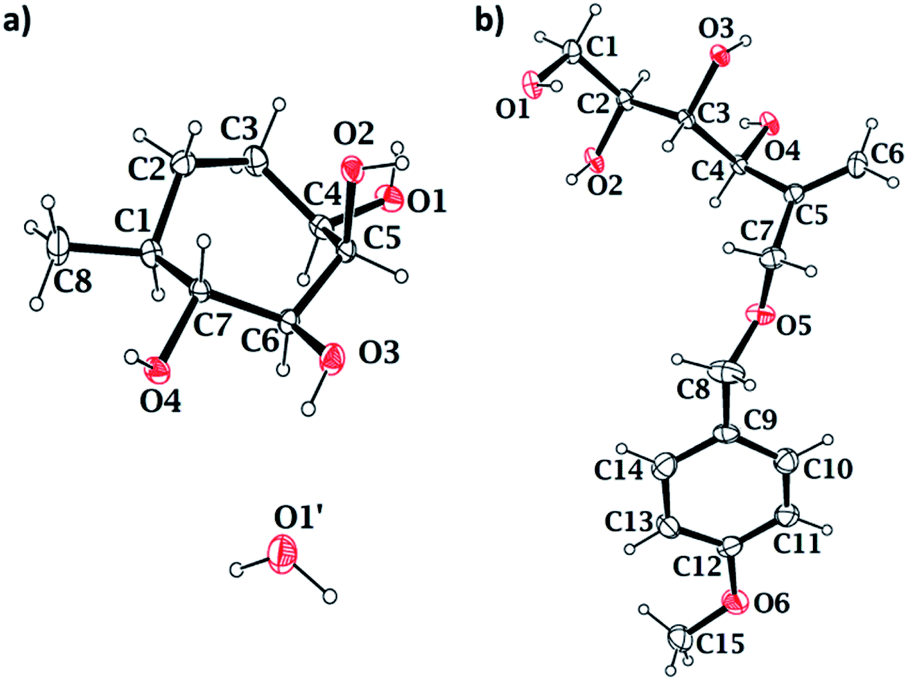 | ||
| Fig. 2 ORTEP diagram of compounds (a) 15 and (b) 23 with 50% probability level. | ||
To obtain compound 4, we oxidised compound 12 using Dess–Martin periodinane to ketone 18 (Scheme 1). Unfortunately, our attempts to debenzylate the cyclic ketone using BCl3, BBr3 or FeCl3 were not successful and resulted in a complex mixture of many inseparable compounds. Under these conditions, such reactions were successful in the case of six-membered carbasugar derivatives with alkene and ketone functional groups.19b,23 This unexpected outcome necessitated a detour in the reaction sequence. Hence we decided to replace benzyl protecting groups in compound 12 with p-methoxybenzyl (PMB), since the latter can be deprotected under mild conditions.
We synthesized diacetal 19 from D-mannitol in an overall yield of 80% over two steps as reported.19b Stannylene-acetal-mediated regioselective alkylation of 19 with p-methoxybenzylchloride gave PMB ether 20 (76% yield) as an oily liquid. Ether 20 on Swern oxidation24 gave ketone 21 in good yield. Wittig reaction on the ketone resulted in alkene 22 in 76% yield. Then, the isopropylidene groups of compound 22 were deprotected using 0.1 M HCl to yield tetraol 23. We confirmed the structure of the tetraol 23 using single crystal X-ray diffraction analysis (Fig. 2b). The primary hydroxyl of the tetraol 23 was selectively tritylated to yield triol 24 in 88% yield. All the free hydroxyl groups of the triol 24 were protected by alkylation using p-methoxybenzyl chloride to get alkene 25. The trityl group was then cleaved by acidic hydrolysis to get compound 26. We then oxidised the free primary hydroxyl group of compound 26 under Swern oxidation conditions to obtain corresponding aldehyde 27. Barbier allylation of aldehyde 27, gave an inseparable mixture of diastereomers 28. The mixture of diastereomers 28 was directly treated with Grubb's second generation catalyst to obtain cyclised product 29 as an inseparable mixture of diastereomers in 83% yield. The free hydroxyl of compound 29 was oxidised using Dess–Martin periodinane25 to obtain ketone 30, which on treatment with a 1% solution of TFA in DCM (v/v) resulted in target compound 4 in 74% yield (Scheme 3). In the NOESY spectrum of compound 4, the peak corresponding to H-7 at 4.37 ppm does not couple with that of H-5 at 3.7 ppm. This confirms that H-7 has anti-relationship with H-5.
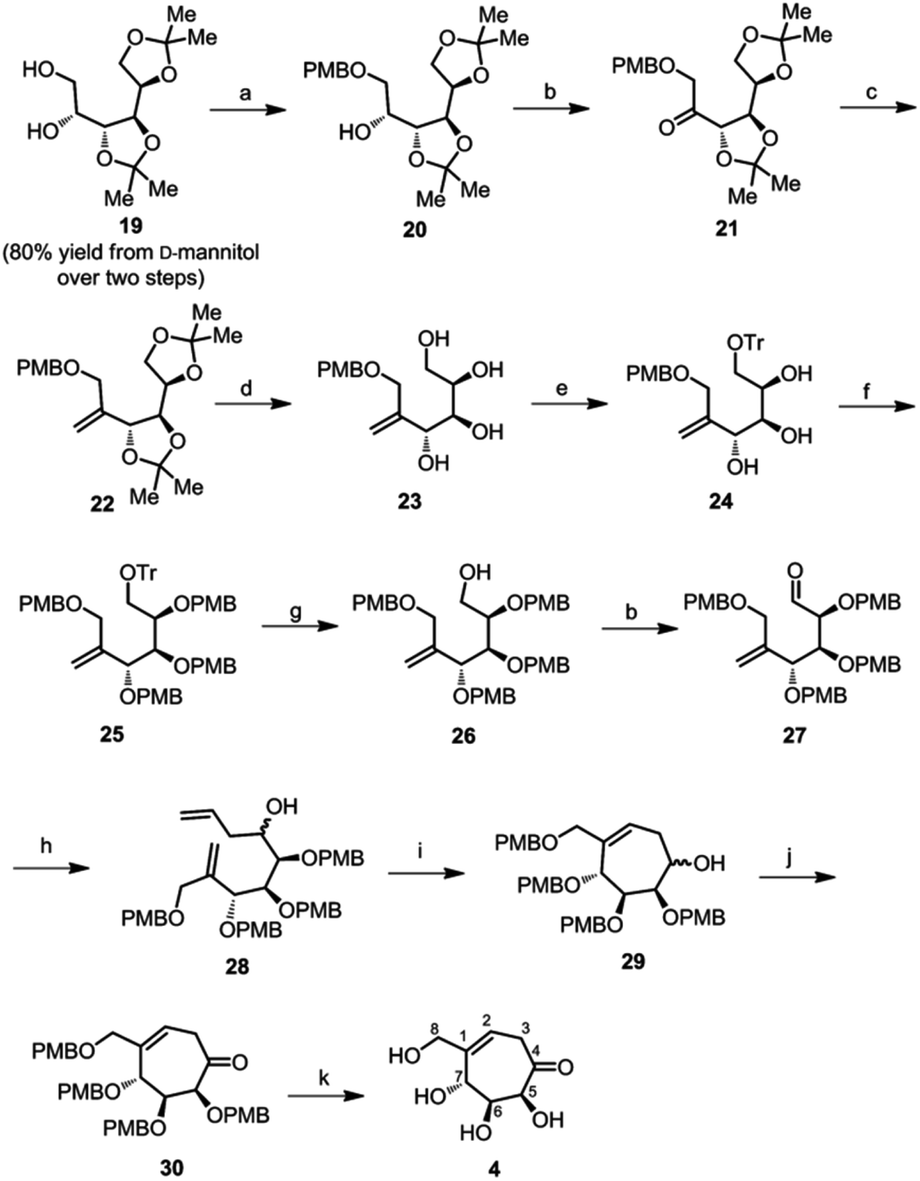 | ||
| Scheme 3 Synthesis of compound 4. Reagents and conditions: (a) Bu2SnO, p-methoxybenzyl chloride, toluene, reflux, 24 h, 76%; (b) oxalyl chloride, DMSO, triethylamine, DCM, −78 °C, 2 h, 75%; (c) PPh3PCH3Br, nBuLi, THF, −78 °C, 3 h, 76%; (d) 0.1 M HCl, methanol, rt, 30 min, 76%; (e) trityl chloride, diisopropylethylamine, DCM, rt, 4 h, 88%; (f) p-methoxybenzyl chloride, NaH, DMF, 0 °C to rt, 3 h, 76%; (g) 0.05 M HCl, methanol/chloroform (3:1, v/v), rt, 30 min, 79%; (h) Zn, allylbromide, THF, 0 °C, 30 min, 70%; (i) Grubb's 2nd generation catalyst, DCM, 50 °C, 12 h, 83%; (j) Dess–Martin periodinane, DCM, rt, 4 h, 84%; (k) 5% TFA, DCM, 0 °C, 3 h, 74%. | ||
We have evaluated the inhibition potential of these novel cyclitols towards various glycosidases (Table 1). Fig. 3 shows a comparison of structural elements of carbasugars that are inhibitors of various enzymes with their natural substrates. Apart from this positional similarity, the 3D conformations of these molecules play major roles in their activity. All these compounds inhibited α-glucosidase from Saccharomyces cerevisiae with IC50 values in the range 1–10 mM. On the other hand, none of these compounds inhibits α-glucosidase from Bacillus stearothermophilus. The difference in the sequence and 3D-structure of bacterial and fungal enzymes could be the reason for this difference in activities. Except compound 5, all other compounds inhibited β-glucosidase from almonds, but to a lesser extent compared to their activity towards α-glucosidases. While compounds 5, 15 and 16 inhibited jack bean α-mannosidase, only compound 4 could inhibit β-mannosidase (from Helix pomatia). The activities of these compounds towards galactosidases varied greatly with the source of the enzyme. While all compounds except 5 inhibited bacterial (E. coli) α-galactosidase, only the compound 6 inhibited α-galactosidase from coffee beans. All the five compounds inhibit mammalian (bovine liver) β-galactosidase, but they are inactive towards bacterial (E. coli) β-galactosidase and only the compound 16 inhibits fungal (Aspergillus oryzae) β-galactosidase. It is clear that minor changes in the structure lead to a huge difference in activities and also the activity varies with the source of enzymes. This is, in fact, advantageous for finding a selective inhibitor for an organism-specific enzyme. Detailed structure–activity relationship studies with various analogs would yield specific inhibitors having therapeutic potentials. In comparison to standard inhibitors, the seven-membered carbasugar analogs exhibited moderate inhibition potential.
| Name of the enzymes | Percentage inhibition (%) at 1 mM concentration of the inhibitor | ||||
|---|---|---|---|---|---|
| 4 | 5 | 6 | 15 | 16 | |
| a Concentration of inhibitor required for 50% inhibition of the enzyme.b NI – no inhibition up to 20 mM concentration. | |||||
| α-Glucosidase from Saccharomyces cerevisiae | 41.1 (IC50a = 1.5 ± 0.1 mM) | 3.3 (IC50 = 11.5 ± 0.1 mM) | 22.1 (IC50 = 2.9 ± 0.4 mM) | 19.5 (IC50 = 9.5 ± 1.8 mM) | 24.3 (IC50 = 3.7 ± 0.6 mM) |
| α-Glucosidase from Bacillus stearothermophilus | NIb | NI | NI | NI | NI |
| β-Glucosidase from almonds | 12.3 | NI | 3.2 | 14.1 | 29.8 |
| α-Mannosidase from jack bean | NI | 12.5 | NI | 10.6 | 35.1 (IC50 = 4.3 ± 1.2 mM) |
| β-Mannosidase from Helix pomatia | 16.2 | NI | NI | NI | NI |
| α-Galactosidase from E. coli | 17.5 | NI | 20.7 (IC50 = 3.5 ± 0.3 mM) | 34.7 (IC50 = 9.4 ± 0.5 mM) | 26.8 |
| α-Galactosidase from green coffee beans | NI | NI | 41.7 (IC50 = 1.3 ± 0.2 mM) | NI | NI |
| β-Galactosidase from E. coli | NI | NI | NI | NI | NI |
| β-Galactosidase from bovine liver | 5.9 | 42.1 (IC50 = 5.6 ± 0.5 mM) | 41.9 (IC50 = 1.9 ± 0.3 mM) | 48.8 (IC50 = 1.9 ± 0.5 mM) | 25.2 (IC50 = 4.6 ± 0.04 mM) |
| β-Galactosidase from Aspergillus oryzae | NI | NI | NI | NI | 12.8 |
 | ||
| Fig. 3 Structural comparison of enzyme-inhibitors of various glycosidases with their natural substrates. | ||
Conclusions
We have synthesised five novel seven-membered carbasugar analogs by adopting a chiral pool approach, utilizing the inexpensive and readily available D-mannitol as the starting material. All the synthesised seven-membered cyclitols exhibited weak inhibition towards various glycosidases. The glycosidase inhibition exhibited by these novel seven-membered cyclitols shows the importance of developing ring expanded cyclitols for various biological explorations. Ring-expanded cyclitols, having more number of modifiable sites, allows the synthesis of a wide variety of derivatives. Extensive research on the synthesis of many such ring-expanded cyclitols and their structure–activity correlation might give many compounds that can unravel the signalling pathways and that are of high therapeutic potentials. This research also showed an unusual deoxygenation of an allyl benzyl ether. This deoxygenation pathway might have been influenced by the conformational features of the seven-membered ring, and this warrants a systematic investigation.Experimental section
General
All reagents and solvents were purchased from commercial sources and used without further purification. Pre-coated TLC silica gel 60 F254 plates were purchased from Merck. TLC analyses were carried out by visualizing the chromatograms under UV light and further by heating the plates after dipping into ceric ammonium molybdate staining solution. Silica gel (200–400 mesh) was used for column chromatography. The 1H NMR, COSY, DEPT, 13C NMR and HMQC spectra were recorded on an Avance III-500 (Bruker) NMR spectrometer. The chemical shifts of NMR spectra are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.0 ppm). 13C NMR spectra were recorded with complete proton decoupling. All NMR data were collected at 25 °C. Elemental analyses were done on Elementar, vario MICRO cube elemental analyzer. High resolution mass spectra were recorded using Q Exactive TM-Benchtop-LC-HRMS. UV absorbance during enzyme assay was measured using multimode plate reader Infinite 200 PRO at 25 °C.:4, v/v) as eluent to remove any impurities and obtain the diastereomeric mixture 11 (9 g, 70% yield) as an yellow oily liquid. The mixture was used for the next reaction without further purification.:2, v/v) as eluent, to yield the cyclized derivative 12 (0.78 g, 81%) as a brown oil, which consists of inseparable mixture of diastereomers in the ratio 1:0.3.Acetylation of derivative 12
To a solution of mixture of diastereomers 12 (0.88 g, 1.37 mmol) in 5 mL anhydrous pyridine at 0 °C, acetic anhydride (0.16 mL, 1.7 mmol) and DMAP (0.017 g, 0.14 mmol) were added and the reaction mixture was stirred at room temperature for 3 h. At complete consumption of the starting material, pyridine was evaporated under reduced pressure. The residue thus obtained was diluted with ethyl acetate (50 mL) and taken into a separating funnel. The organic layer was washed successively with saturated NaHCO3 solution (20 mL × 2), brine and water. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The mixture of diastereomers was separated by column chromatography using acetone/petroleum ether (1:5, v/v) as eluent, to get the isomer 13 (0.65 g, 69% yield, RF 0.3 in acetone/petroleum ether, 1:4, v/v) and 14 (0.2 g, 21% yield, RF 0.25 in acetone/petroleum ether, 1:4, v/v) as yellow oily liquids.
:19, v/v) as eluent. The collected fractions were evaporated under reduced pressure to yield the carbasugar 6 (0.025 g, 84% yield) as a colourless oily liquid: [α] = −24.1 (c 0.108, methanol); 1H NMR (500 MHz, CD3OD) δ: 5.75–5.72 (m, 1H, H-2), 4.63 (d, J = 8.5 Hz, 1H, H-7), 4.21–4.03 (m, 3H, H-5, H-8A and H-8B), 3.65 (d, J = 10 Hz, 1H, H-4), 3.55 (d, J = 8.5 Hz, 1H, H-6), 2.72–2.64 (m, 1H, H-3A), 2.12–2.03 (m, 1H, H-3B); 13C NMR (125 MHz, CD3OD) δ: 143.6 (C-1), 121.1 (C-2), 74.4 (C-5), 73.3 (C-6), 70.6 (C-4), 67.7 (C-7), 64.1 (C-8), 28.0 (C-3); elemental analysis calcd for C8H14O5; C, 50.52; H, 7.42; found: C, 50.71; H, 7.31.:6, v/v) as eluent to give compound 15 (0.02 g, 36% yield, RF 0.3) as a gum which crystallized from a mixture of MeOH–CHCl3 (1:19, v/v), 16 (0.012 g, 22% yield, RF 0.25) and 17 (0.016 g, 27% yield, inseparable mixture of diastereomers, RF 0.2 in MeOH/CHCl3, 1:3, v/v) as yellow oily liquids.:9, v/v) as eluent to yield compound 20 (16.6 g, 76% yield) as a colourless liquid: [α] = +11.9 (c 0.21, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.28 (d, J = 8.5 Hz, 2H, Ar-H), 6.80 (d, J = 8.5 Hz, 2H, Ar-H), 4.55 (d, J = 11.9 Hz, 1H, –CH2Ph), 4.52 (d, J = 11.85 Hz, 1H, –CH2Ph), 4.18–4.14 (m, 1H, H-6A), 4.12–4.07 (m, 1H, H-5), 4.02–3.97 (m, 1H, H-6B), 3.92–3.82 (m, 3H, H-2, H-3 and H-4), 3.81 (s, 3H, –OCH3), 3.7 (dd, J = 10 Hz, 2.5 Hz, 1H, H-1A), 3.57–3.54 (m, 1H, H-1B), 3.39 (s, 1H, –OH), 1.36 (s, 3H, –CH3), 1.3–1.27 (m, 9H, –CH3); 13C NMR (125 MHz, CDCl3) δ: 159.2, 130.3, 129.3, 113.7, 110.0 (–C(CH3)2), 109.5 (–C(CH3)2), 80.6 (C-4), 79.9 (C-3), 76.2 (C-5), 73.1 (–CH2Ph), 71.8 (C-2), 71.0 (C-1), 67.5 (C-6), 55.2 (–OCH3), 27.0 (–CH3), 26.9 (–CH3), 26.4 (–CH3), 25.1 (–CH3); elemental analysis calcd for C20H30O7; C, 62.81; H, 7.91; found: C, 62.72; H, 8.17.:6, v/v) as eluent to yield the ketone 21 (8.9 g, 75% yield) as an oily liquid: [α] = +4.08 (c 0.19, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.22 (d, J = 8.25 Hz, 2H, Ar), 6.81 (d, J = 8.25 Hz, 2H, Ar), 4.51–4.44 (m, 2H, –CH2Ph), 4.39–4.29 (m, 3H, H-1A, H-1B and H-3), 4.16–4.08 (m, 2H, H-4 and H-5), 4.05–4.0 (m, 1H, H-6A), 3.89 (dd, J = 8.5 Hz, 4.8 Hz, 1H, H-6B), 3.74 (s, 3H, –OCH3), 1.36 (s, 3H, –CH3), 1.32 (s, 3H, –CH3), 1.28–1.25 (m, 6H, –CH3); 13C NMR (125 MHz, CDCl3) δ: 204.5 (CO), 158.5, 128.7, 128.1, 112.8, 110.5 (–C(CH3)2), 108.8 (–C(CH3)2), 79.6 (C-3), 77.1 (C-4), 75.2 (C-5), 71.9 (–CH2Ph), 71.5 (C1), 65.5 (C-6), 54.2 (–OCH3), 25.8 (–CH3), 25.4 (–CH3), 25.0 (–CH3), 24.1 (–CH3); elemental analysis calcd for C20H28O7; C, 63.14; H, 7.42; found: C, 63.35; H, 7.28.:9, v/v) as eluent to give compound 22 (5.3 g, 76% yield) as an oily liquid: [α] = −3.54 (c 0.23, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.19 (d, 2H, J = 8.5 Hz, Ar-H), 6.81 (d, J = 8.5 Hz, 2H, Ar-H), 5.33 (s, 1H, H-2A′), 5.27 (s, 1H, H-2B′), 4.43–4.33 (m, 3H, H-3 and –CH2Ph), 4.09–4.05 (m, 2H, H-1A and H-5), 4.02–3.95 (m, 2H, H-1B and H-6A), 3.91–3.85 (m, 2H, H-4 and H-6B), 3.73 (s, 3H, –OCH3), 1.34 (m, 6H, –CH3), 1.30 (s, 3H, –CH3), 1.26 (s, 3H, –CH3); 13C NMR (125 MHz, CDCl3): δ: 159.1 (Ar), 142.8 (C2), 130.3 (Ar), 129.3 (Ar), 115.6 (C-2′), 113.7 (Ar), 109.6 (–C(CH3)2), 109.2 (–C(CH3)2), 80.5 (C-3), 80.3 (C-4), 76.4 (C-5), 71.8 (–CH2Ph), 69.5 (C-1), 66.7 (C-6), 55.2 (–OCH3), 27.0 (–CH3), 26.8 (–CH3), 26.4 (–CH3), 25.2 (–CH3); elemental analysis calcd for C21H30O6; C, 66.65; H, 7.99; found: C, 66.46; H, 8.22.:5, v/v) as eluent to yield triol 24 (1.6 g, 88% yield) as a viscous liquid: [α] = +2.66 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.36 (d, J = 7.4 Hz, 6H, Ar-H), 7.25–7.19 (m, 6H, Ar-H), 7.19–7.16 (m, 3H, Ar-H), 7.13 (d, J = 8.5 Hz, 2H, Ar-H), 6.76 (d, J = 8.5 Hz, 2H, Ar-H), 5.16 (s, 1H, H-2A′), 5.14 (s, 1H, H-2B′), 4.39–4.33 (m, 3H, H-3 and –CH2Ph), 4.05 (d, J = 11.25 Hz, 1H, H-1A), 3.89 (d, J = 11.25 Hz, 1H, H-1B), 3.86–3.8 (m, 1H, H-5), 3.7 (s, 3H, –OCH3), 3.64–3.59 (m, 1H, H-4), 3.31 (dd, J = 9.5 Hz, 4.5 Hz, 1H, H-6A), 3.28–3.23 (m, 2H, OH (C-4) and H-6B), 2.95 (d, J = 6.05 Hz, 1H, OH (C-3)), 2.63 (d, J = 5 Hz, 1H, OH (C-5)); 13C NMR (125 MHz, CDCl3) δ: 158.3, 143.9 (C-2), 142.6, 128.5, 128.2, 127.5, 126.9, 126.1, 116.1 (C-2′), 112.9, 85.9 (–C(Ph)3), 72.3 (C-4), 71.9 (C-3), 71.2 (–CH2Ph), 70.3 (C-5), 69.7 (C-1), 63.8 (C-6), 54.2 (–OCH3); elemental analysis calcd for C34H36O6; C, 75.53; H, 6.71; found: C, 75.55; H, 6.41.:4, v/v) as eluent to yield compound 25 (1.9 g, 76% yield) as a pale yellow liquid: [α] = −10.2 (c 0.16, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.41–7.32 (m, 6H, Ar-H), 7.21–7.09 (m, 13H, Ar-H), 7.08 (d, J = 8 Hz, 2H, Ar-H), 6.81 (d, J = 8 Hz, 2H, Ar-H), 6.78–6.72 (m, 4H, Ar-H), 6.68 (d, J = 8.5 Hz, 2H, Ar-H), 6.6 (d, J = 8.5 Hz, 2H, Ar-H), 5.28 (s, 1H, H-2A′), 5.16 (s, 1H, H-2B′), 4.57 (d, J = 11 Hz, 1H, –CH2Ph), 4.41–4.32 (m, 3H, –CH2Ph), 4.32–4.24 (m, 2H, –CH2Ph), 4.15 (d, 1H, J = 11 Hz, –CH2Ph), 4.12–4.07 (m, 2H, H-3 and –CH2Ph), 3.93 (d, J = 13 Hz, 1H, H-1A), 3.87 (d, 1H, J = 12.9 Hz, H-1B), 3.76–3.63 (m, 14H, H-4, H-5 and –OCH3 × 4), 3.42–3.35 (m, 1H, H-6A), 3.22–3.15 (m, 1H, H-6B); 13C NMR (125 MHz, CDCl3) δ: 159.1, 159.0, 158.9, 144.1, 143.1, 131.1, 130.6, 130.5, 130.4, 129.8, 129.6, 129.1, 128.9, 128.8, 128.6, 127.7, 126.8, 115.4, 113.7, 113.6, 113.3, 86.5 (–C(CH3)3), 79.9 (C-4), 79.7 (C-3), 78.5 (C-5), 74.1 (–CH2Ph), 72.3 (–CH2Ph), 72.0 (–CH2Ph), 70.5 (–CH2Ph), 70.4 (C-1), 63.6 (C-6), 55.3 (–OCH3), 55.2 (–OCH3), 55.1 (–OCH3); elemental analysis calcd for C58H60O9; C, 77.31; H, 6.71; found: C, 77.49; H, 6.60.:7, v/v) as eluent to yield compound 26 (1.15 g, 79% yield) as an yellow oily liquid: [α] = −13.04 (c 0.14, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.19–7.11 (m, 6H, Ar-H), 7.04 (d, J = 8.5 Hz, 2H, Ar-H), 6.78–6.72 (m, 8H, Ar-H), 5.36 (s, 1H, H-2A′), 5.29 (s, 1H, H-2B′), 4.54 (d, J = 10.75 Hz, 1H, –CH2Ph), 4.51 (d, J = 10.8 Hz, 1H, –CH2Ph), 4.45 (d, J = 11.35 Hz, 1H, –CH2Ph), 4.39 (d, J = 11.45 Hz, 1H, –CH2Ph), 4.36 (d, J = 11.45 Hz, 1H, –CH2Ph), 4.21 (d, J = 11.1 Hz, 1H, –CH2Ph), 4.16 (d, J = 11.35 Hz, 1H, –CH2Ph), 4.09–4.03 (m, 2H, H-3 and –CH2Ph), 3.95 (d, J = 12.75 Hz, 1H, H-1A), 3.9 (d, J = 12.75 Hz, 1H, H-1B), 3.74–3.64 (m, 15H, H-4, H-6A and H-6B), 3.52–3.49 (m, 1H, H-5), 2.17 (t, 1H, J = 5.75 Hz, –OH); 13C NMR (125 MHz, CDCl3) δ: 159.2, 159.1, 142.9 (C-2), 130.5, 130.4, 130.2, 129.9, 129.8, 129.2, 116.1 (C-2′), 113.8, 113.7, 113.6, 80.5 (C-3), 80.0 (C-4), 78.7 (C-5), 74.7 (–CH2Ph), 72.4 (–CH2Ph), 71.0 (–CH2Ph), 70.3 (–CH2Ph), 70.2 (C-1), 60.8 (C-6), 55.3 (–OCH3), 55.2 (–OCH3); elemental analysis calcd for C39H46O9; C, 71.10; H, 7.04; found: C, 71.35; H, 6.98.:4, v/v) to yield compound 27 (0.8 g, 81%) as an yellow liquid: [α] = +24.83 (c 0.22, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 9.49 (s, 1H, HCO-), 7.16 (d, 2H, J = 8.5 Hz, Ar-H), 7.13–7.05 (m, 6H, Ar-H), 6.78–6.71 (m, 8H, Ar-H), 5.38 (s, 1H, C-2A′), 5.36 (s, 1H, C-2B′), 4.51 (d, J = 11.05 Hz, 1H, –CH2Ph), 4.45 (d, J = 11.05 Hz, 1H, –CH2Ph), 4.42 (d, J = 11.3 Hz, 1H, –CH2Ph), 4.37 (d, J = 11.5 Hz, 1H, –CH2Ph), 4.33 (d, J = 11.4 Hz, 1H, –CH2Ph), 4.32 (d, J = 11.2 Hz, 1H, –CH2Ph), 4.18 (s, 1H, –CH2Ph), 4.15 (s, 1H, –CH2Ph), 4.11 (d, J = 5.5 Hz, 1H, H-3), 3.9–3.87 (m, 3H, H-5, H-1A and H-1B), 3.86–3.84 (m, 1H, H-4), 3.71 (s, 12H, –OCH3); 13C NMR (125 MHz, CDCl3) δ: 201.8 (HCO), 159.3, 159.2, 142.3 (C-2), 130.27, 130.20, 130.1, 129.8, 129.7, 129.5, 129.2, 117.1 (C-2′), 113.8, 113.7, 113.6, 83.3 (C-5), 81.2 (C-4), 80.5 (C-3), 74.1 (–CH2Ph), 72.4 (–CH2Ph), 72.1 (–CH2Ph), 70.7 (–CH2Ph), 70.0 (C-1), 55.2 (–OCH3); elemental analysis calcd for C39H44O9; C, 71.32; H, 6.75; found: C, 71.35; H, 6.63.:4, v/v) as eluent. Compound 28 (5.2 g, 70% yield) was obtained as an yellow oily liquid, which consists of mixture of diastereomers in the ratio 1:0.2.:2, RF 0.2) as eluent, to yield the cyclic derivative 29 (0.4 g, 83%) as a brown oil, which consists of mixture of isomers in the ratio 1:0.2.:5, v/v) as eluent to yield ketone 30 (84 mg, 84%) as a pale yellow liquid: [α] = −58.2 (c 0.2, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.22–7.17 (m, 2H, Ar-H), 7.12 (d, J = 8.4 Hz, 2H, Ar-H), 7.08 (d, J = 8.45 Hz, 2H, Ar-H), 6.98 (d, J = 8.4 Hz, 2H, Ar-H), 6.81–6.71 (m, 8H, Ar-H), 5.72 (t, J = 5.8 Hz, 1H, H-2), 4.66 (d, J = 11.7 Hz, 1H, –CH2Ph), 4.64–4.60 (m, 2H, H-5 and –CH2Ph), 4.43 (d, J = 11.7 Hz, 1H, –CH2Ph), 4.34 (d, J = 11.1 Hz, 1H, –CH2Ph), 4.29–4.24 (m, 4H, –CH2Ph, H-8A and H-8B), 4.10–4.07 (m, 1H, H-6), 4.02 (d, J = 5.55 Hz, 1H, H-7), 3.96–3.91 (m, 1H, –CH2Ph), 3.77–3.68 (m, 13H, –CH2Ph and –OCH3), 3.28 (dd, J = 16.4 Hz, 4.75 Hz, 1H, H-3A), 3.04 (dd, J = 16.45 Hz, 7.1 Hz, 1H, H-3B); 13C NMR (125 MHz, CDCl3) δ: 202.2 (CO), 158.2, 158.1, 136.2 (C-1), 129.2, 129.0, 128.6, 128.3, 120.1 (C-2), 112.7, 112.5, 83.6 (C-5), 77.8 (C-6), 73.8 (C-7), 73.0 (–CH2Ph), 71.8 (–CH2Ph), 70.9 (–CH2Ph), 70.6 (–CH2Ph), 70.5 (C-8), 54.2 (–OCH3), 40.2 (C-3); elemental analysis calcd for C40H44O9; C, 71.84; H, 6.63; found: C, 72.08; H, 6.55.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KMS thanks Department of Science and Technology, India for a SwarnaJayanti Fellowship.Notes and references
- (a) A. Tamburrini, C. Colombo and A. Bernardi, Med. Res. Rev., 2020, 40, 495 CrossRef CAS; (b) L. Cipolla, B. L. Ferla, C. Airoldi, C. Zona, A. Orsato, N. Shaikh, L. Russo and F. Nicotra, Future Med. Chem., 2010, 2, 587 CrossRef CAS; (c) P. Compain, Molecules, 2018, 23, 1658 CrossRef.
- (a) J. P. Ribeiro, T. Diercks, J. Jimenez-Barbero, S. Andre, H. J. Gabius and F. J. Canada, Biomolecules, 2015, 5, 3177 CrossRef CAS; (b) B. Wu, J. Ge, B. Ren, Z. Pei and H. Dong, Tetrahedron, 2015, 71, 4023 CrossRef CAS.
- (a) M. Kitaoka, S. Ogawa and H. A. Taniguchi, Carbohydr. Res., 1993, 247, 355 CrossRef CAS; (b) L. I. Willems, T. J. M. Beenakker, B. Murray, S. Scheij, W. W. Kallemeijn, R. G. Boot, M. Verhoek, W. E. Donker-Koopman, M. J. Ferraz and E. R. van Rijssel, J. Am. Chem. Soc., 2014, 136, 11622 CrossRef CAS.
- A. R. Sawkar, W.-C. Cheng, E. Beutler, C.-H. Wong, W. E. Balch and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15428 CrossRef CAS.
- S. Roscales and J. Plumet, Int. J. Carbohydr. Chem., 2016, 2016, 4760548 Search PubMed.
- (a) M. Lefoix, A. Tatibouet, S. Cottaz, H. Driguez and P. Rollin, Tetrahedron Lett., 2002, 43, 2889 CrossRef CAS; (b) Q. Gao, C. Zaccaria, M. Tontini, L. Poletti, P. Costantinob and L. Lay, Org. Biomol. Chem., 2012, 10, 6673 RSC; (c) E. Leclerc, X. Pannecoucke, M. Etheve-Quelquejeuc and M. Sollogoub, Chem. Soc. Rev., 2013, 42, 4270 RSC; (d) G. Aydin, T. Savran, S. Baran and A. Baran, Bioorg. Med. Chem. Lett., 2018, 28, 2555 CrossRef CAS.
- (a) A. D. Borthwick, K. Biggadike and J. L. Paternoster, Bioorg. Med. Chem. Lett., 1993, 3, 2577 CrossRef CAS; (b) J. Wang, Y. Jin, K. L. Rapp, M. Bennett, R. F. Schinazi and C. K. Chu, J. Med. Chem., 2005, 48, 3736 CrossRef CAS; (c) J.-s. Yoon, G. Kim, D. B. Jarhad, H.-R. Kim, Y.-S. Shin, S. Qu, P. K. Sahu, H. O. Kim, H. W. Lee, S. B. Wang, Y. J. Kong, T.-S. Chang, N. S. Ogando, K. Kovacikova, E. J. Snijder, C. C. Posthuma, M. J. van Hemert and L. S. Jeong, J. Med. Chem., 2019, 62, 6346 CrossRef CAS.
- M. S. Visser, K. D. Freeman-Cook, S. J. Brickner, K. E. Brighty, P. T. Le, S. K. Wade, R. Monahan, G. J. Martinelli, K. T. Blair and D. E. Moore, Bioorg. Med. Chem. Lett., 2010, 20, 6730 CrossRef CAS.
- W.-L. Ng, H.-C. Li, K. K.-M. Lau, A. K. N. Chan, C. B.-S. Lau and T. K. M. Shing, Sci. Rep., 2017, 7, 5581 CrossRef.
- (a) Y. Usami, I. Takaoka, H. Ichikawa, Y. Horibe, S. Tomiyama, M. Ohtsuka, Y. Imanishi and M. Arimoto, J. Org. Chem., 2007, 72, 6127 CrossRef CAS; (b) M. Li, Y. Li, K. A. Ludwik, Z. M. Sandusky, D. A. Lannigan and G. A. O'Doherty, Org. Lett., 2017, 19, 2410 CrossRef CAS.
- (a) O. Arjona, A. M. Gomez, J. C. Lopez and J. Plumet, Chem. Rev., 2007, 107, 1919 CrossRef CAS; (b) W. W. Kallemeijn, K.-Y. Li, M. D. Witte, A. R. A. Marques, J. Aten, S. Scheij, J. Jiang, L. I. Willems, T. M. Voorn-Brouwer, C. P. A. A. V. Roomen, R. Ottenhoff, R. G. Boot, H. V. D. Elst, M. T. C. Walvoort, B. I. Florea, J. D. C. Codee, G. A. V. D. Marel, J. M. F. G. Aerts and H. S. Overkleeft, Angew. Chem., Int. Ed., 2012, 51, 12529 CrossRef CAS.
- (a) P. Wen, V. Vetvicka and D. Crich, J. Org. Chem., 2019, 84, 5554 CrossRef CAS; (b) Y. Usami, in Studies in Natural Products Chemistry, ed. R. Atta-ur, Elsevier, 2014, vol. 41, p. 287 Search PubMed.
- (a) O. Duclos, M. Mondange, A. Dureault and J. C. Depezay, Tetrahedron Lett., 1992, 33, 8061 CrossRef CAS; (b) C. R. Johnson and S. J. Bis, J. Org. Chem., 1995, 60, 615 CrossRef CAS; (c) T. Faitg, J. Soulie, J.-Y. Lallemand and L. Ricard, Tetrahedron: Asymmetry, 1999, 10, 2165 CrossRef CAS; (d) F.-D. Boyer and I. Hanna, Tetrahedron Lett., 2001, 42, 1275 CrossRef CAS.
- J. Saha and M. W. Peczuh, Adv. Carbohydr. Chem. Biochem., 2011, 66, 121 CrossRef CAS.
- (a) H. Li, Y. Bleriot, C. Chantereau, J.-M. Mallet, M. Sollogoub, Y. Zhang, E. Rodríguez-García, P. Vogel, J. Jiménez-Barbero and P. Sinaÿ, Org. Biomol. Chem., 2004, 2, 1492 RSC; (b) H. Li, C. Schutz, S. Favre, Y. Zhang, P. Vogel, P. Sinaya and Y. Bleriot, Org. Biomol. Chem., 2006, 4, 1653 RSC.
- (a) L. A. Paquette, G. Moura-Letts and G. P. Wang, J. Org. Chem., 2009, 74, 2099 CrossRef CAS; (b) G. Mehta, K. Pallavi and S. Katukojvala, Tetrahedron Lett., 2009, 50, 4519 CrossRef CAS; (c) R. Madsen, Eur. J. Org. Chem., 2007, 399 CrossRef CAS; (d) G. Mehta, R. Mohanrao, S. Katukojvala, Y. Landais and S. Sen, Tetrahedron Lett., 2011, 52, 2893 CrossRef CAS; (e) K. Ecer and E. Salamci, Tetrahedron, 2014, 70, 8389 CrossRef CAS; (f) G. Moura-Letts and L. A. Paquette, J. Org. Chem., 2008, 73, 7663 CrossRef CAS; (g) W. Wang, Y. Zhang, M. Sollogoub and P. Sinay, Angew. Chem., Int. Ed., 2000, 39, 2466 CrossRef CAS; (h) L. A. Paquette and Y. Zhang, J. Org. Chem., 2006, 71, 4353 CrossRef CAS; (i) Y. Zozik, E. Salamci and A. Kilic, Tetrahedron Lett., 2017, 58, 4822 CrossRef CAS; (j) A. Osuch-Kwiatkowska and S. Jarosz, Curr. Org. Chem., 2014, 18, 1674 CrossRef CAS.
- (a) M. E. Sengul, A. Menzek and N. Saracoglu, J. Chem. Res., 2005, 2005, 382 CrossRef; (b) R. Beniazza, V. Desvergnes, G. Mehta, N. Blanchard, F. Robert and Y. Landais, J. Org. Chem., 2011, 76, 791 CrossRef CAS; (c) M. F. El-Mansy, A. Sar, S. Lindeman and W. A. Donaldson, Chem.–Eur. J., 2013, 19, 2330 CrossRef CAS; (d) D. A. Khlevin, S. E. Sosonyuk, M. V. Proskurnina and N. S. Zefirov, Eur. J. Org. Chem., 2013, 2381 CrossRef CAS; (e) A. Pearson and S. Katiyar, Tetrahedron, 2000, 56, 2297 CrossRef CAS; (f) A. A. Yadav, P. S. Sarang, M. Sau, S. Thirumalairajan, G. K. Trivedi and M. M. Salunkhe, Tetrahedron Lett., 2012, 53, 3599 CrossRef CAS.
- (a) Y. Bleriot, A. Giroult, J.-M. Mallet, E. Rodriguez, P. Vogel and P. Sinay, Tetrahedron: Asymmetry, 2002, 13, 2553 CrossRef CAS; (b) E. Sisu, M. Sollogoub, J.-M. Mallet and P. Sinay, Tetrahedron, 2002, 58, 10189 CrossRef CAS; (c) C. Jia, Y. Zhang and L. Zhang, Tetrahedron: Asymmetry, 2003, 14, 2195 CrossRef CAS; (d) J. Marco-Contelles and E. de Opazo, J. Org. Chem., 2002, 67, 3705 CrossRef CAS; (e) P. R. Skaanderup and R. A. Madsen, J. Org. Chem., 2003, 68, 2115 CrossRef CAS; (f) J. Marco-Contelles and E. de Opazo, J. Carbohydr. Chem., 2002, 21, 201 CrossRef CAS; (g) J. Marco-Contelles and E. Opazo, Tetrahedron Lett., 2000, 41, 5341 CrossRef CAS; (h) I. Hanna and L. Ric, Org. Lett., 2000, 2, 2651 CrossRef CAS; (i) J. Marco-Contelles and E. Opazo, J. Org. Chem., 2000, 65, 5416 CrossRef CAS; (j) C. Gravier-Pelletier, W. Maton, T. Dintinger, C. Tellier and Y. L. Merrer, Tetrahedron, 2003, 59, 8705 CrossRef CAS; (k) G. Rassu, L. Auzzas, L. Pinna, V. Zambrano, F. Zanardi, L. Battistini, E. Gaetani, C. Curti and G. Casiraghi, J. Org. Chem., 2003, 68, 5881 CrossRef CAS.
- (a) S. Mondal, A. Prathap and K. M Sureshan, J. Org. Chem., 2013, 78, 7690 CrossRef CAS; (b) A. Vidyasagar and K. M. Sureshan, Eur. J. Org. Chem., 2014, 2349 CrossRef CAS; (c) S. Mondal and K. M. Sureshan, Org. Biomol. Chem., 2014, 12, 7279 RSC; (d) A. Vidyasagar and K. M. Sureshan, Org. Biomol. Chem., 2015, 13, 3900 RSC; (e) S. Mondal and K. M. Sureshan, J. Org. Chem., 2016, 81, 11635 CrossRef CAS; (f) R. Mohanrao, A. Asokan and K. M. Sureshan, Chem. Commun., 2014, 50, 6707 RSC.
- J. H. Dam, P. Fristrup and R. Madsen, J. Org. Chem., 2008, 73, 3228 CrossRef CAS.
- W. Huang, H.-Y. Rong and J. Xu, J. Org. Chem., 2015, 80, 6628 CrossRef CAS.
- S. Dey and N. Jayaraman, Pure Appl. Chem., 2014, 86, 1401 CAS.
- (a) J. Yang, H. Xu, Y. Zhang, L. Bai, Z. Deng and T. Mahmud, Org. Biomol. Chem., 2011, 9, 438 RSC; (b) A. Scaffidi, K. A. Stubbs, D. J. Vocadlo and R. V. Stick, Carbohydr. Res., 2008, 343, 2744 CrossRef CAS; (c) D. H. Mac, R. Samineni, A. Sattar, S. Chandrasekhar, J. S. Yadav and R. Gree, Tetrahedron, 2011, 67, 9305 CrossRef CAS.
- R. Ding, Y. Liu, M. Han, W. Jiao, J. Li, H. Tian and B. Sun, J. Org. Chem., 2018, 83, 12939 CrossRef CAS.
- A. Sonousi, A. Vasella and D. Crich, J. Org. Chem., 2020, 85, 7583 CrossRef CAS.
- A. Sevšek, M. Celan, B. Erjavec, L. Q. van Ufford, J. S. Torano, E. E. Moret, R. J. Pieters and N. I. Martin, Org. Biomol. Chem., 2016, 14, 8670 RSC.
- H. A. Jung, P. Paudel, S. H. Seong, B.-S. Min and J. S. Choi, Bioorg. Med. Chem. Lett., 2017, 27, 2274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures and full characterization of compounds by 1H NMR, 13C NMR. CCDC 2046641 and 2046642. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00804h |
| This journal is © The Royal Society of Chemistry 2021 |