
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst-free one-pot, four-component approach for the synthesis of di- and tri-substituted N-sulfonyl formamidines†
Ai-Ran Liua,
Lei Zhanga,
Jiao Lia and
Abudureheman Wusiman *ab
*ab
aSchool of Chemistry and Chemical Engineering, Xinjiang Normal University, Urumqi, Xinjiang 830054, P. R China. E-mail: arahman@xjnu.edu.cn
bXinjiang Key Laboratory of Energy Storage and Photoelectrocatalytic Materials, Urumqi 830054, China
First published on 27th April 2021
Abstract
A straightforward one-pot, multicomponent approach was developed to synthesize di- and tri-substituted N-sulfonyl formamidines from sulfonyl chlorides, NaN3, ethyl propiolate, and primary/secondary amines under mild conditions without catalysts or additives. Structural analysis of the di-substituted sulfonyl formamidines indicated formation of the E-syn/anti isomeric form. Tri-substituted analogues only formed E-isomers.
Amidines are important nitrogen-containing organic compounds owing to their unique structural and chemical properties.1 N-Sulfonyl amidines are a special class of amidines, which serve as essential intermediates in numerous important organic reactions,2 and can be useful building blocks for synthesizing heterocyclic compounds3 or chelating ligands for transition metals.4 In addition, sulfonyl amidines have wide applications in biopharmaceutical molecular design and the exploration of lead compounds in drug discovery.5 Over the past decade, research regarding the formation of N-sulfonyl amidines has led to remarkable progress.6
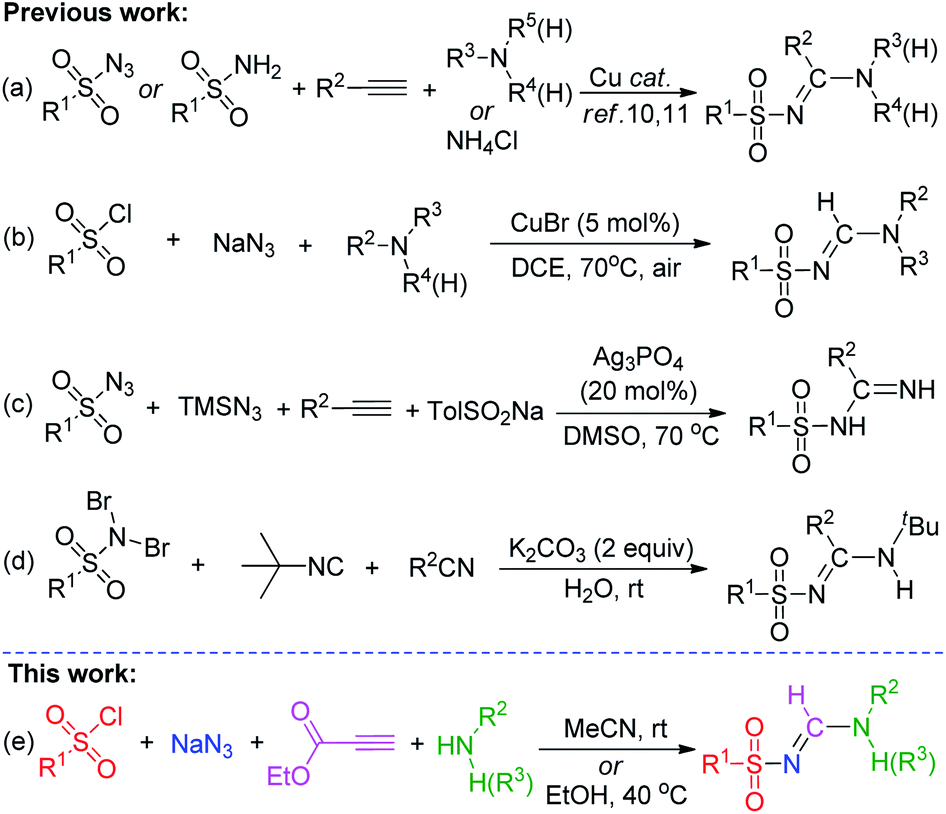
Recently, multicomponent reaction (MCR) methods have attracted considerable attention for their potential ability to access biologically active compounds7 and molecules relevant to drug discovery.8 N-Sulfonylamidines fit these criteria because they are found in numerous biologically active natural products and important biopharmaceuticals.5 Various elegant MCR methods involving the formation of N-sulfonylamidines have already been developed. For example, a Cu-catalyzed three-component tandem reaction between (i) sulfonyl azides9/amides,10 (ii) alkynes, and (iii) primary, secondary, or tertiary amines or ammonium salts has been described (Scheme 1a). Another recently reported Cu-catalyzed three-component approach employed sulfonyl chlorides, sodium azides, and amines (Scheme 1b).11 Additionally, Bi and co-workers12 described a silver-catalyzed, one-pot, four-component reaction involving terminal alkynes reacting directly with trimethylsilyl azide (TMSN3), sodium sulfinate, and sulfonyl azide (Scheme 1c). Phukan et al.13 described a metal-free strategy for the synthesis of di-substituted sulfonyl amidines via a one-pot reaction between tert-butylisonitrile and N,N-dibromoaryl sulfonamides in the presence of a nitrile compound in aqueous media (Scheme 1d), and other groups have applied similar methods.14 However, most protocols still typically require special reagents, including transition metal catalysts or expensive and potentially explosive sulfonyl azides, and they often proceed at elevated temperatures. Therefore, the development of an efficient and practical method for the synthesis of N-sulfonylamidine derivatives is critical. Although tri-substituted sulfonyl(form)amidines have been widely explored,6a–r the synthesis of di-substituted N-sulfonyl formamidines is rare in the literature; to our knowledge, only one research paper (from Jacobson and co-workers, 1977)15 has described the synthesis of N,N′-di-substituted sulfonyl formamidines using sulfonamide and isocyanides in the presence of a copper catalyst.
 | ||
| Scheme 1 Multicomponent reactions for the synthesis of N-sulfonylamidines. | ||
In recent years, the reaction of enamines and azides has received increasing interest due to their diversified chemical reactions.16 More recently, Wan and co-workers developed methods for the direct synthesis of tri-substituted,6o and NH2-featured6t sulfonyl (form)amidines by using N,N-disubstituted enaminoesters and NH2-functionalized enaminone with sulfonylazide. However, NH-functionalized enaminone did not undergo this reaction. In addition these reactions require 5.0 and 2.5 equiv. of sulfonyl azides, respectively, and the desired tri-substituted N-sulfonyl formamidine was obtained in a lower yield. We hypothesized that sulfonyl chlorides, NaN3, primary (secondary) amine, and ethyl propiolate might react under same conditions to generate N-sulfonyl formamidines via in situ formation of corresponding NH-mono and N,N-disubstituted enaminoesters17 and sulfonyl azides.18 On the basis of our previous work regarding the synthesis of N-sulfonyl formamidine,19 we herein describe a mild and simple one-pot multicomponent method for the synthesis of di- and tri-substituted N-sulfonyl formamidines (Scheme 1e). This catalyst-free cascade approach avoids any metals and additives, all of the substrates are commercially available, inexpensive and easily handled. In addition these reactions can be carried out in an open atmosphere at room temperature.
To evaluate the feasibility of this hypothesis, we initially considered the formation of di-substituted sulfonyl formamidines, and tosyl chloride (TsCl; 1a), NaN3 (2), ethyl propiolate (3), and butylamine (4a) were chosen as model substrates to optimize the reaction conditions (Table 1). The reaction proceeded smoothly in ethanol (EtOH) at room temperature and afforded di-substituted sulfonylformamidine 5a, in 38% yield (with nearly a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 syn/anti isomeric ratio) together with N-butyltosyl amide as by-product (entry 1). Following investigations with other solvents (entries 1–9), it was determined that acetonitrile (MeCN) led to the best yield (entry 3) among the tested solvents (i.e., toluene, EtOAc, DMF, THF, H2O, and MeOH). Closer inspection of the reaction (monitored using thin-layer chromatography; TLC) revealed that the intermediate TsN3 (generated from 1a and 2), was consumed easily. Probably the excess of sulfonyl azide play a crucial role on the reaction yield.12 Therefore we thought to increase the substrate loading of 1a and 2 under the same reaction conditions, when both the 1a and 2 dosages were increased to 1.5 equiv. the highest yield (67%) of desired 5a and 24% of N-butyltosyl amide (by-product) were obtained, respectively (entry 10). With addition of more 1a and 2, the yield did not improve further (entry 11). When the reaction temperature was increased to 40 °C or decreased to 0 °C, the yield was reduced to 60% or 30%, respectively (entries 12 and 13). When, 3-butyn-2-one was used in place of ethyl propiolate (3), and the same product was isolated in 30% yield (entry 14). Whereas, under O2 or N2 atmosphere, the yields were no longer improved, and afforded 65% and 68% yields of desired product, respectively (entries 15 and 16). Therefore, the optimal reaction conditions (entry 11) were set as follows: TsCl (1.5 equiv.), NaN3 (1.5 equiv.), ethyl propiolate (1.0 equiv.), and BuNH2 (1.0 equiv.) in MeCN at room temperature.
:3 syn/anti isomeric ratio) together with N-butyltosyl amide as by-product (entry 1). Following investigations with other solvents (entries 1–9), it was determined that acetonitrile (MeCN) led to the best yield (entry 3) among the tested solvents (i.e., toluene, EtOAc, DMF, THF, H2O, and MeOH). Closer inspection of the reaction (monitored using thin-layer chromatography; TLC) revealed that the intermediate TsN3 (generated from 1a and 2), was consumed easily. Probably the excess of sulfonyl azide play a crucial role on the reaction yield.12 Therefore we thought to increase the substrate loading of 1a and 2 under the same reaction conditions, when both the 1a and 2 dosages were increased to 1.5 equiv. the highest yield (67%) of desired 5a and 24% of N-butyltosyl amide (by-product) were obtained, respectively (entry 10). With addition of more 1a and 2, the yield did not improve further (entry 11). When the reaction temperature was increased to 40 °C or decreased to 0 °C, the yield was reduced to 60% or 30%, respectively (entries 12 and 13). When, 3-butyn-2-one was used in place of ethyl propiolate (3), and the same product was isolated in 30% yield (entry 14). Whereas, under O2 or N2 atmosphere, the yields were no longer improved, and afforded 65% and 68% yields of desired product, respectively (entries 15 and 16). Therefore, the optimal reaction conditions (entry 11) were set as follows: TsCl (1.5 equiv.), NaN3 (1.5 equiv.), ethyl propiolate (1.0 equiv.), and BuNH2 (1.0 equiv.) in MeCN at room temperature.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. | t (h) | Yieldb (%) |
| a Reactions were performed with 1a (1.2 mmol), 2 (1.2 mmol), 3 (1.0 mmol), and 4a (1.0 mmol) in 4 mL of solvent at room temperature under open-air conditions, unless otherwise noted.b Isolated yield after column chromatography.c 1a and 2 were 1.5 mmol.d N-Butyltosyl amide was isolated in 24% yield.e 1a and 2 were 1.8 mmol.f 3-Butyn-2-one was used instead of ethyl propiolate (3).g Under O2 (1 atm) atmosphere.h Under N2 atmosphere. | ||||
| 1 | EtOH | rt | 6 | 38 |
| 2 | DCM | rt | 12 | 36 |
| 3 | MeCN | rt | 8 | 56 |
| 4 | Toluene | rt | 18 | 12 |
| 5 | EtOAc | rt | 18 | 10 |
| 6 | DMF | rt | 18 | Trace |
| 7 | THF | rt | 8 | 11 |
| 8 | H2O | rt | 8 | 7 |
| 9 | MeOH | rt | 8 | 18 |
| 10c | MeCN | rt | 8 | 67d |
| 11e | MeCN | rt | 8 | 66 |
| 12 | MeCN | 40 °C | 5 | 60 |
| 13 | MeCN | 0 °C | 12 | 30 |
| 14f | MeCN | rt | 5 | 30 |
| 15g | MeCN | rt | 6 | 65 |
| 16h | MeCN | rt | 6 | 68 |
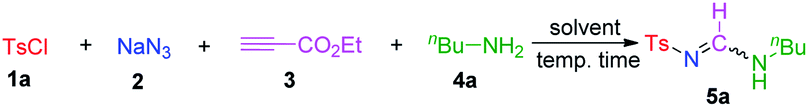
Applying the optimized reaction conditions, we explored the substrate scope of this reaction (Table 2). The aromatic sulfonyl chlorides were first investigated using n-butylamine, and it was observed that electron-donating or electron-withdrawing groups on the benzene ring (5a–5e) were tolerated well by this reaction, generating the desired products in moderate to good yields with 7:3 E-syn/anti isomeric ratios. With electron-withdrawing groups, such as NO2, Cl, and Br, the yields were relatively higher than those obtained with substrates containing electron-donating groups. The best yield was obtained (5d) when using 2-chlorobenzenesulfonyl chloride, which revealed that the substituent on the benzene ring had a significant effect on the reaction yield. Then, naphthalene sulfonyl chloride and 3-pyridinyl chloride were employed under the standard conditions, and 79% and 75% yields were isolated, respectively (5f and 5g). Unfortunately, this methodology did not work with aliphatic sulfonyl chloride; when n-butyl sulfonyl chloride was used as a reaction partner, the desired product (5h) was not detected. Interestingly, the 4-ethoxycarbonyl-1H-1,2,3-triazole was isolated with 70% yield. In addition, benzyl sulfonyl chloride was tested, and the same product was observed in 24% yield under identical reaction conditions. A similar product was synthesised from enaminone and tosyl azide.16d
| a Reactions were performed with 1 (1.5 mmol), 2 (1.5 mmol), 3 (1 mmol), and 4 (1 mmol) in 2.0 mL of solvent at room temperature under open-air conditions, unless otherwise noted.b Isolated yield after column chromatography.c The ratio of the E-syn and E-anti isomers is given in parentheses.d Gram scale reaction after 7 h.e 4-Ethoxycarbonyl-1H-1,2,3-triazole was isolated in 70% yield.f Methylamine aqueous solution was used.g Reaction was performed in an MeCN/H2O (3:1) solvent mixture at 80 °C. |
|---|
 |
Next, the scope of linear, branched, and cyclic primary aliphatic amines was studied, and it was determined that the amine structure did not appreciably influence the reaction yield because moderate to good yields were obtained with similar isomeric ratios (5i–5o). When tert-butylamines were used as substrates, satisfactory yields of the desired products were observed (5p–5r). Interestingly, the syn/anti rotameric ratios of the products changed to 3:7, and this observation was likely attributed to the effect of the bulky tert-butyl group on the molecular configuration.
It is worth mentioning that only a trace amount of 5s was observed when aniline was treated under standard conditions. However, the same reaction yield (43%) of the desired products (with a 1:1 syn/anti rotameric ratio) was obtained in a MeCN/H2O solvent mixture at elevated temperature with a longer reaction time. This special condition is likely required because of the poor reactivity between aniline and ethyl propiolate.20
To further clarify the structural properties of these sulfonyl formamidines, the nuclear magnetic resonance (NMR) spectra and X-ray single crystal structural data were examined. All of the products had two sets of signals in both their 1H- and 13C-NMR spectra (see the ESI†). In the 1H-NMR spectra, we observed that the imide (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) proton appeared as two doublets, with different intensities and coupling constants (J ≈ 13 Hz and 6.0 Hz). The signals corresponding to the N–H proton also appeared as two doublets or broad peaks with different intensities. These results confirmed the formation of di-substituted sulfonyl formamidines as either the Z-syn/anti or E-syn/anti-isomeric/rotameric forms (Fig. 1a).15
N) proton appeared as two doublets, with different intensities and coupling constants (J ≈ 13 Hz and 6.0 Hz). The signals corresponding to the N–H proton also appeared as two doublets or broad peaks with different intensities. These results confirmed the formation of di-substituted sulfonyl formamidines as either the Z-syn/anti or E-syn/anti-isomeric/rotameric forms (Fig. 1a).15
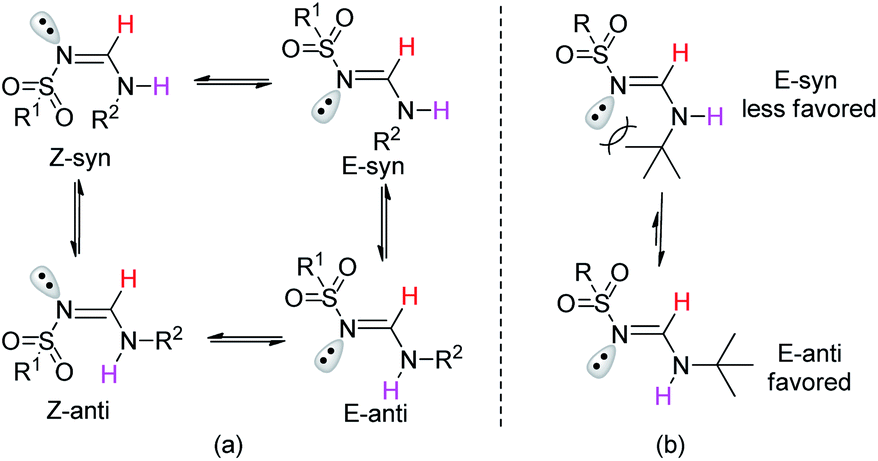 | ||
| Fig. 1 (a) Geometrical/rotational isomers of N-alkyl-N′-sulfonyl formamidine. (b) The effect of the tert-butyl group on the configuration. | ||
Additionally, on the basis of the 1H-NMR spectra, we observed that the syn/anti rotameric ratios were nearly 7:3 for linear and less branched N-alkyl-N′-sulfonyl formamidines; in contrast, the N-tert-butyl-N′-sulfonyl formamidines had syn/anti ratios of 3:7 (see the ESI† and Fig. 2). This result was attributed to the fact that in the syn configuration, the bulky tert-butyl group introduces more steric hindrance, causing this form to be relatively less favored (Fig. 1b).
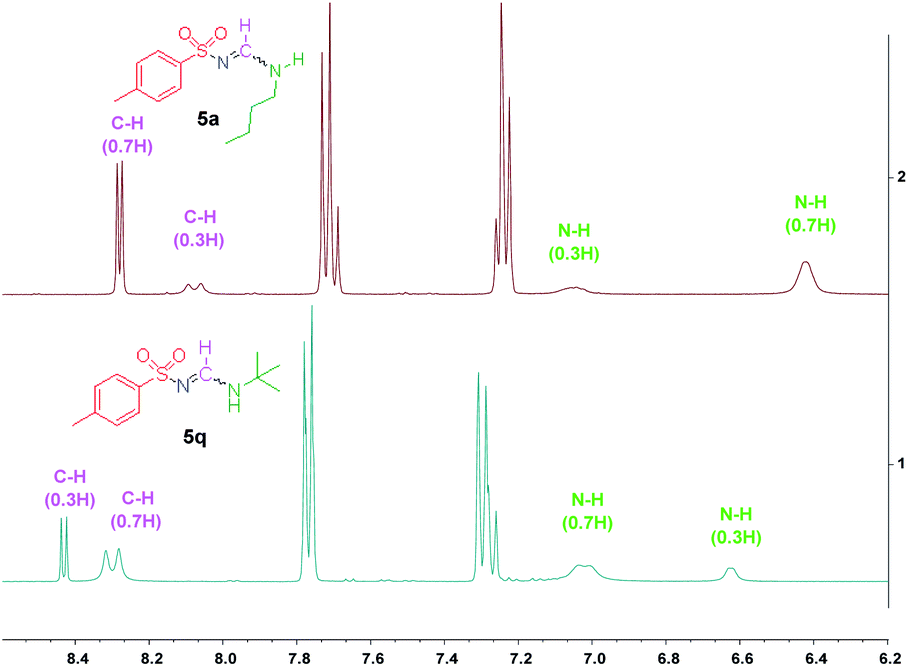 | ||
| Fig. 2 1H NMR spectrum of compounds 5a and 5q. | ||
The NMR results are consistent with the X-ray single crystal data.21 For example, 5c was mainly produced in the E-syn configuration (Fig. 3), but 5r was predominantly generated in the E-anti form (Fig. 4). Therefore, we concluded that the linear and less branched N-alkyl-N′-sulfonyl formamidines exist mainly as E-syn isomers, and the N-tert-butyl-N′-sulfonyl formamidines exist predominantly in the E-anti isomeric form.
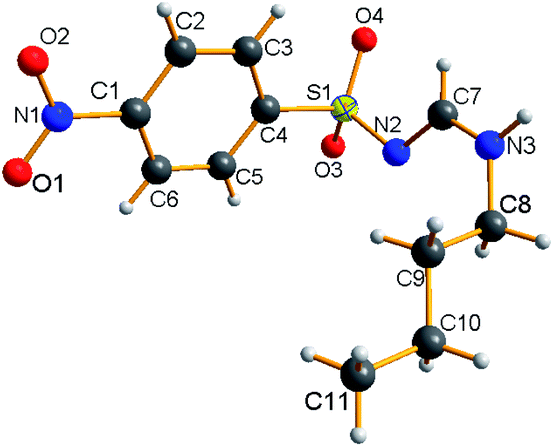
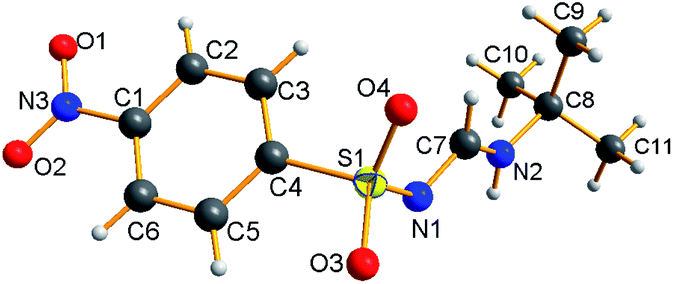
To elucidate the reaction mechanism, a series of control experiments were carried out (Scheme 2). First, the tosyl chloride (1a) and NaN3 (2) were conducted under standard conditions and isolated 90% yield of tosylazide (A) (Scheme 2a). After that the reaction of ethyl propiolate (3) and butylamine (4a) gave 93% yield of enaminoester (B) under same conditions (Scheme 2b). This indicates that established reaction conditions are suitable for the formation of two intermediates. Next the tosylazide (A) was treated with enaminoester (B) and isolated 73% of desired 5a together with 17% of N-butyltosyl amide and 15% of 4-ethoxycarbonyl-1H-1,2,3-triazole, respectively (see the ESI, S6†). This result revealed that the 1,2,3-triazole and N-substituted sulfonamide were eliminated after formation of two intermediates.
 | ||
| Scheme 2 Control experiments. | ||
On the basis of the experimental results obtained in the present study and a literature survey,16 a potential mechanism for the MCR was proposed (Scheme 3). First, the ethylpropiolate and amine reacted to generate enaminoester (B),17 while sulfonylazide (A)18 was simultaneously formed from sulfonyl chloride and NaN3 under the same conditions. Then, the triazoline intermediate (C) was produced from active components A and B through a 1,3-dipolar cycloaddition reaction.22 The subsequent cycloreversion of the intermediate C and release of one molecule of ethyldiazoacetate afforded the desired product 5 (route, a).23 The elimination of corresponding sulfonamide from C yields 4-ethoxycarbonyl-1H-1,2,3-triazole (route, b).24
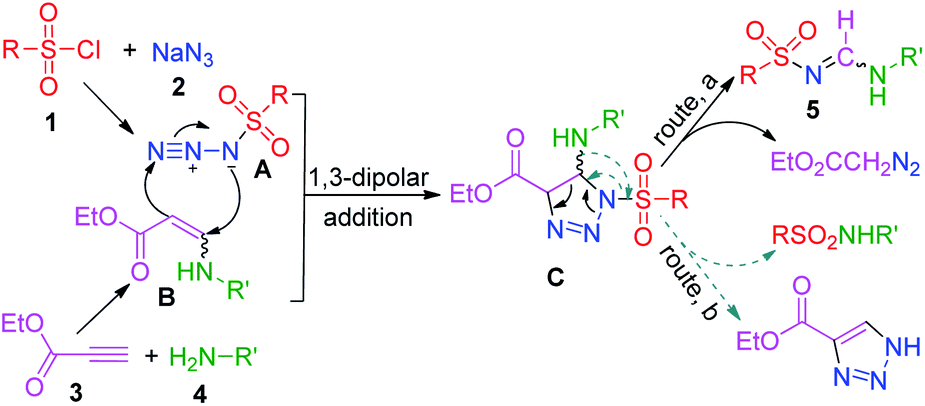 | ||
| Scheme 3 Proposed reaction mechanism. | ||
After a thorough examination of the synthesis of di-substituted N-sulfonyl formamidines, the developed MCR was further extended to the synthesis of tri-substituted sulfonyl formamidines by using secondary amines. First, the reaction between TsCl (1a), NaN3 (2), ethyl propiolate (3), and diethylamine (6a) was optimized (for details, see ESI,† p. S3), and the desired tri-substituted sulfonyl formamidine (7a) was isolated in 91% yield under the optimized conditions. The substrate scopes of the reactions were explored, and the results are presented in Table 3. First, we investigated the reactivity of various sulfonyl chlorides under the optimized conditions. Arylsulfonyl chlorides bearing electron-donating or electron-withdrawing groups on their benzene rings were well tolerated by this reaction process, and generated the desired products in good yields (7a–7d). Moreover, with naphthalene sulfonyl chloride, the reaction proceeded smoothly, and the desired product 7e was obtained in a satisfactory yield. Hetero aromatic sulfonyl chlorides were also tolerated by this reaction, and the best yield (7g) was obtained using 3,5-dimethylisoxazole-4-sulfonyl chloride. Notably, the aliphatic sulfonyl chlorides, i.e., benzylsulfonyl chloride and butylsulfonyl chloride, afforded the products 7h and 7i in 73% and 89% isolated yields, respectively. Interestingly, when aliphatic sulfonyl chlorides and secondary amine were tested, the 4-ethoxycarbonyl-1H-1,2,3-triazole was not observed. This indicates that the N,N-disubstituted triazoline intermediate (see the Scheme 3) preferentially undergoes cycloreversion and eliminate ethyldiazoacetate to form sulfonyl amidine.
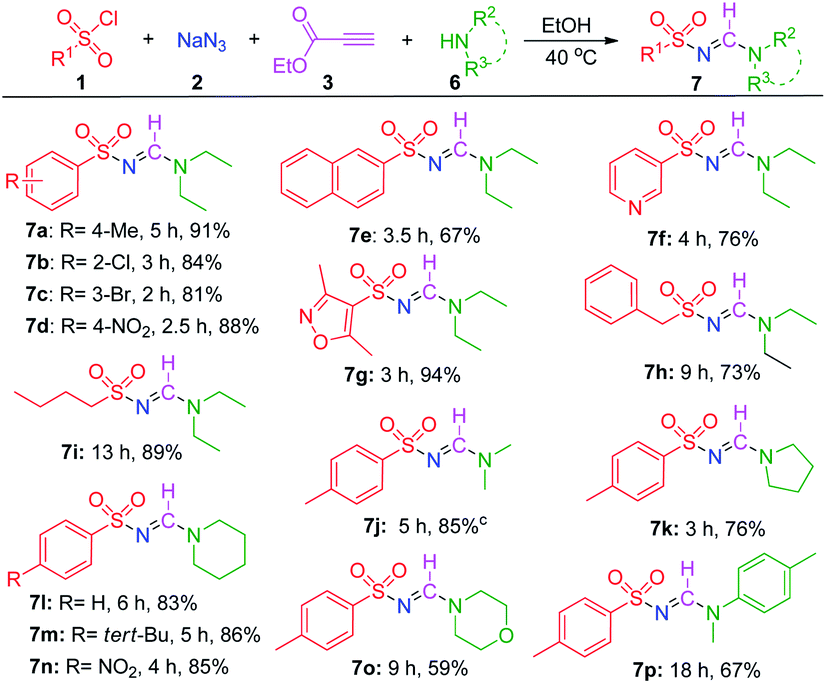
Next, dimethylamine was tested, and the desired product, 7j, was isolated in good yield (85%). When the cyclic and heterocyclic secondary amines, such as pyrrolidine, piperidine, and morpholine were used as the starting materials, the corresponding sulfonylamidines (7k–7o) were obtained with yields between 59% and 86%. Finally, N-methylaniline reacted slowly and led to a 67% isolated yield (7p).
On the basis of the 1H and 13C-NMR spectra of the tri-substituted N-sulfonyl formamidines (one set of signals in both types of spectra, see the ESI†), it was determined that these reactions produced only the E-isomer; this result is consistent with earlier reports.25
Finally, aqueous ammonia was used as a reaction partner, but unfortunately, the expected mono-substituted N-sulfonyl formamidine was not detected.
Conclusions
In summary, a straightforward, one-pot, multicomponent method was developed for the synthesis of di- and tri-substituted N-sulfonyl formamidines using readily accessible substrates under very mild conditions free of catalysts or other additives. This protocol is inexpensive to carry out and step-economic, so it provides a potential route for the construction of diverse N-sulfonyl formamidines in moderate to high yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21762044), and Innovation Training Project of Xinjiang Undergraduate Education (No: S202010762017).Notes and references
- S. Patai, The Chemistry of Amidines and Imidiares, Wiley, New York, 1975 Search PubMed.
- T. Kumamoto and I. Ishikawa, Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organoctalysts, John Wiley & Sons Press, West Sussex, 2009, p. 295 Search PubMed.
- (a) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed; (b) Y.-F. Wang, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 3679 CrossRef CAS PubMed; (c) M. A. McGowan, C. Z. McAvoy and S. L. Buchwald, Org. Lett., 2012, 14, 3800 CrossRef CAS PubMed.
- (a) J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219 CrossRef CAS; (b) A. Wusiman, X. Tusun and C. D. Lu, Eur. J. Org. Chem., 2012, 16, 3088 CrossRef.
- (a) M. Y. Lee, M. H. Kim, J. Kim, S. H. Kim, B. T. Kim, I. H. Jeong, S. Chang, S. H. Kim and S.-Y. Chang, Bioorg. Med. Chem. Lett., 2010, 20, 541 CrossRef CAS PubMed; (b) A. Goubet, A. Chardon, P. Kumar, P. K. Sharma and R. N. Veedu, Bioorg. Med. Chem. Lett., 2013, 23, 761 CrossRef CAS PubMed.
- Selective literatures for the synthesis of sulfonyl amidines. (a) X. L. Xu, Z. C. Ge, D. P. Cheng, L. Ma, C. S. Lu, Q. F. Zhang, N. Yao and X. N. Li, Org. Lett., 2010, 12, 897 CrossRef CAS PubMed; (b) L. Zhang, J. H. Su, S. J. Wang, C. F. Wan, Z. G. Zha, J. F. Z. Du and Y. Wang, Chem. Commun., 2011, 47, 5488 RSC; (c) M. Aswad, J. Chiba, T. Tomohiro and Y. Hatanaka, Chem. Commun., 2013, 49, 10242 RSC; (d) S. Chen, Y. Xu and X. Wan, Org. Lett., 2011, 13, 6152 CrossRef CAS PubMed; (e) N. Chandna, N. Chandak, P. Kumar, J. K. Kapoorb and P. K. Sharma, Green Chem., 2013, 15, 2294 RSC; (f) K. Hajibabaei and H. Zali-Boeini, Synlett, 2014, 25, 2044 CrossRef CAS; (g) J. Chen, Y. P. Guo, M. H. Sun, G. T. Fan and L. Zhou, Chem. Commun., 2014, 50, 12367 RSC; (h) L. Dianova, V. Berseneva, T. Beryozkina, I. Efimov, M. Kosterina, O. Eltsov, W. Dehaen and V. Bakulev, Eur. J. Org. Chem., 2015, 31, 6917 CrossRef; (i) J. Chen, W. Long, S. Fang, Y. Yang and X. Wan, Chem. Commun., 2017, 53, 13256 RSC; (j) S. Y. Chow and L. R. Odell, J. Org. Chem., 2017, 82, 2515 CrossRef CAS PubMed; (k) J. Chen, W. Long, Y. Yang and X. Wan, Org. Lett., 2018, 20, 2663 CrossRef CAS PubMed; (l) Y. Weiguang, H. Dayun, Z. Xiaobao, L. Dongping, W. Xinyan and H. Yuefei, Chem. Commun., 2018, 54, 8222 RSC; (m) J. Gui, H. Xie, H. Jiang and W. Zeng, Org. Lett., 2019, 21, 2804 CrossRef CAS PubMed; (n) R. Ding, H. Chen, Y. Xu, H.-T. Tang, Y. Chen and Y. Pan, Adv. Synth. Catal., 2019, 361, 3656 CrossRef; (o) X. Zheng and J. P. Wan, Adv. Synth. Catal., 2019, 361, 5690 CrossRef CAS; (p) F. Yi, Q. Sun, J. Sun, C. Fu and W. Yi, J. Org. Chem., 2019, 84, 6780 CrossRef CAS PubMed; (q) B. Kaboudin, S. Torabi, F. Kazemi and H. Aoyama, RSC Adv., 2020, 10, 26701 RSC; (r) W. Xia, B. Huang, C. Yang and J. zhou, Chem. Commun., 2020, 56, 5010 RSC; (s) Q. Gou, Z. Liu, T. Cao, X. Tan, W. Shi, M. Ran and J. Qin, J. Org. Chem., 2020, 85, 2092 CrossRef CAS PubMed; (t) G.-D. Wang, Y.-H. Guo and J.-P. Wan, Chin. J. Org. Chem., 2020, 40, 645 CrossRef; (u) X.-X. Zheng, Y.-Y. Liu and J.-P. Wan, Chin. J. Org. Chem., 2020, 40, 1891 CrossRef and references cited therein..
- (a) A. Dömling, Chem. Rev., 2006, 106, 17 CrossRef PubMed; (b) I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol., 2008, 12, 324 CrossRef CAS PubMed.
- B. B. Touré and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef PubMed.
- Selective literatures for the Synthesis of sulfonyl amidines by Cu-catalyzed MCRs. (a) I. Bae, H. Han and S. Chang, J. Am. Chem. Soc., 2005, 127, 2038 CrossRef CAS PubMed; (b) S. Mandal, H. M. Gauniyal, K. Pramanik and B. Mukhopadhyay, J. Org. Chem., 2007, 72, 9753 CrossRef CAS PubMed; (c) J. Kim, S. Y. Lee, J. Lee, Y. Do and S. Chang, J. Org. Chem., 2008, 73, 9454 CrossRef CAS PubMed; (d) I. Yavari, S. Ahmadian, M. Ghazanfarpur-Darjani and Y. Solgi, Tetrahedron Lett., 2011, 52, 668 CrossRef CAS; (e) J. Y. Kim, S. H. Kim and S. Chang, Tetrahedron Lett., 2008, 49, 1745 CrossRef CAS; (f) J. Wang, P. Lu and Y. Wang, Org. Chem. Front., 2015, 2, 1346 RSC; (g) T. D. Suja, K. V. L. Divya, L. V. Naik, A. R. Kumar and A. Kamal, Bioorg. Med. Chem. Lett., 2016, 26, 2072 CrossRef CAS PubMed; (h) X. He, Y. Shang, J. Hu, K. Ju, W. Jiang and S. Wang, Sci. China Chem., 2012, 55, 214 CrossRef CAS; (i) M. Xu, C. Kuang, Z. Wang and Q. Yang, Synlett, 2010, 17, 2664 Search PubMed.
- J. Kim, S. S. Stahl, J. Kim and S. S. Stahl, J. Org. Chem., 2015, 80, 2448 CrossRef CAS PubMed.
- W. Z. Bi, W. J. Zhang, Z. J. Li, X. Y. Xia, X. L. Chen, L. B. Qu and Y. F. Zhao, Eur. J. Org. Chem., 2019, 35, 6071 CrossRef.
- B. Liu, Y. Ning, M. Virelli, G. Zanoni, E. A. Anderson and X. Bi, J. Am. Chem. Soc., 2019, 141, 1593 CrossRef CAS PubMed.
- D. Mishra, A. J. Borah, P. Phukan, D. Hazarika and P. Phukan, Chem. Commun., 2020, 56, 8408 RSC.
- (a) S. Shojaei, Z. Ghasemi and A. Shahrisa, Tetrahedron Lett., 2017, 58, 3957 CrossRef CAS; (b) M. Jagadale, P. Bhange, R. Salunkhe, D. Bhange, M. Rajmane and G. Rashinkar, Appl. Catal., A, 2006, 511, 95 CrossRef; (c) M. J. Kim, B. R. Kim, C. Y. Lee and J. Kim, Tetrahedron Lett., 2016, 57, 4070 CrossRef CAS; (d) T. Yang, H. Cui, C. Zhang, L. Zhang and C. Y. Su, Inorg. Chem., 2013, 52, 9053 CrossRef CAS PubMed; (e) Y. Huang, W. Yi, Q. Sun and F. Yi, Adv. Synth. Catal., 2018, 360, 3074 CrossRef CAS; (f) J. Wang, J. Liu, H. Ding, J. Wang, P. Lu and Y. Wang, J. Org. Chem., 2015, 80, 5842 CrossRef CAS PubMed; (g) J. Chen, W. Long, Y. Zhao, H. Li, Y. Zheng, P. Lian and X. Wan, Chinese J. Chem., 2018, 36, 857 CrossRef CAS; (h) B. Yao, C. Shen, Z. Liang and Y. Zhang, J. Org. Chem., 2014, 79, 936 CrossRef CAS PubMed; (i) Y. Zhang and Z. Chen, Tetrahedron Lett., 2018, 59, 4183 CrossRef CAS; (j) Z. Zhang, B. Huang, G. Qiao, L. Zhu, F. Xiao, F. Chen and Z. Zhang, Angew. Chem., Int. Ed., 2017, 56, 4320 CrossRef CAS PubMed.
- P. Jakobsen and S. Treppendahl, Tetrahedron, 1977, 33, 3137 CrossRef CAS.
- (a) V. A. Bakulev, T. Beryozkina, J. Thomas and W. Dehaen, Eur. J. Org. Chem., 2018, 262 CrossRef CAS and references cited therein.; (b) J.-P. Wan, S. Cao and Y. Liu, Org. Lett., 2016, 18, 6034 CrossRef CAS PubMed; (c) A. De Nino, P. Merino, V. Algieri, M. Nardi, M. L. Di Gioia, B. Russo, M. A. Tallarida and L. Maiuolo, Catalysts, 2018, 8, 364 Search PubMed; (d) L. Yang, Y. Wu, Y. Yang, C. Wen and J.-P. Wan, Beilstein J. Org. Chem., 2018, 14, 2348 CrossRef CAS PubMed; (e) L. Maiuolo, B. Russo, V. Algieri, M. Nardi, M. L. Di Gioia, M. A. Tallarida and A. D. Nino, Tetrahedron Lett., 2019, 60, 672 CrossRef CAS.
- M. Regitz, J. Hocker and A. Liedhegener, Org. Synth., 1973, 48, 36 Search PubMed.
- (a) X.-Y. Chen, L. Zhang, Y. Tang, S. Yuan, B. Zhu, G. Chen and X. Cheng, Synlett, 2020, 31, 878 CrossRef CAS; (b) Y. I. Él'natanov and R. G. Kostyanovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1988, 37, 302 CrossRef; (c) N. A. Randive, V. Kumar and V. A. Nair, Mon. Chem., 2010, 141, 1329 CrossRef CAS; (d) G. Choudhary and R. K. Peddinti, Green Chem., 2011, 13, 3290 RSC.
- (a) A. Rouzi, R. Hudabaierdi and A. Wusiman, Tetrahedron, 2018, 74, 2475 CrossRef CAS; (b) A. Wusiman and R. Hudabaierdi, Synth. Comm., 2017, 47, 2015 CrossRef CAS.
- D. R. Chisholm, R. Valentine, E. Pohl and A. Whiting, J. Org. Chem., 2016, 81, 7557 CrossRef CAS PubMed.
- CCDC 2055587 (5c) and 2055586 (5r) contain the supplementary crystallographic data for this paper.†.
- R. Fusco, G. Bianchetti, D. Pocar and R. Ugo, Chem. Ber., 1963, 96, 802 CrossRef CAS.
- (a) T. Gao, M. Zhao, X. Meng, C. Li and B. Chen, Synlett, 2011, 1281 CAS; (b) Y. Xu, Y. Wang and S. Zhu, J. Fluorine Chem., 2000, 104, 195 CrossRef CAS; (c) A. Contini, E. Erba and S. Pellegrino, Synlett, 2012, 23, 1523 CrossRef CAS.
- (a) A. S. Maiorana, D. Pocar and P. Dalla Croce, Tetrahedron Lett., 1966, 7, 6043 CrossRef; (b) P. Zanirato, J. Chem. Soc., Perkin Trans. 1, 2002, 1420 RSC; (c) N. Kato, Y. Hamada and T. Shioiri, Chem. Pharm. Bull., 1984, 32, 2496 CrossRef CAS; (d) N. Kawai, N. Kato, Y. Hamada and T. Shioiri, Chem. Pharm. Bull., 1983, 31, 3139 CrossRef CAS.
- S. Wang, Z. Wang and X. Zheng, Chem. Comm., 2009, 47, 7372 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2055586 and 2055587. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00772f |
| This journal is © The Royal Society of Chemistry 2021 |