
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of microstructural variations on morphology and separation properties of polybutadiene-based polyurethanes†
Ali Pournaghshband Isfahani ab,
Mahdi Shahroozc,
Takuma Yamamotoab,
Ansori Muchtarab,
Masateru M. Itoab,
Daisuke Yamaguchiab,
Mikihito Takenakaad,
Easan Sivaniah*ab and
Behnam Ghalei*ab
ab,
Mahdi Shahroozc,
Takuma Yamamotoab,
Ansori Muchtarab,
Masateru M. Itoab,
Daisuke Yamaguchiab,
Mikihito Takenakaad,
Easan Sivaniah*ab and
Behnam Ghalei*ab
aInstitute for Integrated Cell-Materials Sciences (iCeMS), Kyoto University, Yoshida-Honmachi, Sakyo-ku, 606-8501 Kyoto, Japan. E-mail: esivaniah@icems.kyoto-u.ac.jp; bghalei@icems.kyoto-u.ac.jp
bDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Nishikyo-ku, 615-8510 Kyoto, Japan
cInstitute for Sustainable Industries and Liveable Cities, Victoria University, 14428 Melbourne, VIC, Australia
dInstitute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan
First published on 26th April 2021
Abstract
Polybutadiene-based polyurethanes with different cis/trans/1,2-vinyl microstructure contents are synthesized. The phase morphology and physical properties of the polymers are investigated using spectroscopic analysis (FTIR and Raman), differential scanning calorimetry (DSC), X-ray scattering (WAXD and SAXS) and atomic force microscopy (AFM). In addition, their gas transport properties are determined for different gases at 4 bar and 25 °C. Thermodynamic incompatibility and steric hindrance of pendant groups are the dominant factors affecting the morphology and properties of the PUs. FTIR spectra, DSC, and SAXS analysis reveal a higher extent of phase mixing in high vinyl-content PUs. Moreover, the SAXS analysis and AFM phase images indicate smaller microdomains by increasing the vinyl content. Smaller permeable soft domains as well as the lower phase separation of the PUs with higher vinyl content create more tortuous pathways for gas molecules and deteriorate the gas permeability of the membranes.
Introduction
Knowledge about the structure–property relationships of block copolymers is necessary for selecting proper materials for any specific application. Most properties of block copolymers, such as polyurethanes (PUs), are directly related to their multiphase morphology.1–3 From the theoretical perspective, the difference between the polarity of the urethane hard segment and polyol soft segment (i.e., thermodynamic incompatibility) leads to a phase-separated structure.4 Besides, higher chain mobility (i.e., kinetic factor) of either soft or hard segments can improve phase separation.5,6 Nevertheless, complete phase segregation rarely takes place in PU systems. The intra- and inter-segmental hydrogen bonding in the hard and soft domains determine the block structures. Besides, the crystallinity and glass transition temperature of each block determine the morphology.7PU morphology can be manipulated by the chemistry of its components (e.g., polyol and diisocyanates), thermal annealing,8,9 and synthesis methods.10 The extent of phase segregation was found to have a major impact on the mechanical and thermal properties of PUs.10 The dissolution of soft segments in the hard domains disrupts hydrogen bonding and the molecular order in urethane chains that could lead to lower mechanical properties.11–13 It was shown that using symmetrical monomers in PU synthesis can considerably improve phase separation and mechanical properties.14–16 Moreover, gas permeation in PU membranes is significantly influenced by the degree of phase separation between the hard and soft segments. Since the soft domains are the main diffusion pathway for gas molecules in PU membranes, phase intermixing can adversely affect gas transport properties. A wide range of gas permeability has been found for different PUs, ranging from 10 to 1000 barrer (1 barrer = 10−10 cm3 (STP) cm cm−2 s−1 cmHg−1), which discloses the impact of morphology on the separation properties of PU membranes.17,18
The degree of phase separation can be altered by the extent of hydrogen bonding, steric hindrance and incompatibility between the two blocks. The urethane-urea bonds or crosslinking the hard segments favor phase segregation.19,20 Moreover, moderate phase mixing was observed in polyether-based PUs because of the association of oxygen groups in polyether with urethane –NH groups in the hard segments. Polyethylene glycol (PEG) and polypropylene glycol (PPG) are among the most common polyethers in the PU industry. Compared to PPG, PEG has a higher tendency to mix with the urethane hard segments due to its lower hydrophobicity. Therefore, the gas permeability of PEG-based PU membranes is inferior due to their lower phase separation and semi-crystalline structure as compared to the PPG-based membranes with amorphous soft domains.21 In addition to thermodynamic parameters, it was claimed that steric hindrance and chain conformation play an important role in the disruption of hydrogen bonding between the hard and soft segments.22–24 In our previous publication, a hydroxyl-terminated PEG-b-PPG block copolymer was used for PU synthesis.21 The methyl side group in PPG spatially hindered the interaction between the PEG and the hard segment. The membranes showed a ca. 400% improvement in CO2 permeability compared to the PEG-based PU membrane. Although polyols with methyl and/or fluorine pendant groups would enhance phase segregation,25,26 the presence of bulky side groups in the chain extender has the opposite effect.10 Moreover, tuning the morphology of block copolymer membranes for a better gas separation performance was accomplished by the incorporation of nanoparticles in the polymer matrix.27–31 Interaction of fillers with the functional groups of either soft or hard blocks reduces phase-intermixing in the PU matrix.
The development of PU systems with low miscibility between hard and soft segments is desirable in gas separation membranes. Polydimethylsiloxane (PDMS) and hydroxyl-terminated polybutadiene (HTPBD) are well-known polyols in this regard. Their lower solubility parameter compared to other polyols results in a well-phase-separated PU structure, the properties of which are entirely independent of the hard segment chemistry.32 Additionally, the PUs based on PDMS and HTPBD soft segments exhibit high gas permeability, excellent hydrophobicity and good thermal and oxidation stability.17,33 The large polarity difference between the urethane and PBD creates isolated hard domains with a nearly sharp interface boundary.
The morphology of polybutadiene-based PUs has been studied over the past years.34–36 Nevertheless, the impact of structural configuration (i.e., 1,2-vinyl, cis, trans) on morphology and properties of PUs has not yet been investigated. The pendant 1,2-vinyl moieties in PBD can inhibit the interactions between the two segments and also can suppress the PU crystallinity. In contrast, the viscosity and Tg of the polyol, as two critical parameters in the determination of PU morphology, increase with vinyl content. Also, the polarity and chain mobility of PUs can be influenced by the cis/trans ratio in the polyol.37 In this study, hydroxyl-terminated PBDs with different vinyl contents (7%, 20%, 65%, and 90%) were utilized for PU synthesis. Subsequently, the effect of cis and trans microstructures on the properties of PUs was evaluated by X-ray scattering methods and thermo-mechanical analysis. The gas transport properties of the synthesized membranes were also explored for a variety of gases.
Experimental
Materials
Four types of hydroxyl-terminated polybutadiene with different vinyl contents (7%, 20%, 65%, and 90%) were kindly provided by Nippon Soda Co., Japan, and Cray Valley Co., USA. All polyols were dried under vacuum at 80 °C for 48 h to remove residual water before synthesis. The chemical structure and properties of HTPBDs were shown in Table 1. Isophorone diisocyanate (IPDI), super dehydrated dimethylacetamide (DMA), super dehydrated tetrahydrofuran (THF), and dimethylformamide (DMF) were obtained from Wako Pure Chemical Industries, Ltd., Japan and used as received. Octanediamine (ODA, Tokyo Chemical Industry) and dibutyltin dilaurate (DBTDL, Wako Pure Chemical Industries) were used as chain extender and catalyst, respectively, after being dried over 4 Å molecular sieve (Nacalai Tesque, Japan).| Sample | Polyol | Chemical structure | Mn (g mol−1) | 1-2,Vinyl% | Trans% | Cis% | Tg of polyol (°C) |
|---|---|---|---|---|---|---|---|
| PU(0,0,7) | GI-3000® | 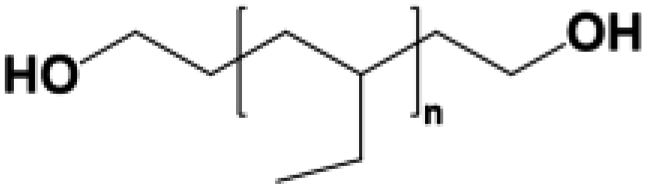 |
3100 | 7 | — | — | −38.0 |
| PU(60,20,20) | Poly bd®R-45HTL | 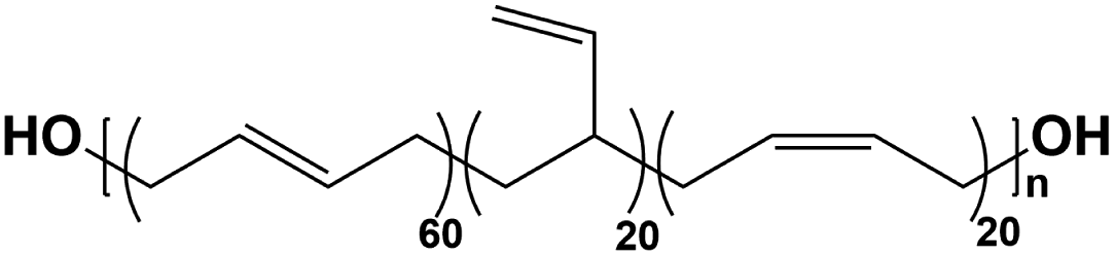 |
2800 | 20 | 60 | 20 | −80.0 |
| PU(22.5,12.5,65) | Krasol®LBH-P3000 | 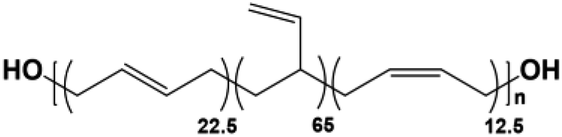 |
3200 | 65 | 22.5 | 12.5 | −48.0 |
| PU(0,0,90) | G-3000® | 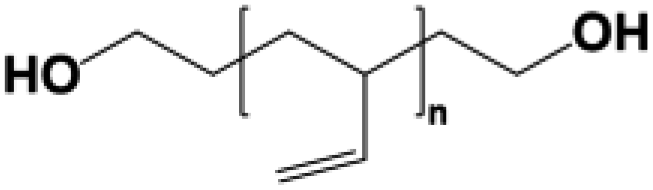 |
3000 | 90 | — | — | −17.0 |
Polymer synthesis
The chemical reaction was shown in Fig. S1.† An exact amount of IPDI (10 mmol) was added dropwise into the stirred HTPBD solution (5 mmol) in 20 mL DMAc/THF (1/1 vol%). 0.1 mL DBTDL was added to the reactor, and the mixture was stirred under Ar atmosphere at 70 °C for 2 h to obtain the NCO-terminated prepolymer. The polymerization was proceeded by the addition of 5 mmol ODA (dissolved in 20 mL THF) into the cooled-room temperature reaction mixture and stirred few minutes. After the completion of the reaction, an exact amount of the mixture was cast into Teflon Petri-dishes, and the membrane films were formed under controlled evaporation at 50 °C for 24 h. The prepared membranes with the thickness of 60–70 μm were further dried under vacuum at 80 °C for 24 h before testing. The synthesized polymers were denoted PU(t,c,v), wherein t, c, and v respectively stand for trans, cis and vinyl isomer contents in HTPBD.Characterization
The chemical structure of the synthesized PUs was determined by Raman (Horiba XploRA, Japan) and FTIR (Shimadzu IR Tracer-100 spectrometer, Japan) spectroscopies. Raman spectra were collected in the range of 3100–400 cm−1 with a resolution of 1 cm−1 and a laser excitation wavelength of 532 nm. ATR-FTIR spectra were obtained in the range of 4000–600 cm−1 with a resolution of 4 cm−1 and 128 scans. The morphology of the samples was determined by wide-angle X-ray diffraction (WAXD, Rigaku RINT XRD, Japan) using CuKα radiation (λ = 1.54 Å) and the scattering range of 2θ = 5–40°. Small-angle X-ray scattering (SAXS, NANO-Viewer, Rigaku, Co, Ltd., Japan) was performed using a 1.2 kW rotating-anode X-ray generator (40 kV, 30 mA). The X-ray wavelength and sample to detector distance were 1.54 Å and 1300 mm, respectively. Thermal transition temperatures of the PUs were detected by differential scanning calorimetry (DSC, Bruker DSC 3100SA, Germany) under N2 atmosphere in the range of −100 °C to 200 °C and at a scanning rate of 10°C min−1. Thermogravimetric analysis (TGA, Rigaku Thermo Plus EVO 2, Japan) was performed under an air atmosphere at the heating rate of 10°C min−1 up to 800 °C. Phase morphological analysis was carried out by atomic force microscopy (AFM, Nano wizard 2, JPK Instruments, Japan) under AC mode with a 6 N m−1 cantilever.Gas permeation test
Pure gas permeability of the PU membranes to H2, O2, N2, CH4, and CO2 was measured using the constant volume-variable pressure method at 4 bar and 25 °C. The permeate pressure and temperature were monitored by a sensor (Keller PAA 33X) connected to Labview software. The permeability of each membrane was measured three times to assess the reproducibility of the data. Gas transport across polymeric membranes is described by the solution-diffusion mechanism, where the permeability is the product of diffusivity coefficient (D) and solubility coefficient (S): P = D × S. The gas permeability is given by the following equation:
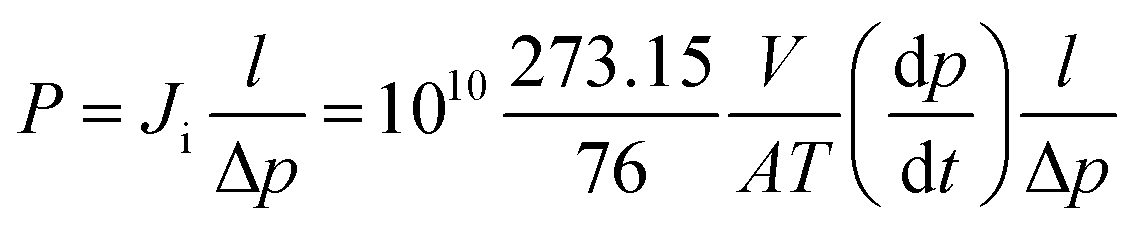 | (1) |
| αi/j = Pi/Pj | (2) |
Results and discussion
Spectroscopic analysis
Fig. 1a represents evidence for the existence of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and carbonyl groups in the synthesized PUs. The disappearance of the NCO peak in FTIR spectra at 2300 cm−1 indicates the completion of the reaction. The bands appearing at 907 cm−1, 995 cm−1, and 3070 cm−1 are assigned to the CC bending vibrations and stretching mode of 1,2-vinyl substitutes,38 which are absent in the PU(0,0,7) sample. Besides, the band at 965 cm−1 refers to the bending CC vibrations of the trans-1,4-PBD microstructure,38,39 where the PU(60,20,20) with 60% trans isomer exhibits the most prominent peak than other samples. The obvious peaks at 1640 cm−1 and 1710 cm−1 correspond to the bonded and free carbonyl groups, respectively, which are helpful in comprehending the phase separation of PUs.40 The presence of pendant functional groups impedes the formation of hydrogen bonding between the hard and soft segments. Nevertheless, the thermodynamic parameter is the dominant factor for the lower phase separation of PU(22.5,12.5,65) compared to that of PU(60,20,20). The R45-HT polyol is less polar than LBHP-3000 due to its higher density of trans-isomer. Therefore, R45-HT has a lower tendency to mix with the polar urethane segments. The steric effect of the pendant functional groups is significant in similar PU(0,0,90) and PU(0,0,7) structures. The higher phase separation of PU(0,0,7) is associated with a higher content of bulky ethyl groups, which effectively disrupt the hydrogen bonding between the urethane –NH groups and the polyol. The A value which is a well-known parameter for the evaluation of the bulkiness of functional groups is 1.75 kcal mol−1 and 1.35 kcal mol−1 for ethyl and 1,2-vinyl substitutes, respectively.41,42
C bonds and carbonyl groups in the synthesized PUs. The disappearance of the NCO peak in FTIR spectra at 2300 cm−1 indicates the completion of the reaction. The bands appearing at 907 cm−1, 995 cm−1, and 3070 cm−1 are assigned to the CC bending vibrations and stretching mode of 1,2-vinyl substitutes,38 which are absent in the PU(0,0,7) sample. Besides, the band at 965 cm−1 refers to the bending CC vibrations of the trans-1,4-PBD microstructure,38,39 where the PU(60,20,20) with 60% trans isomer exhibits the most prominent peak than other samples. The obvious peaks at 1640 cm−1 and 1710 cm−1 correspond to the bonded and free carbonyl groups, respectively, which are helpful in comprehending the phase separation of PUs.40 The presence of pendant functional groups impedes the formation of hydrogen bonding between the hard and soft segments. Nevertheless, the thermodynamic parameter is the dominant factor for the lower phase separation of PU(22.5,12.5,65) compared to that of PU(60,20,20). The R45-HT polyol is less polar than LBHP-3000 due to its higher density of trans-isomer. Therefore, R45-HT has a lower tendency to mix with the polar urethane segments. The steric effect of the pendant functional groups is significant in similar PU(0,0,90) and PU(0,0,7) structures. The higher phase separation of PU(0,0,7) is associated with a higher content of bulky ethyl groups, which effectively disrupt the hydrogen bonding between the urethane –NH groups and the polyol. The A value which is a well-known parameter for the evaluation of the bulkiness of functional groups is 1.75 kcal mol−1 and 1.35 kcal mol−1 for ethyl and 1,2-vinyl substitutes, respectively.41,42
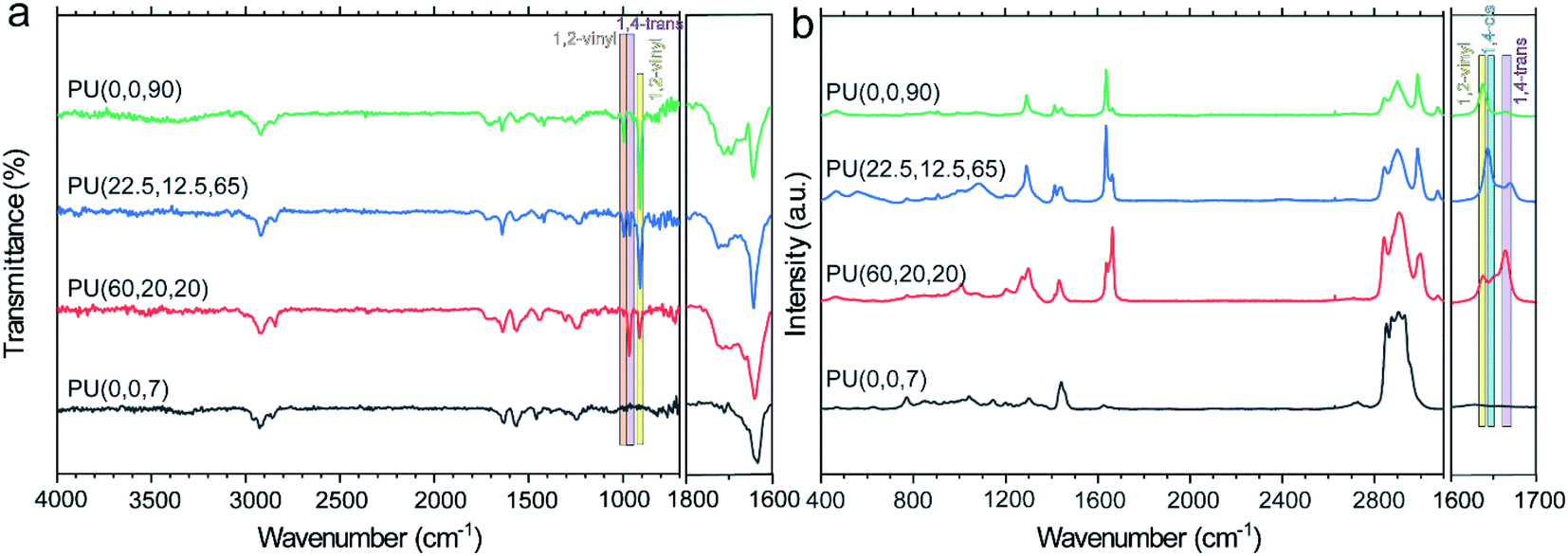 | ||
| Fig. 1 FTIR (a) and Raman (b) spectra of the synthesized polyurethanes. | ||
The degree of phase separation can be evaluated by hydrogen bonding index (HBI), calculated from eqn (3):
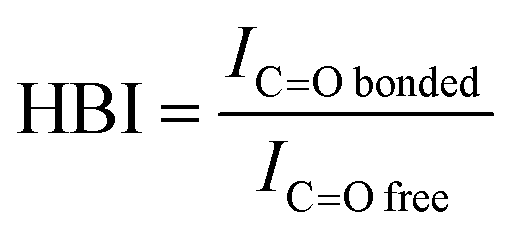 | (3) |
O,bonded and ICO,free correspond to the absorbance intensity of bonded and free carbonyl peaks. The HBI value of the synthesized PUs decreased as follows: PU(0,0,7)(5.6) > PU(60,20,20)(3.0) > PU(22.5,12.5,65)(2.3) > PU(0,0,90)(1.4), which is in agreement with the above discussion. The disruption of hydrogen bonding due to the steric effect of bulky functional groups gives rise to a larger HBI value.
Understanding the PU characteristics depends on a comprehensive study of the polybutadiene structure; thus, it helps analyze the different isomers in PBD quantitatively. Raman spectroscopy detects the CC stretching vibrations in the trans unit since they are infrared inactive.43 As shown in Fig. 1b, the peaks at 1631 cm−1, 1651 cm−1 and 1663 cm−1 are ascribed to the CC stretching modes of 1,2-vinyl, cis and trans in the polybutadiene-based PUs.44,45 The molar content of each configuration can be evaluated by the integration of the corresponding peak. For example, the vinyl content of PU(0,0,90), PU(22.5,12.5,65), PU(60,20,20) and PU(0,0,7) was calculated to be around 80%, 61%, 25%, and 0%, respectively, which are in a good agreement with the pure polyol specifications reported in Table 1.
Thermomechanical properties
Thermal transitions in block copolymers provide useful information about their morphology. DSC thermograms were collected for each sample in the second heating cycle to erase the thermal history (Fig. 2a). Glass transition temperature (Tg) of the soft segment is an indication of the phase separation; however, the synthesized polymers display neither melting peaks nor Tg for the hard segments because of the asymmetric structure of IPDI.19 Tg of the PUs was interpreted in comparison with its value for the pure polyols in Table S1.† The highest Tg was observed at −2 °C for the PU(0,0,90), which also has a significant deviation from the Tg of GI-3000 (ΔTg = 15 °C, Table S1†). In contrast, Tgs of PU(0,0,7) and PU(60,20,20) are about the same as their pure polyols, GI-3000 and R45-HT, indicating high purity of the soft domains (i.e., greater phase separation). Besides, the slope of the Tgs of PU(0,0,7) and PU(60,20,20) is sharper than that of the other samples. This can be attributed to the lower phase mixing which can lead to a lower restriction on chain mobility. The trend of phase segregation observed in DSC was supported by FTIR, where PU(0,0,90) and PU(0,0,7) have the highest and lowest phase mixing, respectively.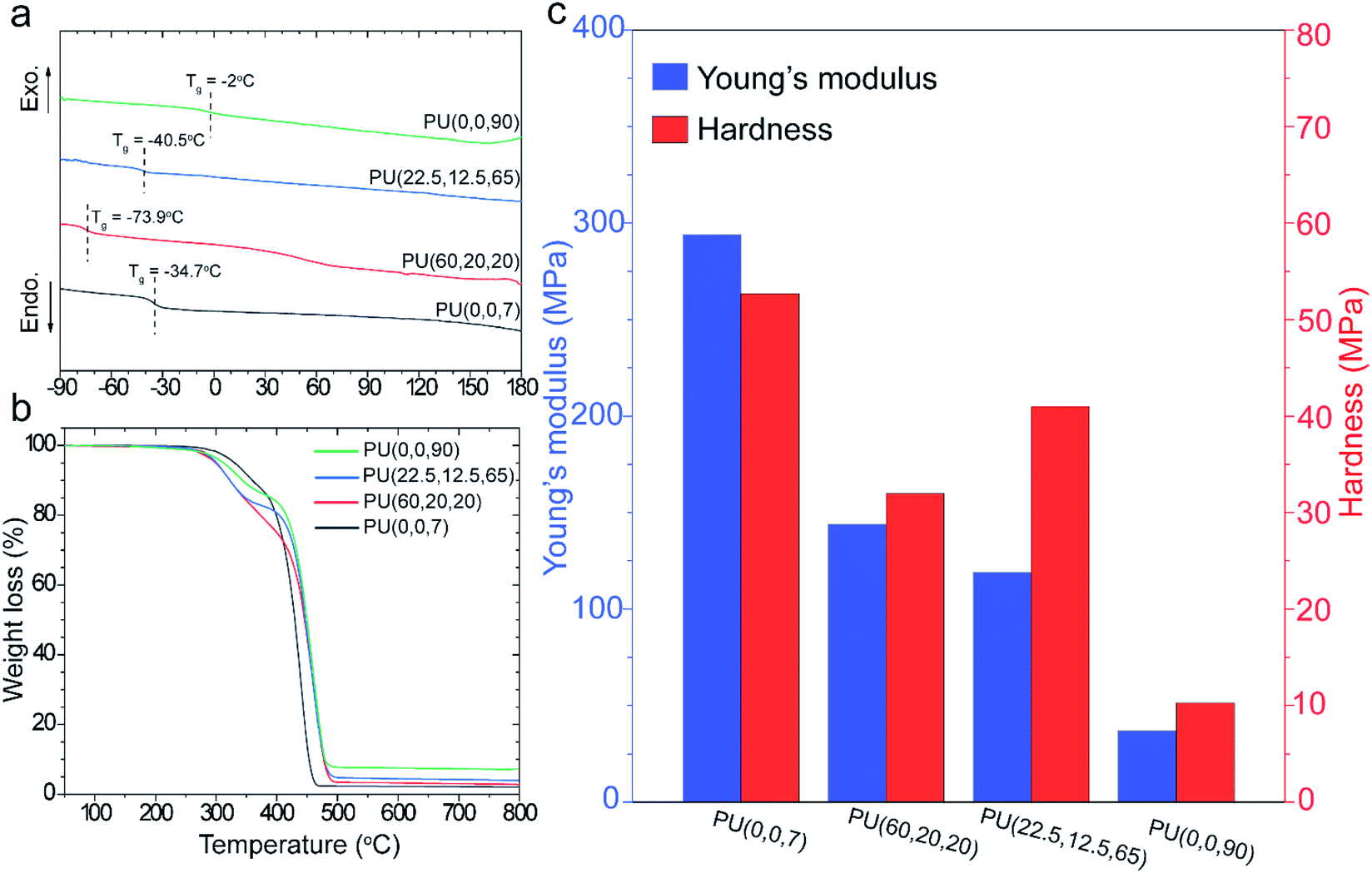 | ||
| Fig. 2 DSC thermograms of the synthesized PUs (a); thermogravimetrical analysis of the synthesized PUs under air atmosphere (b); mechanical properties of the prepared membranes using nanoindentaion method (c). | ||
Thermal stability of the synthesized PUs was studied by TGA in Fig. 2b. Typically, PUs undergo a two-step degradation process; the decomposition commences by cleavage of urethane bonds in the hard segments at around 300 °C. Although the polymers consist of the same hard segment chemistry, the temperature and decomposition rate are different in each sample. For example, T10% (the temperature at which 10% weight loss occurs) of PU(0,0,90) at 330 °C improves to 362 °C for PU(0,0,7) (Table S1†). It is supposed that PU(0,0,7) benefits from stronger hydrogen bonds in the hard domains since it has lower interferences with the polyols. Therefore, the energy required for decomposition is larger than other samples with lower phase separation. This behavior was also reported in previous works.20,46 Although degradation of the hard segments starts earlier for the PUs with unsaturated 1,2-vinyl groups, the decomposition of these samples, i.e., PU(60,20,20), PU(22.5,12.5,65) and PU(0,0,90), at higher temperature (above 400 °C) were improved compared to PU(0,0,7). The crosslinking reactions proceeded via 1,2-vinyl groups at high temperatures delayed the degradation. The higher residual char yield of the unsaturated PUs confirms this hypothesis (Table S1†).
Type and content of microstructures (vinyl, cis, trans) in polybutadienes determine the mechanical properties of the synthesized PUs. Fig. 2c compares elastic modulus (E) and hardness (H) of the samples, measured by the nanoindentation method. An overall decay in the mechanical properties was observed by increasing the vinyl content. For example, the elastic modulus of saturated PU(0,0,7) decreased from 293.9 MPa to 36.2 MPa for PU(0,0,90), a significant loss of about 85%. It is believed that the lower phase separation, higher Tg and the distorted structure of the PUs (refer to XRD section) in the presence of vinyl pendant groups are responsible for this decline.13 In addition, the higher modulus of PU(60,20,20) (E = 151 MPa) than PU(22.5,12.5,65) (E = 102 MPa) can be explained by higher trans-microstructure content in the R-45HT butadiene backbone.47 Nevertheless, we are uncertain about the influence of 1,2-vinyl groups on the hardness of the PUs. It seems a lower hardness of PU(60,20,20) and PU(22.5,12.5,65) compared to other samples is related to the higher flexibility of these polymers due to the presence of trans isomers.
Morphology and microstructure
AFM phase images of the PUs display remarkable changes in the phase separation. The difference in modulus of the hard and soft domains manifests itself in the color profile of the captured images, where darker and lighter areas refer to the hard-rich and soft-rich domains, respectively. Fig. 3a shows smaller and more phase-mixed structures for the samples with higher 1,2-vinyl contents. The average size of the soft domain for PU(0,0,90) was 10–30 nm, while it increased to 50–70 nm in PU(0,0,7). The larger domain size of the PU(0,0,7) compared to other samples was also confirmed by SAXS data (ESI†). It seems the self-assembly of the soft segments to form soft domains is restricted more by increasing the vinyl content. The vinylic groups can act as anchors in the polymer backbone, which decreases the chain mobility (i.e., higher viscosity).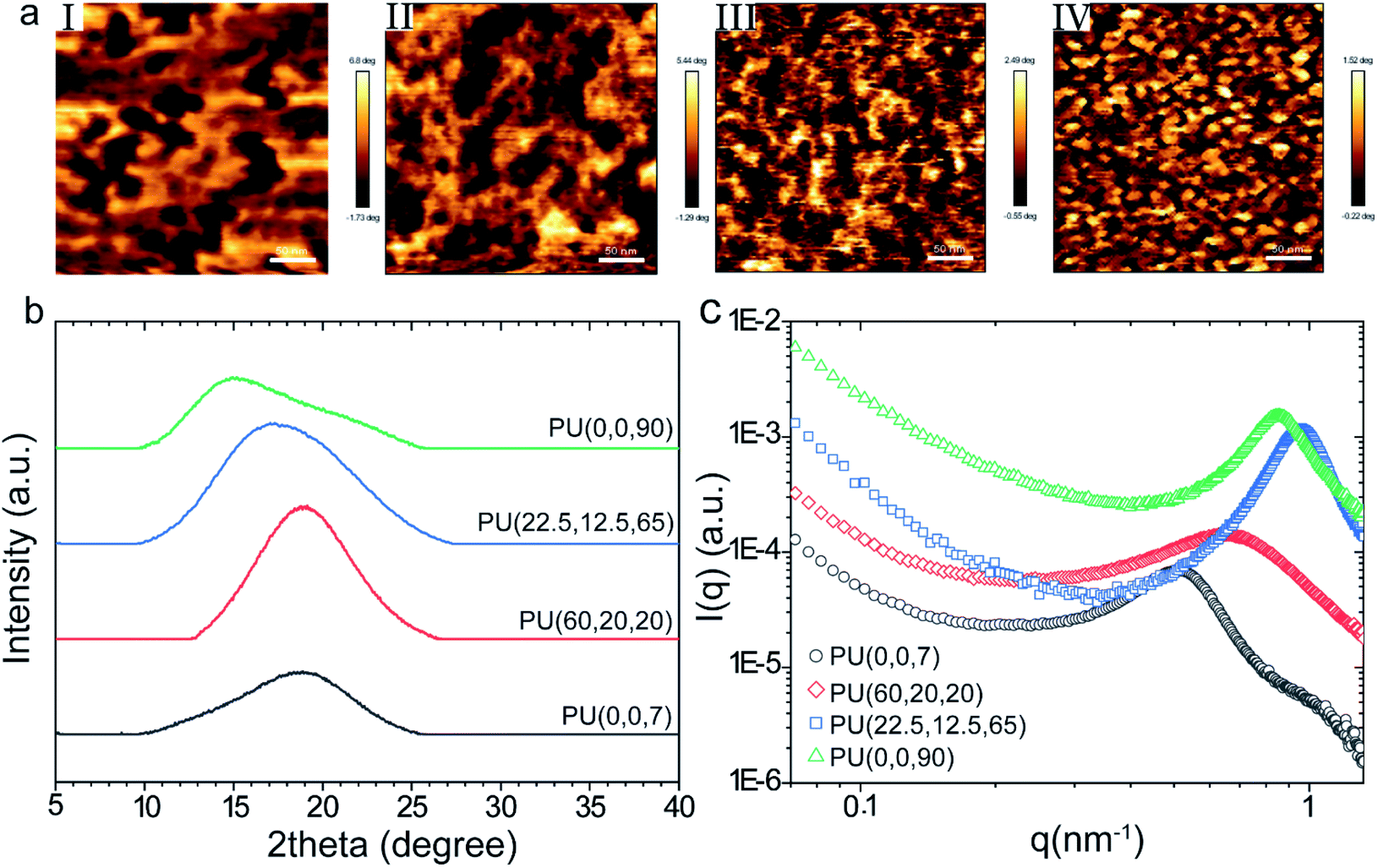 | ||
| Fig. 3 AFM phase images (a) for the prepared membranes: (I)PU(0,0,7), (II) PU(60,20,20), (III) PU(22.5,12.5,65), (IV) PU(0,0,90), scale bar is 50 nm; WAXD (b) and SAXS (c) diffraction patterns of the synthesized PUs. | ||
The influence of vinyl groups on the microstructure of the synthesized PUs was investigated by WAXD, as shown in Fig. 3b. A broad amorphous peak was observed at 2θ = 15–20° for all samples. As observed in Fig. 3b, the peak shifts to lower frequencies by increasing the vinyl content. This increase in chain spacing by vinyl content can be explained by the repulsion effect due to the dipole–dipole interactions between the vinyl and urethane CO groups.48,49
The average size of the microdomains can be evaluated from the SAXS patterns (shown in Fig. 3c) using the peak analysis of the Lorentz-corrected curves (Fig. S2 and Table S2†). The estimated volume fraction of the microdomains obtained from the analysis of the one-dimensional correlation function.50–52 (described in ESI†) is higher than the nominal volume fraction of hard or soft segments derived from the molar volumes of each monomer (Table S2†), which reveals the occurance of phase mixing in the hard microdomains of the synthesized polymers.53
Gas permeation
Table 2 demonstrates the separation performance of the PU membranes for CO2, N2, CH4, H2, and O2 at 4 bar at 25 °C. CO2 permeability of the membranes is higher than the permeability of H2 despite its larger gas molecular size. The same result was also seen for CH4 permeation with a bigger dynamic diameter (dk = 3.8 Å) than N2 (dk = 3.64 Å). The order of gas permeability through the PU membranes is P(CO2) > P(H2) > P(O2) > P(CH4) > P(N2), which is a common trend in rubbery materials such as PDMS and Pebax.54,55 In general, the gas transport properties in PU membranes strongly rely on the gas solubility, which is controlled by the critical temperature (Tc) of the gas: CO2 (304.2 K) > CH4 (190.9 K) > O2 (154.6) > N2 (126.3 K) > H2 (33.2 K).56| Membrane | Permeability (barrer) | Selectivity | ||||||
|---|---|---|---|---|---|---|---|---|
| H2 | CO2 | O2 | N2 | CH4 | CO2/N2 | CO2/CH4 | O2/N2 | |
| PU(0,0,7) | 28.8 ± 0.8 | 34.7 ± 1.8 | 8.4 ± 0.3 | 2.7 ± 0.1 | 4.7 ± 0.3 | 12.9 ± 0.8 | 7.4 ± 0.6 | 3.1 ± 0.2 |
| PU(60,20,20) | 23.4 ± 0.6 | 79.8 ± 3.0 | 10.8 ± 0.5 | 3.5 ± 0.1 | 11.6 ± 0.7 | 22.8 ± 1.1 | 6.9 ± 0.5 | 3.1 ± 0.2 |
| PU(22.5,12.5,65) | 29.2 ± 0.5 | 65.2 ± 3.2 | 9.5 ± 0.3 | 3.0 ± 0.2 | 9.3 ± 0.5 | 21.7 ± 1.8 | 7.0 ± 0.5 | 3.2 ± 0.2 |
| PU(0,0,90) | 19.4 ± 0.4 | 28.1 ± 0.8 | 4.1 ± 0.2 | 1.1 ± 0.1 | 2.7 ± 0.1 | 25.5 ± 2.4 | 10.4 ± 0.5 | 3.7 ± 0.4 |
The differences in morphology of the hard and soft microdomains across the chemically-similar samples can lead to different gas permeability. As can be observed from Table 2, the gas permeability of the samples with lower vinyl content is generally higher than their high-vinyl-content counterparts. This can be due to the higher chain flexibility of samples with lower vinyl content, which leads to a higher fractional free volume in the soft domains. It is commonly observed in rubbery polymers that a decrease in Tg leads to a higher gas diffusivity.57 In addition, volume fraction, size and purity of the microdomains can affect gas permeability of the samples with different vinyl contents.
In comparing PU(0,0,7) and PU(0,0,90), the reduction in the gas permeability with vinyl content can also be attributed to a lower purity of the soft microdomains in PU(0,0,90) (i.e., lower phase separation), as confirmed by DSC and FTIR results. Besides, the size of soft microdomains (Fig. 3a and Table S2†) in PU(0,0,90) is smaller than that in PU(0,0,7). Therefore, gas molecules face higher tortuosity in the permeable soft phase, leading to a lower gas permeability of PU(0,0,90).
Regarding the morphology of PU(60,20,20) and PU(22.5,12.5,65), similar to above, the size of soft microdomains (Fig. 3a and Table S2†) decreased with an increase in vinyl content. Additionally, based on DSC and FTIR results, the purity of the soft domains is only slightly affected in these samples by increasing the vinyl content. Consequently, the higher gas permeability of PU(60,20,20) can also be attributed to the larger microdomains and the lower Tg of the soft segments in this sample.
As can be observed in Table 2, the CO2/N2 and CO2/CH4 selectivities of PU(0,0,90) are larger than those of PU(0,0,7), which can be attributed to a higher phase mixing and a more tortuous pathway for gas species in the former sample compared to the latter, as discussed earlier. Conversely, the CO2/N2 and CO2/CH4 selectivities are almost unchanged among PU(60,20,20) and PU(22.5,12.5,65). This result can be attributed to the similarity in the extent of phase mixing in the soft microdomains of these samples. It can also be observed in Table 2 that O2/N2 selectivity is almost the same across all samples, being slightly higher in PU(0,0,90). The higher O2/N2 selectivity of PU(0,0,90) can be associated with the higher sieving ability of the membrane due to its smaller microdomains and more rigidified structure compared to other samples (higher soft domain Tg ∼ −2 °C).
Conclusion
A series of polybutadiene-based polyurethane structures with different amounts of trans, cis and vinyl structural configurations were synthesized. The effect of microstructural variations on the phase morphology, thermo-mechanical properties and gas transport behavior of the PU films were studied. The thermal stability of the PUs was slightly improved (up to 25 °C) by increasing the vinyl contents. However, the elastic modulus and hardness deteriorated more than 80% for the PUs with a large amount of vinyl groups. Higher trans content in PU(60,20,20) improved its chain mobility (Tg = −73.9 °C) and phase separation compared to other samples. The CO2 permeability of PU(60,20,20) ∼74 barrer was more than twice the permeability of the PUs without trans-microstructure, PU(0,0,7) and PU(0,0,90). SAXS results and AFM phase images revealed smaller microdomains for the samples with higher vinyl content, which created a more tortuous diffusion pathway for gas molecules within the membranes. Besides, the intermixing of hard and soft segments increased by increasing the vinyl contents (i.e., lower phase separation).Author contributions
Ali Pournaghshband: conceptualization, methodology, investigation, data analysis, writing-review & editing. Mahdi Shahrooz: data analysis, investigation, software, writing-review & editing. Takuma Yamamoto: experiments & investigation, data analysis. Ansori Muchtar: experiments & investigation, data analysis. Masateru M. Ito: investigation, data analysis. Daisuke Yamaguchi: data analysis, software, validation. Mikihito Takenaka: data analysis, validation. Easan Sivaniah: supervision; conceptualization; validation; writing-review & editing. Behnam Ghalei: supervision; conceptualization; validation; writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. I. gratefully acknowledges the kakenhi Grants-in-Aid for Scientific Research (Grant number JP19F19043) from the Japan Society for the Promotion of Science. E. S. acknowledges JST-Mirai project funding. iCeMS is supported by the World Premier International Research Initiative (WPI).Notes and references
- A. Fakhar, M. Sadeghi, M. Dinari, M. Zarabadipoor and R. Lammertink, Eur. Polym. J., 2020, 122, 109346 CrossRef CAS.
- M. Radjabian and V. Abetz, Prog. Polym. Sci., 2020, 101219 CrossRef CAS.
- Z. Wang, A. P. Isfahani, K. Wakimoto, B. B. Shrestha, D. Yamaguchi, B. Ghalei and E. Sivaniah, ChemSusChem, 2018, 11, 2744–2751 CrossRef CAS PubMed.
- A. Fakhar, M. Sadeghi, M. Dinari and R. Lammertink, J. Membr. Sci., 2019, 574, 136–146 CrossRef CAS.
- M. A. Hood, B. Wang, J. M. Sands, J. J. La Scala, F. L. Beyer and C. Y. Li, Polymer, 2010, 51, 2191–2198 CrossRef CAS.
- Y. Li, T. Gao and B. Chu, Macromolecules, 1992, 25, 1737–1742 CrossRef CAS.
- Y. Li, T. Gao, J. Liu, K. Linliu, C. R. Desper and B. Chu, Macromolecules, 1992, 25, 7365–7372 CrossRef CAS.
- J. T. Koberstein and A. F. Galambos, Macromolecules, 1992, 25, 5618–5624 CrossRef CAS.
- N. Hossieny, V. Shaayegan, A. Ameli, M. Saniei and C. Park, Polymer, 2017, 112, 208–218 CrossRef CAS.
- P. Krol, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef CAS.
- Q. Tian, G. Yan, L. Bai, X. Li, L. Zou, L. Rosta, A. Wacha, Q. Li, I. Krakovský and M. Yan, Polymer, 2018, 147, 1–7 CrossRef CAS.
- J. Koberstein, A. Galambos and L. Leung, Macromolecules, 1992, 25, 6195–6204 CrossRef CAS.
- L. M. Leung and J. T. Koberstein, Macromolecules, 1986, 19, 706–713 CrossRef CAS.
- C. Prisacariu, Polyurethane elastomers: from morphology to mechanical aspects, Springer Science & Business Media, 2011 Search PubMed.
- M. Sadeghi, M. A. Semsarzadeh, M. Barikani and B. Ghalei, J. Membr. Sci., 2011, 385, 76–85 CrossRef.
- S. Sami, E. Yildirim, M. Yurtsever, E. Yurtsever, E. Yilgor, I. Yilgor and G. L. Wilkes, Polymer, 2014, 55, 4563–4576 CrossRef CAS.
- H. B. Park, C. K. Kim and Y. M. Lee, J. Membr. Sci., 2002, 204, 257–269 CrossRef CAS.
- A. Pournaghshband Isfahani, M. Sadeghi, K. Wakimoto, B. B. Shrestha, R. Bagheri, E. Sivaniah and B. Ghalei, ACS Appl. Mater. Interfaces, 2018, 10, 17366–17374 CrossRef CAS PubMed.
- A. P. Isfahani, B. Ghalei, R. Bagheri, Y. Kinoshita, H. Kitagawa, E. Sivaniah and M. Sadeghi, J. Membr. Sci., 2016, 513, 58–66 CrossRef CAS.
- A. P. Isfahani, B. Ghalei, K. Wakimoto, R. Bagheri, E. Sivaniah and M. Sadeghi, J. Mater. Chem. A, 2016, 4, 17431–17439 RSC.
- A. P. Isfahani, M. Sadeghi, K. Wakimoto, A. H. Gibbons, R. Bagheri, E. Sivaniah and B. Ghalei, J. Membr. Sci., 2017, 542, 143–149 CrossRef CAS.
- C. W. Meuse, X. Yang, D. Yang and S. L. Hsu, Macromolecules, 1992, 25, 925–932 CrossRef CAS.
- J.-L. Hong, C. P. Lillya and J. C. Chien, Polymer, 1992, 33, 4347–4351 CrossRef CAS.
- M. M. Coleman, G. J. Pehlert and P. C. Painter, Macromolecules, 1996, 29, 6820–6831 CrossRef CAS.
- P. J. Flory and M. Volkenstein, Biopolymers, 1969, 8, 699–700 CrossRef.
- W. P. Chen, D. J. Kenney, K. C. Frisch, S. W. Wong and R. Moore, J. Polym. Sci., Part B: Polym. Phys., 1991, 29, 1513–1524 CrossRef CAS.
- A. A. Shamsabadi, A. P. Isfahani, S. K. Salestan, A. Rahimpour, B. Ghalei, E. Sivaniah and M. Soroush, ACS Appl. Mater. Interfaces, 2020, 12, 3984–3992 CrossRef CAS PubMed.
- G. Huang, A. P. Isfahani, A. Muchtar, K. Sakurai, B. B. Shrestha, D. Qin, D. Yamaguchi, E. Sivaniah and B. Ghalei, J. Membr. Sci., 2018, 565, 370–379 CrossRef CAS.
- A. P. Isfahani, M. Sadeghi, S. Nilouyal, A. Muchtar, G. Huang, M. Ito, D. Yamaguchi, E. Sivaniah and B. Ghalei, J. Mater. Chem. A, 2020, 8, 9382–9391 RSC.
- M. Sadeghi, A. P. Isfahani, A. A. Shamsabadi, S. Favakeh and M. Soroush, J. Appl. Polym. Sci., 2020, 137, 48704 CrossRef CAS.
- S. H. Yuan, A. P. Isfahani, T. Yamamoto, A. Muchtar, C. Y. Wu, G. Huang, Y. C. You, E. Sivaniah, B. K. Chang and B. Ghalei, Small Methods, 2020, 2000021 CrossRef CAS.
- B. Bengtson, C. Feger, W. MacKnight and N. Schneider, Polymer, 1985, 26, 895–900 CrossRef CAS.
- S.-L. Huang and J.-Y. Lai, J. Membr. Sci., 1995, 105, 137–145 CrossRef CAS.
- C. Li, S. L. Goodman, R. M. Albrecht and S. L. Cooper, Macromolecules, 1988, 21, 2367–2375 CrossRef CAS.
- I. Krakovský, H. Urakawa and K. Kajiwara, Polymer, 1997, 38, 3645–3653 CrossRef.
- I. Fink, B. Eling, E. Pöselt and G. A. Luinstra, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1162–1172 CrossRef CAS.
- Z. Cao, Q. Zhou, S. Jie and B.-G. Li, Ind. Eng. Chem. Res., 2016, 55, 1582–1589 CrossRef CAS.
- Y.-J. Li, N. Nakamura, Y.-F. Wang, M. Kodama and T. Nakaya, Chem. Mater., 1997, 9, 1570–1577 CrossRef CAS.
- N. Akram, K. M. Zia, R. Sattar, S. Tabassum and M. Saeed, J. Appl. Polym. Sci., 2019, 136, 47289 CrossRef.
- M. Sadeghi, A. A. Shamsabadi, A. Ronasi, A. P. Isfahani, M. Dinari and M. Soroush, Chem. Eng. Sci., 2018, 192, 688–698 CrossRef CAS.
- D. P. White, J. C. Anthony and A. O. Oyefeso, J. Org. Chem., 1999, 64, 7707–7716 CrossRef CAS.
- J. A. Hirsch, Top. Stereochem., 1967, 1, 199 CAS.
- S. Cornell and J. Koenig, Macromolecules, 1969, 2, 540–545 CrossRef CAS.
- S. Poshyachinda, H. G. Edwards and A. F. Johnson, Polymer, 1991, 32, 338–342 CrossRef CAS.
- D. J. Nagle, M. Celina, L. Rintoul and P. M. Fredericks, Polym. Degrad. Stab., 2007, 92, 1446–1454 CrossRef CAS.
- S.-L. Huang, P.-H. Chang, M.-H. Tsai and H.-C. Chang, Sep. Purif. Technol., 2007, 56, 63–70 CrossRef CAS.
- R. M. Shankar, T. K. Roy and T. Jana, J. Appl. Polym. Sci., 2009, 114, 732–741 CrossRef CAS.
- J. A. Brydson, Plastics materials, Elsevier, 1999 Search PubMed.
- P. Liu, P. McCarren, P. H.-Y. Cheong, T. F. Jamison and K. Houk, J. Am. Chem. Soc., 2010, 132, 2050–2057 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small angle X-ray scattering, Academic press, 1982 Search PubMed.
- M. Shahrooz, M. Sadeghi, R. Bagheri and M. Laghaei, Macromolecules, 2016, 49, 4220–4228 CrossRef CAS.
- G. Strobl and M. Schneider, J. Polym. Sci., Polym. Phys. Ed., 1980, 18, 1343–1359 CrossRef CAS.
- J. Sewell, J. Appl. Polym. Sci., 1973, 17, 1741–1747 CrossRef CAS.
- S. Matteucci, Y. Yampolskii, B. D. Freeman and I. Pinnau, Mater. Sci. Membr. Gas Vap. Sep., 2006, 1, 1–2 Search PubMed.
- A. A. Shamsabadi, F. Seidi, E. Salehi, M. Nozari, A. Rahimpour and M. Soroush, J. Mater. Chem. A, 2017, 5, 4011–4025 RSC.
- Q. Song, S. Nataraj, M. V. Roussenova, J. C. Tan, D. J. Hughes, W. Li, P. Bourgoin, M. A. Alam, A. K. Cheetham and S. A. Al-Muhtaseb, Energy Environ. Sci., 2012, 5, 8359–8369 RSC.
- B. Freeman, Y. Yampolskii and I. Pinnau, Materials science of membranes for gas and vapor separation, John Wiley & Sons, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAXS data, analysis of one-dimensional correlation function, schematic representation of polyurethane synthesis, and thermal analysis data. See DOI: 10.1039/d1ra00764e |
| This journal is © The Royal Society of Chemistry 2021 |