DOI:
10.1039/D1RA00747E
(Paper)
RSC Adv., 2021,
11, 10393-10399
Dissolving microneedles delivering cancer cell membrane coated nanoparticles for cancer immunotherapy†
Received
28th January 2021
, Accepted 3rd March 2021
First published on 11th March 2021
Abstract
Recently, a variety of tumor vaccines and immune system stimulators such as toll-like receptor (TLR) agonists have been widely investigated for cancer immunotherapy via transdermal delivery. Despite these great research efforts, low efficiency and discomfort remain a huge technical hurdle for the development of immunotherapeutics. Here, we design a facile method to deliver drugs to the skin through microneedles (MNs) to stimulate the immune system in two ways. As one of the tumor vaccines, cancer cell membrane proteins can act as tumor-specific antigens that are presented to antigen presenting cells (APCs) to activate the immune system. In addition, a toll-like receptor 7 (TLR7) agonist of imiquimod (R837) can suppress cancer cell growth by inhibiting angiogenesis. Using poloxamer 407 (F127) as a nanocarrier, F127 nanoparticles (F127 NPs) are loaded with R837 and then coated with cancer cell membranes (M). These F127–R837@M NPs are loaded in rapidly dissolving MNs and delivered through the skin. MNs loaded with F127–R837@M NPs show significant inhibition of cancer cell growth in both prophylactic vaccination and antitumor immunotherapy in vivo. The dual immune system stimulating F127–R837@M NPs could be effectively used for cancer immunotherapy.
1. Introduction
Cancer immunotherapy has been extensively investigated to suppress malignant tumor growth.1–3 Using patients' own immune systems, cancer immunotherapy has higher specificity and therapeutic effect with fewer side effects than other cancer therapies (e.g., chemotherapy).4,5 Antigen presenting cells (APCs), such as dendritic cells (DCs), B cells, and macrophage cells, play an important role in recognizing antigens invading from outside and delivering them to T cells to activate the immune system.6–8 These APCs are activated by various factors such as cancer cell originated DNA, mRNA, peptides, and proteins.9,10 In addition to stimulating APCs with specific antigens, the immune system can be activated by sending signals directly to immune cell toll-like receptors (TLRs). R837 is a cancer cell lysate which can activate the individual's immune system.8 R837 is one of the representative TLR stimulators. It binds to TLR7, up-regulates IL-18 and down-regulates MMP-9, inhibiting the angiogenesis of cancer and controlling the tumor growth.11–11 In particular, cytotoxic T lymphocytes recognize cancer cells through cell surface proteins and present antigens.14–16 Accordingly, cancer cell membrane (M) proteins can be effectively used to enhance the vaccination effect.
Microneedles (MNs) are one of the most effective means of delivering immune drugs. MNs do not cause pain when inserted into the skin, and some immunotherapies through skin have been reported to be more efficient than injections.17–19 MN tips, which are hundreds of micrometers long, make tiny holes in the outermost layers of the skin, effectively delivering drugs to APCs and resident T cells.20–24 Among many types of MNs including solid MNs, coated MNs and dissolving MNs, dissolving MNs can be effectively used for the highest amount of drug loading in the entire tips without hazardous waste.18–20 Since dissolving MNs are made of water-soluble polymers, they cannot generally deliver hydrophobic drugs.17,26,27 To overcome this problem, we prepared amphiphilic block copolymer NPs that can transport hydrophobic drugs in water-soluble polymer MNs.
Fig. 1 shows a schematic illustration of transdermal immunotherapy using dissolving MNs to deliver cancer cell membrane coated NPs for the treatment of cancers. We chose polyvinylpyrrolidone (PVP) as a MN matrix material considering its biocompatibility, rapid dissolution, mechanical strength and water solubility.27,28 PVP dissolving MNs that deliver dual immune drugs to APCs were prepared by mixing F127 triblock copolymers and R837 topical immune response modifiers and coating with cancer cell membranes (F127–R837@M). The prepared MNs had sufficient mechanical strength to penetrate the mouse skin and were found to be effectively absorbed to the body when inserted into the skin. F127–R837@M NPs loaded on MNs were then presented to APCs to secret immunofactors and to activate the immune network of lymphatic vessels. After cytokines release tests by ELISA, in vivo cancer immunotherapy was carried out to assess the cancer cell growth suppression. Finally, the dual cancer immunotherapy using the MNs was discussed as a promising platform for the treatment of skin cancers.
 |
| | Fig. 1 Schematic illustration for transdermal immunotherapy using dissolving microneedles to deliver cancer cell membrane protein coated F127–R837 nanoparticles. | |
2. Materials and methods
2.1 Materials
Poloxamer 407 (pluronic F127), polyvinylpyrrolidone (PVP), dimethyl sulfoxide (DMSO), dichloromethane (DCM), 5-aminofluorescein and dialysis tube were purchased from Sigma-Aldrich. Imiquimod (R837) was purchased from Macrogen (Seoul, Korea). Dulbecco's modified eagle's medium (DMEM)-high glucose, fetal bovine serum (FBS), antibiotics were obtained from Invitrogen (Carlsbad, CA). Cell lysis buffer was used as received. Vectashield antifade mounting medium with DAPI was purchased from Vector Laboratories (Burlingame, CA). Human colorectal carcinoma cell line (HCT116) and murine macrophage cell line (RAW 264.7) were purchased from Korean Cell Line Bank (Seoul, Korea). ELISA kits were purchased from Cusabio (Wuhan, Hubei, China). Six-week-old male BALB/c mice were purchased and bred in a pathogen-free facility at the Pohang University of Science and Technology (POSTECH). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of POSTECH and approved by the Institutional Animal Care and Use Committee (IACUC). All chemicals were used without further purification.
2.2 Synthesis and characterization of F127–R837@M NPs
The F127–R837 NPs were synthesized by the oil-in-water emulsion method as described elsewhere.29 Briefly, F127 (100 mg) and R837 (1 mg) were dissolved in DCM (1 mL) and added dropwise into distilled water. The solution was sonicated for 10 min and vaporized with a rotary evaporator to remove the DCM solvent. The solution was dialyzed against distilled water and ethanol (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) using a 10 kDa MWCO snake skin dialysis tube with stirring for 3 days. After dialysis, the solution was filtered through a 0.45 μm PVDF membrane filter to remove aggregates. HCT116 membrane was obtained from alive HCT116 cells by using the cell lysis buffer.30 HCT116 cells were washed with a cold PBS buffer and the tissue culture flask was kept on ice bath for 10 min. The PBS buffer was aspirated and the cell lysis buffer at 4 °C was added and incubated for 20 min. After that, the HCT116 cells were scraped from the tissue culture flask using a rubber spatula. The cell solution was centrifuged at 1000 × g for 3 min and the supernatant was collected to a fresh tube. The HCT116 membrane was mixed with F127–R837 NPs, as prepared before, at 4 °C overnight. The size of F127–R837@M NPs was measured with a dynamic light scattering instrument (DLS, Malvern, UK). The morphology of F127–R837@M NPs was characterized by transmission electron microscopy (TEM, H-7650, Hitachi, Japan). The coating of cancer cell membrane onto NPs was confirmed by bicinchonic acid (BCA) assay. The stability of R837 was assessed by HPLC using C18 column at the flow rate of 1 mL min−1 and the UV absorption wavelength of 245 nm. R837 quantification was calculated by comparing the area according to the graph intensity.
:3) using a 10 kDa MWCO snake skin dialysis tube with stirring for 3 days. After dialysis, the solution was filtered through a 0.45 μm PVDF membrane filter to remove aggregates. HCT116 membrane was obtained from alive HCT116 cells by using the cell lysis buffer.30 HCT116 cells were washed with a cold PBS buffer and the tissue culture flask was kept on ice bath for 10 min. The PBS buffer was aspirated and the cell lysis buffer at 4 °C was added and incubated for 20 min. After that, the HCT116 cells were scraped from the tissue culture flask using a rubber spatula. The cell solution was centrifuged at 1000 × g for 3 min and the supernatant was collected to a fresh tube. The HCT116 membrane was mixed with F127–R837 NPs, as prepared before, at 4 °C overnight. The size of F127–R837@M NPs was measured with a dynamic light scattering instrument (DLS, Malvern, UK). The morphology of F127–R837@M NPs was characterized by transmission electron microscopy (TEM, H-7650, Hitachi, Japan). The coating of cancer cell membrane onto NPs was confirmed by bicinchonic acid (BCA) assay. The stability of R837 was assessed by HPLC using C18 column at the flow rate of 1 mL min−1 and the UV absorption wavelength of 245 nm. R837 quantification was calculated by comparing the area according to the graph intensity.
2.3 Fabrication and characterization of PVP MNs
PVP solution (200 mg mL−1) and the PVP solution with F127–R837@M NPs (20 mg mL−1) were prepared, respectively. The NPs containing PVP solution was pipetted onto the female PDMS mold (1 cm × 1 cm) with a 10 × 10 array of conical cavities (base diameter: 200 μm, tip height: 500 μm) and the solution completely filled the tips of the mold in a vacuum chamber at room temperature.13,31 After removal of all the bubbles, the PVP solution was pipetted onto the PDMS mold to form a backbone and the solvent was evaporated in a vacuum chamber overnight. After drying, PVP MNs were carefully peeled off from the mold and stored in the vacuum chamber to prevent MNs from melting in the air. The morphology of MNs was examined using an optical microscope (Eclipse TS100, Nikon, Japan) and a scanning electron microscope (Philips electron optics B.V., Netherland). The penetration capability of the MNs was tested ex vivo on a porcine skin and in vivo on a mouse back skin. The fat layer on a slice of porcine skin produced during the delivery process was washed out with ethanol. A piece of porcine skin was cut off and cleaned with PBS. The piece of porcine skin was affixed and the MN was gently pressed with a thumb into the skin for 1 min. Afterward, the MN was peeled off. To obtain a cross-sectional image of the porcine skin after MN insertion, the perforated portion of the skin was cut vertically. The image was observed with an optical microscope. In vivo mouse skin penetration test of MNs was also performed as described above.
2.4 MN-assisted transdermal delivery of FITC-labeled F127–R837@M NPs
Six-week-old BALB/c mice were anesthetized by subcutaneously injecting a mixture of 40 μL of ketamine and 10 μL of rompun in 200 μL of PBS.26 After 20 min, the mice were anesthetized and the hair of the back portion to be inserted with a MN was removed with an electrical clipper and hair removal cream. FITC-labeled NPs were synthesized by the reaction of FITC and F127–NH2. Briefly, F127–NH2 was dissolved in THF, then 2 molar excess of FITC was added. The mixture was stirred for 2 h and purified by dialysis and lyophilization. The MN loaded with FITC-labeled NPs was inserted into the back of mice. After 1 and 24 h, the MNs were removed, respectively, the mice were sacrificed, and the back skin tissues were dissected. The back skin tissues were observed by two-photon fluorescence microscopy (TCS SP5II MP SMD FLIM, Leica, Deerfield, IL). The images were obtained by skin tissue tomography (Z-stacks), 512 × 512 pixels, and reconstructed with LAS AF Lite 2.6.1 (Leica) and ImageJ program.
2.5 Cellular uptake and cell viability test
The 5 × 105 murine macrophage cells (RAW 264.7) were incubated in a 6-well microplate for 24 h. After adhesion onto a microplate, the culture medium was replaced with a serum-free medium. FITC labeled F127–R837@M NPs (10 μg) were dropped into each well for 24 h. To label the nucleus of RAW 264.7, aqueous mounting medium was dropped for 4 h. The cells were washed with PBS and fixed in a 10% formaldehyde solution. The cells were observed by confocal microscopy (TCS SP5II MP SMD FLIM, Leica, Deerfield, IL). Further cellular uptake test was performed by measuring the amount of R837 in the cell media with HPLC. After 1 and 24 h, each cell media was aspirated and analyzed. Cell viability test was performed by MTT assay at various concentration of NPs (1, 5, 10 and 20 μg mL−1) using the same procedure with the cellular uptake experiment.
2.6 Tumor challenge and immune response test
BALB/c mice (n = 9) were divided into 4 groups for vaccination. Each group was inserted by MNs loaded with blank (control), R837, F127–R837, and F127–R837@M, respectively. The R837 was administered at 20 μg per each mouse. 106 HCT116 cells were dispersed in 100 μL of PBS and administered by subcutaneous injection (day 0). The tumor size measurements were conducted at day 10 post-injection of tumor cells. Tumors were regarded as ellipses and the tumor volume was determined as length × (width)2/2 by measuring its long radius and short diameter. At day 7, the MNs were inserted onto site of tumor of the anesthetized mouse skin and gently pressed with a thumb for 60 s. After 1 h, the remaining MNs were removed from the mouse skin. The four groups of mice were injected with MNs six times in two days apart (days 7, 9, 11, 13, 15, and 17). Seven days post-injection, from day 26, it was judged that the tumor was too large and the mice were sacrificed according to the ethical rules of animal experiments. The IFN-γ and TNF-α secretion was analyzed by ELISA at day 19 using the sample collected by eye bleeding.
2.7 In vivo toxicity test
To assess the toxicity of F127–R837@M NPs, MNs were injected intravenously into BALB/c mice (n = 3) and compared with the control group intravenously injected with PBS. One week later, blood samples were collected from the mice heart and centrifuged to obtain serum for the blood analysis. The blood biochemical analysis was performed with a chemistry analyzer (BS-390, Mindary, SZ).
2.8 Statistical analysis
All data are expressed in means ± standard deviations (SDs). Statistical analysis was performed with an unpaired student's t-test. P values lower than 0.05 were considered statistically significant.
3. Results and discussion
3.1 Synthesis and characterization of F127–R837@M NPs
In order for cancer immunotherapy, we designed NPs that co-stimulate both TLR-7 and APCs directly. F127 NPs (ca. 40 nm) were prepared by the oil-in-water emulsion process (Fig. 2a). F127–R837 NPs (ca. 50 nm) were prepared by mixing R837 and F127 solutions. Cancer cell membrane was obtained by cell lysate protocol and coated on the F127–R837 NPs.25 After coating, the size of NPs was significantly increased to ca. 60 nm. To confirm the cancer cell membrane coating, we performed the BCA assay (Fig. 2b). F127 and F127–R837 NPs had almost no absorption at the wavelength of 562 nm, but the protein content in F127–R837@M NPs increased with increasing concentration of NPs. Fig. 2c and d show the TEM images of F127–R837 and F127–R837@M NPs, respectively. We observed the round shape of F127–R837 NPs with an average size of ca. 50 nm. The average size of F127–R837@M NPs was ca. 60 nm, which was larger than that of F127–R837. From these characterization data, we confirmed the successful preparation of F127–R837@M NPs. Fig. 2e shows the release of R837 at F127 NPs over time. It is confirmed that the drug was released at less than 2% even after one day, and about 4% after 36 hours. This result can prove that the NPs we synthesized have sufficient stability to be delivered into the body. To further evaluate cellular uptake, R837 remaining in the cell media was quantitatively analyzed by HPLC to determine how much R837 was absorbed into the cells (Fig. 2f). F127–R837@M (10 μg of R837) was applied under the same conditions as the previously experimented cells and incubated for 1 and 24 h. Then, each aspirated cell media was analyzed by HPLC. When incubated for 1 h, R837 was hardly absorbed by the cells in the media, whereas it was confirmed that almost no R837 remained in the cell media after incubation for 24 hours. Through this, we could confirm that the phagocytosis of F127–R837@M NPs is effectively performed by RAW 264.7 macrophages.
 |
| | Fig. 2 (a) Particle size by DLS and (b) Bradford's assay of F127, F127–R837 and F127–R837@M NPs (n = 3, ***P < 0.001). TEM images of (c) F127–R837 and (d) F127–R837@M NPs (scale bar = 300 nm). (e) In vitro release of R837 from F127–R837 NPs analyzed by HPLC. (f) The amount of R837 uptaken by RAW 264.7 macrophages. | |
3.2 Fabrication and characterization of F127–R837@M loaded PVP MNs
To deliver the F127–R837@M NPs in a non-invasive manner, we fabricated dissolving PVP MNs using a PDMS female mold. PVP MNs showed sufficient mechanical strength to penetrate the skin after complete drying and dissolved quickly in the skin with high biocompatibility.32 As shown in Fig. 3a, PVP MN containing F127–R837@M NPs was successfully fabricated with a 100 tips on a 1 cm × 1 cm backbone with a tip to tip distance of 1 mm. Fig. 3b shows the microneedle penetration in the porcine skin. The MNs were gently applied to the animal's skin with a thumb. The MNs penetrated through the skin without bending or twisting (Fig. 3c), indicating that the MNs had sufficient strength to penetrate the skin. The mechanical strength of MNs were also measured by compression test (Fig. S3†). The inserted MNs melted 50% in 10 s and more than 90% in 60 s, which might be caused by the strong hygroscopic property of PVP (Fig. 3d–f). As shown in Fig. S1b and c,† the stability of F127–R837@M NPs was assessed over time. The size and the surface charge of F127–R837@M NPs were maintained even after 3 days according to the dynamic light scattering (DLS) analysis. In addition, in vitro release of cell membrane protein was not so much significant from F127–R837@M NPs for 3 days, reflecting the stability of F127–R837@M NPs.
 |
| | Fig. 3 (a) OM images of F127–R837@M loaded PVP MNs. Photographs of (b) a porcine skin after insertion of MNs. (c) Side view image of the porcine skin. (d–f) OM images of MNs after dissolving for 0, 10 and 60 s, respectively (scale bar = 300 μm in (a) and (c–f) and 1 mm in (b)). | |
3.3 Transdermal delivery of F127–R837@M loaded PVP MNs
To evaluate the drug delivery of MNs, MNs loaded with FITC-labeled F127–R837@M NPs were inserted into the back of mouse skin. Drug absorption was assessed after inserting MNs into the mouse skin and confirmed by comparing the back skin images 1 and 24 h post-treatment. Fig. 4a shows the Z-stack images of two-photon microscopy for FITC-labeled NPs loaded MNs penetrating the mouse skin at 30 μm intervals. Green fluorescence of FITC was not observed in the control group without MNs. However, FITC fluorescence was clearly observed in the image of mouse skin tissue at 1 and 24 h post-treatment. The drug appeared to be delivered through the hole in the middle of images. MNs were also dissolved in the periphery of the hole. After 1 h post-treatment, the fluorescence could be detected at 150 μm, which was decreased with increasing depth. However, after 24 h post-treatment, a shimmer of fluorescence could be detected, which disappeared at 90 μm. From the results, we confirmed that the MNs were dissolved in the skin with increasing time, and the dissolved MN tips and drugs diffused around the skin. In addition, it could be observed that the size of the hole by the MN tips did not become sharper as it penetrated deeper, matching with the shape of the tip, but did not appear to have a distinct tendency. This is because the MN tips do not penetrate the skin by removing the skin's flesh, but pierces and enters the skin cells. Therefore, when the MN tips disappeared from the skin, the skin cells return to their place due to the elasticity of the skin. Fig. 4b shows the side images of mouse skin tissue at 1 h and 24 h post-treatment. The blue fluorescence corresponds to the signal generated by the second harmonic generation (SHG) of collagen on the two-photon micrograph. FITC was highly observed near the injection sites. From these results, it was confirmed that MNs successfully penetrated the mouse skin and delivered the drug into the body.
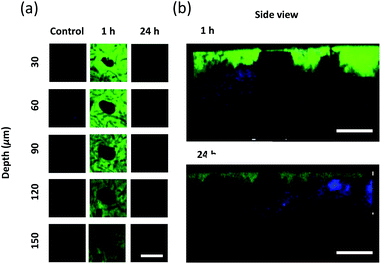 |
| | Fig. 4 Two-photon fluorescence images of FITC-labeled F127–R837@M NPs delivered to the mouse skin via microneedles. (a) Z-stack images of FITC-labeled F127–R837@M NPs with increasing depth and time. (b) Side view images of mouse skin after delivery of FITC-labeled F127–R837@M NPs via microneedles (scale bar = 300 μm). | |
To confirm that the drugs were properly delivered to APCs, a cellular uptake test was performed with RAW 264.7 macrophage cells. For in vitro tracking, FITC labeled F127–R837@M NPs were added to cultured RAW 264.7 cells for 12 h. Afterwards, the nuclei were stained with DAPI to show the location of cells and visualized by confocal microscopy. Fig. 5a and b show the cells and FITC labeled drugs, respectively. The two merged images confirmed that the drug was clearly absorbed to the macrophage cells (Fig. 5c). To assess the biocompatibility of F127–R837@M NPs, MTT assay was conducted with increasing concentration of F127–R837@M NPs in comparison with R837 and F127–R837 at the concentrations of 1, 5, 10 and 20 μg mL−1, respectively (Fig. 5d). From the results, we could confirm the cytocompatibility of F127–R837@M NPs with insignificant cytotoxicity.
 |
| | Fig. 5 (a) Hoechst, (b) amino-fluorescein and (c) merged images by confocal microscopy of RAW 264.7 cells treated with F127–R837@M NPs. F127–R837@NPs were loaded with amino-fluorescein (yellow channel) and nucleus was stained with DAPI (blue channel) (scale bar = 100 μm). (d) Macrophage cell viability with increasing concentration of R837, F127–R837 or F127–R837@M after 12 h. | |
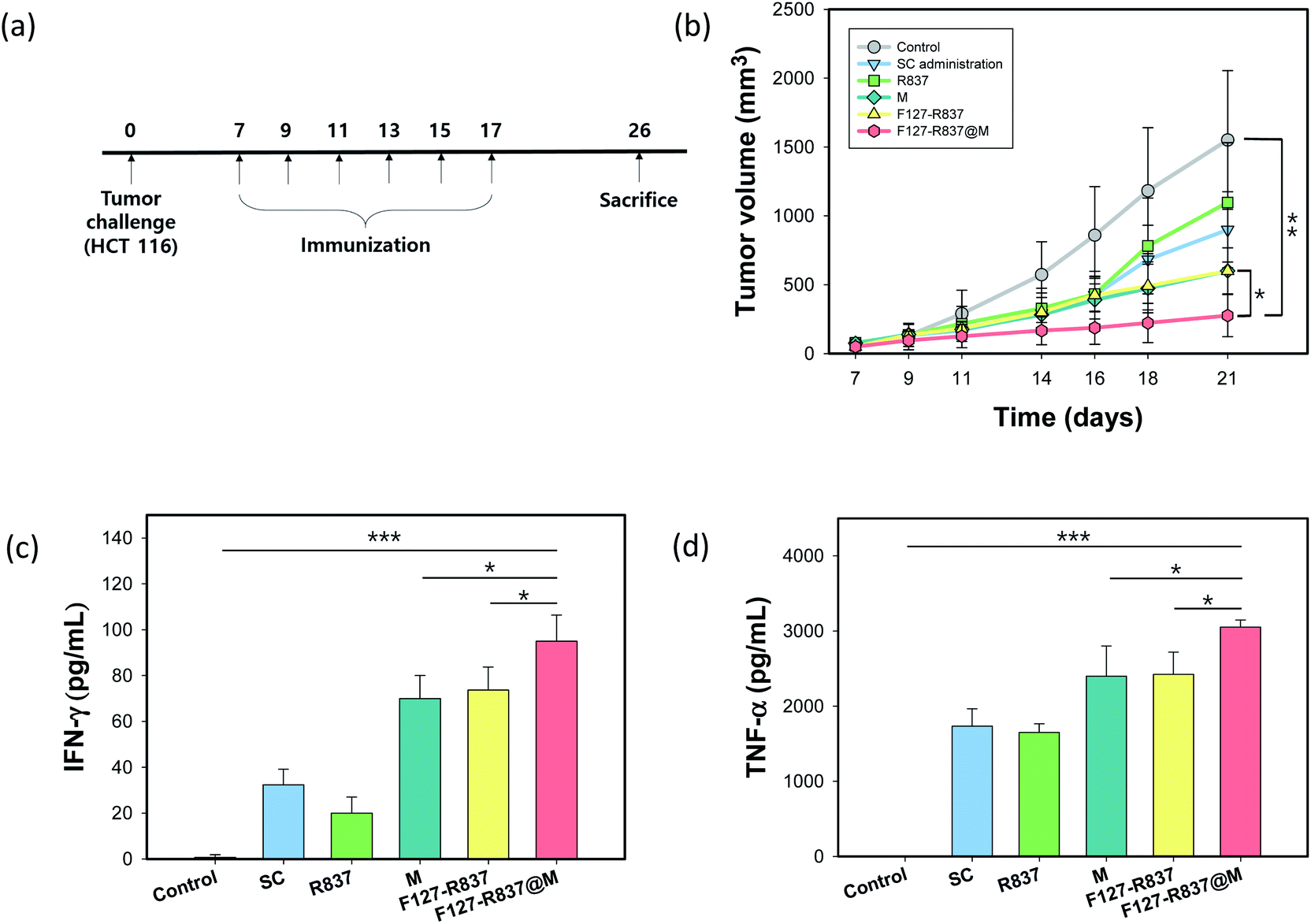
3.4 Tumor challenge and immune responses
The toxicity of MN was assessed by the blood biochemistry assay for total protein (TP), albumin (ALB), alkaline phosphatase (ALP), aspartate transaminase (AST), and alanine transaminase (ALT). The MNs were inserted into the mouse skin for 1 h and the blood samples of the mice were collected and used to obtain the plasma by centrifugation after 1 week. The analytical factors were similar to that of the control mice without inserting MNs for all items, which confirmed the biocompatibility of F127–R837@M NPs (Fig. 6a and b). In addition, after tumor challenge, we investigated the immunization effect of F127–R837@M NPs encapsulated in the MNs for the treatment of cancer (Fig. 7a). HCT116 (106) cells were injected into the mouse skin. After 10 days, the MN immunization was performed 6 times every other day. Mice were sacrificed at day 26 according to the POSTECH ethical guidelines of animal experiments. As shown in Fig. 7b, the tumor growth after immunization with MNs was significantly retarded with a tumor volume 8-fold smaller than that of the control group at day 21, clearly showing the therapeutic effect on tumor growth suppression. Although MNs containing only cancer cell membrane protein and MNs containing F127–R837 NPs showed a meaningful decrease in the tumor volume, the therapeutic effect of MNs containing F127–R837@M NPs was the most statistically significant by the dual immunization. In addition, the subcutaneous administration of F127–R837@M NPs resulted in the insignificant inhibitory effect on the tumor growth. From the results, we could confirm the remarkable tumor growth inhibition of MNs containing F127–R837@M NPs by the effective immunization possibly via resident T cells and APCs in the skin. At day 17 after 6 times MNs treatment, blood was collected for the cytokine analysis. Fig. 7c and d show the enhanced production of cytokine factors of IFN-γ and TNF-α after treatment with F127–R837@M NPs in MNs in consistent with the tumor growth in Fig. 7b. The results of the ELISA were well matched with the tumor growth suppression in Fig. 7b. Taken together, we could confirm the feasibility of F127–R837@M NPs encapsulated in MNs for cancer immunotherapy.
 |
| | Fig. 6 In vivo biocompatibility of F127–R837@M NPs assessed by the blood biochemistry assay for (a) TP and ALB, and (b) ALP, AST and ALT. | |
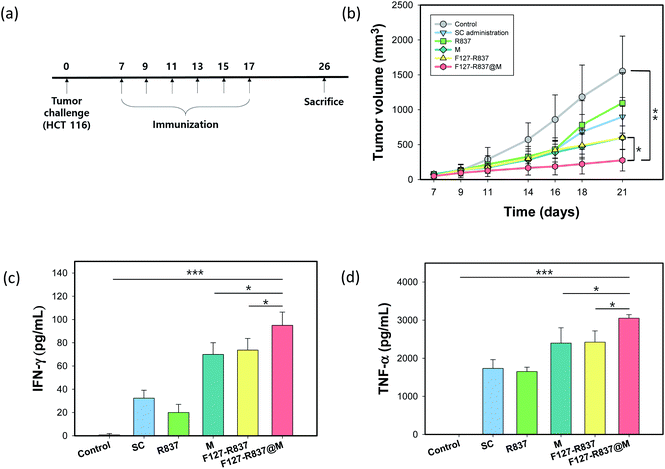 |
| | Fig. 7 Antitumor immunization with F127–R837@M NPs encapsulated in MNs after tumor challenge. (a) Schedule of the cancer immunotherapy. (b) Tumor growth after treatment with F127–R837@M NPs encapsulated in MNs (n = 9, **P < 0.01, *P < 0.05). (c and d) The concentration of (c) IFN-γ and (d) TNF-α in the serum by ELISA (***P < 0.001, **P < 0.01, *P < 0.05). | |
4. Conclusions
We successfully developed a dissolving MN containing cancer cell membrane coated NPs for cancer immunotherapy. The NPs were prepared by coating cancer cell membranes on F127 micelles loaded with R837. The minimally invasive dissolving MNs were inserted into the skin and delivered the immunogenic F127–R837@M NPs to skin-resident APCs. The facile immunization of MNs resulted in significantly inhibited tumor growth with the cytokine production of IFN-γ and TNF-α by stimulating the immune systems in the mice. The blood biocompatibility assessment confirmed the biocompatibility of dissolving MNs containing NPs stimulating both TLR7 and APCs directly. From all these results, F127–R837@M NPs encapsulated in MNs would be successfully harnessed for cancer immunotherapy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Basic Science Research Program (2020R1A2C3014070), Development of Medical Devices (2020M3E5D8105732) and the Engineering Research Center (ERC) Program (NRF-2017R1A5A1014708) of the National Research Foundation (NRF) funded by the Ministry of Science and ICT, Korea.
Notes and references
- Z. Urban-Wojciuk, M. M. Khan, B. L. Oyler, R. Fåhraeus, N. Marek-Trzonkowska, A. Nita-Lazar, T. R. Hupp and D. R. Goodlett, Front. Immunol., 2019, 10, 1–10 CrossRef PubMed.
- M. J. Braunstein, J. Kucharczyk and S. Adams, Target. Oncol., 2018, 13, 583–598 CrossRef PubMed.
- L. Huang, H. Xu and G. Peng, Cell. Mol. Immunol., 2018, 15, 428–437 CrossRef CAS PubMed.
- R. A. Lake and B. W. S. Robinson, Nat. Rev. Cancer, 2005, 5, 397–405 CrossRef CAS PubMed.
- J. Wang, W. Xu, N. Zhang, C. Yang, H. Xu, Z. Wang, B. Li, J. Ding and X. Chen, J. Controlled Release, 2021, 332, 1–9 CrossRef CAS PubMed.
- M. O. Dellacherie, B. R. Seo and D. J. Mooney, Nat. Rev. Mater., 2019, 4, 379–397 CrossRef.
- J. Wang, Z. Li, Z. Wang, Y. Yu, D. Li, B. Li and J. Ding, Adv. Funct. Mater., 2020, 30, 1–25 Search PubMed.
- X. Feng, W. Xu, Z. Li, W. Song, J. Ding and X. Chen, Adv. Sci., 2019, 6, 1900101 CrossRef PubMed.
- A. V. Kroll, R. H. Fang, Y. Jiang, J. Zhou, X. Wei, C. L. Yu, J. Gao, B. T. Luk, D. Dehaini, W. Gao and L. Zhang, Adv. Mater., 2017, 29, 1–9 CrossRef PubMed.
- G. N. Shi, C. N. Zhang, R. Xu, J. F. Niu, H. J. Song, X. Y. Zhang, W. W. Wang, Y. M. Wang, C. Li, X. Q. Wei and D. L. Kong, Biomaterials, 2017, 113, 191–202 CrossRef CAS PubMed.
- H. Y. Kim, M. Kang, Y. W. Choo, S. H. Go, S. P. Kwon, S. Y. Song, H. S. Sohn, J. Hong and B. S. Kim, Nano Lett., 2019, 19, 5185–5193 CrossRef CAS PubMed.
- A. Bartholomeusz and S. Locarnini, Antiviral Ther., 2006, 55, 52–55 Search PubMed.
- V. W. Li, W. W. Li, K. E. Talcott and A. W. Zhai, J. Drugs Dermatol., 2005, 4, 708–717 Search PubMed.
- L. Maatouk, A. C. Compagnion, M. A. C. De Sauvage, A. P. Bemelmans, S. Leclere-Turbant, V. Cirotteau, M. Tohme, A. Beke, M. Trichet, V. Bazin, B. N. Trawick, R. M. Ransohoff, F. Tronche, B. Manoury and S. Vyas, Nat. Commun., 2018, 9, 1–15 CrossRef PubMed.
- Y. Wang, Y. X. Lin, J. Wang, S. L. Qiao, Y. Y. Liu, W. Q. Dong, J. Wang, H. W. An, C. Yang, M. Mamuti, L. Wang, B. Huang and H. Wang, ACS Nano, 2019, 13, 7568–7577 CrossRef CAS PubMed.
- S. Li, X. Feng, J. Wang, W. Xu, M. A. Islam, T. Sun, Z. Xie, C. Wang, J. Ding and X. Chen, Adv. Funct. Mater., 2019, 29, 1–11 Search PubMed.
- N. W. Kim, S. Y. Kim, J. E. Lee, Y. Yin, J. H. Lee, S. Y. Lim, E. S. Kim, H. T. T. Duong, H. K. Kim, S. Kim, J. E. Kim, D. S. Lee, J. Kim, M. S. Lee, Y. T. Lim and J. H. Jeong, ACS Nano, 2018, 1–8 Search PubMed.
- Y. Ye, C. Wang, X. Zhang, Q. Hu, Y. Zhang, Q. Liu, D. Wen, J. Milligan, A. Bellotti, L. Huang, G. Dotti and Z. Gu, Sci. Immunol., 2017, 2, 1–13 Search PubMed.
- H. Kim, K. Y. Seong, J. H. Lee, W. Park, S. Y. Yang and S. K. Hahn, ACS Biomater. Sci. Eng., 2019, 5, 5150–5158 CrossRef CAS PubMed.
- J. Xu, B. Xu, J. Tao, Y. Yang, Y. Hu and Y. Huang, Small, 2017, 13, 1–13 Search PubMed.
- J. H. Kim, J. U. Shin, S. H. Kim, J. Y. Noh, H. R. Kim, J. Lee, H. Chu, K. Y. Jeong, K. H. Park, J. D. Kim, H. K. Kim, D. H. Jeong, T. S. Yong, J. W. Park and K. H. Lee, Biomaterials, 2018, 150, 38–48 CrossRef CAS PubMed.
- N. W. Kim, M. S. Lee, K. R. Kim, J. E. Lee, K. Lee, J. S. Park, Y. Matsumoto, D. G. Jo, H. Lee, D. S. Lee and J. H. Jeong, J. Controlled Release, 2014, 179, 11–17 CrossRef CAS PubMed.
- M. Zaric, O. Lyubomska, C. Poux, M. L. Hanna, M. T. McCrudden, B. Malissen, R. J. Ingram, U. F. Power, C. J. Scott, R. F. Donnelly and A. Kissenpfennig, J. Invest. Dermatol., 2015, 135, 425–434 CrossRef CAS PubMed.
- G. Ma and C. Wu, J. Controlled Release, 2017, 251, 11–23 CrossRef CAS PubMed.
- A. F. Moreira, C. F. Rodrigues, T. A. Jacinto, S. P. Miguel, E. C. Costa and I. J. Correia, Pharmacol. Res., 2019, 148, 104438 CrossRef CAS PubMed.
- A. Mohammed and H. A. Yelwa, Small Rumin. Res., 1993, 12, 107–113 CrossRef.
- L. Guo, J. Chen, Y. Qiu, S. Zhang, B. Xu and Y. Gao, Int. J. Pharm., 2013, 447, 22–30 CrossRef CAS PubMed.
- S. P. Sullivan, D. G. Koutsonanos, M. Del Pilar Martin, J. W. Lee, V. Zarnitsyn, S. O. Choi, N. Murthy, R. W. Compans, I. Skountzou and M. R. Prausnitz, Nat. Med., 2010, 16, 915–920 CrossRef CAS PubMed.
- H. Choi, W. Choi, J. Kim, W. H. Kong, K. S. Kim, C. Kim and S. K. Hahn, Biomacromolecules, 2019, 20, 3767–3777 CrossRef CAS PubMed.
- E. Kinoshita-Kikuta, E. Kinoshita, A. Yamada, M. Endo and T. Koike, Proteomics, 2006, 6, 5088–5095 CrossRef CAS PubMed.
- K. Y. Seong, M. S. Seo, D. Y. Hwang, E. D. O'Cearbhaill, S. Sreenan, J. M. Karp and S. Y. Yang, J. Controlled Release, 2017, 265, 48–56 CrossRef CAS PubMed.
- M. Leone, J. Mönkäre, J. A. Bouwstra and G. Kersten, Pharm. Res., 2017, 34, 2223–2240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00747e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Keum Yong Seongb,
Hye Hyeon Hana,
Seung Yun Yangb and
Sei Kwang Hahn
a,
Keum Yong Seongb,
Hye Hyeon Hana,
Seung Yun Yangb and
Sei Kwang Hahn